

Abstract
Lynch syndrome (LS) confers inherited cancer predisposition due to germline mutations in a DNA mismatch repair (MMR) gene, e.g. MSH2. MMR is a repair pathway for removal of base mismatches and insertion/deletion loops caused by endogenous and exogenous factors. Loss of MMR through somatic alteration of the wild‐type allele in LS results in defective MMR (dMMR). Lifestyle/environmental factors can modify colorectal cancer risk in sporadic and LS patients. Ethanol and its metabolite acetaldehyde are classified as group one carcinogens, and acetaldehyde causes a range of DNA lesions. However, DNA repair pathways responsible for correcting most of such DNA lesions remain uncharacterised. We hypothesised that MMR plays a role in protecting colorectal epithelium from ethanol/acetaldehyde‐induced DNA damage. Here, an LS mouse model (intestinal epithelial conditional‐knockout for Msh2) was used to determine if there is a gene–environment interaction between dMMR and ethanol/acetaldehyde that accelerates colorectal tumourigenesis in LS. Mice underwent either long‐term ethanol treatment or water treatment. Most ethanol‐treated mice demonstrated colonic hyperproliferation and adenoma formation (with some invasive adenocarcinomas) within 6 months (15/23, 65%), compared with one colonic tumour after 15 months in water‐treated mice (1/23, 4%) (p < 0.0001, Fisher's exact test). A significantly greater number of dMMR colonic crypt foci precursors were observed in ethanol‐treated compared with water‐treated mice (p = 0.0029, Student's t‐test). Moreover, increased plasma acetaldehyde levels were detected in ethanol‐treated compared with water‐treated mice (p = 0.0019, Mann–Whitney U‐test), along with significantly increased DNA damage response in the colonic epithelium. Long‐term ethanol treatment was associated with significantly increased colonic epithelial proliferation and markedly reduced apoptosis in dMMR adenomas, consistent with enhanced survival of aberrant dMMR relative to MMR‐proficient colonic epithelium. In conclusion, there is strong evidence for a gene–environment interaction between dMMR and acetaldehyde, causing acceleration of dMMR‐driven colonic tumour formation in this LS model, indicating that advice to limit alcohol consumption should be considered for LS patients. © 2021 The Authors. The Journal of Pathology published by John Wiley & Sons, Ltd. on behalf of The Pathological Society of Great Britain and Ireland.
Keywords: Lynch syndrome, mismatch repair, Msh2, ethanol, acetaldehyde, colorectal, adenoma
Introduction
Lynch syndrome (LS) is caused by constitutional (mostly germline heterozygous) pathogenic mutations in one of the DNA mismatch repair (MMR) genes (MSH2, MLH1, MSH6, and PMS2). In LS patients, an inherited MMR gene variant, when combined with an acquired second inactivating event (mutation or deletion) of the wild‐type allele of the same MMR gene, results in complete loss of MMR pathway function in the affected cells. MMR deficiency (dMMR) leads to hypermutability, with an increase in the mutation rate by 100‐ to 1000‐fold due to uncorrected base mismatches, and to microsatellite instability (MSI) due to variation in the lengths of repetitive microsatellite sequences (e.g. AAAAAAA … or CACACACA … or similar) due to uncorrected insertion/deletion loops that are prone to occur as DNA replication errors in repetitive sequences [1].
LS patients have a higher lifetime risk for several cancer types, mainly in the large intestine (LI) and endometrium but also in the small intestine (SI), stomach, hepatobiliary tract, pancreas, skin (sebaceous tumours), and several other organs [1, 2, 3]. Colonic adenomas are found in up to 50% (lifetime risk) of LS cases, mostly in the proximal colon, compared with 26% for sporadic adenomas that are mostly in the distal colorectum [4]. In addition, proximal LS adenomas progress to high‐grade dysplasia more frequently than distal LS adenomas, and are also more often highly dysplastic than larger distal adenomas [4].
The variable expression of cancer predisposition phenotypes amongst LS patients suggests important effects of allelic variation, genetic modifiers, sex differences, and environmental and/or lifestyle factors, together with complex genetic and environmental interactions. Therefore, it is important to investigate relevant environmental/lifestyle risk factors that may influence cancer risk in LS patients, in order to provide appropriate cancer prevention advice, surveillance, and care. Several lifestyle factors are associated with increased risk of sporadic colorectal cancer (CRC), and these may have similar or enhanced effects in LS patients. Several studies and systematic reviews provided evidence that smoking, diet, and obesity are linked with an increased risk of sporadic CRC [5, 6, 7, 8, 9, 10, 11]. Worldwide, a total of approximately 389 000 cancers, representing 3.9% of all cancers, derive from chronic alcohol consumption [12]. Chronic alcohol consumption is associated with cancers of the upper aerodigestive tract, liver, breast, and colorectum, and ethanol has been declared a group 1 carcinogen by the International Agency for Research on Cancer (IARC) [7]. Hence, it is appropriate to investigate the potential implications of alcohol consumption on LS patients' CRC susceptibility using a model system, as there have been very few studies on whether alcohol affects CRC risk in LS patients; for example, a recent cross‐sectional multicentre Japanese study that showed that alcohol consumption is significantly correlated with increased risk of early‐onset CRC in LS patients [13]. Carcinogenic effects of ethanol are related mainly to its metabolite acetaldehyde [7]. Acetaldehyde is a highly reactive molecule able to induce a wide range of DNA lesions. However, the DNA repair pathways responsible for repairing many of these DNA lesions remain unknown and a potential role for MMR is currently uncertain. The MMR pathway helps to maintain genomic stability via DNA post‐replicative error repair. Here, MMR is involved in cellular responses to DNA damage induced by exogenous and endogenous chemical carcinogens [14], by coordinating the cell's response to MMR‐recognised DNA alterations through cell cycle arrest and/or apoptosis [1, 15, 16, 17].
Wojciechowicz et al generated a mouse model of LS by intercrossing the Lgr5CreERT2 transgene (created by Barker et al) with Msh2‐null (Msh2 +/− ) and Msh2‐floxed (Msh2 flox/flox ) alleles [18, 19, 20, 21]. In these Lgr5CreERT2;Msh2 flox/− (Msh2‐LS) mice, tamoxifen is used to induce Cre recombinase expression and activity in scattered intestinal Lgr5+ stem cells and this mediates recombination at LoxP sites, causing somatic loss of the second (floxed) Msh2 allele. This generates crypts (populated by the Msh2‐null stem cells) that are defective for mismatch repair (dMMR). This occurs in scattered foci along the small and large intestines of the mice forming dMMR crypts and crypt foci, which mimic the pattern of scattered dMMR crypts and crypt foci in LS patients [21]. Wojciechowicz et al showed that tamoxifen‐induced Msh2‐LS mice developed intestinal tumours (adenomas and adenocarcinomas) after around 19 months following induction [21]. Here, Msh2‐LS mice were used to investigate the role of MMR to protect intestinal epithelium from some types of ethanol/acetaldehyde‐induced DNA damage and to test the hypothesis that a gene–environment interaction between dMMR and ethanol/acetaldehyde can accelerate colorectal tumour development and progression.
