

ABSTRACT
Huntington disease is an autosomal dominant inherited brain disorder that typically becomes manifest in adulthood. Juvenile‐onset Huntington disease refers to approximately 5% of patients with symptom onset before the age of 21 years. The causal factor is a pathologically expanded CAG repeat in the Huntingtin gene. Age at onset is inversely correlated with CAG repeat length. Juvenile‐onset patients have distinct symptoms and signs with more severe pathology of involved brain structures in comparison with disease onset in adulthood. The aim of this review is to compare clinical and pathological features in juvenile‐ and adult‐onset Huntington disease and to explore which processes potentially contribute to the observed differences. A specific focus is placed on molecular mechanisms of mutant huntingtin in early neurodevelopment and the interaction of a neurodegenerative disease and postnatal brain maturation. The importance of a better understanding of pathophysiological differences between juvenile‐ and adult‐onset Huntington disease lies in development and implementation of new therapeutic strategies. © 2021 The Authors. Movement Disorders published by Wiley Periodicals LLC on behalf of International Parkinson and Movement Disorder Society
Keywords: juvenile Huntington disease, pediatric Huntington disease, pediatric neurodegenerative disease, disease mechanisms, neurodevelopment
Huntington disease (HD) is an autosomal dominant progressive brain disorder caused by a pathological CAG repeat expansion coding for huntingtin (HTT gene), with an elongated polyglutamine tract. 1 The length of the CAG repeat shows an inverse correlation with the age at onset. 2 Symptoms become manifest at a mean age of 45 (range 2–87) years. 3 , 4 , 5 All patients with symptom onset before the age of 21 years, irrespective of their present age, are referred to as juvenile‐onset Huntington disease (JHD), which is seen in 4% to 10% of all HD cases. 6 , 7 The term pediatric Huntington disease (PHD) is reserved for all patients with manifest disease who are still below the age of 18 years. 7 Based on clinical signs, further distinction is made between childhood (<10 years) and adolescent onset (10−18 years). 2 , 8 In approximately 50% of JHD cases the CAG expansion is ≥60, exceeding 80 repeats in childhood onset. 8 , 9 About 80% of JHD patients inherit the repeat expansion via paternal transmission. 8 , 10
JHD patients are often difficult to diagnose. 11 , 12 This is mainly due to psychiatric and cognitive complaints that are easily misdiagnosed. 9 , 11 , 12 , 13 Apart from the atypical clinical presentation, disease progression in childhood‐onset HD patients is faster and survival shorter compared to adult‐onset HD (AHD). 8 Furthermore, morphological changes in JHD brains are generally more severe than in AHD brains. 14 These phenotypical and pathological differences raise the hypothesis of aberrant pathomechanisms. Detangling pathophysiological differences between JHD and AHD is important for the successful treatment of pediatric patients. Various treatments are currently under investigation in AHD patients, yet JHD patients are excluded from most therapeutic trials.
This review aims to highlight differences in clinical, neuropathological, and imaging features between JHD and AHD. Suggestions for further studies are made by explaining these differences in the light of HD pathophysiology and brain development.
Clinical Features
HD is characterized by motor and cognitive dysfunction and by psychiatric and behavioral changes, leading to loss of independence and eventually death. The median disease duration after motor onset in childhood JHD is 9 years, compared to 18 years in adolescent and adult HD. 5 , 8
Motor Symptoms
Between 42% and 94% of JHD patients develop postural instability, bradykinesia, and rigidity, in combination with dystonia in the initial stages of motor onset. 8 , 12 , 15 , 16 , 17 , 18 In contrast, motor onset with bradykinesia and rigidity is seen in only 20% of AHD patients, yet most AHD patients may become hypokinetic and rigid at the end of their disease. 19 , 20 Chorea, which is the initial motor sign in 80% of AHD cases, is rarely seen in early JHD, but gradually evolves with disease progression in a subset of adolescent HD. 8 , 18 , 21 , 22 Motor signs more often seen as initial signs in JHD include dysarthria and loss of dexterity, such as writing. 8 , 12 , 21 Oral dyskinesias, 8 , 11 , 18 tics, 8 , 21 , 22 and myoclonus 8 , 12 , 15 , 21 are more frequently seen in later stages of JHD. Of note are conflicting reports on the appearance of ataxia, 8 , 15 , 21 which is probably a definition problem. Ataxia, imbalance, incoordination, and unsteady walking are words probably that refer to the same common early signs in JHD, which can also be seen in early stages of AHD. 23
Cognitive Symptoms
Cognitive deficits are reported as initial disease signs in 30%–83% of JHD patients, before motor onset is apparent. 8 , 21 , 24 , 25 This wide range might be attributed to differences in description but emphasizes the notion that cognitive deficits are prominent in the initial stage of JHD. Similarly, in about one‐third of AHD cases, early cognitive disturbance, typically related to psychomotor speed and deterioration of executive functions such as attention, planning, and flexibility of mind, are present years before the first motor symptoms appear. 25 , 26 More specific for JHD is the prevalence of developmental delay. This feature is used to describe delays in cognitive, motor, language, and social development, thus referring to a broader range of neurological features than just cognition. An early cohort study reported that 6 of 33 JHD cases present with some form of developmental delay. 18 An additional description of three unrelated JHD patients (CAG repeats of 93, 100, and 120) mentioned a developmental delay in speech and language, followed by a delay in social and motor skills. 27 Several other publications confirmed the presence of developmental delay in the JHD population and showed that these delays are particularly seen in patients with childhood disease onset. 8 , 21 Of note is the lack of data delineating the neurological base of the observed delays (eg, cognition, motor, and/or social) as well as comparative data with AHD.
