

Abstract
Objective
The classical description of Dravet syndrome, the prototypic developmental and epileptic encephalopathy, is of a normal 6‐month‐old infant presenting with a prolonged, febrile, hemiclonic seizure and showing developmental slowing after age 1 year. SCN1A pathogenic variants are found in >80% of patients. Many patients have atypical features resulting in diagnostic delay and inappropriate therapy. We aimed to provide an evidence‐based definition of SCN1A‐Dravet syndrome in readiness for precision medicine trials.
Methods
Epilepsy patients were recruited to the University of Melbourne Epilepsy Genetics Research Program between 1995 and 2020 by neurologists from around the world. Patients with SCN1A pathogenic variants were reviewed and only those with Dravet syndrome were included. Clinical data, including seizure and developmental course, were analyzed in all patients with SCN1A‐Dravet syndrome.
Results
Two hundred and five patients were studied at a median age of 8.5 years (range 10 months to 60 years); 25 were deceased. The median seizure‐onset age was 5.7 months (range 1.5–20.6 months). Initial seizures were tonic‐clonic (52%) and hemiclonic (35%), with only 55% being associated with fever. Only 34% of patients presented with status epilepticus (seizure lasting ≥30 minutes). Median time between first and second seizure was 30 days (range 4 hours to 8 months), and seven patients (5%) had at least 6 months between initial seizures. Median ages at onset of second and third seizure types were 9.1 months (range 3 months–25.4 years) and 15.5 months (range 4 months–8.2 years), respectively. Developmental slowing occurred prior to 12 months in 27%.
Significance
An evidence‐based definition of SCN1A‐Dravet syndrome is essential for early diagnosis. We refine the spectrum of Dravet syndrome, based on patterns of seizure onset, type, and progression. Understanding of the full spectrum of SCN1A‐Dravet syndrome presentation is essential for early diagnosis and optimization of treatment, especially as precision medicine trials become available.
Keywords: developmental and epileptic encephalopathy, Dravet syndrome, genetics, phenotypic spectrum, SCN1A
Key Points.
Dravet syndrome has broader phenotypic limits than often recognized, highlighting the need for evidence‐based epilepsy syndrome definitions.
The initial seizure in Dravet syndrome is febrile in only 55% of patients, and 3% of patients never have seizures with fever.
More than 50% of patients with Dravet syndrome present with a tonic‐clonic seizure, whereas only 35% present with a hemiclonic seizure.
Only one‐third of patients present with status epilepticus, and 7% of patients never have status epilepticus.
Age at onset in Dravet syndrome can occur up to 19 months of age in patients with heterozygous SCN1A pathogenic variants.
1. INTRODUCTION
With the promise of precision medicine now becoming a reality, prompt diagnosis for patients with the most severe group of epilepsies, the developmental and epileptic encephalopathies (DEEs), is vital. 1 Dravet syndrome (OMIM 607208), due to pathogenic SCN1A variants in >80% patients, is the prototypic DEE. 1 , 2 , 3 , 4 The tantalizing success of emerging gene therapy for Scn1a murine models highlights the urgent need for prompt diagnosis of patients with Dravet syndrome. 5 Earlier diagnosis informs the optimization of anti‐seizure medicines and identifies patients in readiness for trials of precision therapies, which hold the promise of altering the trajectory of this devastating disorder. 5
A diagnosis of Dravet syndrome is frequently delayed, even as late as 3 years of age, as clinicians are cautious about making such a serious diagnosis until all the clinical features emerge. This often means the wrong anti‐seizure medicines are prescribed, which causes seizure exacerbation and adversely affects development. 6 With the advent of next‐generation sequencing, earlier molecular diagnosis can be achieved, even in the first year of life. However, although molecular identification of pathogenic variants in SCN1A is possible, it is not sufficient for a diagnosis of Dravet syndrome. Genotype‐phenotype correlation is essential given the broad spectrum of SCN1A‐related epilepsy syndromes, ranging from the self‐limited phenotypes within genetic epilepsy with febrile seizures plus (GEFS+) 7 to the profound impairment of early infantile SCN1A‐DEE. 8 Patients with GEFS+ would not warrant inclusion in a high‐risk precision medicine trial. Conversely, neither would patients with early infantile SCN1A‐DEE, which is caused by a gain‐of‐function mutation, 9 whereas Dravet syndrome is associated with loss‐of‐function mutations. Incorrect use of a Dravet precision medicine could be life‐threatening for these profoundly impaired patients.
