

Abstract
Introduction
Analysis of sequence data in high‐risk pedigrees is a powerful approach to detect rare predisposition variants.
Methods
Rare, shared candidate predisposition variants were identified from exome sequencing 19 Alzheimer's disease (AD)‐affected cousin pairs selected from high‐risk pedigrees. Variants were further prioritized by risk association in various external datasets. Candidate variants emerging from these analyses were tested for co‐segregation to additional affected relatives of the original sequenced pedigree members.
Results
AD‐affected high‐risk cousin pairs contained 564 shared rare variants. Eleven variants spanning 10 genes were prioritized in external datasets: rs201665195 (ABCA7), and rs28933981 (TTR) were previously implicated in AD pathology; rs141402160 (NOTCH3) and rs140914494 (NOTCH3) were previously reported; rs200290640 (PIDD1) and rs199752248 (PIDD1) were present in more than one cousin pair; rs61729902 (SNAP91), rs140129800 (COX6A2, AC026471), and rs191804178 (MUC16) were not present in a longevity cohort; and rs148294193 (PELI3) and rs147599881 (FCHO1) approached significance from analysis of AD‐related phenotypes. Three variants were validated via evidence of co‐segregation to additional relatives (PELI3, ABCA7, and SNAP91).
Discussion
These analyses support ABCA7 and TTR as AD risk genes, expand on previously reported NOTCH3 variant identification, and prioritize seven additional candidate variants.
Keywords: ABCA7, Alzheimer's disease, genetic analysis, high‐risk pedigree, NOTCH3, rare variant analysis, TTR, Utah Population Database, whole exome sequence
1. INTRODUCTION
Genetic variation significantly impacts Alzheimer's disease (AD) risk and is estimated to account for 53% of total trait variance. 1 To date, genome‐wide association studies (GWAS) for AD have identified common variants occurring in more than 30 genes. 1 , 2 Despite this progress, it is estimated that more than 40% of the genetic variance of AD remains uncharacterized. 3 While rare variation contributing to AD is difficult to detect via GWAS, it may be discoverable in pedigree‐based designs. It is well recognized that high‐risk pedigree studies are a powerful and efficient method for identification of rare predisposition variants and should be performed when these rare resources are available. 4 , 5 , 6 Pedigree‐based studies are ideal for rare variant identification because rare variants can occur at a higher‐than‐population rate among related affected individuals, thereby enhancing statistical power. 7 , 8 Additionally, pedigree analyses limit locus heterogeneity (i.e., different gene loci or gene loci interactions causing a similar phenotype) because the rare variants are inherited by a common ancestor with a common haplotype. Although familial locus heterogeneity may exist, the success rate at identifying causal variants in large pedigrees can be much higher than in clinical settings. 9 Pedigree‐based designs have been successfully applied to gene discovery for many phenotypes 10 , 11 , 12 including AD, where notable examples are PLD3, 13 NOTCH3, 14 and RAB10. 15 Here, a pedigree‐based analysis using exome sequences from 19 AD‐affected cousin pairs belonging to pedigrees with a statistical excess of AD mortality was performed to identify genetic variants associated with AD risk. Candidate variants arising from this analysis were then further prioritized in publicly available, large‐scale datasets to further evaluate their risk of AD. Prioritized variants were then tested for co‐segregation in additional previously sampled relatives of the index cousin pairs.
2. MATERIAL AND METHODS
2.1. Utah Population Database (UPDB)
The UPDB includes a genealogy of Utah, representing the founders in the mid‐1800s to their modern‐day descendants. It originated from three‐generation genealogy records 16 and is kept current with Utah vital statistics data, which includes approximately 50,000 largely unrelated founders of European descent. More than three million individuals with at least three generations of genealogy connecting to Utah founders in the UPDB were analyzed here. The UPDB has been linked to various phenotypic data including the Utah Cancer Registry from 1966 and Utah death certificates from 1904 to 2014. This unique combination of genealogy with phenotypes has contributed to the identification, recruitment, consent, and sampling of over 30,000 individuals in thousands of high‐risk pedigrees representing many disorders. Previous analyses of the UPDB genealogy have reported that founder effects are not present among the Utah population and indicate low inbreeding levels similar to other places in the United States. 17 , 18
RESEARCH IN CONTEXT
Systematic review: Despite recent advances in identifying common genetic variants associated with Alzheimer's disease (AD), it is estimated that more than 40% of the genetic variance of AD remains uncharacterized. High‐risk pedigree studies offer more statistical power for identification of rare predisposition variants, and they have previously been used to identify variants in PLD3, NOTCH3, and RAB10.
Interpretation: Pedigree‐based analyses using exome sequences from 19 AD‐affected cousin pairs with extended segregation assays were performed. These analyses add support to ABCA7 and TTR as AD risk genes. Furthermore, seven other candidate variants are prioritized in addition to NOTCH3 variants that were previously reported.
Future directions: Our analyses provide support for rare variant prioritization through pedigree‐based analyses. Additional inquiries into ABCA7 and TTR as AD risk genes are warranted. Moreover, these analyses suggest rare variants in NOTCH3, PIDD1, SNAP91, COX6A2, MUC16, PELI3, and FCHO1 may contribute to AD risk.
2.2. Identification of sampled high‐risk AD mortality pedigrees
Among the individuals with stored DNA from Utah pedigrees were 199 subjects whose death certificates indicated AD as a cause of death. These 199 individuals were related in 102 independent, descending pedigrees including between two and six sampled AD mortality cases. Each pedigree was tested for an excess of AD deaths among descendants as described elsewhere. 15 using all available genealogy and death certificate data. Twenty‐four high‐risk pedigrees including two to four sampled AD cases were identified. Of those pedigrees, 19 unrelated pedigrees were selected for this analysis that included at least one sampled AD‐affected approximate‐cousin pair, ranging in relationship from avuncular (n = 1; expected shared DNA = 25%) to third cousin (n = 1; expected shared DNA = 0.78%).
2.3. Exome sequencing of AD cousin pairs
Two AD‐affected cousins from each of the 19 high‐risk pedigrees were exome sequenced at the Huntsman Cancer Institute's Genomics Core Facility. The Agilent SureSelect XT Human All Exon + UTR (v5) capture kit was used to prepare the DNA library from 2 μg of DNA per sample. Paired‐end reads of up to 150 base pairs were sequenced on the Illumina HiSeq 2000 sequencer. BWA‐MEM 19 , 20 mapped raw reads to the human genome v37 (GRCh37) reference genome. The Genome Analysis Toolkit 3.5.0 (GATK) 21 software called variants using the Broad Institute's best practices guidelines. Variants were removed if they occurred outside the exon capture kit intended area of coverage, and the remaining variants were annotated with ANNOVAR 22 for their predicted pathogenicity.
Pathogenicity predictions were conducted in ANNOVAR using various algorithms because deleterious predictions made by multiple prediction methods are more likely to be reliable. 23 Table S1 in supporting information shows scores reported by SIFT, 24 , 25 Polyphen‐2, 26 LRT, 23 MutationTaster2, 27 FATHMM, 28 PROVEAN, 29 VEST3, 30 MetaSVM, MetaLR, M‐CAP, 31 REVEL, 32 CADD, 33 DANN, 34 EIGEN, 35 GenoCanyon, 36 and GERP++. 37 Prioritized variants were predicted to be deleterious by at least two of those functional prediction algorithms.