Psychiatric Symptoms
About 30% of JHD patients present with some form of psychiatric or behavioral disturbances before motor onset, which increases to 75% during disease progression. 8 , 12 , 15 , 21 , 22 , 25 These numbers largely resemble those in adult‐onset disease. 25 , 26 , 28 However, the nature of psychiatric complaints in JHD patients differs from those seen in AHD patients. Obsessive–compulsive behaviors are more common in adolescent onset (50%–73%) compared to AHD (26%). The prevalence of behavioral deficits is higher in adolescent‐onset HD patients compared with childhood‐onset HD. 8 , 28 , 29 Psychotic symptoms are reported in 17% to 39% of JHD patients, 8 , 22 and only in about 4% of AHD cases. 28
Other Symptoms
The most remarkable difference in clinical appearance between JHD and AHD is the higher prevalence of epileptic seizures in the JHD population, estimated to be 30% to 35%. Observational studies show it is far more common in childhood‐onset HD patients than in adolescent‐onset HD. 8 , 13 , 30 In AHD, the prevalence of epileptic seizures is comparable to population risk. Furthermore, sleep disturbance, pain, and itching are explicitly or more commonly mentioned in the juvenile population. 22 Unintended weight loss and hypermetabolic state is seen in both AHD and JHD cases, yet its severity correlates with an increase in CAG repeat length and thus is more severe in JHD patients. 31 , 32
Neuropathology and Imaging
In HD pathology, various cell types and brain regions are affected, and although there are shared characteristics between JHD and AHD, subtle pathological differences between the two forms exist. There is a lack of systematic assessments of differences between JHD and AHD brains. What is known, mainly based on small sample sizes from either pathology or imaging studies, will be discussed here and illustrated in Fig. 1.
FIG 1.
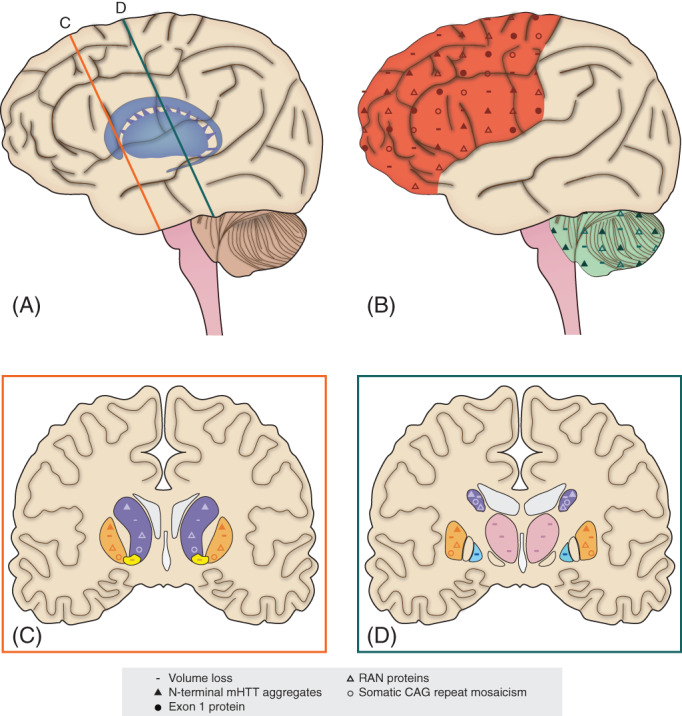
Schematic illustration showing differences in juvenile‐onset Huntington disease (JHD) neuropathology in comparison with adult‐onset Huntington disease (AHD) neuropathology. More severe pathological hallmarks of JHD brains are seen in A, inset C and D: the subcortical grey matter structures and B: the frontoparietal cortex and, to a lesser extent, the cerebellum. Volume loss (−) is more pronounced in the frontoparietal cortex (red), cerebellum (green), caudate nucleus (purple), putamen (orange), nucleus accumbens (yellow), internal segment of the globus pallidus (blue), and the thalamus (pink). N‐terminal mutant huntingtin (mHTT) aggregates (▴) are more abundant in the frontal cortex, caudate nucleus, putamen, and, to a lesser extent, cerebellum of JHD brains. Exon 1 protein (●) is more abundant in the frontoparietal cortex and hippocampus (not displayed) of JHD brains. Repeat‐associated non‐ATG nuclear (RAN) proteins () are more abundant in the striatum, frontal cortex, and cerebellum of JHD brains. Somatic CAG repeat mosaicism () is greater in the neocortex, caudate nucleus, and putamen of JHD brains. [Color figure can be viewed at wileyonlinelibrary.com]
Subcortical Structures
In HD, progressive neostriatal (eg, putamen and caudate nucleus) loss of medium spiny projection neurons and concomitant reactive increase of astroglial cells (eg, astrogliosis) are the most prominent neuropathological changes observed, and determine mainly the neuropathological grading (see Box 1). 33 Neostriatal volume loss is generally more severe in JHD brains when compared with AHD brains and follows a linear correlation with CAG repeat length (see Table 1 and Fig. 1). 14 , 33 , 34 , 35 , 36 Of note is the importance of disease severity and duration of illness at autopsy in the interpretation of volume loss between the two forms, which is only sporadically taken into account in pathological comparisons. Magnetic resonance imaging (MRI) studies confirm these findings by comparing neostriatal volume loss and disease severity between patient groups with varying CAG repeat lengths. 37 Furthermore, other pathological hallmarks like mutant huntingtin (mHTT) aggregates and somatic CAG repeat mosaicism (see Box 2) are more severe in the neostriatum of JHD brains when compared to AHD brains (Fig. 1). 38 , 39 , 40 , 41 In addition to neostriatal pathology, other subcortical regions, such as the internal segment of the globus pallidus, the nucleus accumbens, and thalamus, are more often affected in post‐mortem JHD brains when compared to AHD brains (see Fig. 1). 34 Thalamic and pallidal volume loss in JHD patients was also observed in a recent age‐matched cross‐sectional MRI study (N = 19). 42 An inverse relationship between CAG repeat length and thalamic volume loss was found within this same cohort. Taken together, these findings show more severe pathology of subcortical structures in JHD patients.
Box 1. Vonsattel Grading System.
This is a five‐scale neuropathological classification (0–4) based on the sequential pathology of striatal regions. 33 The assignment of a grade is based on macroscopic and microscopic findings, the latter on three standardized coronal sections of the striatum.