Materials and methods
Ethanol treatment of Msh2‐LS mice
Forty‐six Msh2‐LS mice (provided by Hein te Riele, The Netherlands Cancer Institute, Amsterdam; on a mixed background C57BL/6J and FVB as described elsewhere [20, 21]), aged 7–9 weeks (24 males and 22 females), received intraperitoneal (i.p.) injections of 0.15 mg tamoxifen/g body weight (BW) on day 1 and 0.1 mg tamoxifen/g BW on days 2, 3, and 4. From day 5, mice were provided with either 20% ethanol in drinking water or standard drinking water. Ethanol was administered to both induced and non‐induced Msh2‐LS mice to simulate the pathological effects of ethanol in humans; because ethanol clearance is five times faster in mice than in humans, we used 20% ethanol in drinking water as previously validated [22, 23, 24]. None of the mice showed abnormal behaviour or reduced body weight initially, indicating good acceptance of the ethanol regime. Long‐term ethanol treatment of Msh2‐LS mice followed two different approaches. First, ten water‐treated mice and ten ethanol‐treated mice were monitored until either any one of the clinical signs indicative of intestinal tumour formation (see supplementary material, Table S1) were visible or they displayed >20% BW loss compared with the initial BW. Second, water‐ and ethanol‐treated mice (13 mice each) were monitored; when an ethanol‐treated mouse had to be culled for signs of intestinal tumour formation or >20% loss of BW, an age‐matched and treatment duration‐matched water‐treated control mouse was culled at the same time point. The results from both approaches were collected and presented together. A further 24 Msh2‐LS control mice, aged 7–9 weeks, received i.p. injections of 0.15 mg corn oil/g BW (without tamoxifen, so there was no induction of Cre recombinase activity) on day 1 and 0.1 mg corn oil/g BW on days 2, 3, and 4; from day 5, mice were provided with either 20% ethanol in drinking water (12 mice) or standard drinking water (12 mice). Both water‐ and ethanol‐treated non‐induced age‐matched mice were sacrificed when they reached the same end‐time points as the ethanol‐treated induced Msh2‐LS mice that were culled for clinical signs indicative of intestinal tumour formation or >20% BW loss.
Tissue collection and analysis
At necropsy, blood was collected and the SI, LI, caecum, stomach, liver, lungs, spleen, thymus, lymph nodes, and any other organ or tissue showing abnormalities were collected and fixed in 10% neutral‐buffered formalin for 24 h. Tissues were processed and paraffin‐embedded to blocks, sectioned, and stained with haematoxylin and eosin (H&E). Immunohistochemistry (IHC) was performed using anti‐MSH2 (ab70270, 1:4000; Abcam, Cambridge, UK), anti‐MSH6 (ab92471, 1:500, Abcam), anti‐MLH1 (ab92312, 1:250, Abcam), anti‐Ki‐67 (ab16667, 1:500, Abcam), anti‐β‐catenin (610154, 1:100; BD Transduction Laboratories, San José, CA, USA), anti‐cCas3 (9664, 1:50; Cell Signaling Technologies, Danvers, MA, USA), anti‐γH2AX (2577, 1:100, Cell Signaling Technologies), and anti‐p53 (CM5, 1:100; provided by Dr Phil Coates, Masaryk Memorial Cancer Institute, Brno, Czechia) antibodies, as previously described [22]. Image analysis software QuPath v0.2.0 was used to analyse immunohistochemically detected MSH2, Ki67, γ‐H2AX, and p53 protein expression [25].
Acetaldehyde assay
Blood was fractionated by centrifugation at 3000 × g for 15 min at 4 °C. Plasma was collected and immediately snap‐frozen in liquid nitrogen and stored at −80 °C. Plasma acetaldehyde concentrations were determined using an acetaldehyde assay kit (K‐ACHYD; Megazyme, Co. Wicklow, Ireland), according to the manufacturer's instructions.
Statistical analysis
Data were analysed using GraphPad Prism v7.0 software (GraphPad Software Inc, San Diego, CA, USA) and are shown as mean with SD error bars. Group data were compared using unpaired, two‐tailed Student's t‐test or Mann–Whitney U‐test as appropriate, or two‐way ANOVA with Bonferroni's multiple comparisons correction test. Association between two categories was assessed by two‐sided Fisher's test. Percentage of survival was analysed by the log‐rank (Mantel–Cox) test. Differences between groups were considered statistically significant if p < 0.05.
Results
Ethanol causes increased colonic tumour development in Msh2‐LS mice
After several months of ethanol treatment, most of the induced Msh2‐LS mice displayed either anal prolapse or >20% reduction in BW and were culled for necropsy. In total, 15/23 (65%) ethanol‐treated induced Msh2‐LS mice demonstrated colonic adenoma formation, and in six cases, invasive adenocarcinoma was present, all within an average of 6 months (Figures 1 and 2). All 15 ethanol‐treated induced Msh2‐LS tumour‐bearing mice showed zones of colonic crypt hyperproliferative changes mainly in either the proximal colon or the mid‐colon, within which adenoma formation was observed. Invasive adenocarcinomas were observed developing from adenomas in 26% (five in the colon and one in the caecum) of the 23 ethanol‐treated induced Msh2‐LS mice (40% of adenoma‐bearing mice). No small intestinal tumours or hyperproliferative zones were observed in any of these mice. Additionally, two of the 15 colonic tumour‐bearing mice developed cutaneous sebaceous adenomas, a tumour type that frequently occurs in LS patients (supplementary material, Figure S1). Eight of 23 ethanol‐treated induced Msh2‐LS mice did not show any intestinal tumour formation, but a uterine endometrial adenocarcinoma was found in one mouse after 12 months of ethanol treatment; this tumour type is also frequently seen in LS [1, 3]. The water‐treated induced Msh2‐LS control mice showed longer intestinal tumour‐free survival compared with the ethanol‐treated mice, with only 1 of 23 (4%) mice showing large intestinal tumour formation in the proximal colon, involving the caecum, after 15 months (Figure 2). No intestinal abnormalities were observed in any of the other 22 water‐treated induced Msh2‐LS control mice; however, uterine endometrial invasive adenocarcinomas were observed in two mice (after 12 and 15 months of water treatment) (supplementary material, Figure S2).
Figure 1.
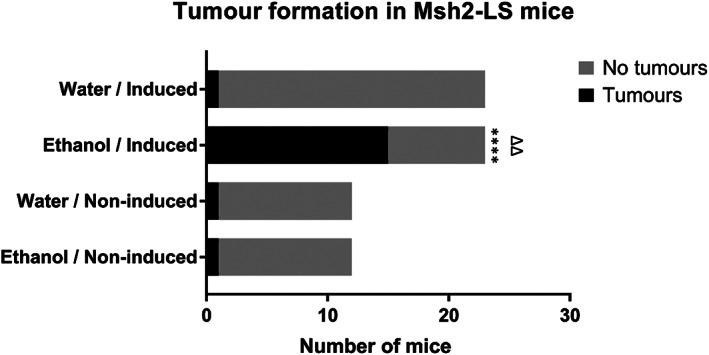
Tumour formation in Msh2‐LS mice. Bar chart of the numbers of Msh2‐LS mice that developed large intestinal tumours (both adenomas and adenocarcinomas) after receiving either 20% ethanol or standard drinking water. 15/23 (65%) ethanol‐treated induced Msh2‐LS mice developed large intestinal tumours compared with 1/23 (4%) water‐treated induced Msh2‐LS mice; Fisher's exact test, ****p < 0.0001. In both groups of non‐induced Msh2‐LS mice (water‐treated and ethanol‐treated), 1/12 (8.4%) non‐induced Msh2‐LS control mice developed colonic neoplasms; Fisher's exact test, no significant differences observed. Comparison of the tumour‐bearing ethanol‐treated induced Msh2‐LS mice with ethanol‐treated non‐induced Msh2‐LS mice showed a significant difference in the development of large intestinal tumours; Fisher's exact test, ΔΔ p = 0.0016.