2.4. The Alzheimer's Disease Genetics Consortium Dataset
Some variant prioritization (described below) was based on comparisons to the Alzheimer's Disease Genetics Consortium (ADGC) dataset. ADGC contains imputed single‐nucleotide polymorphism (SNP) array data for 28,730 subjects (11,967 males and 16,760 females), including 10,486 AD cases and 10,168 healthy controls. Of the shared rare exonic variants in the cousin pairs, 291 were sequenced or imputed in the ADGC dataset.
2.5. The Knight cerebrospinal fluid dataset
The Knight Alzheimer's Disease Research Center at Washington University School of Medicine (Knight ADRC) cerebrospinal fluid (CSF) dataset was used for analyses of association between candidate variants and levels of AB42, Tau, or PTau. This cohort consisted of 3963 subjects (1895 males and 1675 females), including 1479 AD cases and 1370 healthy controls. All samples were genotyped using the Illumina 610 or OmniExpress chip. Of the shared rare variants in the cousin pairs, only 12 appeared in the Knight ADRC dataset. Additional imputation on the Knight ADRC dataset would likely lead to a higher false discovery rate because rare variant imputation is much less accurate than common variant imputation. 38 Therefore, those 12 variants were used only for additional variant prioritization on the rare variants identified in the high‐risk pedigrees.
2.6. Wellderly dataset
The Wellderly study 39 is an ongoing study that includes elderly people (age 80–105) who are cognitively healthy without medical interventions. Six hundred Wellderly individuals had their whole genomes sequenced using the Complete Genomics platform. These genomes were used to perform additional variant prioritization because functionally relevant rare genetic variants associated with AD should not be present in an elderly population that does not exhibit cognitive decline.
2.7. Assessing common genetic variants
Polygenic risk scores for each cousin were calculated and compared to ADGC cases and controls to ensure that the excess risk for AD was not caused by an excess of common disease‐associated variants (see Note S1 in supporting information). The AD polygenic risk scores calculated from Lambert, Ibrahim‐Verbaas, 2 and apolipoprotein E (APOE) genotype for each cousin are shown in Table S2 in supporting information. A Welch's two‐sample t‐test shows that the mean AD risk from common variants in the high‐risk cousin pairs is significantly less than the mean AD risk for ADGC cases (P‐value = 2.836 × 10−9; see Figure S1 in supporting information) and controls (P‐value = 9.486 × 10−4; see Figure S2 in supporting information). Therefore, the propensity of AD in these high‐risk pedigrees is likely caused by rare genetic variants that can be prioritized using the pipeline shown below.
2.8. Initial variant prioritization
Candidate variants were initially required to be present in both AD‐affected cousin pairs from at least 1 of the 19 high‐risk AD pedigrees. Further analysis in Ingenuity Variant Analysis software (QIAGEN, Inc.) ensured variants were rare by removing variants with a population minor allele frequency (MAF) greater than 0.01 in 1000 Genomes, 40 Exome Aggregation Consortium (ExAC) non‐Finnish European, 41 The Genome Aggregation Database (gnomAD), 41 and NHLBI GO Exome Sequencing Project (ESP), Seattle, WA (http://evs.gs.washington.edu/EVS/) [March 2018].
Ingenuity Variant Analysis then prioritized variants based on their predicted pathogenicity. Variants considered “Pathogenic,” “Likely Pathogenic,” or “Unknown” by the American College of Medical Genetics (ACMG), 42 or resulted in either a loss or gain of gene function by in silico functional prediction algorithms were prioritized.
2.9. Prioritization in external datasets
Four distinct strategies were used to further prioritize candidate variants in external datasets related to AD. Each strategy was evaluated independently of the others. Variants that met any one of these criteria were prioritized as candidate variants for AD. Variants were assumed to be likely causing the excess of AD mortality in the pedigrees if they were (1) a known AD risk variant, (2) a CSF biomarker of AD with a P‐value less than .1, (3) not present in the Wellderly dataset and more prevalent in ADGC AD cases than ADGC controls, or (4) observed in more than one AD‐affected cousin pair, as described below.
2.9.1. Known AD risk variant
A literature search of all shared rare variants was conducted in Ingenuity Variant Analysis to determine the extent to which these variants were previously implicated in AD pathology, indicating additional support from independent studies. Variants with publications supporting their association with AD were prioritized.
2.9.2. Increased AD risk in CSF dataset
Three linear regressions were conducted on the Knight ADRC CSF data using PLINK 43 : one for each of the phenotypes of interest (AB42, Tau, and PTau), with age, APOE status, sex, and two principal components 2 , 44 as covariates. The threshold for prioritizing candidate variants was P‐value less than .1 for any of the three phenotypes. The significance threshold was relaxed because variants had a low minor allele frequency and a small sample size.
2.9.3. AD risk gradient
Variants positively affecting AD mortality are expected to be more prevalent in diseased elderly cohorts than healthy elderly cohorts. The Wellderly dataset was used to ensure that prioritized variants were not present in a healthy longevity cohort. Furthermore, the MAF of the variant in ADGC controls needed to be less than or equal to the MAF in ADGC AD cases to ensure that the variant was more prevalent in AD cases than controls.
2.9.4. Multiple hit pedigrees
Variants that were identified in more than one AD‐affected cousin pair were also prioritized as candidates. This prioritization provided additional evidence for the candidate variant as a potential AD risk factor because of its prevalence in AD subjects from multiple high‐risk pedigrees. Because all variants analyzed in this study are rare variants (MAF < 0.01), it is not likely that the same rare variant would be present in two independent pedigrees and shared by both affected cousins by random chance.
2.10. Evidence for segregation with AD risk in additional affected sampled relatives
A set of 199 individuals previously sampled for Utah high‐risk disease studies whose Utah death certificate included AD as a cause of death and had at least three generations of genealogy linking to Utah founders, was assayed for 9 of the 11 variants. An assay was not available for PIDD1 rs200290640, and the assay for PIDD1 rs199752248 failed due to too many homologous regions in the genome to specify the right location. The AD cases from all affected cousin pairs were included, and all assays correctly identified the observed carriers for each of the nine candidate variants. Evidence of co‐segregation to additional affected relatives was evaluated with the RVsharing program. 45 The RVsharing calculates the probability of an observed configuration of carriers and affection status in a pedigree having occurred by chance transmission, assuming that the variant is rare (MAF < 0.01) and has entered the pedigree only once. The RVsharing program provides a test of co‐segregation (e.g., linkage) in which the strength of evidence against the null hypothesis of no co‐segregation between the disease and a variant is expressed as an exact probability (P‐value) for a given pedigree structure and disease configuration. Because the pedigrees were pre‐screened for a statistical excess of AD, rare variants that were shared among the related individuals likely contribute to the excess in AD cases. Institutional Review Board approval was in place for all reported analyses.