Grade 0: no macro‐ and microscopic alterations are seen, yet subtle neuronal cell loss of the head of the caudate nucleus can be quantified when compared with non‐neurological control brains; grade 1: there are no major macroscopic changes, but neuronal loss and astrogliosis can be reliably observed microscopically in the caudate nucleus and to a smaller extent in the putamen; grade 2: caudate nucleus volume loss can be observed macroscopically (medial outline into the lateral ventricle slightly convex); grade 3: gross caudate nucleus volume loss extents in a straight or concave line with the lateral ventricle; and grade 4: outline of caudate nucleus is concave, as is the anterior limb of the internal capsule, and 95% of neostriatal neurons are lost microscopically.
TABLE 1.
| Vonsattel grading system | JHD brains (N = 50) | AHD brains (N = 1300) |
|---|---|---|
| Grade 0 | Not reported | 1% |
| Grade 1 | 10% a | 4% |
| Grade 2 | Not reported | 16% |
| Grade 3 | 26% | 53% |
| Grade 4 | 64% | 28% |
Ninety percent of grade 1 juvenile‐onset Huntington disease (JHD) brains were from JHD patients who committed suicide and therefore do not reflect end‐stage disease. For the adult‐onset Huntington disease (AHD) brains, these percentages are unknown/not provided.
Box 2. Box.
Aberrant protein expression and aggregates are well known pathological hallmarks in neurodegenerative disease and can be found in both intra‐ and extracellular compartments. In HD, N‐terminal mHTT aggregates are found in neurons of the neocortex, neostriatum, hippocampal area, and brainstem and, to a lesser extent, in glial cells. 39 , 40 , 41 , 43 , 44 Also, other mHTT protein species are selectively expressed in HD brains, such as aberrantly spliced exon 1 protein and repeat‐associated non‐ATG (RAN) proteins. 39 , 45 , 46 As to the relative toxicity of mHTT protein species, HD cell and animal models show that, in particular, exon 1 and certain RAN protein species have more detrimental effects on cell function and death than full‐length mHTT protein. 46 , 47 Another pathological hallmark in HD is the degree of somatic CAG repeat mosaicism. With increasing CAG repeat length, the occurrence of both germline and somatic expansion of the trinucleotide length increases. HD pathology is positively correlated with the extent of CAG repeat mosaicism. 38 , 48 , 49
Neocortex
Neocortical volume and pyramidal neuron loss is also found in AHD and PHD brains. In general, atrophy is most pronounced in frontal and parietal regions and is most often seen in Vonsattel grade 3 and 4 brains. 33 Based on macroscopy, frontal and parietal atrophy is more commonly observed in post‐mortem JHD brains when compared with AHD brains (see Fig. 1), like the higher Vonsattel grades in JHD brains. 34 More widespread cortical volume loss and faster volume loss over time has also been observed in a longitudinal MRI analysis of 2 JHD patients who carried CAGs higher then 55, when compared with 34 AHD patients with repeat lengths between 40 and 55. 50 , 51 However, cross‐sectional analysis of cortical volume loss and CAG repeat length in the same cohort failed to replicate this result. 52 In addition, structural brain MRI in 19 JHD patients showed relatively preserved cortical volumes when compared with age‐matched controls, 42 suggesting that cortical volume loss in JHD brains is related to end‐stage disease. Other pathological hallmarks such as N‐terminal mHTT ‐, exon 1 ‐, and RAN protein aggregates and somatic CAG repeat mosaicism, are more highly expressed in the neocortex of JHD brains when compared with AHD brains, as shown in Fig. 1. 39 , 40 , 41 , 45 , 46 The need for more refined comparison of JHD and AHD post‐mortem brains, with extensive longitudinal MRI data, analyzing and comparing JHD and AHD patients, will be essential to clarify the relative involvement of cortical pathology in the two forms.
Cerebellum
The hypothesis of more pronounced cerebellar pathology in JHD brains remains a matter of controversy. Severe macroscopic cerebellar atrophy is described in a subset of neuropathological and MRI studies of JHD brains, all of them from childhood‐onset cases (see Fig. 1). 53 , 54 , 55 , 56 , 57 , 58 However, of concern is the cause of cerebellar atrophy in these JHD brains, since most of the examined cases (11 out of 13) were known to have epilepsy, which could potentially cause hypoxic–ischemic events in this area. In both JHD and AHD brains, subtle macroscopic cerebellar atrophy is seen together with extensive striatal degeneration (Vonsattel grades 3 and 4). 33 Cross‐sectional imaging studies of cerebellar volume loss in both JHD and AHD patients have failed to replicate group differences in the amount of cerebellar atrophy. 51 , 52 Of particular interest are two recent in vivo MRI studies that revealed relative enlargement of anterior cerebellar compartments in AHD and JHD subjects when compared with age‐matched controls. 42 , 59 Additional functional investigations have suggested the cerebellum is a compensatory brain structure for pathological basal ganglia changes in early HD disease stages. 60 , 61 Studies of the presence of mHTT and RAN protein aggregates in HD cerebelli found evidence that such pathological hallmarks are selectively or more prominently found in JHD cerebelli (Fig. 1). 45 , 46 , 56 In particular, the higher expression of RAN protein species suggests that toxicological processes are more likely to take place in JHD cerebelli when compared to AHD cerebelli. Additional research into the role of the cerebellum in HD pathophysiology is needed to further clarify discrepant in vivo and post‐mortem findings and to address its relation to JHD and AHD.
White Matter
While substantial evidence suggests (microstructural) white matter changes in HD, the relative involvement of white matter pathology in JHD as compared to AHD is as yet unknown. 62 In JHD patients, scarce quantitative data of white matter involvement are available and diffusion tensor imaging (DTI) measures have never been published. Hedjoudje and others showed subtle cerebral white matter volume decrease in three JHD siblings with childhood onset of disease, carrying CAG repeats >120, when compared to age‐matched controls. 63 Tereshchenko and others replicated this finding in a sample of 19 JHD patients (CAG repeat range 54 to 96) and revealed more pronounced white matter volume decrease in JHD patients with longer repeats, suggesting a CAG‐dependent relation. 42 More comparative imaging data in both JHD and AHD patients is needed to reliably interpret possible differences in white matter involvement between the two forms.