Figure 2.
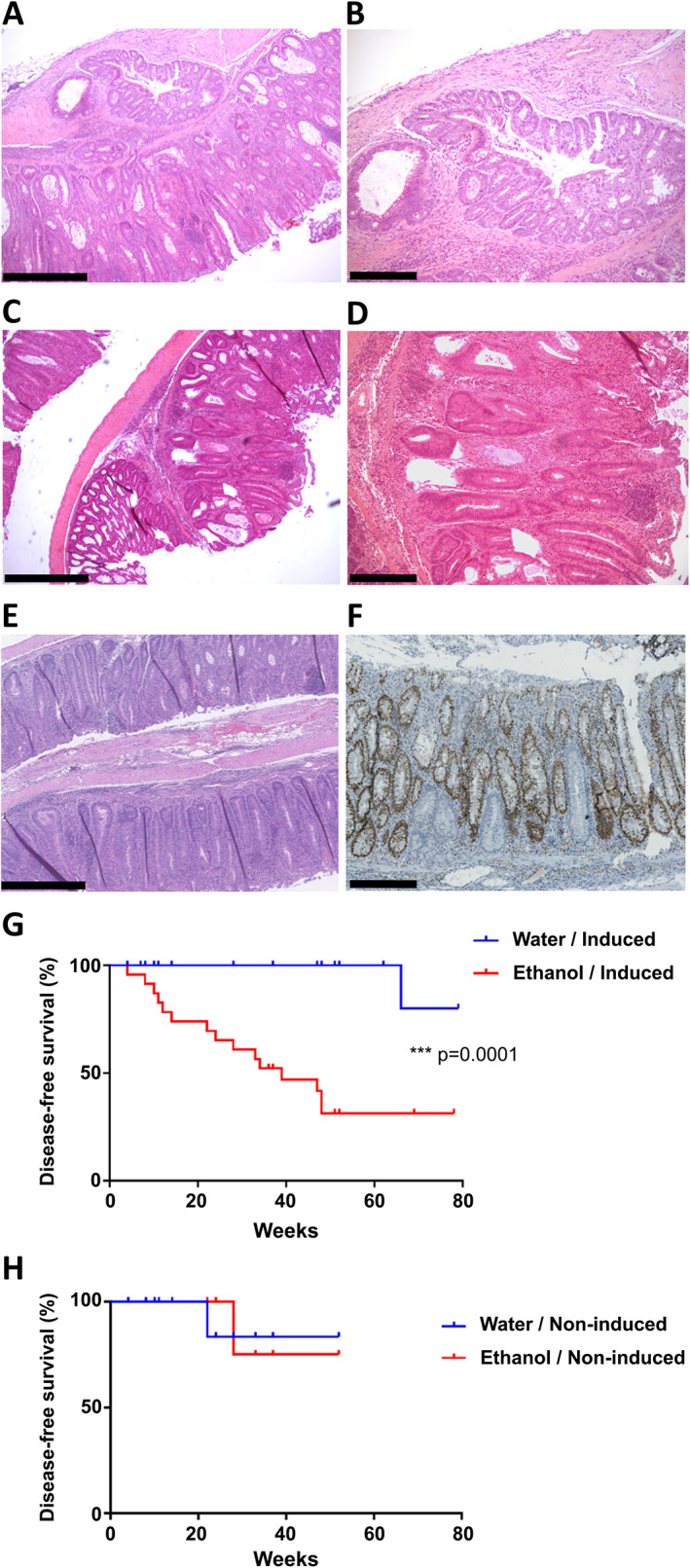
Large intestinal tumours and survival analysis. (A–D) Representative images of proximal colonic invasive adenocarcinoma (A, B) and proximal colonic adenoma (C, D) in ethanol‐treated induced Msh2‐LS mice, respectively. (E) Representative image of colonic crypt epithelial hyperproliferative changes in ethanol‐treated induced Msh2‐LS mice, demonstrating the increase in length of the affected crypts. (F) Representative image of MSH2 immunostaining of murine colonic hyperproliferative zone with some MSH2‐negative crypts from ethanol‐treated induced Msh2‐LS mice. Original magnification: 50× (A, C, E); 100× (B, D, F). Scale bars: 500 μm (A, C, E); 250 μm (B, D, F). (G) Tumour incidence shown as survival curves in induced Msh2‐LS mice treated with either 20% ethanol (red) or water (blue); log‐rank (Mantel–Cox) test, ***p = 0.0001. (H) Tumour incidence shown as survival curves in non‐induced Msh2‐LS control mice (with no Cre activation and thus no MSH2‐negative cells or crypts) treated with either 20% ethanol (red) or water (blue); log‐rank (Mantel–Cox) test, no significant differences observed.
Three of 12 (25%) ethanol‐treated non‐induced Msh2‐LS mice showed zones of colonic crypt epithelial hyperproliferation, involving mostly the mid‐colon, but no intestinal adenomas were present. One of 12 (8.4%) ethanol‐treated non‐induced Msh2‐LS mice showed a proximal colonic invasive adenocarcinoma (after 28 weeks). Eight of 12 (66.6%) ethanol‐treated non‐induced Msh2‐LS mice showed no intestinal adenomas, cancers, colonic hyperproliferation, or other abnormalities. In the water‐treated non‐induced Msh2‐LS mice, only 1 of 12 (8.4%) showed colonic adenoma formation in the proximal colon (after 22 weeks of water treatment). No tumours were observed in the SI, stomach, anal canal, liver, hepatobiliary tract, pancreas, spleen, lymph nodes or thymus, in any of the induced Msh2‐LS experimental mice or in any of the non‐induced Msh2‐LS control mice, either water‐treated or ethanol‐treated. The dominant neoplasm in this model was colonic adenoma (with some progressing to adenocarcinomas) and their incidence was statistically significantly greater in ethanol‐treated induced Msh2‐LS mice (p < 0.0001, Fisher's exact test; p = 0.0001, Mantel–Cox test for survival difference) compared with water‐treated induced Msh2‐LS mice, and either ethanol‐treated or water‐treated non‐induced Msh2‐LS mice (p = 0.0016 for both comparisons, Fisher's exact test) (Figures 1 and 2).
Loss of MSH2 protein expression in Msh2‐LS mice confirms defective MMR in adenomas and colonic crypt foci
Immunostaining for MSH2 showed that all colonic adenomas tested from ethanol‐treated induced Msh2‐LS mice had MSH2‐negative dysplastic glands, surrounded by or admixed with MSH2‐positive crypts showing normal, reactive or hyperproliferative changes (Figure 3). The percentage of MSH2‐negative crypts in non‐tumour‐bearing colonic mucosa was statistically significantly higher in ethanol‐treated induced Msh2‐LS mice compared with that from water‐treated induced Msh2‐LS mice (p = 0.0029, Student's t‐test) (Figures 3 and 4). The percentage of MSH2‐negative crypts was also significantly higher in the SI of ethanol‐treated than in that of water‐treated induced Msh2‐LS mice, and there were more MSH2‐negative crypts in the SI than in the colon in both water‐treated and ethanol‐treated induced Msh2‐LS mice (supplementary material, Figure S3). However, no tumours were observed in the SI in any mice. No MSH2‐negative crypts were observed in either small or large intestinal mucosal epithelium of non‐induced Msh2‐LS mice, consistent with lack of induction of Cre activity resulting in continued expression of protein from the floxed Msh2 allele (supplementary material, Figure S4). Immunostaining for MSH6 and MLH1 confirmed secondary loss of MSH6 in MSH2‐negative adenomas and MSH2‐negative dMMR crypt foci, but normal expression of MLH1 (supplementary material, Figures S5 and S6).