3. RESULTS
3.1. Variant prioritization
Initial variant prioritization of rare variants shared by at least one cousin pair identified 400 rare variants spanning 470 genes. Ingenuity Variant Analysis subsequently prioritized 382 variants in 447 genes that were pathogenic, likely pathogenic, uncertain significance, or associated with a gain or loss of gene function. Of those shared rare variants, 117 variants in 138 genes had a biological interaction with genes implicated in AD. The numbers of variants included after each analysis are shown in Figure 1, and each variant is listed in File S1 in supporting information. That list of 117 variants was then used by four independent prioritization screens to identify 11 rare variants spanning 10 genes with the strongest support for increasing AD risk in these high‐risk pedigrees (see Table 1). Four rare variants previously associated with increased AD risk were identified using a literature search in Ingenuity Variant Analysis. The Knight ADRC CSF dataset identified two additional variants (Note: of the 117 prioritized variants, only 11 appeared in the CSF dataset) that were associated with increased risk for AB42, Tau, or Ptau in CSF. Additionally, the AD Risk Gradient identified three variants that were not present in the Wellderly dataset and were more prevalent in ADGC cases than ADGC controls. Finally, two variants were present in two independent high‐risk AD pedigrees.
FIGURE 1.
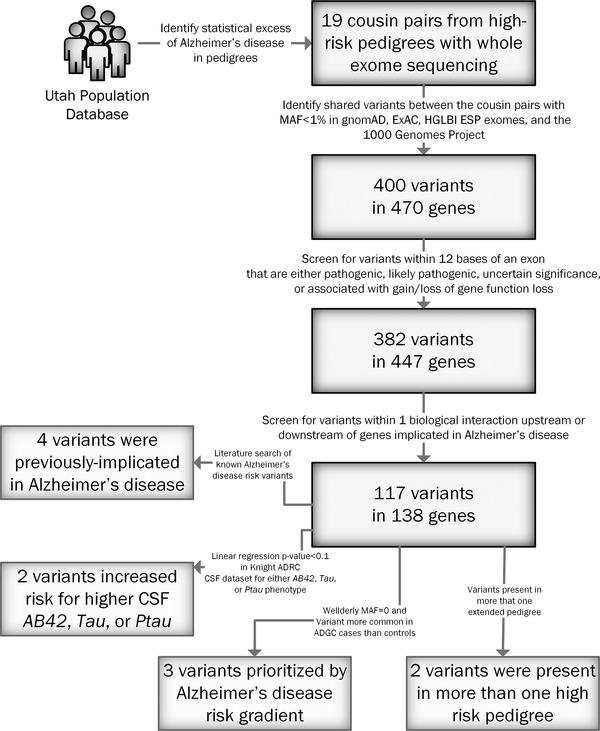
Flowchart depicting variant prioritization. The number of rare variants and genes remaining at each level are shown. ADRC, Alzheimer's Disease Research Center; CSF, cerebrospinal fluid; MAF, minor allele frequency.
TABLE 1.
Prioritized variants
| Accession number | Gene | HGVS variant | Impact | Final prioritization |
|---|---|---|---|---|
| rs201665195 | ABCA7 | NM_019112.3:c.302T > G | Missense | Known AD risk variant 46 , 47 |
| rs28933981 | TTR | NM_000371.3:c.416C > T | Missense | Known AD risk variant 48 |
| rs141402160 | NOTCH3 | NM_000435.3:c.743G > C | Missense | Known AD risk variant (reported using these pedigrees in Patel et al. 49 ) |
| rs140914494 | NOTCH3 | NM_000435:c.593C > T | Missense | Known AD risk variant (reported using these pedigrees in Patel et al. 49 ) |
| rs148294193 | PELI3 | NM_001243135.1:c.115G > C | Missense | CSF biomarker P‐value = .0553 |
| rs147599881 | FCHO1 | NM_001161357.1:c.557G > A | Missense | CSF biomarker P‐value = .0624 |
| rs61729902 | SNAP91 | NM_00124279.1:c.2113C > T | Missense | AD risk gradient |
| rs140129800 | COX6A2 | NM_005205.3:c.34T > G | Missense | AD risk gradient |
| rs191804178 | MUC16 | NM_024690.2:c.10900C > T | Missense | AD risk gradient |
| rs200290640 | PIDD1 | NM_145886.3:c.2044C > T | Missense | Prioritized in multiple pedigrees |
| rs199752248 | PIDD1 | NM_145886.3:c.2042‐2A > G | Splicing (intronic) | Prioritized in multiple pedigrees |
Notes: These variants are most likely to affect AD pathology in the high‐risk pedigrees. The accession number, affected gene, the Human Genome Variation Society (HGVS) variant annotation, impact on translation, and the final prioritization option that identified the variant are shown.
Abbreviations: AD, Alzheimer's disease; CSF, cerebrospinal fluid.
3.2. Evidence for co‐segregation with AD risk in additional affected sampled relatives
Five candidate variants assayed for segregation evidence had no additional carriers beyond the original cousin pair, including: MUC16 rs191804178, NOTCH3 rs141402160, NOTCH3 rs140914494, FCHO1 rs147599881, and COX6A2 rs140129800. An additional carrier was identified among the 199 assayed AD cases for TTR rs28933981, but the additional carrier was not related to the original AD‐affected cousin pair sharing the variant. Additional related AD‐affected carriers were observed among the 199 assayed cases for four of the nine assayed candidate variants. The other five newly identified AD case carriers were not related to any of the other carriers or to each other. Five AD‐affected carriers were observed for PELI3 rs148294193, including the original cousin pair. One of the newly identified case carriers was a cousin (and avuncular) to the original cousin pair (rare variant sharing P‐value = .006), and the other two newly identified case carriers were unrelated to all other carriers. Four AD‐affected carriers were observed for ABCA7 rs201665195 including the original cousin pair. One of the newly identified case carriers was a sibling to one of the original cousin carriers (rare variant sharing P‐value = .027), and one carrier was not related to any other carriers. Three AD‐affected carriers were observed for SNAP91 rs61729902, including the original cousin pair. The newly identified case carrier was a sibling of one of the original cousins (rare variant sharing P‐value = .027).
4. DISCUSSION
Analysis of exomes from 19 AD‐affected cousin pairs identified 400 shared rare candidate AD predisposition variants. Initial prioritization with Ingenuity Variant Analysis on likely pathogenicity and biological context reduced this list to 117 rare variants occurring in 138 genes. Further prioritization in one of four independent datasets, further prioritized 11 variants in 10 genes (Table 1). Four of these variants in three genes (ABCA7, TTR, and NOTCH3) represent replications of previous associations to AD, while the remaining eight variants are better classified as candidate variants still requiring validation, although each exhibited some level of replication through one of the four independent prioritization strategies. Finally, variants in ABCA7, SNAP91, and PELI3 showed significant evidence of segregation to other related AD cases in the high‐risk pedigrees in which they were identified.
Four identified rare variants were previously reported in the literature as associated with increasing AD risk (known AD risk variants). The first variant, NM_019112.3:c.302T > G (rs201665195), is found in the ABCA7 gene on chromosome 19. Unfortunately, this variant was not included in the ADGC validation datasets, so it could not be independently validated beyond the cousin pairs. Le Guennec et al. 46 observed this variant, along with other rare ABCA7 variants, in AD cases and confirmed that rare, loss of function, and predicted damaging missense variants in ABCA7 are more common in patients with AD. Vardarajan et al. 47 also identified this rare variant in two of their three late‐onset AD (LOAD) cohorts, and this variant was not seen in unaffected individuals. The cousin pairs analysis, and evidence for segregation, adds additional support to ABCA7 as a gene that impacts AD.