Differences in Clinicopathology and Causal Factors
Summarizing the previous paragraphs, JHD shows a faster disease progression and a shorter survival, as compared to AHD, with some distinct clinical symptoms and signs, like hypokinetic‐ and rigid syndrome, developmental delay, behavioral disorder, epilepsy, and psychosis. This is accompanied by increased pathological hallmarks, such as subcortical volume loss and gliosis and selective or higher number of mHTT aggregates, exon 1 and RAN proteins, and somatic CAG repeat mosaicism (Fig. 1). Differences in these clinicopathological measures can be explained by environmental, biological, and pathophysiological factors. Although the upbringing of JHD patients in a family with an affected HD parent might well affect certain cognitive and psychiatric measures, differences in biological and pathophysiological factors are important to consider in the light of treatment opportunities.
General HD pathophysiology involves both cell dysfunction and cell loss, with clinical symptoms and signs as a result. Contributing factors to cell dysfunction and loss are a gain of toxic mHTT protein function and a loss of normal huntingtin protein function, and include RNA toxicity, transcriptional dysregulation, mitochondrial dysfunction, excitotoxicity, and inflammation. 64 Neurodegenerative pathomechanisms in HD follow a linear correlation with CAG repeat length and age at disease onset and are an important contributor to the clinicopathological differences between JHD and AHD patients. Apart from neurodegeneration, aberrant neurodevelopment is also thought to contribute to HD pathophysiology as studies in diverse HD models and post‐mortem HD brain material have shown defects in cell differentiation, migration, and maturation. 65 , 66 A recent in vivo study in children carrying a HTT gene expansion − whom will develop HD clinical characteristics later in life − substantiates these preclinical data and showed structural and functional changes in the striatum and cerebellum as young as 6 years. 37 , 60 As to how these pathological changes during brain development relate to clinical measures later in life (eg, AHD) is unknown. In this regard, JHD patients not only have clinical symptoms during brain maturation, they also have a higher incidence of clinical characteristics that relate to abnormal neurodevelopment. It is therefore likely that postnatal brain maturation and neurodevelopmental pathomechanisms also contribute to the clinicopathological differences between AHD and JHD patients. One could argue that neurodevelopmental defects exceed a certain threshold for normal brain function in JHD patients, whereas these defects only act on a subclinical level in AHD patients. In the following paragraph we will highlight certain neurodevelopmental pathomechanisms and postnatal brain maturation processes and relate these to distinct clinical characteristics in JHD patients.
Neurodevelopmental Defects
Brain development involves overlapping processes of: (1) neurogenesis and cell differentiation; (2) neuronal migration; (3) synaptogenesis; (4) neural circuitry formation; and (5) synaptic pruning and myelination (see Fig. 2). The first two processes are mainly established before birth (prenatal neurodevelopment), and the latter three continue to change well into early adulthood (postnatal brain maturation). 67 As mentioned earlier, various preclinical studies have revealed aberrant neurogenesis and cell differentiation in relation to pathologically expanded CAG repeat lengths, as is nicely reviewed by Wiatr and others. 65 Two such studies, using HD stem cells with increasing CAG repeat lengths, have revealed defective progenitor cell differentiation, abnormal multinucleated neuron morphology, chromosomal instability, and changes in cytokinesis in a CAG‐dependent manner. 68 , 69 This suggests greater neurogenesis and cell differentiation defects in JHD when compared with AHD. Greater defects in early developmental processes can cause distinct clinical characteristics in the JHD population, such as developmental delay (see pink, green, and blue lines in Fig. 2), as is similarly seen in a large heterogeneous group of neurodevelopmental disorders. 70 Another study in R6/2 mice highlighted changes in neuronal migration and arborization (see green and blue lines in Fig. 2) that are similar to those found in focal cortical dysplasia (FCD) type 1. 71 Changes in cortical development are a common etiology of developmental delay and epileptic seizures in a variety of neurodevelopmental disorders. Future studies should determine if there are FCD‐like changes in JHD and AHD patient material.
FIG 2.
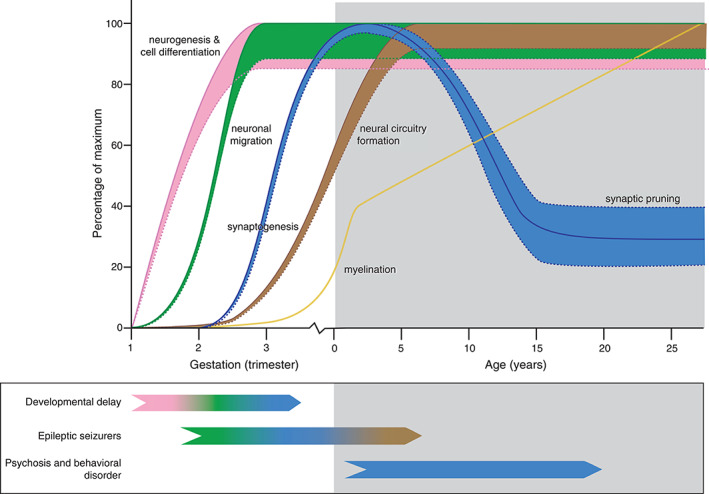
Model for potential effects of mutant huntingtin (mHTT) in the developing brain of juvenile‐onset Huntington disease (JHD) patients. Normal brain development involves overlapping processes of neurogenesis and cell differentiation (pink line), neuronal migration (green line), neural circuitry formation by dendrite branching (brown line), and synaptogenesis (first part of blue line), followed by selective synaptic pruning (second part of blue line) and myelination (yellow line). Straight lines represent physiological brain development, dotted lines potentially altered neurodevelopment due to a dosage effect of CAG repeat length or interplay with brain maturation in JHD patients. Distinct clinical characteristics in JHD patients include developmental delay, epileptic seizures, psychosis, and behavioral disorders that could relate to various defects in neurodevelopment or brain maturation (see color code of the representative lines). Note the potential bidirectional effect on synapse abundance (blue dotted lines) on developmental delay (associated with impaired prenatal synaptogenesis) and psychosis and behavioral disorders (associated with impaired postnatal synaptic pruning). [Color figure can be viewed at wileyonlinelibrary.com]