Figure 3.
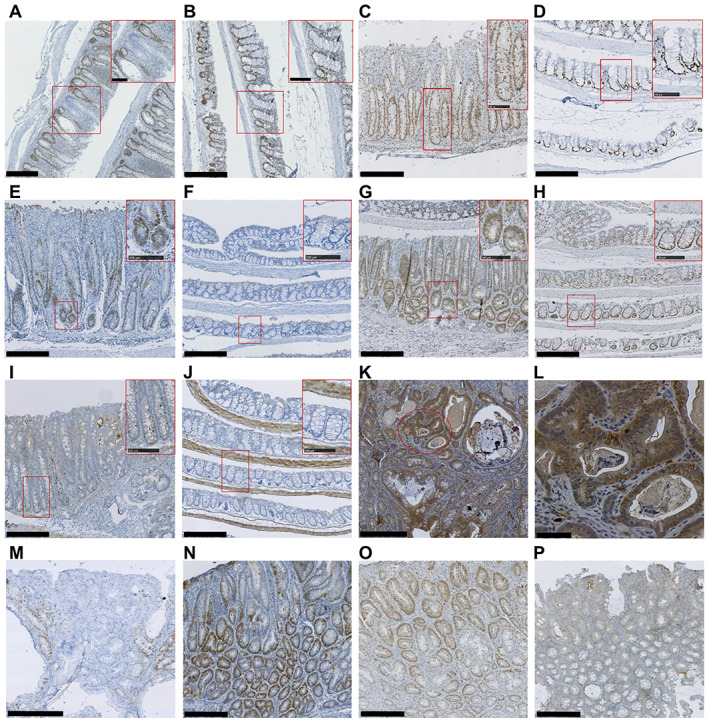
Representative images of IHC analysis of induced Msh2‐LS murine colonic mucosal epithelium and adenomas. (A, B) MSH2 immunostaining of murine colon from ethanol‐treated induced Msh2‐LS mice (A) and water‐treated induced Msh2‐LS mice (B). (C, D) Ki‐67 immunostaining of murine colon from ethanol‐treated induced Msh2‐LS mice (C) and water‐treated induced Msh2‐LS mice (D). (E, F) Immunostaining for γ‐H2AX in ethanol‐treated induced Msh2‐LS mice (E) and water‐treated induced Msh2‐LS mice (F). (G, H) Immunostaining for p53 in ethanol‐treated induced Msh2‐LS mice (G) and water‐treated induced Msh2‐LS mice (H). (I, J) Immunostaining for cCas3 in ethanol‐treated induced Msh2‐LS mice (I) and water‐treated induced Msh2‐LS mice (J). All main panels show original magnification of 100× (with further magnification of 200× in the upper right inset red rectangles). (K, L) Representative image of β‐catenin immunostaining in colonic adenoma from ethanol‐treated induced Msh2‐LS mouse (K), with selected area (red oval) within image K (original magnification 100×) further magnified to 400× in image L. (M–P) Representative images of colonic adenomas from ethanol‐treated induced Msh2‐LS mice immunostained for MSH2 (M), γ‐H2AX (N), p53 (O), and cCas3 (P). Original magnification: 100× (M–P). Scale bars: 250 μm (A–K, M–P, main panel); 100 μm (A–J, inset rectangles); 50 μm (L, main panel).
Figure 4.
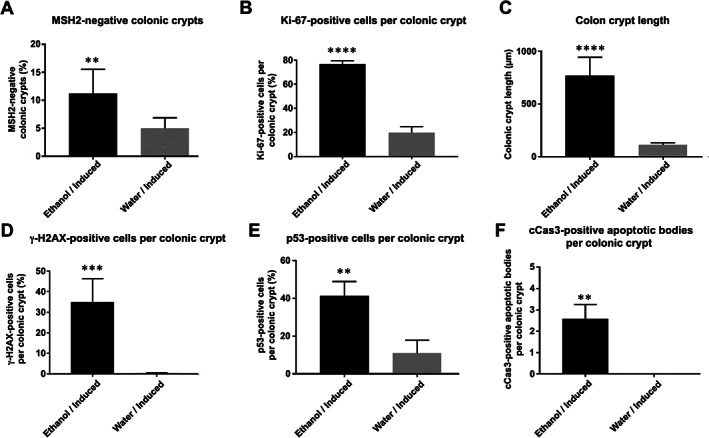
Quantitative analyses of immunohistochemical stains of murine colonic mucosa from ethanol‐ (black bar) and water‐treated (grey bar) induced Msh2‐LS mice. (A) Percentage of MSH2‐negative colonic crypts; paired Student's t‐test, **p = 0.0029 versus water. (B) Percentage of Ki‐67‐positive cells per colonic crypt; paired Student's t‐test, ****p < 0.0001 versus water. (C) Colonic crypt lengths (μm); Mann–Whitney U‐test, ****p < 0.0001. (D) Percentage of γ‐H2AX‐positive cells per colonic crypt; paired Student's t‐test, ***p < 0.0009 versus water. (E) Percentage of p53‐positive cells per colonic crypt; paired Student's t‐test, **p < 0.0011 versus water. (F) Number of cleaved caspase‐3 (cCas3)‐positive apoptotic bodies per colonic crypt; paired Student's t‐test, **p = 0.0026 versus water (data shown as mean ± SD error bars, n = 6 for all comparisons).
Expression of β‐catenin protein was investigated by IHC in colonic tumours from ethanol‐treated induced Msh2‐LS mice, which showed a heterogeneous pattern with variable numbers of adenoma cells showing moderately to strongly positive β‐catenin nuclear immunostaining due to accumulation and translocation of β‐catenin into tumour nuclei (Figure 3), indicating activation of the Wnt signalling pathway in the adenomas compared with low levels of β‐catenin expression in normal mucosal epithelium (supplementary material, Figure S7).
Ethanol induces colonic epithelial proliferation
To evaluate intestinal crypt epithelium proliferative activity, expression of the proliferation marker Ki‐67 was assessed immunohistochemically and by crypt length measurements (Figures 2, 3, 4). The proportion of Ki‐67‐positive cells per crypt was significantly greater in colons from ethanol‐treated induced Msh2‐LS mice compared with water‐treated induced Msh2‐LS mice (p < 0.0001, Student's t test). This confirmed the presence of large zones of colonic mucosal crypt epithelial hyperproliferation observed morphologically and quantified by crypt length measurements in association with long‐term ethanol treatment (Figures 2 and 4). The percentage of Ki‐67‐positive cells per crypt in the SI of ethanol‐treated induced Msh2‐LS mice was only slightly but significantly higher than that in the SI of water‐treated induced Msh2‐LS mice (supplementary material, Figure S8).
Ethanol is associated with colonic DNA damage response and epithelial apoptosis
To investigate the potential for ethanol‐induced DNA alterations, IHC was performed for DNA damage response biomarkers γ‐H2AX and p53. γ‐H2AX immunostaining showed high expression levels in the ethanol‐treated induced Msh2‐LS large intestinal adenomas (both colonic and caecal). The percentage of γ‐H2AX‐positive cells was significantly higher in ethanol‐treated induced Msh2‐LS non‐tumour‐bearing colonic mucosal epithelium (35%) compared with water‐treated induced Msh2‐LS colonic epithelium (0.4%) (p < 0.0009, Student's t‐test) (Figures 3 and 4). By contrast, very few or no γ‐H2AX‐positive cells were observed in both ethanol‐treated induced Msh2‐LS and water‐treated induced Msh2‐LS small intestinal mucosal epithelium (supplementary material, Figure S9).