Variant NM_000371.3:c.416C > T (rs28933981) is located in the TTR gene on chromosome 18. Although this variant was sequenced in the ADGC validation datasets, it did not pass quality control thresholds, so it could not be used for validation. Sassi et al. 48 observed this variant in 6 out of 332 AD cases (1.8% of cases), and this variant had a strong effect size (odds ratio = 6.19, 95% confidence interval = 1.099–63.091). It was also observed in two of their 676 cognitive‐normal control samples (0.30% of controls). TTR is known to be involved in amyloid beta (Aβ) catabolism and has no homologous proteins, suggesting that subtle changes to this protein could have strong functional implications. 48 This analysis of high‐risk cousin pairs adds additional support to TTR as a gene that impacts AD.
Patel et al. 49 reported missense mutations NM_000435.3:c.743G > C (rs141402160) and NM_000435:c.593C > T (rs140914494) in NOTCH3 using the pedigrees from this analysis combined with other population resources. However, these variants were not sequenced in the validation datasets. NOTCH3 plays a key role in neural development and is known to cause cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). The same region of NOTCH3 was also linked to AD in a Turkish family. 14 . NOTCH3 is known to bind PSEN1 50 , 51 and PSEN2. 50 Furthermore, NOTCH3 had a P‐value of 1 × 10−4 from the gene‐based testing of AD risk in the ADGC dataset, implying that multiple variants in the gene may play a role in AD risk.
Two variants approached significance for increased AD risk in the Knight ADRC CSF dataset. A missense mutation in PELI3 (NM_001243135.1:c.115G > C; rs148294193) has a suggestive positive influence on AD risk (P‐value = .0553). PELI3 binds UBC 52 and APP. 53 UBC is significantly downregulated in AD brains suggesting that decreased UBC function may be important in AD pathogenesis including increased neuronal death and non‐regulated APP production. 54 APP is a well‐studied AD risk gene, and many mutations in this gene are known to cause early‐onset AD. APP is cleaved into Aβ peptides, which are a major component of the amyloid plaques deposited in the brains of AD patients. 55 PELI3 encodes a scaffold protein and an intermediate signaling protein in the innate immune response pathway. Segregation of the PELI3 variant to an additional AD‐affected relative in the original high‐risk pedigree was observed.
A missense mutation NM_001161357.1:c.557G > A (rs147599881) in FCHO1 has an AD risk P‐value of .0624 in the CSF with a positive direction. FCHO1 also binds UBC 56 and APP. 53 FCHO1 is involved in vesicle‐mediated transportation and clathrin‐mediated endocytosis.
Three variants were not present in the Wellderly dataset and were more prevalent in ADGC cases than ADGC controls. Missense variant NM_00124279.1:c.2113C > T (rs61729902) is located in SNAP91, which encodes a protein that binds UBC. 57 SNAP91 is involved in vesicle‐mediated transportation and clathrin‐mediated endocytosis. SNAP91 is a paralog of PICALM, a known top 10 AD risk gene, 58 and they both have similarities between their functions of clathrin‐mediated endocytosis. The SNAP91 variant was observed in an additional AD case in the original high‐risk pedigree sequenced.
Another missense mutation, NM_005205.3:c.34T > G (rs140129800), was found in the COX6A2 gene, which encodes a protein that binds APP. 53 COX6A2 is a terminal enzyme in the mitochondrial respiratory chain and is part of the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway for AD. 59 , 60 , 61
Missense mutation NM_024690.2:c.10900C > T (rs191804178) in MUC16 affects binding of UBC, 62 and MUC16 forms a protective mucous barrier on the apical surfaces of the epithelia. Mutations in MUC16 are associated with ovarian cancer, endometriosis, Pseudo‐Meigs syndrome, serous cystadenocarcinoma, and bronchogenic cysts. MUC16 has not previously been implicated in AD.
4.1. Variants observed in more than one AD‐affected cousin pair
Three variants were observed to be shared by both members of the AD‐affected cousin pair in more than one high‐risk pedigree. Missense variant NM_145886.3:c.2044C > T (rs200290640) and splice site variant NM_145886.3:c.2042‐2A > G (rs199752248) both affect the PIDD1 gene, which is shown to bind APOE ε4. 63 Neither of these variants were sequenced in the ADGC validation datasets. PIDD1 contains a death domain, interacts with other death domain proteins, and is suggested to be an effector of p53‐dependent apoptosis. Mutations in PIDD1 are associated with poikiloderma with neutropenia (PN) disorder that affects the skin and the immune system.
4.2. Strengths and limitations
Rare variant‐sharing in AD‐affected relatives who are members of validated high‐risk pedigrees is central to prioritize candidate variants. Similar approaches capitalizing on shared genetics between related individuals in high‐risk pedigrees have been used successfully in Utah for decades to identify rare variants contributing to common diseases, 10 , 64 , 65 as well as more recent adaptations. 12 , 66 This study design is limited by a relatively small available sample size (19 pedigrees with 38 index cases), the absence of other ethnicities besides Whites, and the fact that AD phenotyping was based solely on death certificate data. However, the approach raises statistical power by increasing the relative allele frequencies of rare variants, which allows a single pedigree in the sample set to identify rare candidate variants, a key advantage in the presence of locus heterogeneity. Replication in external datasets can be difficult to achieve, given the rare nature of such variants. However, the absence of these limitations in external dataset validations (i.e., if all variants were present in ADGC, Knight ADRC, or the Wellderly dataset) would likely lead to more candidate variants identified through this approach. The prioritization criteria were very conservative and report only the most supported variants that likely affect AD within these high‐risk pedigrees. Additional variants that may affect AD within these pedigrees may have been de‐prioritized because of the stringent nature of the analysis. All genetic variants identified at each prioritization level are reported for future research to assess the relative support of each variant. The most supported variants were also assessed for co‐segregation using a small number of additional AD‐affected relatives with the AD phenotype within the original high‐risk pedigrees. Although the UPDB population genealogy data include the possibility of undocumented relationships among subjects and inadequate phenotyping strategies, previous research shows that founder effects and inbreeding are no greater in the UPDB than in the general population, 17 , 18 and the UPDB has been used extensively in previous disease studies.
4.3. Conclusion
The presence of rare variants identified here may explain the prevalence of AD mortality in 19 of the 36 AD‐affected individuals from high‐risk pedigrees (see Table S2). The excess AD mortality observed in the remaining individuals might be due to complex interactions, heterogeneity in the pedigree, misdiagnosis of AD, non‐coding variants, or variants that were removed due to stringent prioritization criteria. Because the initial prioritization included only variants in genes known or predicted to affect AD pathology, the analysis is dependent on current understanding of AD pathology and may not encompass all disease‐causing variants. However, the purpose of this study was to identify highly supported candidate variants associated with AD risk. To that end, these analyses provide additional support to previous studies that implicate ABCA7 and TTR with AD mortality. Because many known AD risk variants were prioritized in these analyses, other prioritized variants also likely affect AD mortality, and all 11 variants spanning 9 genes should be prioritized in future analyses. These outcomes indicate that a high‐risk pedigree approach can achieve sufficient power to detect rare variants, particularly when coupled with external datasets that contain meaningful data about disease risk.