Changes in postnatal brain maturation could also contribute to the clinical picture in JHD. Neural circuitry formation by dendrite branching and formation of new synapses starts during prenatal neurodevelopment but continues to expand into childhood (see brown line in Fig. 2). A longitudinal imaging study in asymptomatic children carrying an HTT gene expansion showed an incremental effect of CAG repeat length on functional circuitry adaptations. 37 Resilience of brain regions such as the cerebellum is thought to functionally compensate for early developmental changes, with symptoms occurring as soon as this resilience cannot overcome the accumulating toxic effects of mHTT. 61 A higher burden of toxic RAN and mHTT protein species in JHD cerebelli could explain early loss of resilience and symptom onset, as well as the early occurrence of clinical characteristics, such as postural instability, ataxia, dysarthria, hand dexterity, and dystonia. Furthermore, significant changes in neurotransmitter systems and ion channels during neural circuitry formation render the immature brain more prone to an imbalance between neuronal (GABA‐ergic) inhibition and (glutamergic) excitation, in favor of the latter. 72 This imbalance is an important contributor to the higher incidence of epileptic seizures in childhood neurodevelopmental disorders. The simultaneous dysfunction and loss of GABA‐ergic medium spiny neurons of the striatum as well as prefrontal neurons in JHD patients with childhood‐onset of disease renders these patients prone to such an imbalance. This could provide another explanation for selective occurrence of epileptic seizures in JHD patients. Changes in synapse abundance and pruning could also be involved in JHD pathophysiology (blue line in Fig. 2). By the age of 2 years, infant brain contains about 150% of synaptic connections compared to adult brain. During adolescence, a steep decline in synaptic connections is determined by the amount and timing of neural activity, which is further regulated by elements of the immune system such as microglial and complement function. 73 , 74 Changes in synapse abundance play an important role in the pathogenesis of neurodevelopmental disorders such as schizophrenia, obsessive–compulsive disorder, and autism. 73 , 75 As mentioned earlier, functional circuitries are known to be altered in HD patients. Furthermore, reactive and cell‐autonomous effects of HD microglia induce a pro‐inflammatory transcription profile and reduced fractalkine expression. 76 , 77 The latter protein plays an important role in microglial capacity for synaptic pruning. Since psychosis and obsessive–compulsive disorder are far more prevalent in adolescent‐onset HD, aberrant synaptic pruning is an interesting target for future studies.
Discussion and Future Directions
Clinicopathological differences between JHD and AHD exist and can be explained by pathological, biological, and environmental factors. A dosage effect of CAG repeat length on neurodegenerative and neurodevelopmental defects, as well as interaction of pathology with ongoing brain maturational processes in JHD patients, may be responsible for the observed difference. Due to the low prevalence of JHD there is still a huge lack of data linking CAG repeat length or age at disease onset with the underlying pathophysiology. In addition, many (pre)clinical studies focus on JHD or AHD and fail to structurally compare the two forms. Unraveling possible pathophysiological differences between JHD and AHD is important for the development of therapeutics designed to reduce symptoms or alter disease progression. For instance, due to differences in symptoms that affect brain areas and pathomechanisms, JHD patients might benefit from therapeutics which have been shown to be ineffective in adult patients. Conversely, therapeutics that target mutant huntingtin might be disproportionally damaging in JHD patients due to its effect on concurrent brain maturation. Furthermore, neurodevelopmental pathomechanisms largely take place in utero and are more difficult to influence once symptoms present themselves. Patients with a complex disorder such as HD might therefore benefit from a combination of therapies in a personalized way rather than a generalized one. Although the severity of JHD will lead clinicians to treat JHD patients as soon as viable therapeutic options are identified in the adult population, lack of well‐established and reliable outcome measures in the pediatric population will further complicate successful implementation of therapeutic strategies. Therefore, future studies should focus on inclusion of both JHD and AHD patients, longitudinal study designs, structural comparison of CAG repeat lengths, or age at disease onset, the specification of JHD read‐out parameters and possible interactions with postnatal brain development. Furthermore, international collaboration is necessary due to the rarity of JHD, and ethical and legal issues in pediatric studies must be overcome. Finally, the implementation of highly standard translational research methods will greatly enhance our knowledge of JHD.
Author Roles
H.S.B. was responsible for the primary conception and organization of the review content and writing the first draft. Both R.A.C.R. and W.M.C.v.R.‐M. reviewed and critiqued the manuscript. S.T.d.B. supervised the conception and organization of the review content and reviewed and critiqued the manuscript.
Financial Disclosures of All Authors
H.S.B.: The LUMC institution received a grant from the European Huntington Disease Network (EHDN), participates in an EU Horizon 2020 project: Innovative Medicines Initiative (IMI) 2 (IDEA_FAST), and participates in clinical trials sponsored by Hoffmann‐LaRoche, Prilenia, and by the Cure Huntington Disease Initiative (CHDI). R.A.C.R.: None. W.M.C.v.R.‐M.: Intellectual Property Rights: Published Patents: 1. Main inventor on published patent WO2012/018257. 2. Main inventor on published patent WO2015/053624. 3. Co‐inventor on published patent application SCA3 WO 2017/053781. Consultancies: BridgeBio Pharma, Inc, a Delaware corporation. Expert Testimony: MRC Expert Review Group for UK Dementia Research Institute ‐ NMHB (UKRI DRI QQR). Advisory Boards: Medical Advisory Board of the Dutch Ataxia Patient Association (ataxie.nl). Employment: Leiden University Medical Center ‐ Department of Human Genetics. Grants: Ongoing Research Support: 1. 2019–2025: ZonMw grant. 2. 2020–2022: Campagne Team Huntington grant. 3. 2019–2020: Service Contract with UniQure. 4. 2018–2020: Campagne Team Huntington grant. 5. 2018–2019: Hersenstichting/SCA1 dedicated fund. 6. 2018–2022: COST Action CA17103. 7. 2018–2021: AFM Telethon. 8. 2017–2020: ZonMw grant. 9. 2017–2019: Campagne Team Huntington grant. 10. 2016–2020: Research contract with ProQR, The Netherlands. S.T.d.B.: The LUMC institution received a grant from the European Huntington Disease Network (EHDN), participates in an EU Horizon 2020 project: Innovative Medicines Initiative (IMI) 2 (IDEA_FAST), and participates in clinical trials sponsored by Hoffmann‐LaRoche, Prilenia, and by the Cure Huntington Disease Initiative (CHDI).