The presence of DNA damage induces p53 pathway activation that can be demonstrated immunohistochemically as a greater proportion of cells containing moderate to high (but variable) nuclear staining of p53 in individual cells (‘wild‐type pattern’, distinguishable from the mutated p53‐associated ‘overexpression’ or ‘complete absence’ patterns seen in some neoplasms) [26, 27]. Ethanol‐treated induced Msh2‐LS colonic adenomas showed widespread variably high p53 expression reflecting the ‘wild‐type pattern’ in response to ethanol‐induced genotoxic damage. No tumours showed either the ‘overexpression’ or the ‘null’ patterns associated with Tp53 mutation. A significantly higher proportion of p53‐positive cells with high to moderate nuclear staining was observed in ethanol‐treated induced Msh2‐LS colonic mucosal epithelium (41.3%) compared with that from water‐treated induced Msh2‐LS mice (10.8%) (p < 0.0011, Student's t‐test) (Figures 3 and 4). The percentage of p53‐positive cells in small intestinal mucosal epithelium was greater in ethanol‐treated induced Msh2‐LS mice than in water‐treated induced Msh2‐LS mice (supplementary material, Figure S10).
MMR is involved in a signalling cascade that leads to cell cycle arrest and/or apoptosis when severe DNA damage has occurred [15]. To detect apoptotic events, IHC analysis of cleaved caspase‐3 (cCas3) was performed. Ethanol‐treated induced Msh2‐LS murine colonic adenomas showed no or almost no detectable cCas3+ apoptotic bodies, indicating rare to no apoptotic events in dMMR colonic tumours (Figure 3). IHC of cCas3 showed significantly higher numbers of cCas3+ apoptotic bodies in ethanol‐treated induced Msh2‐LS non‐tumour‐bearing colon compared with no or almost no detectable cCas3+ apoptotic bodies in water‐treated induced Msh2‐LS colon (p = 0.0026, Student's t‐test), consistent with increased colonic epithelial apoptosis associated with exposure to ethanol or its metabolites.
The increased DNA damage response observed in ethanol‐treated induced Msh2‐LS murine colonic mucosal epithelium is consistent with the higher levels of circulating acetaldehyde detected by the plasma acetaldehyde assay, which showed statistically significantly higher plasma acetaldehyde levels in ethanol‐treated than in water‐treated induced Msh2‐LS mice (p = 0.0019, Mann–Whitney U‐test) (Figure 5). There was no statistically significant difference in the plasma acetaldehyde levels between ethanol‐treated and water‐treated non‐induced Msh2‐LS mice.
Figure 5.
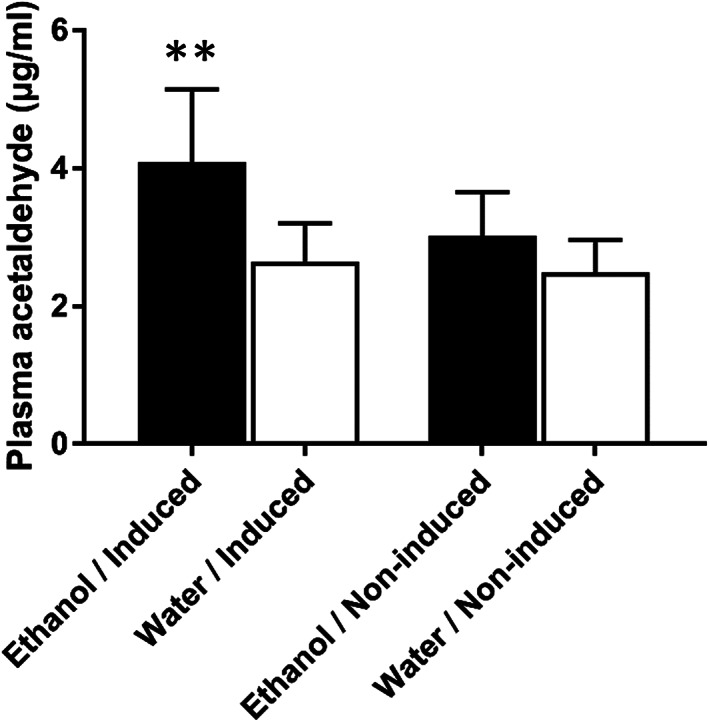
Plasma acetaldehyde concentrations in ethanol‐ (black bar) and water‐treated (white bar) induced Msh2‐LS mice and non‐induced Msh2‐LS mice; Mann–Whitney U‐test, **p = 0.0019 (data shown as mean ± SD error bars, n = 4–6).
Discussion
In this mouse model of LS, long‐term ethanol consumption (relative to water consumption) caused acceleration of dMMR‐driven colonic tumour formation with increased numbers of adenomas and some adenocarcinomas. Ethanol‐treated Msh2‐LS mice showed evidence of large intestinal hyperproliferation and adenoma formation (with six adenocarcinomas) mostly in the proximal and mid‐colon in 65% animals, within an average of 6 months of ethanol treatment. This is in stark contrast to the water‐treated induced Msh2‐LS control mice, none of which developed large intestinal tumours over the same time period, with only one colonic adenoma observed at 15 months (4%). Non‐induced Msh2‐LS mice were treated with ethanol as a control to test for unknown variables that might affect tumour formation in the absence of loss of Msh2: 66.6% (8/12) of the ethanol‐treated mice together with 91.6% (11/12) of the water‐treated mice showed no colonic neoplasms. No small intestinal adenomas were seen in any of the ethanol‐treated or water‐treated induced Msh2‐LS mice, despite scattered Lgr5‐Cre activation and generation of dMMR crypt foci in the small intestinal mucosa. These findings indicate that these mice represent a very good model of human LS with colorectal tumours as the major tumour predisposition with proximal and mid‐colonic location. Even without induced inactivation of the second Msh2 allele, some of the Msh2‐LS mice (with one deleted Msh2 and one functional Msh2 allele as seen in human LS) developed occasional skin sebaceous adenomas and endometrial adenocarcinomas, both typical tumours of human LS [3, 4].
Our investigation of potential precursor dMMR lesions in the Msh2‐LS murine intestines showed that induced Cre activation caused loss of MSH2 expression in Lgr5+‐expressing crypt epithelial stem cells scattered along the entire small and large intestines. The Lgr5+ stem cells located at the crypt base generate daughter cells that can expand to fill the entire crypt–villus epithelium in the SI or the entire crypt in the colon by monoclonal conversion, as observed in these mice using MSH2 IHC that showed MSH2‐negative entire crypts, confirming previous observations [18, 21]. Statistically significantly higher proportions of such dMMR crypt foci precursors in both the small and the large intestines were detected in ethanol‐treated compared with water‐treated induced Msh2‐LS mice. All ethanol‐treated induced Msh2‐LS murine colonic adenomas showed MSH2‐negative dysplastic glands, consistent with colonic adenomas arising from MSH2‐negative (dMMR) crypt foci precursors. This confirms data from human LS studies that dMMR crypt foci are tumour precursors and is consistent with observations that LS patients' risk of colonic tumour formation correlates with the size of MMR‐deficient crypt clusters that increase over time in LS patients [21, 28, 29].