Historically, epilepsy syndromes are described in an initial small cohort of patients and, as more patients are recognized, the phenotypic spectrum of that syndrome expands. 1 , 4 , 10 Despite many patients having typical clinical presentations, there are patients who have atypical features, often leading to a delay in diagnosis. These observations emphasize the need for evidence‐based diagnostic criteria based on large cohorts of patients.
We aimed to define the full phenotypic spectrum of SCN1A‐positive Dravet syndrome based on deep phenotyping of a large cohort of patients. These data will inform evidence‐based diagnostic criteria to enable early recognition of Dravet syndrome and facilitate involvement in precision medicine trials.
2. PATIENTS AND METHODS
2.1. Patients
Patients with SCN1A‐related epilepsies were recruited to the University of Melbourne Epilepsy Genetics Research Program between 1995 and 2020, referred by neurologists from Australia and internationally. Patients with SCN1A‐epilepsies, including Dravet syndrome, GEFS+, myoclonic‐atonic epilepsy, and early infantile SCN1A‐DEE, were reviewed by the authors who are all experts in diagnosis of Dravet syndrome. Only patients with Dravet syndrome were included, whereas patients with other phenotypes were excluded from the study. 7 , 8 We searched the literature using the term “Dravet syndrome” and the Medical Subject Headings (MeSH) thesaurus terms “phenotype” or “syndrome” before September 10, 2020 to identify published patients with Dravet syndrome associated with mosaic SCN1A pathogenic variants and patients with SCN1A mutations with onset after age 12 months.
The Austin Health Human Research Ethics Committee approved the study. Written informed consent was obtained from the patients, or their parents or legal guardians if the patients were younger than 18 years of age or had intellectual disability. Written patient consent forms are archived at the senior author's institution. Individual‐level data and clinical data have been deidentified for publication. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
2.2. Study design
We analyzed patient data by detailed in‐person clinical consultation where possible, and also interviewed parents using a validated seizure questionnaire 11 together with medical record review; prospective and retrospective data were analyzed. We determined the age, nature, duration, and temperature of the first seizure for each patient; the timing and duration of the first five consecutive seizures; and age at onset of additional seizure types that evolved over the patient's life, where possible. Seizure types were classified according to the 2017 International League Against Epilepsy (ILAE) classification. 1 , 12 Intellectual outcome was determined by neuropsychological assessment or from reports of developmental milestones and school performance. The age at which developmental plateauing or regression occurred was ascertained according to parental and clinician report. The pathogenicity of SCN1A variants was classified according to the American College of Medical Genetics guidelines. 13 Frequency calculations were determined using all patients with the available information for each feature to account for missing data.
3. RESULTS
Our research program included 261 patients with SCN1A pathogenic variants. Of these, 209 had Dravet syndrome, 44 had GEFS+, 7 one had epileptic spasms, 14 and 7 had early infantile SCN1A‐DEE. 8
We identified 205 of 209 patients with SCN1A‐Dravet syndrome with clinical data available for analysis (115 female, 56%). The cohort had a median age at study of 8.5 years (range 10 months to 60 years); 25 patients (12%) were deceased (Table S1).
3.1. Age at first seizure
Our cohort had a median age at seizure onset of 5.7 months (range 1.5 to 20.6 months, Figure 1A). Onset before 3 months of age occurred in 10 patients, including the youngest two patients at 6 to 7 weeks.
FIGURE 1.