Acknowledgments
The authors thank G. Kracht for illustrating the figures of this educational review.
Relevant conflicts of interest/financial disclosures: Nothing to report.
Funding agencies: Leiden University Medical Center.
Data Availability Statement
Data sharing not applicable ‐ no new data generated, or the article describes entirely theoretical research
References
- 1. The Huntington Disease Collaborative Research Group . A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington disease chromosomes. Cell 1993;72(6):971–983. [DOI] [PubMed] [Google Scholar]
- 2. Squitieri F, Frati L, Ciarmiello A, Lastoria S, Quarrell O. Juvenile Huntington disease: does a dosage‐effect pathogenic mechanism differ from the classical adult disease? Mech Ageing Dev 2006;127(2):208–212. [DOI] [PubMed] [Google Scholar]
- 3. Milunsky JM, Maher TA, Loose BA, Darras BT, Ito M. XL PCR for the detection of large trinucleotide expansions in juvenile Huntington disease. Clin Genet 2003;64(1):70–73. [DOI] [PubMed] [Google Scholar]
- 4. Warren JD, Firgaira F, Thompson EM, Kneebone CS, Blumbergs PC, Thompson PD. The causes of sporadic and ‘senile’ chorea. Aust N Z J Med 1998;28(4):429–431. [DOI] [PubMed] [Google Scholar]
- 5. Roos RA. Huntington disease: a clinical review. Orphanet J Rare Dis 2010;5:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Quarrell O, O'Donovan KL, Bandmann O, Strong M. The prevalence of juvenile Huntington disease: a review of the literature and meta‐analysis. PLoS Curr 2012;4:e4f8606b8742ef8603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Quarrell OWJ, Nance MA, Nopoulos P, et al. Defining pediatric Huntington disease: time to abandon the term juvenile Huntington disease? Mov Disord 2019;4:584–585. [DOI] [PubMed] [Google Scholar]
- 8. Fusilli C, Migliore S, Mazza T, et al. Biological and clinical manifestations of juvenile Huntington disease: a retrospective analysis. Lancet Neurol 2018;17(11):986–993. [DOI] [PubMed] [Google Scholar]
- 9. Quarrell OWJ, Nance M. Juvenile Huntington Disease and Other Trinucleotide Repeat Disorders. The Diagnostic Challenge. Oxford: Oxford University Press; 2012. [Google Scholar]
- 10. Went LN, Vegter‐van der Vlis M, Bruyn GW. Parental transmission in Huntington disease. Lancet 1984;1(8386):1100–1102. [DOI] [PubMed] [Google Scholar]
- 11. Gonzalez‐Alegre P, Afifi AK. Clinical characteristics of childhood‐onset (juvenile) Huntington disease: report of 12 patients and review of the literature. J Child Neurol 2006;21(3):223–229. [DOI] [PubMed] [Google Scholar]
- 12. Ribai P, Nguyen K, Hahn‐Barma V, et al. Psychiatric and cognitive difficulties as indicators of juvenile Huntington disease onset in 29 patients. Arch Neurol 2007;64(6):813–819. [DOI] [PubMed] [Google Scholar]
- 13. Achenbach J, Thiels C, Lücke T, Saft C. Clinical manifestation of juvenile and pediatric HD patients: a retrospective case series. Brain Sci 2020;10(6):340–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vonsattel JPG, Cortes EP, Keller CE. Juvenile Huntington Disease and Other Trinucleotide Repeat Disorders. Juvenile Huntington Disease: Neuropathology. Oxford: Oxford University Press; 2012. [Google Scholar]
- 15. Siesling S, Vegter‐van der Vlis M, Roos RA. Juvenile Huntington disease in The Netherlands. Pediatr Neurol 1997;17(1):37–43. [DOI] [PubMed] [Google Scholar]
- 16. Brackenridge CJ. Factors influencing dementia and epilepsy in Huntington disease of early onset. Acta Neurol Scand 1980;62(5):305–311. [DOI] [PubMed] [Google Scholar]
- 17. van Dijk JG, van der Velde EA, Roos RA, Bruyn GW. Juvenile Huntington disease. Hum Genet 1986;73(3):235–239. [DOI] [PubMed] [Google Scholar]
- 18. Nance MA. Genetic testing of children at risk for Huntington disease. US Huntington disease genetic testing group. Neurology 1997;49(4):1048–1053. [DOI] [PubMed] [Google Scholar]
- 19. Jacobs M, Hart EP, van Zwet EW, et al. Progression of motor subtypes in Huntington disease: a 6‐year follow‐up study. J Neurol 2016;263(10):2080–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. van de Zande NA, Massey TH, McLauchlan D, et al. Clinical characterization of dystonia in adult patients with Huntington disease. Eur J Neurol 2017;24(9):1140–1147. [DOI] [PubMed] [Google Scholar]
- 21. Cronin T, Rosser A, Massey T. Clinical presentation and features of juvenile‐onset Huntington disease: a systematic review. J Huntington Dis 2019;8(2):171–179. [DOI] [PubMed] [Google Scholar]
- 22. Moser AD, Epping E, Espe‐Pfeifer P, et al. A survey‐based study identifies common but unrecognized symptoms in a large series of juvenile Huntington disease. Neurodegener Dis Manag 2017;7(5):307–315. [DOI] [PubMed] [Google Scholar]
- 23. Franklin GL, Camargo CHF, Meira AT, et al. Is ataxia an underestimated symptom of Huntington disease? Front Neurol 2020;11:571843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Poniatowska R, Habib N, Krawczyk R, Rakowicz M. Correlation between magnetic resonance and genetic, clinical, neurophysiological and neuropsychological studies of patients with juvenile form of Huntington disease. Case Rep Clin Pract Rev 2001;146:1633–1645. [Google Scholar]
- 25. McAllister B, Gusella JF, Landwehrmeyer GB, et al. Timing and impact of psychiatric, cognitive, and motor abnormalities in Huntington disease. Neurology 2021;96(19):e2395–e2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Craufurd D, Snowden J. Neuropsychiatry and neuropsychology. Huntington Dis 2014;4:36–65. [PubMed] [Google Scholar]