Colorectal neoplasm formation often involves dysregulation of Wnt signalling and this was assessed by immunohistochemical staining of β‐catenin, the key Wnt‐signalling intermediate [30]. Normal colorectal mucosal crypt epithelium exhibited mild to moderate membranous and weak cytoplasmic staining for β‐catenin, but absent nuclear localisation. In contrast, both colorectal adenomas and adenocarcinomas from induced Msh2‐LS mice showed variably moderately to strongly positive nuclear β‐catenin localisation, indicating Wnt pathway activation occurring in adenoma formation in this model similar to that observed in human LS, in which homozygous or hemizygous β‐catenin mutations are more prevalent than in sporadic CRC [31].
Ki‐67 is a biomarker that is present in proliferating cells but absent in quiescent cells and may be predictive of some tumour behaviour [32]. In normal large and small intestinal mucosa, Ki‐67 is expressed only in the proliferating cells around the base and lower third of crypts [33] and this was confirmed in this study. High levels of Ki‐67 expression were observed in ethanol‐treated murine proximal and mid‐colons within the zones of mucosal crypt hyperproliferation (morphologically identified as showing elongated crypt length). Hyperproliferative zones were only seen in ethanol‐treated induced (and non‐induced) murine colonic epithelial mucosa; they were not observed in the SI of these mice, or in the LI or SI of water‐treated induced Msh2‐LS mice or water‐treated non‐induced mice. This is consistent with ethanol‐induced large intestinal crypt epithelial hyperproliferation that has previously been described by our group (and others) after long‐term ethanol treatment of wild‐type mice and ALDH1b1‐depleted mice [22, 34, 35].
Ethanol is rapidly absorbed from the gastrointestinal tract [35] and is metabolised by alcohol dehydrogenases (ADHs) to highly reactive acetaldehyde, which is further oxidised to acetate by acetaldehyde dehydrogenases (ALDHs). Acetaldehyde is very reactive and can cause a range of DNA modifications: DNA adducts, single and double strand breaks, point mutations, increased sister chromatid exchanges, DNA–protein crosslinks or DNA interstrand crosslinks (ICLs) [36]. Langevin et al demonstrated that the Fanconi anaemia (FA) DNA repair pathway has a crucial role in counteracting acetaldehyde‐induced genotoxicity in mice, as the FA DNA repair pathway is essential for the repair of DNA ICLs [37, 38]. We hypothesised that the DNA MMR repair system plays a role in protecting cells from some types of ethanol/acetaldehyde‐induced DNA damage. In addition to hypermutability and MSI, loss of MMR pathway function results in dMMR with reduced susceptibility to cell cycle arrest and apoptosis that would normally be induced by those types of DNA damage recognised by the MMR pathway [1, 15, 39]. Statistically significantly higher plasma acetaldehyde levels were detected in ethanol‐treated induced Msh2‐LS mice compared with water‐treated induced Msh2‐LS mice.
DNA damage response was evaluated by immunohistochemical staining for γ‐H2AX, which showed significantly increased levels in ethanol‐treated compared with water‐treated induced Msh2‐LS murine colonic mucosal epithelium, but there were fewer differences in the SI. This increased colonic γ‐H2AX is consistent with DNA damage brought about by ethanol/acetaldehyde, demonstrating that ethanol exposure has a significant mutagenic effect mainly on dMMR colonic mucosal epithelium but less so on dMMR small intestinal mucosa. This suggests that MSH2 plays a key role in protecting the MMR‐proficient colonic epithelial cells against this type of DNA damage, but there may be additional mechanisms protecting small intestinal epithelial cells from ethanol/acetaldehyde‐induced DNA damage. The presence of an elevated colonic DNA damage response was confirmed by the high percentage of p53‐positive cells observed in the ethanol‐treated compared with the water‐treated induced Msh2‐LS mice. MMR is involved in a signalling cascade that leads to either cell cycle arrest or apoptosis if severe DNA damage has occurred. We previously showed that MSH2‐dependent apoptosis is primarily mediated through a p53‐dependent pathway, and that MSH2 is required to signal delayed p53‐independent death [15]. This is applicable to human colorectal carcinogenesis in Lynch syndrome as LS‐associated CRCs have a low frequency of TP53 mutations [1, 2, 3, 4]. It has been observed that MMR‐deficient cells fail to recruit ATM and ATR proteins, preventing p53 phosphorylation in response to DNA damage [40, 41]. MMR‐deficient cells show predisposition to malignancy by failing to repair DNA damage (of MMR‐recognised type) and are unable to engage apoptosis to delete such DNA‐damaged cells [15, 17]. Whereas ethanol exposure induced significantly increased apoptosis of predominantly MMR‐proficient (pMMR) normal‐appearing colonic epithelium in ethanol‐treated compared with water‐treated induced Msh2‐LS mice, almost no cCas3+ apoptotic bodies were observed in ethanol‐treated induced Msh2‐LS murine dMMR colonic adenomas, indicating almost complete failure of ethanol/acetaldehyde‐mediated DNA damage to induce apoptosis in dMMR colonic tumours. Thus, ethanol/acetaldehyde may select for preferential dMMR stem cell survival relative to adjacent pMMR stem cells (which can still die from pMMR‐induced apoptosis) providing a selective competitive advantage, as shown for dMMR stem cells in another model system [42], promoting monoclonal conversion of crypts by dMMR cells (further investigations of dMMR crypt development dynamics are ongoing).
In conclusion, the ethanol‐treated Msh2‐LS mouse model closely recapitulates the pattern seen in human LS patients for both precursor dMMR colonic crypt foci and colonic adenomas with (in some cases) progression to adenocarcinoma. Ethanol treatment was shown to induce zones of hyperproliferation of the colonic, but not the small intestinal, mucosal epithelium and this appears to contribute to intestinal adenoma formation by acting as a tumour promoter, which occurs mostly in those parts of the colon (proximal and mid‐colon) affected by hyperproliferation in this Msh2‐LS mouse model. Ethanol and acetaldehyde were shown to cause colonic mucosal epithelial DNA damage responses, but less so in the SI, by IHC for both γ‐H2AX and p53. We propose an explanatory model, that in dMMR colonic epithelial stem cells, modelled in these Msh2‐LS mice, ethanol/acetaldehyde causes DNA damage that is not recognised or repaired due to defective MMR following inactivation of the second Msh2 allele that leaves these cells unable to activate the MMR signalling pathway. Hence, dMMR cells fail to induce apoptosis and thus show inappropriate survival of ethanol/acetaldehyde‐damaged cells, compared with MMR‐proficient cells (Figure 6). These aberrantly surviving DNA‐damaged dMMR colonic epithelial cells are proposed to be stimulated to proliferate by ethanol consumption, leading to ethanol/acetaldehyde‐mediated selection for expansion of the number of dMMR foci and an increased probability of acquisition and fixation of DNA mutations, due to both ongoing ethanol/acetaldehyde‐mediated DNA damage and dMMR hypermutability, explaining both the increased dMMR crypt precursors and the accelerated colonic adenoma formation, with some adenomas evolving to adenocarcinoma (Figure 6). Hence, there is strong evidence that the DNA MMR system plays a role in protecting colonic epithelial cells from some types of ethanol/acetaldehyde‐induced DNA damage and that there is a gene–environment interaction between dMMR and ethanol/acetaldehyde exposure that accelerates colorectal tumour development and progression in this mouse model. This dMMR–ethanol interaction is highly likely to apply to human LS patients, indicating that appropriate lifestyle advice concerning limiting alcohol consumption should be considered for LS patients.
Figure 6.