Partial support for all data sets within the Utah Population Database (UPDB) was provided by Huntsman Cancer Institute, University of Utah and the Huntsman Cancer Institute's Cancer Center Support grant, P30 CA42014 from National Cancer Institute. LACA receives partial support from the Huntsman Cancer Institute's Cancer Center Support grant, P30 CA42014 from National Cancer Institute.
Acknowledgment is made to the donors of Alzheimer's Disease Research, a program of the BrightFocus Foundation for support of this research through grant #A2020118F (PI: Miller).
This study is part of the NHLBI Grand Opportunity Exome Sequencing Project (GO‐ESP). Funding for GO‐ESP was provided by NHLBI grants RC2 HL103010 (HeartGO), RC2 HL102923 (LungGO), and RC2 HL102924 (WHISP). The exome sequencing was performed through NHLBI grants RC2 HL102925 (BroadGO) and RC2 HL102926 (SeattleGO). HeartGO gratefully acknowledges the following groups and individuals who provided biological samples or data for this study. DNA samples and phenotypic data were obtained from the following studies supported by the NHLBI: the Atherosclerosis Risk in Communities (ARIC) study, the Coronary Artery Risk Development in Young Adults (CARDIA) study, Cardiovascular Health Study (CHS), the Framingham Heart Study (FHS), the Jackson Heart Study (JHS), and the Multi‐Ethnic Study of Atherosclerosis (MESA).
Data from Alzheimer's Disease Genetics Consortium (ADGC) were appropriately downloaded from dbGaP (accession: phs000372.v1.p1). We acknowledge the contributions of the members of the ADGC listed in Appendix: Alzheimer's Disease Genetics Consortium Collaborators.
Supporting information
Supporting Information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
We appreciate the contributions of Brigham Young University in supporting this research. This research is supported by RF1AG054052 (PI: Kauwe) and U01AG052411 (PI: Goate).
Alzheimer's Disease Genetics Consortium Collaborators
The members of the Alzheimer's Disease Genetics Consortium (ADGC) are: Marilyn S. Albert1, Roger L. Albin2‐4, Liana G. Apostolova5, Steven E. Arnold6, Clinton T. Baldwin7, Robert Barber8, Michael M. Barmada9, Lisa L. Barnes10, 11, Thomas G. Beach12, Gary W. Beecham13, 14, Duane Beekly15, David A. Bennett10, 16, Eileen H. Bigio17, Thomas D. Bird18, Deborah Blacker19,20, Bradley F. Boeve21, James D. Bowen22, Adam Boxer23, James R. Burke24, Joseph D. Buxbaum25, 26, 27, Nigel J. Cairns28, Laura B. Cantwell29, Chuanhai Cao30, Chris S. Carlson31, Regina M. Carney13, Minerva M. Carrasquillo33, Steven L. Carroll34, Helena C. Chui35, David G. Clark36, Jason Corneveaux37, Paul K. Crane38, David H. Cribbs39, Elizabeth A. Crocco40, Carlos Cruchaga41, Philip L. De Jager42,43, Charles DeCarli44, Steven T. DeKosky45, F. Yesim Demirci9, Malcolm Dick46, Dennis W. Dickson33, Ranjan Duara47, Nilufer Ertekin‐Taner33,48, Denis Evans49, Kelley M. Faber50, Kenneth B. Fallon34, Martin R. Farlow51, Lindsay A. Farrer7,52,76,77,83, Steven Ferris53, Tatiana M. Foroud50, Matthew P. Frosch54, Douglas R. Galasko55, Mary Ganguli56, Marla Gearing57,58, Daniel H. Geschwind59, Bernardino Ghetti60, John R. Gilbert13,14, Sid Gilman2, Jonathan D. Glass61, Alison M. Goate41, Neill R. Graff‐Radford33,48, Robert C. Green62, John H. Growdon63, Jonathan L. Haines64, 65, Hakon Hakonarson66, Kara L. Hamilton‐Nelson13, Ronald L. Hamilton67, John Hardy68, Lindy E. Harrell36, Elizabeth Head69, Lawrence S. Honig70, Matthew J. Huentelman37, Christine M. Hulette71, Bradley T. Hyman63, Gail P. Jarvik72,73, Gregory A. Jicha74, Lee‐Way Jin75, Gyungah Jun7,76,77, M. Ilyas Kamboh9,78, Anna Karydas23, John S.K. Kauwe79, Jeffrey A. Kaye80,81, Ronald Kim82, Edward H. Koo55, Neil W. Kowall83,84, Joel H. Kramer85, Patricia Kramer80,86, Walter A. Kukull87, Frank M. LaFerla88, James J. Lah61, Eric B. Larson38,89, James B. Leverenz90, Allan I. Levey61, Ge Li91, Andrew P. Lieberman92, Chiao‐Feng Lin29, Oscar L. Lopez78, Kathryn L. Lunetta76, Constantine G. Lyketsos93, Wendy J. Mack94, Daniel C. Marson36, Eden R. Martin13,14, Frank Martiniuk95, Deborah C. Mash96, Eliezer Masliah55,97, Richard Mayeux70, 109, 110, Wayne C. McCormick38, Susan M. McCurry98, Andrew N. McDavid31, Ann C. McKee83,84, Marsel Mesulam99, Bruce L. Miller23, Carol A. Miller100, Joshua W. Miller75, Thomas J. Montine90, John C. Morris28, 101, Jill R. Murrell50, 60, Amanda J. Myers40, Adam C. Naj13, John M. Olichney44, Vernon S. Pankratz102, Joseph E. Parisi103,104, Margaret A. Pericak‐Vance13, 14, Elaine Peskind91, Ronald C. Petersen21, Aimee Pierce39, Wayne W. Poon46, Huntington Potter30, Joseph F. Quinn80, Ashok Raj30, Murray Raskind91, Eric M. Reiman37,105‐107, Barry Reisberg53,108, Christiane Reitz70,109,110, John M. Ringman5, Erik D. Roberson36, Ekaterina Rogaeva111, Howard J. Rosen23, Roger N. Rosenberg112, Mary Sano26, Andrew J. Saykin50,113, Gerard D. Schellenberg29, Julie A. Schneider10,114, Lon S. Schneider35,115, William W. Seeley23, Amanda G. Smith30, Joshua A. Sonnen90, Salvatore Spina60, Peter St George‐Hyslop111,116, Robert A. Stern83, Rudolph E. Tanzi63, John Q. Trojanowski29, Juan C. Troncoso117, Debby W. Tsuang91, Otto Valladares29, Vivianna M. Van Deerlin29, Linda J. Van Eldik118, Badri N. Vardarajan7, Harry V. Vinters5,119, Jean Paul Vonsattel120, Li‐San Wang29, Sandra Weintraub99, Kathleen A. Welsh‐Bohmer24, 121, Jennifer Williamson70, Randall L. Woltjer122, Clinton B. Wright123, Steven G. Younkin33, Chang‐En Yu38, Lei Yu10
1Department of Neurology, Johns Hopkins University, Baltimore, Maryland; 2Department of Neurology, University of Michigan, Ann Arbor, Michigan; 3Geriatric Research, Education and Clinical Center (GRECC), VA Ann Arbor Healthcare System (VAAAHS), Ann Arbor, Michigan; 4Michigan Alzheimer Disease Center, Ann Arbor, Michigan; 5Department of Neurology, University of California Los Angeles, Los Angeles, California; 6Department of Psychiatry, University of Pennsylvania Perelman School of Medicine, Philadelphia, Pennsylvania; 7Department of Medicine (Genetics Program), Boston University, Boston, Massachusetts; 8Department of Pharmacology and Neuroscience, University of North Texas Health Science Center, Fort Worth, Texas; 9Department of Human Genetics, University of Pittsburgh, Pittsburgh, Pennsylvania; 10Department of Neurological Sciences, Rush University Medical Center, Chicago, Illinois; 11Department of Behavioral Sciences, Rush University Medical Center, Chicago, Illinois; 12Civin Laboratory for Neuropathology, Banner Sun Health Research Institute, Phoenix, Arizona; 13The John P. Hussman Institute for Human Genomics, University of Miami, Miami, Florida; 14Dr. John T. Macdonald Foundation