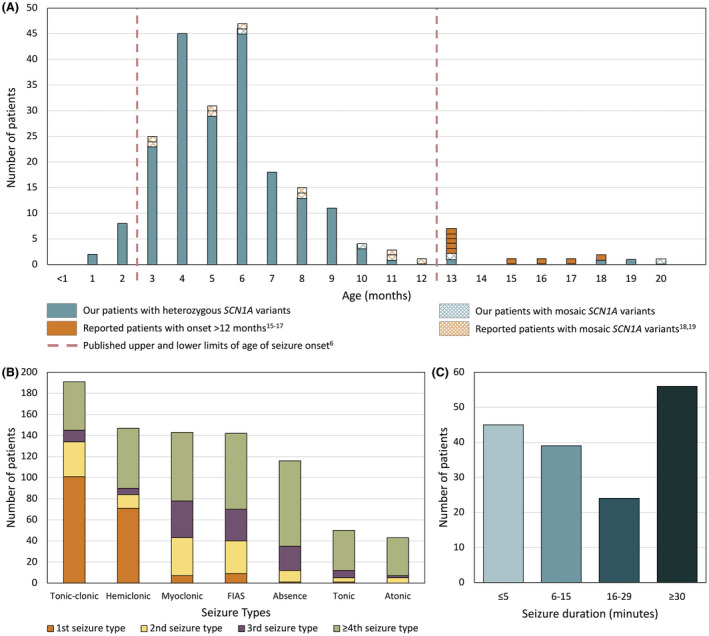
(A) Age at seizure onset in our cohort of 205 patients with SCN1A‐Dravet syndrome together with published patients with onset after 12 months or with pathogenic mosaic SCN1A variants. (B) First, second, third, and fourth or more seizure type to develop in patients. (C) Duration of first seizure in 164 patients with SCN1A‐Dravet syndrome
Five patients had seizure onset after 12 months: at 13, 13, 18, 19, and 20 months (Table S1). The patient with the latest onset at 20 months was mosaic for her SCN1A pathogenic variant (Patient 171; 22% p.Val1390Met allele frequency in lymphocyte DNA). In contrast, our three other patients with mosaicism had seizure onset at 6, 10, and 13 months.
Our literature search revealed an additional nine patients with onset between age 13 months and 18 months. 15 , 16 , 17 Conversely, in two articles reporting 10 patients with SCN1A mosaicism (13%–40% allele frequency) associated with Dravet syndrome, all had onset by age 12 months. 18 , 19 Taken together, our findings extend the seizure‐onset age in Dravet syndrome to 19 months in patients with heterozygous pathogenic SCN1A variants.
3.2. Initial seizure type
In 195 of 205 patients (95%), information regarding the initial seizure type was available. Contrary to the classical description of onset with hemiclonic seizures, the most common presentation was a tonic‐clonic seizure in 101 of 195 (52%, Table 1, Figure 1B). The second most frequent presentation was a hemiclonic seizure in 69 of 195 patients (35%), followed by rare patients with focal impaired awareness seizures (FIAS) (10/195, 5%), myoclonic seizures (7/195, 4%), tonic seizures (1/195, 0.5%), or absence seizures (1/195, 0.5%). Multiple seizure types occurred at onset in 6 of 195 patients (3%).
TABLE 1.