Schematic diagram of the proposed model of the MMR/ethanol/acetaldehyde gene–environment interactions in both MMR‐proficient (upper panel) and MMR‐deficient (lower panel) colonic epithelial cells. Upon ethanol/acetaldehyde exposure, in some colonic epithelial stem cells there is DNA base damage that normally would be recognised and repaired by the MMR system, or if unrepaired this base damage may induce replication errors, such as base mismatches or insertion/deletion loops (InDels), during S‐phase of the cell cycle. Here, the MMR‐proficient cell is able to activate DNA mismatch repair of the (MMR‐recognised) base damage, bringing about either cell cycle arrest in the context of mild DNA damage to allow DNA repair or cell death by apoptosis for more severe DNA damage. By contrast, the MMR‐deficient cell (dMMR due to mutant Msh2) is unable to activate the MMR signalling pathway and so there is neither cell cycle arrest nor apoptosis, resulting in aberrant survival of DNA‐damaged cells that can undergo ethanol‐induced subsequent proliferation. The proliferating dMMR stem cells populate the colonic crypt and expand further to form dMMR crypt foci. Stimulated by ethanol to undergo increased proliferation, these cells form hyperproliferative crypts whilst remaining subject to ongoing DNA damage from continued exposure to ethanol/acetaldehyde. These dMMR cells can accumulate mutations reflecting dMMR genomic instability and are consequently at increased risk of tumour formation, thus explaining the acceleration of colonic adenoma formation and increased probability of evolution to adenocarcinoma.
Author contributions statement
MJA, DJA and GC conceived the experiments. GC, YZ and MM carried out experiments. MJA and GC carried out histopathological analysis. GC, DJA and MJA analysed the data and produced the figures. GC and MJA wrote the manuscript. All the authors reviewed and agreed to the final version of the manuscript.
Supporting information
Figure S1. Tumour distribution in ethanol‐treated induced Msh2‐LS tumour‐bearing mice
Figure S2. Representative images of skin sebaceous adenoma and endometrial invasive adenocarcinoma from ethanol‐treated induced Msh2‐LS mice
Figure S3. Immunohistochemical analysis of MSH2 protein expression in small intestinal mucosal epithelium of Msh2‐LS mice treated with either 20% ethanol in drinking water or standard drinking water
Figure S4. Immunohistochemical analysis of MSH2 protein expression in large and small intestinal mucosal epithelium of non‐induced Msh2‐LS control mice treated with either 20% ethanol in drinking water or standard drinking water
Figure S5. Immunohistochemical analysis of MSH6 protein expression in intestinal mucosal epithelium
Figure S6. Immunohistochemical analysis of MLH1 protein expression in intestinal mucosal epithelium
Figure S7. Immunohistochemical analysis of β‐catenin protein expression and localisation in large intestinal mucosal adenoma and adjacent normal epithelium from a positive‐control Apc Min mouse
Figure S8. Immunohistochemical analysis of Ki‐67 protein expression in small intestinal mucosa of Msh2‐LS mice treated with either 20% ethanol in drinking water or standard drinking water
Figure S9. Immunohistochemical analysis of γ‐H2AX protein expression in small intestinal mucosa of Msh2‐LS mice treated with either 20% ethanol in drinking water or standard drinking water
Figure S10. Representative images of immunohistochemical analysis of p53 protein expression in small intestinal mucosal epithelium of Msh2‐LS mice treated with either 20% ethanol in drinking water or standard water
Table S1. Clinical scoring system for monitoring mice for signs of intestinal tumour development
Acknowledgements
We thank The Pathological Society of Great Britain and Ireland for funding a PhD sponsorship grant supporting this work, Hein te Riele (The Netherlands Cancer Institute, Amsterdam, The Netherlands) for providing the Msh2‐LS mouse model, Phil Coates for providing the anti‐p53 CM1 antibody, and Helen Caldwell and Elaine McLay for technical support and sample processing.
Conflict of interest statement: MJA is currently President of The Pathological Society, owners of this Journal. No other conflicts of interest were declared.
References
- 1. Poulogiannis G, Frayling IM, Arends MJ. DNA mismatch repair deficiency in sporadic colorectal cancer and Lynch syndrome. Histopathology 2010; 56: 167–179. [DOI] [PubMed] [Google Scholar]
- 2. Bellizzi AM, Frankel WL. Colorectal cancer due to deficiency in DNA mismatch repair function: a review. Adv Anat Pathol 2009; 16: 405–417. [DOI] [PubMed] [Google Scholar]
- 3. Frankel W, Arends M, Frayling IM, et al. Lynch syndrome: genetic tumour syndromes of the digestive system. In World Health Organization Classification of Tumours of the Digestive System (5th edn), Nagtegaal ID, Odze RD, Klimstra D, et al. (eds). IARC Press: Lyon, 2019; Chapter 14. [Google Scholar]
- 4. Rijcken FE, Koornstra JJ, van der Sluis T, et al. Early carcinogenic events in HNPCC adenomas: differences with sporadic adenomas. Dig Dis Sci 2008; 53: 1660–1668. [DOI] [PubMed] [Google Scholar]
- 5. Hannan LM, Jacobs EJ, Thun MJ. The association between cigarette smoking and risk of colorectal cancer in a large prospective cohort from the United States. Cancer Epidemiol Biomarkers Prev 2009; 18: 3362–3367. [DOI] [PubMed] [Google Scholar]
- 6. Leufkens AM, Van Duijnhoven FJ, Siersema PD, et al. Cigarette smoking and colorectal cancer risk in the European Prospective Investigation into Cancer and Nutrition Study. Clin Gastroenterol Hepatol 2011; 9: 137–144. [DOI] [PubMed] [Google Scholar]
- 7. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans . Alcohol consumption and ethyl carbamate. IARC Monogr Eval Carcinog Risks Hum 2010; 96: 3–1383. [PMC free article] [PubMed] [Google Scholar]
- 8. Dai Z, Xu YC, Li N. Obesity and colorectal cancer risk: a meta‐analysis of cohort studies. World J Gastroenterol 2007; 13: 4199–4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ning Y, Wang L, Giovannucci EL. A quantitative analysis of body mass index and colorectal cancer: findings from 56 observational studies. Obes Rev 2010; 11: 19–30. [DOI] [PubMed] [Google Scholar]
- 10. van Duijnhoven FJ, Botma A, Winkels R, et al. Do lifestyle factors influence colorectal cancer risk in Lynch syndrome? Fam Cancer 2013; 12: 285–293. [DOI] [PubMed] [Google Scholar]
- 11. Bouvard V, Loomis D, Guyton KZ, et al. Carcinogenicity of consumption of red and processed meat. Lancet Oncol 2015; 16: 1599–1600. [DOI] [PubMed] [Google Scholar]
- 12. Rehm J, Room R, Monteiro M, et al. Alcohol as a risk factor for global burden of disease. Eur Addict Res 2003; 9: 157–164. [DOI] [PubMed] [Google Scholar]
- 13. Miguchi M, Hinoi T, Tanakaya K, et al. Alcohol consumption and early‐onset risk of colorectal cancer in Japanese patients with Lynch syndrome: a cross‐sectional study conducted by the Japanese Society for Cancer of the Colon and Rectum. Surg Today 2018; 48: 810–814. [DOI] [PubMed] [Google Scholar]