Department of Human Genetics, University of Miami, Miami, Florida; 15National Alzheimer's Coordinating Center, University of Washington, Seattle, Washington; 16Rush Alzheimer's Disease Center, Rush University Medical Center, Chicago, Illinois; 17Department of Pathology, Northwestern University, Chicago, Illinois; 18Department of Neurology, University of Washington, Seattle, Washington; 19Department of Epidemiology, Harvard School of Public Health, Boston, Massachusetts; 20Department of Psychiatry, Massachusetts General Hospital/Harvard Medical School, Boston, Massachusetts; 21Department of Neurology, Mayo Clinic, Rochester, Minnesota; 22Swedish Medical Center, Seattle, Washington; 23Department of Neurology, University of California San Francisco, San Francisco, California;, 24Department of Medicine, Duke University, Durham, North Carolina; 25Department of Neuroscience, Mount Sinai School of Medicine, New York, New York; 26Department of Psychiatry, Mount Sinai School of Medicine, New York, New York; 27Departments of Genetics and Genomic Sciences, Mount Sinai School of Medicine, New York, New York; 28Department of Pathology and Immunology, Washington University, St. Louis, Missouri; 29Department of Pathology and Laboratory Medicine, University of Pennsylvania Perelman School of Medicine, Philadelphia, Pennsylvania; 30USF Health Byrd Alzheimer's Institute, University of South Florida, Tampa, Florida; 31Fred Hutchinson Cancer Research Center, Seattle, Washington; 32Department of Psychiatry, Vanderbilt University, Nashville, Tennessee; 33Department of Neuroscience, Mayo Clinic, Jacksonville, Florida; 34Department of Pathology, University of Alabama at Birmingham, Birmingham, Alabama; 35Department of Neurology, University of Southern California, Los Angeles, California; 36Department of Neurology, University of Alabama at Birmingham, Birmingham, Alabama; 37Neurogenomics Division, Translational Genomics Research Institute, Phoenix, Arizona; 38Department of Medicine, University of Washington, Seattle, Washington; 39Department of Neurology, University of California Irvine, Irvine, California; 40Department of Psychiatry and Behavioral Sciences, Miller School of Medicine, University of Miami, Miami, Florida; 41Department of Psychiatry and Hope Center Program on Protein Aggregation and Neurodegeneration, Washington University School of Medicine, St. Louis, Missouri, 42Program in Translational NeuroPsychiatric Genomics, Institute for the Neurosciences, Department of Neurology & Psychiatry, Brigham and Women's Hospital and Harvard Medical School, Boston, Massachusetts; 43Program in Medical and Population Genetics, Broad Institute, Cambridge, Massachusetts; 44Department of Neurology, University of California Davis, Sacramento, California; 45University of Virginia School of Medicine, Charlottesville, Virginia; 46Institute for Memory Impairments and Neurological Disorders, University of California Irvine, Irvine, California; 47Wien Center for Alzheimer's Disease and Memory Disorders, Mount Sinai Medical Center, Miami Beach, Florida; 48Department of Neurology, Mayo Clinic, Jacksonville, Florida; 49Rush Institute for Healthy Aging, Department of Internal Medicine, Rush University Medical Center, Chicago, Illinois; 50Department of Medical and Molecular Genetics, Indiana University, Indianapolis, Indiana; 51Department of Neurology, Indiana University, Indianapolis, Indiana; 52Department of Epidemiology, Boston University, Boston, Massachusetts; 53Department of Psychiatry, New York University, New York, New York; 54C.S. Kubik Laboratory for Neuropathology, Massachusetts General Hospital, Charlestown, Massachusetts; 55Department of Neurosciences, University of California San Diego, La Jolla, California; 56Department of Psychiatry, University of Pittsburgh, Pittsburgh, Pennsylvania; 57Department of Pathology and Laboratory Medicine, Emory University, Atlanta, Georgia; 58Emory Alzheimer's Disease Center, Emory University, Atlanta, Georgia; 59Neurogenetics Program, University of California Los Angeles, Los Angeles, California; 60Department of Pathology and Laboratory Medicine, Indiana University, Indianapolis, Indiana; 61Department of Neurology, Emory University, Atlanta, Georgia; 62Division of Genetics, Department of Medicine and Partners Center for Personalized Genetic Medicine, Brigham and Women's Hospital and Harvard Medical School, Boston, Massachusetts; 63Department of Neurology, Massachusetts General Hospital/Harvard Medical School, Boston, Massachusetts; 64Department of Molecular Physiology and Biophysics, Vanderbilt University, Nashville, Tennessee; 65Vanderbilt Center for Human Genetics Research, Vanderbilt University, Nashville, Tennessee; 66Center for Applied Genomics, Children's Hospital of Philadelphia, Philadelphia, Pennsylvania; 67Department of Pathology (Neuropathology), University of Pittsburgh, Pittsburgh, Pennsylvania; 68Institute of Neurology, University College London, Queen Square, London; 69Sanders‐Brown Center on Aging, Department of Molecular and Biomedical Pharmacology, University of Kentucky, Lexington, Kentucky; 70Taub Institute on Alzheimer's Disease and the Aging Brain, Department of Neurology, Columbia University, New York, New York; 71Department of Pathology, Duke University, Durham, North Carolina; 72Department of Genome Sciences, University of Washington, Seattle, Washington; 73Department of Medicine (Medical Genetics), University of Washington, Seattle, Washington; 74Sanders‐Brown Center on Aging, Department Neurology, University of Kentucky, Lexington, Kentucky; 75Department of Pathology and Laboratory Medicine, University of California Davis, Sacramento, California; 76Department of Biostatistics, Boston University, Boston, Massachusetts; 77Department of Ophthalmology, Boston University, Boston, Massachusetts; 78University of Pittsburgh Alzheimer's Disease Research Center, Pittsburgh, Pennsylvania; 79Department of Biology, Brigham Young University, Provo, Utah; 80Department of Neurology, Oregon Health & Science University, Portland, Oregon; 81Department of Neurology, Portland Veterans Affairs Medical Center, Portland, Oregon; 82Department of Pathology and Laboratory Medicine, University of California Irvine, Irvine, California; 83Department of Neurology, Boston University, Boston, Massachusetts; 84Department of Pathology, Boston University, Boston, Massachusetts; 85Department of Neuropsychology, University of California San Francisco, San Francisco, California; 86Department of Molecular & Medical Genetics, Oregon Health & Science University, Portland, Oregon; 87Department of Epidemiology, University of Washington, Seattle, Washington; 88Department of