- 27. Yoon G, Kramer J, Zanko A, et al. Speech and language delay are early manifestations of juvenile‐onset Huntington disease. Neurology 2006;67(7):1265–1267. [DOI] [PubMed] [Google Scholar]
- 28. van Duijn E, Craufurd D, Hubers AA, et al. Neuropsychiatric symptoms in a European Huntington disease cohort (REGISTRY). J Neurol Neurosurg Psychiatry 2014;85(12):1411–1418. [DOI] [PubMed] [Google Scholar]
- 29. Langbehn KE, Cochran AM, van der Plas E, et al. Behavioral deficits in juvenile onset Huntington disease. Brain Sci 2020;10(8):543–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cloud LJ, Rosenblatt A, Margolis RL, et al. Seizures in juvenile Huntington disease: frequency and characterization in a multicenter cohort. Mov Disord 2012;27(14):1797–1800. [DOI] [PubMed] [Google Scholar]
- 31. Aziz NA, van der Burg JM, Landwehrmeyer GB, et al. Weight loss in Huntington disease increases with higher CAG repeat number. Neurology 2008;71(19):1506–1513. [DOI] [PubMed] [Google Scholar]
- 32. Tereshchenko A, McHugh M, Lee JK, et al. Abnormal weight and body mass index in children with juvenile Huntington disease. J Huntington Dis 2015;4(3):231–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vonsattel JP, Keller C, Cortes Ramirez EP. Huntington disease ‐ neuropathology. Handb Clin Neurol 2011;100:83–100. [DOI] [PubMed] [Google Scholar]
- 34. Vonsattel J, Cortes EP, Keller CE. Juvenile Huntington Disease: Neuropathology. New York, NY: Oxford University Press; 2009. [Google Scholar]
- 35. Rosenblatt A, Margolis RL, Becher MW, et al. Does CAG repeat number predict the rate of pathological changes in Huntington disease? Ann Neurol 1998;44(4):708–709. [DOI] [PubMed] [Google Scholar]
- 36. Penney JB Jr, Vonsattel JP, MacDonald ME, Gusella JF, Myers RH. CAG repeat number governs the development rate of pathology in Huntington disease. Ann Neurol 1997;41(5):689–692. [DOI] [PubMed] [Google Scholar]
- 37. van der Plas E, Langbehn DR, Conrad AL, et al. Abnormal brain development in child and adolescent carriers of mutant huntingtin. Neurology 2019;93(10):e1021–e1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kennedy L, Evans E, Chen CM, et al. Dramatic tissue‐specific mutation length increases are an early molecular event in Huntington disease pathogenesis. Hum Mol Genet 2003;12(24):3359–3367. [DOI] [PubMed] [Google Scholar]
- 39. DiFiglia M, Sapp E, Chase KO, et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997;277(5334):1990–1993. [DOI] [PubMed] [Google Scholar]
- 40. Maat‐Schieman ML, Dorsman JC, Smoor MA, et al. Distribution of inclusions in neuronal nuclei and dystrophic neurites in Huntington disease brain. J Neuropathol Exp Neurol 1999;58(2):129–137. [DOI] [PubMed] [Google Scholar]
- 41. Becher MW, Kotzuk JA, Sharp AH, et al. Intranuclear neuronal inclusions in Huntington disease and dentatorubral and pallidoluysian atrophy: correlation between the density of inclusions and IT15 CAG triplet repeat length. Neurobiol Dis 1998;4(6):387–397. [DOI] [PubMed] [Google Scholar]
- 42. Tereshchenko A, Magnotta V, Epping E, et al. Brain structure in juvenile‐onset Huntington disease. Neurology 2019;92(17):e1939–e1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rüb U, Hentschel M, Stratmann K, et al. Huntington disease (HD): degeneration of select nuclei, widespread occurrence of neuronal nuclear and axonal inclusions in the brainstem. Brain Pathol 2014;24(3):247–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jansen AH, van Hal M, Op den Kelder IC, et al. Frequency of nuclear mutant huntingtin inclusion formation in neurons and glia is cell‐type‐specific. Glia 2017;65(1):50–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Neueder A, Landles C, Ghosh R, et al. The pathogenic exon 1 HTT protein is produced by incomplete splicing in Huntington disease patients. Sci Rep 2017;7(1):1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bañez‐Coronel M, Ayhan F, Tarabochia AD, et al. RAN translation in Huntington disease. Neuron 2015;88(4):667–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Benn CL, Landles C, Li H, et al. Contribution of nuclear and extranuclear polyQ to neurological phenotypes in mouse models of Huntington disease. Hum Mol Genet 2005;14(20):3065–3078. [DOI] [PubMed] [Google Scholar]
- 48. Telenius H, Kremer B, Goldberg YP, et al. Somatic and gonadal mosaicism of the Huntington disease gene CAG repeat in brain and sperm. Nat Genet 1994;6(4):409–414. [DOI] [PubMed] [Google Scholar]
- 49. de Rooij KE, de Koning Gans PA, Roos RA, van Ommen GJ, den Dunnen JT. Somatic expansion of the (CAG)n repeat in Huntington disease brains. Hum Genet 1995;95(3):270–274. [DOI] [PubMed] [Google Scholar]
- 50. Ruocco HH, Bonilha L, Li LM, Lopes‐Cendes I, Cendes F. Longitudinal analysis of regional grey matter loss in Huntington disease: effects of the length of the expanded CAG repeat. J Neurol Neurosurg Psychiatry 2008;79(2):130–135. [DOI] [PubMed] [Google Scholar]
- 51. Ruocco HH, Lopes‐Cendes I, Laurito TL, Li LM, Cendes F. Clinical presentation of juvenile Huntington disease. Arq Neuropsiquiatr 2006;64(1):5–9. [DOI] [PubMed] [Google Scholar]