- 14. Stojic L, Mojas N, Cejka P, et al. Mismatch repair‐dependent G2 checkpoint induced by low doses of SN1 type methylating agents requires the ATR kinase. Genes Dev 2004; 18: 1331–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Toft NJ, Winton DJ, Kelly J, et al. Msh2 status modulates both apoptosis and mutation frequency in the murine small intestine. Proc Natl Acad Sci U S A 1999; 96: 3911–3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tomlinson I, Bodmer W. Selection, the mutation rate and cancer: ensuring that the tail does not wag the dog. Nat Med 1999; 5: 11–12. [DOI] [PubMed] [Google Scholar]
- 17. Cerretelli G, Ager A, Arends MJ, et al. Molecular pathology of Lynch syndrome. J Pathol 2020; 250: 518–531. [DOI] [PubMed] [Google Scholar]
- 18. Barker N, van Es JH, Kuipers J, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5 . Nature 2007; 449: 1003–1007. [DOI] [PubMed] [Google Scholar]
- 19. Claij N, te Riele H. Msh2 deficiency does not contribute to cisplatin resistance in mouse embryonic stem cells. Oncogene 2004; 23: 260–266. [DOI] [PubMed] [Google Scholar]
- 20. de Wind N, Dekker M, Berns A, et al. Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell 1995; 82: 321–330. [DOI] [PubMed] [Google Scholar]
- 21. Wojciechowicz K, Cantelli E, Van Gerwen B, et al. Temozolomide increases the number of mismatch repair‐deficient intestinal crypts and accelerates tumorigenesis in a mouse model of Lynch syndrome. Gastroenterology 2014; 147: 1064–1072.e5. [DOI] [PubMed] [Google Scholar]
- 22. Müller MF, Zhou Y, Adams DJ, et al. Effects of long‐term ethanol consumption and Aldh1b1 depletion on intestinal tumourigenesis in mice. J Pathol 2017; 241: 649–660. [DOI] [PubMed] [Google Scholar]
- 23. Dole VP, Gentry RT. Toward an analogue of alcoholism in mice: scale factors in the model. Proc Natl Acad Sci U S A 1984; 81: 3543–3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Holmes RS, Duley JA, Algar EM, et al. Biochemical and genetic studies on enzymes of alcohol metabolism: the mouse as a model organism for human studies. Alcohol Alcohol 1986; 21: 41–56. [PubMed] [Google Scholar]
- 25. Bankhead P, Loughrey MB, Fernández JA, et al. QuPath: open source software for digital pathology image analysis. Sci Rep 2017; 7: 16878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Köbel M, Piskorz AM, Lee S, et al. Optimized p53 immunohistochemistry is an accurate predictor of TP53 mutation in ovarian carcinoma. J Pathol Clin Res 2016; 2: 247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lakin ND, Jackson SP. Regulation of p53 in response to DNA damage. Oncogene 1999; 18: 7644–7655. [DOI] [PubMed] [Google Scholar]
- 28. Kloor M, Huth C, Voigt AY, et al. Prevalence of mismatch repair‐deficient crypt foci in Lynch syndrome: a pathological study. Lancet Oncol 2012; 13: 598–606. [DOI] [PubMed] [Google Scholar]
- 29. Shia J, Stadler ZK, Weiser MR, et al. Mismatch repair deficient‐crypts in non‐neoplastic colonic mucosa in Lynch syndrome: insights from an illustrative case. Fam Cancer 2015; 14: 61–68. [DOI] [PubMed] [Google Scholar]
- 30. Lugli A, Zlobec I, Minoo P, et al. Prognostic significance of the wnt signalling pathway molecules APC, β‐catenin and E‐cadherin in colorectal cancer: a tissue microarray‐based analysis. Histopathology 2007; 50: 453–464. [DOI] [PubMed] [Google Scholar]
- 31. Arnold A, Tronser M, Sers C, et al. The majority of β‐catenin mutations in colorectal cancer is homozygous. BMC Cancer 2020; 20: 1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gerdes J, Lemke H, Baisch H, et al. Cell cycle analysis of a cell proliferation‐associated human nuclear antigen defined by the monoclonal antibody Ki‐67. J Immunol 1984; 133: 1710–1715. [PubMed] [Google Scholar]
- 33. Johnston PG, O'Brien MJ, Dervan PA, et al. Immunohistochemical analysis of cell kinetic parameters in colonic adenocarcinomas, adenomas, and normal mucosa. Hum Pathol 1989; 20: 696–700. [DOI] [PubMed] [Google Scholar]
- 34. Balbo S, Brooks PJ. Implications of acetaldehyde‐derived DNA adducts for understanding alcohol‐related carcinogenesis. Adv Exp Med Biol 2015; 815: 71–88. [DOI] [PubMed] [Google Scholar]
- 35. Brooks PJ, Zakhari S. Acetaldehyde and the genome: beyond nuclear DNA adducts and carcinogenesis. Environ Mol Mutagen 2014; 55: 77–91. [DOI] [PubMed] [Google Scholar]
- 36. Seitz HK, Stickel F. Molecular mechanisms of alcohol‐mediated carcinogenesis. Nat Rev Cancer 2007; 7: 599–612. [DOI] [PubMed] [Google Scholar]
- 37. Kim H, D'Andrea AD. Regulation of DNA cross‐link repair by the Fanconi anemia/BRCA pathway. Genes Dev 2012; 26: 1393–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Langevin F, Crossan GP, Rosado IV, et al. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 2011; 475: 53–58. [DOI] [PubMed] [Google Scholar]
- 39. Seth S, Ager A, Arends MJ, et al. Lynch syndrome – cancer pathways, heterogeneity and immune escape. J Pathol 2018; 246: 129–133. [DOI] [PubMed] [Google Scholar]
- 40. Levine AJ. p53, the cellular gatekeeper for growth and division. Cell 1997; 88: 323–331. [DOI] [PubMed] [Google Scholar]
- 41. Williams AB, Schumacher B. p53 in the DNA‐damage‐repair process. Cold Spring Harb Perspect Med 2016; 6: a026070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hollenbach JP, Resch AM, Palakodeti D, et al. Loss of DNA mismatch repair imparts a selective advantage in Planarian adult stem cells. PLoS One 2011; 6: e21808. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Tumour distribution in ethanol‐treated induced Msh2‐LS tumour‐bearing mice
Figure S2. Representative images of skin sebaceous adenoma and endometrial invasive adenocarcinoma from ethanol‐treated induced Msh2‐LS mice
Figure S3. Immunohistochemical analysis of MSH2 protein expression in small intestinal mucosal epithelium of Msh2‐LS mice treated with either 20% ethanol in drinking water or standard drinking water
Figure S4. Immunohistochemical analysis of MSH2 protein expression in large and small intestinal mucosal epithelium of non‐induced Msh2‐LS control mice treated with either 20% ethanol in drinking water or standard drinking water
Figure S5. Immunohistochemical analysis of MSH6 protein expression in intestinal mucosal epithelium
Figure S6. Immunohistochemical analysis of MLH1 protein expression in intestinal mucosal epithelium
Figure S7. Immunohistochemical analysis of β‐catenin protein expression and localisation in large intestinal mucosal adenoma and adjacent normal epithelium from a positive‐control Apc Min mouse
Figure S8. Immunohistochemical analysis of Ki‐67 protein expression in small intestinal mucosa of Msh2‐LS mice treated with either 20% ethanol in drinking water or standard drinking water
Figure S9. Immunohistochemical analysis of γ‐H2AX protein expression in small intestinal mucosa of Msh2‐LS mice treated with either 20% ethanol in drinking water or standard drinking water
Figure S10. Representative images of immunohistochemical analysis of p53 protein expression in small intestinal mucosal epithelium of Msh2‐LS mice treated with either 20% ethanol in drinking water or standard water
Table S1. Clinical scoring system for monitoring mice for signs of intestinal tumour development