Neurobiology and Behavior, University of California Irvine, Irvine, California; 89Group Health Research Institute, Group Health, Seattle, Washington; 90Department of Pathology, University of Washington, Seattle, Washington; 91Department of Psychiatry and Behavioral Sciences, University of Washington, Seattle, Washington; 92Department of Pathology, University of Michigan, Ann Arbor, Michigan; 93Department of Psychiatry, Johns Hopkins University, Baltimore, Maryland; 94Department of Preventive Medicine, University of Southern California, Los Angeles, California; 95Department of Medicine ‐ Pulmonary, New York University, New York, New York; 96Department of Neurology, University of Miami, Miami, Florida; 97Department of Pathology, University of California San Diego, La Jolla, California; 98School of Nursing Northwest Research Group on Aging, University of Washington, Seattle, Washington; 99Cognitive Neurology and Alzheimer's Disease Center, Northwestern University, Chicago, Illinois; 100Department of Pathology, University of Southern California, Los Angeles, California; 101Department of Neurology, Washington University, St. Louis, Missouri; 102Department of Biostatistics, Mayo Clinic, Rochester, Minnesota; 103Department of Anatomic Pathology, Mayo Clinic, Rochester, Minnesota; 104Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, Minnesota; 105Arizona Alzheimer's Consortium, Phoenix, Arizona; 106Department of Psychiatry, University of Arizona, Phoenix, Arizona; 107Banner Alzheimer's Institute, Phoenix, Arizona; 108Alzheimer's Disease Center, New York University, New York, New York; 109Gertrude H. Sergievsky Center, Columbia University, New York, New York; 110Department of Neurology, Columbia University, New York, New York; 111Tanz Centre for Research in Neurodegenerative Disease, University of Toronto, Toronto, Ontario; 112Department of Neurology, University of Texas Southwestern, Dallas, Texas; 113Department of Radiology and Imaging Sciences, Indiana University, Indianapolis, Indiana; 114Department of Pathology (Neuropathology), Rush University Medical Center, Chicago, Illinois; 115Department of Psychiatry, University of Southern California, Los Angeles, California; 116Cambridge Institute for Medical Research and Department of Clinical Neurosciences, University of Cambridge, Cambridge; 117Department of Pathology, Johns Hopkins University, Baltimore, Maryland; 118Sanders‐Brown Center on Aging, Department of Anatomy and Neurobiology, University of Kentucky, Lexington, Kentucky; 119Department of Pathology & Laboratory Medicine, University of California Los Angeles, Los Angeles, California; 120Taub Institute on Alzheimer's Disease and the Aging Brain, Department of Pathology, Columbia University, New York, New York; 121Department of Psychiatry & Behavioral Sciences, Duke University, Durham, North Carolina; 122Department of Pathology, Oregon Health & Science University, Portland, Oregon; 123Evelyn F. McKnight Brain Institute, Department of Neurology, Miller School of Medicine, University of Miami, Miami, Florida
Teerlink CC, Miller JB, Vance EL, et al. Analysis of high‐risk pedigrees identifies 11 candidate variants for Alzheimer's disease. Alzheimer's Dement. 2022;18:307–317. 10.1002/alz.12397
Craig C. Teerlink and Justin B. Miller wish it to be known that, in their opinion, the first two authors should be regarded as co‐first authors.
Lisa A. Cannon‐Albright and John S.K. Kauwe wish it to be known that, in their opinion, the last two authors should be regarded as co‐last authors.
Correction added on June 29, 2021, after first online publication: Article title was updated.
REFERENCES
- 1. Nacmias B, Bagnoli S, Piaceri I, Sorbi S. Genetic heterogeneity of Alzheimer's disease: embracing research partnerships. J Alzheimer Dis. 2018;62:903‐911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lambert JC, Ibrahim‐Verbaas CA, et al. Meta‐analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013;45:1452‐1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ridge PG, Hoyt KB, Boehme K, et al. Assessment of the genetic variance of late‐onset Alzheimer's disease. Neurobiol Aging. 2016;41:200. e13‐e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747‐753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wijsman EM. The role of large pedigrees in an era of high‐throughput sequencing. Hum Genet. 2012;131:1555‐1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ott J, Wang J, Leal SM. Genetic linkage analysis in the age of whole‐genome sequencing. Nat Rev Genet. 2015;16:275‐284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Clerget‐Darpoux F, Elston RC. Are linkage analysis and the collection of family data dead? Prospects for family studies in the age of genome‐wide association. Hum Hered. 2007;64:91‐96. [DOI] [PubMed] [Google Scholar]
- 8. Ott J, Kamatani Y, Lathrop M. Family‐based designs for genome‐wide association studies. Nat Rev Genet. 2011;12:465‐474. [DOI] [PubMed] [Google Scholar]
- 9. Rehman AU, Santos‐Cortez RL, Drummond MC, et al. Challenges and solutions for gene identification in the presence of familial locus heterogeneity. Eur J Hum Genet. 2015;23:1207‐1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Miki Y, Swensen J, Shattuck‐Eidens D, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science (New York, NY). 1994;266:66. [DOI] [PubMed] [Google Scholar]
- 11. Wooster R, Neuhausen SL, Mangion J, et al. Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12‐13. Science (New York, NY). 1994;265:2088‐2090. [DOI] [PubMed] [Google Scholar]
- 12. Teerlink CC, Huff C, Stevens J, et al. A nonsynonymous variant in the GOLM1 gene in cutaneous malignant melanoma. J Natl Cancer Inst. 2018;110:1380‐1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cruchaga C, Karch CM, Jin SC, et al. Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer's disease. Nature. 2014;505:550‐554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guerreiro RJ, Lohmann E, Kinsella E, et al. Exome sequencing reveals an unexpected genetic cause of disease: nOTCH3 mutation in a Turkish family with Alzheimer's disease. Neurobiol Aging. 2012;33:1008.e17‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ridge PG, Karch CM, Hsu S, et al. Linkage, whole genome sequence, and biological data implicate variants in RAB10 in Alzheimer's disease resilience. Genome Med. 2017;9:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Skolnick M. The Utah genealogical database: a resource for genetic epidemiology. In: Cairns J., Lyon J. L., Skolnick M. (eds.), Banbury Report No. 4: Cancer Incidence in Defined Populations, pp. 285‐297. New York: Cold Spring Harbor Laboratory Press, 1980.