- 52. Ruocco HH, Lopes‐Cendes I, Li LM, Santos‐Silva M, Cendes F. Striatal and extrastriatal atrophy in Huntington disease and its relationship with length of the CAG repeat. Braz J Med Biol Res 2006;39(8):1129–1136. [DOI] [PubMed] [Google Scholar]
- 53. Jervis GA. Huntington chorea in childhood. Arch Neurol 1963;9(3):244–257. [DOI] [PubMed] [Google Scholar]
- 54. Markham CH, Knox JW. Observations on Huntington chorea in childhood. J Pediatr 1965;67(1):46–57. [DOI] [PubMed] [Google Scholar]
- 55. Byers RK, Gilles FH, Fung CJN. Huntington disease in children: neuropathologic study of four cases. Neurology 1973;23(6):561–561. [DOI] [PubMed] [Google Scholar]
- 56. Latimer CS, Flanagan ME, Cimino PJ, et al. Neuropathological comparison of adult onset and juvenile Huntington disease with cerebellar atrophy: a report of a father and son. J Huntington Dis 2017;6(4):337–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nicolas G, Devys D, Goldenberg A, et al. Juvenile Huntington disease in an 18‐month‐old boy revealed by global developmental delay and reduced cerebellar volume. Am J Med Genet A 2011;155a(4):815–818. [DOI] [PubMed] [Google Scholar]
- 58. Sakazume S, Yoshinari S, Oguma E, et al. A patient with early onset Huntington disease and severe cerebellar atrophy. Am J Med Genet A 2009;149a(4):598–601. [DOI] [PubMed] [Google Scholar]
- 59. de Azevedo PC, Guimarães RP, Piccinin CC, et al. Cerebellar gray matter alterations in Huntington disease: a voxel‐based Morphometry study. Cerebellum 2017;16(5–6):923–928. [DOI] [PubMed] [Google Scholar]
- 60. Tereshchenko AV, Schultz JL, Bruss JE, Magnotta VA, Epping EA, Nopoulos PC. Abnormal development of cerebellar‐striatal circuitry in Huntington disease. Neurology 2020;94(18):e1908–e1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. van der Plas E, Schultz JL, Nopoulos PC. The neurodevelopmental hypothesis of Huntington disease. J Huntington Dis 2020;9(3):217–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Casella C, Lipp I, Rosser A, Jones DK, Metzler‐Baddeley C. A critical review of white matter changes in Huntington disease. Mov Disord 2020;35(8):1302–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hedjoudje A, Nicolas G, Goldenberg A, et al. Morphological features in juvenile Huntington disease associated with cerebellar atrophy ‐ magnetic resonance imaging morphometric analysis. Pediatr Radiol 2018;48(10):1463–1471. [DOI] [PubMed] [Google Scholar]
- 64. Ross CA, Tabrizi SJ. Huntington disease: from molecular pathogenesis to clinical treatment. Lancet Neurol 2011;10(1):83–98. [DOI] [PubMed] [Google Scholar]
- 65. Wiatr K, Szlachcic WJ, Trzeciak M, Figlerowicz M, Figiel M. Huntington disease as a neurodevelopmental disorder and early signs of the disease in stem cells. Mol Neurobiol 2018;55(4):3351–3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Barnat M, Capizzi M, Aparicio E, et al. Huntington disease alters human neurodevelopment. Science 2020;369(6505):787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lee Gregory M, Burton VJ, Shapiro BK. Chapter 3 ‐ developmental disabilities and metabolic disorders. In: Zigmond MJ, Rowland LP, Coyle JT, eds. Neurobiology of Brain Disorders. San Diego, CA: Academic Press; 2015:18–41. [Google Scholar]
- 68. Conforti P, Besusso D, Bocchi VD, et al. Faulty neuronal determination and cell polarization are reverted by modulating HD early phenotypes. Proc Natl Acad Sci U S A 2018;115(4):E762–e771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ruzo A, Croft GF, Metzger JJ, et al. Chromosomal instability during neurogenesis in Huntington disease. Development 2018;145(2). [DOI] [PubMed] [Google Scholar]
- 70. van Bokhoven H. Genetic and epigenetic networks in intellectual disabilities. Annu Rev Genet 2011;45:81–104. [DOI] [PubMed] [Google Scholar]
- 71. Cepeda C, Oikonomou KD, Cummings D, et al. Developmental origins of cortical hyperexcitability in Huntington disease: review and new observations. J Neurosci Res 2019;97(12):1624–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wong M. Advances in the pathophysiology of developmental epilepsies. Semin Pediatr Neurol 2005;12(2):72–87. [DOI] [PubMed] [Google Scholar]
- 73. Sakai J. Core concept: how synaptic pruning shapes neural wiring during development and, possibly, in disease. Proc Natl Acad Sci U S A 2020;117(28):16096–16099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Schafer DP, Lehrman EK, Kautzman AG, et al. Microglia sculpt postnatal neural circuits in an activity and complement‐dependent manner. Neuron 2012;74(4):691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Paolicelli RC, Ferretti MT. Function and dysfunction of microglia during brain development: consequences for synapses and neural circuits. Front Synap Neurosci 2017;9:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Creus‐Muncunill J, Ehrlich ME. Cell‐autonomous and non‐cell‐autonomous pathogenic mechanisms in Huntington disease: insights from in vitro and in vivo models. Neurotherapeutics 2019;16(4):957–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kim A, García‐García E, Straccia M, et al. Reduced fractalkine levels lead to striatal synaptic plasticity deficits in Huntington disease. Front Cell Neurosci 2020;14:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable ‐ no new data generated, or the article describes entirely theoretical research