- 17. Jorde LB. Inbreeding in the Utah Mormons: an evaluation of estimates based on pedigrees, isonymy, and migration matrices. Ann Hum Genet. 1989;53:339‐355. [DOI] [PubMed] [Google Scholar]
- 18. O'Brien E, Rogers AR, Beesley J, Jorde LB. Genetic structure of the Utah Mormons: comparison of results based on RFLPs, blood groups, migration matrices, isonymy, and pedigrees. Hum Biol. 1994;66:743‐759. [PubMed] [Google Scholar]
- 19. Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754‐1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM. ArXiv. 2013:1303. [Google Scholar]
- 21. McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res. 2010;20:1297‐1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res. 2010;38:e164‐e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chun S, Fay JC. Identification of deleterious mutations within three human genomes. Genome Res. 2009;19:1553‐1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sim N‐L, Kumar P, Hu J, et al. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012;40:W452‐W7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ng PC, Henikoff SSIFT. Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812‐3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248‐249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep‐sequencing age. Nat Methods. 2014;11:361‐362. [DOI] [PubMed] [Google Scholar]
- 28. Shihab HA, Gough J, Cooper DN, et al. Predicting the functional consequences of cancer‐associated amino acid substitutions. Bioinformatics. 2013;29:1504‐1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics (Oxford, England). 2015;31:2745‐2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Carter H, Douville C, Stenson PD, et al. Identifying Mendelian disease genes with the variant effect scoring tool. BMC Genomics. 2013;14(Suppl 3):S3‐S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jagadeesh KA, Wenger AM, Berger MJ, et al. M‐CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet. 2016;48:1581‐1586. [DOI] [PubMed] [Google Scholar]
- 32. Ioannidis NM, Rothstein JH, Pejaver V, et al. REVEL: an Ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. 2016;99:877‐885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kircher M, Witten DM, Jain P, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Quang D, Chen Y, Xie X. DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics (Oxford, England). 2015;31:761‐763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ionita‐Laza I, McCallum K, Xu B, Buxbaum JD. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat Genet. 2016;48:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lu Q, Hu Y, Sun J, et al. A statistical framework to predict functional non‐coding regions in the human genome through integrated analysis of annotation data. Sci Rep. 2015;5:10576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Davydov EV, Goode DL, Sirota M, et al. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLOS Comput Biol. 2010;6:e1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zheng HF, Ladouceur M, Greenwood CM, Richards JB. Effect of genome‐wide genotyping and reference panels on rare variants imputation. J Genet Genomics. 2012;39:545‐550. [DOI] [PubMed] [Google Scholar]
- 39. Erikson GA, Bodian DL, Rueda M, et al. Whole‐genome sequencing of a healthy aging cohort. Cell. 2016;165:1002‐1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Auton A, Brooks LD, Durbin RM, et al. A global reference for human genetic variation. Nature. 2015;526:68‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein‐coding genetic variation in 60,706 humans. Nature. 2016;536:285‐291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chang CC, Chow CC, Tellier LC, et al. Second‐generation PLINK: rising to the challenge of larger and richer datasets. GigaScience. 2015;4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:e190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bureau A, Younkin SG, Parker MM, et al. Inferring rare disease risk variants based on exact probabilities of sharing by multiple affected relatives. Bioinformatics (Oxford, England). 2014;30:2189‐2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Le Guennec K, Nicolas G, Quenez O, et al. ABCA7 rare variants and Alzheimer disease risk. Neurology. 2016;86:2134‐2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vardarajan BN, Ghani M, Kahn A, et al. Rare coding mutations identified by sequencing of Alzheimer disease genome‐wide association studies loci. Ann Neurol. 2015;78:487‐498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sassi C, Ridge PG, Nalls MA, et al. Influence of coding variability in APP‐Abeta metabolism genes in sporadic Alzheimer's disease. PLoS One. 2016;11:e0150079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Patel D, Mez J, Vardarajan BN, et al. Association of rare coding mutations with Alzheimer disease and other dementias among adults of European ancestry. JAMA Netw Open. 2019;2:e191350‐e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Saxena MT, Schroeter EH, Mumm JS, Kopan R. Murine notch homologs (N1‐4) undergo presenilin‐dependent proteolysis. J Biol Chem. 2001;276:40268‐40273. [DOI] [PubMed] [Google Scholar]
- 51. Mizutani T, Taniguchi Y, Aoki T, et al. Conservation of the biochemical mechanisms of signal transduction among mammalian Notch family members. Proc Natl Acad Sci U S A. 2001;98:9026‐9031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Giegerich AK, Kuchler L, Sha LK, et al. Autophagy‐dependent PELI3 degradation inhibits proinflammatory IL1B expression. Autophagy. 2014;10:1937‐1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Olah J, Vincze O, Virok D, et al. Interactions of pathological hallmark proteins: tubulin polymerization promoting protein/p25, beta‐amyloid, and alpha‐synuclein. J Biol Chem. 2011;286:34088‐34100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stieren ES, El Ayadi A, Xiao Y, et al. Ubiquilin‐1 is a molecular chaperone for the amyloid precursor protein. J Biol Chem. 2011;286:35689‐35698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hunter S, Brayne C. Understanding the roles of mutations in the amyloid precursor protein in Alzheimer disease. Mol Psychiatry. 2018;23:81‐93. [DOI] [PubMed] [Google Scholar]
- 56. Zhang X, Smits AH, van Tilburg GB, et al. An interaction landscape of ubiquitin signaling. Mol Cell. 2017;65:941‐955. [DOI] [PubMed] [Google Scholar]
- 57. Na CH, Jones DR, Yang Y, et al. Synaptic protein ubiquitination in rat brain revealed by antibody‐based ubiquitome analysis. J Proteome Res. 2012;11:4722‐4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rosenberg RN, Lambracht‐Washington D, Yu G, Xia W. Genomics of Alzheimer disease: a review. JAMA Neurology. 2016;73:867‐874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kanehisa M. Toward understanding the origin and evolution of cellular organisms. Prot Sci. 2019;28:1947‐1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kanehisa M, Sato Y, Furumichi M, et al. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019;47:D590‐d5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nathan JA, Kim HT, Ting L, et al. Why do cellular proteins linked to K63‐polyubiquitin chains not associate with proteasomes?. EMBO J. 2013;32:552‐565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Theendakara V, Peters‐Libeu CA, Spilman P, et al. Direct transcriptional effects of apolipoprotein E. J Neurosci. 2016;36:685‐700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tavtigian SV, Simard J, Rommens J, et al. The complete BRCA2 gene and mutations in chromosome 13q‐linked kindreds. Nat Genet. 1996;12:333‐337. [DOI] [PubMed] [Google Scholar]
- 65. Kamb A, Shattuck‐Eidens D, Eeles R, et al. Analysis of the p16 gene (CDKN2) as a candidate for the chromosome 9p melanoma susceptibility locus. Nat Genet. 1994;8:22‐26. [DOI] [PubMed] [Google Scholar]
- 66. Thompson B, Snow A, Koptiuch C, et al. A novel ribosomal protein S20 variant in a family with unexplained colorectal cancer and polyposis. Clin Genet. 2020;97:943‐944. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information