Clinical features of 205 patients with SCN1A‐Dravet syndrome
| Percentage | Number of patients | Number of patients with data available | ||
|---|---|---|---|---|
| Female | 56% | 115 | 205 | |
| Characteristics of first seizure | ||||
| Tonic‐clonic | 52% | 101 | 195 | |
| Hemiclonic | 35% | 69 | 195 | |
| Focal impaired awareness | 5% | 10 | 195 | |
| Myoclonic | 4% | 7 | 195 | |
| Tonic‐clonic + myoclonic | 2% | 4 | 195 | |
| Hemiclonic + myoclonic | 1% | 2 | 195 | |
| Absence | 0.5% | 1 | 195 | |
| Tonic | 0.5% | 1 | 195 | |
| Febrile | 55% | 102 | 184 | |
| Status epilepticus (≥30 min) | 34% | 56 | 164 | |
| Duration 5 min or shorter | 27% | 45 | 164 | |
| >1 seizure in 24 h | 11% | 21 | 188 | |
| Additional seizure types and median age of onset (range) | ||||
| Tonic‐clonic | 6 m (1.6 m−15 y) | 96% | 191 | 200 |
| Convulsive status epilepticus | 7.5 m (3 m−11.2 y) | 89% | 178 | 199 |
| Hemiclonic: | 6.1 m (1.5 m−9.8 y) | 73% | 147 | 201 |
| Alternating sides | 84% | 71 | 84 | |
| Always the same side | 16% | 13 | 84 | |
| Myoclonic | 12 m (2.5 m−10 y) | 72% | 143 | 200 |
| Focal impaired awareness | 18 m (3 m−35.7 y) | 72% | 142 | 196 |
| Absence | 20 m (6 m−7.6 y) | 58% | 116 | 200 |
| Tonic | 3 y (3.7 m−25.4 y) | 26% | 50 | 190 |
| Nonconvulsive status epilepticus | 3.6 y (3 m−25.5 y) | 24% | 43 | 178 |
| Atonic | 2 y (5 m−7 y) | 22% | 43 | 194 |
| Fever sensitive seizures | 97% | 174 | 179 | |
| Number of different seizure types | ||||
| One | 1.5% | 3 | 205 | |
| Two | 11% | 22 | 205 | |
| Three | 19% | 38 | 205 | |
| Four | 31% | 64 | 205 | |
| Five | 27% | 56 | 205 | |
| Six | 9% | 19 | 205 | |
| Seven | 1.5% | 3 | 205 | |
| Developmental characteristics | ||||
| Normal development before seizures | 98% | 194 | 198 | |
| Ambulant | 99% | 185 | 187 | |
| Verbal | 97% | 184 | 190 | |
| No intellectual disability | 1% | 1 | 143 | |
| Borderline IQ | 6% | 8 | 143 | |
| Mild intellectual disability | 22% | 32 | 143 | |
| Moderate intellectual disability | 27% | 38 | 143 | |
| Severe intellectual disability | 45% | 64 | 143 | |
| Autism spectrum disorder | 38% | 39 | 103 | |
| First epileptiform EEG abnormalities recorded | ||||
| Generalized spike‐wave | 44% | 61 | 139 | |
| Focal/multifocal | 42% | 59 | 139 | |
| Generalized spike‐wave + focal/multifocal | 14% | 19 | 139 | |
Abbreviations: EEG, electroencephalography; h, hours; IQ, intelligence quotient; m, months; min, minutes; y, years.
Fever is considered a hallmark of the initial seizure in Dravet syndrome; however, only 102 of 184 (55%) had a fever with their first seizure based on parent or clinician report. Formal documentation of the temperature by parents or medical staff was available for 52 of 205 patients (25%) and 36 of 52 (69%) had a temperature of ≥38 degrees Celsius. Vaccination‐related seizure onset (within 2 days of vaccination) occurred in 55 of 119 patients (46%), of whom 29 (53%) were febrile and 19 (34%) were afebrile; 7 (13%) did not have data regarding temperature at onset. It is important to note that 5 of 179 individuals (3%) never had seizures triggered by fever.
Infants with Dravet syndrome classically present with status epilepticus. We found that the first seizure lasted 30 minutes or more in only 56 of 164 (34%) of our cohort (Figure 1C, Table 1). The median duration of the first seizure across the cohort was 15 minutes (ranging from a few seconds to 3 hours), with 45 of 164 patients (27%) having initial seizures lasting 5 minutes or less (28/164, 17% patients ≤3 minutes). Of the remaining cohort, 39 of 164 (24%) had seizures lasting 6–15 minutes, whereas 24 of 164 (15%) had seizures lasting 16–29 minutes. One hundred and sixty‐seven of 188 patients (89%) had a single seizure in the first 24 hours.
3.3. Time between first five seizures
The median time between the first and second seizure was 30 days (data available for 132/205, 64%), with the time interval ranging from 4 hours to 8 months (Figure 2A). For the second to third seizure, the median interval was also 30 days (101/205, 49%), with a similarly broad range of 2 days to 11 months. The interval between the third and fourth seizure then decreased to a median of 21 days (79/205, 39%; range <1 day to 5.6 months), and 24 days between the fourth and fifth seizure (70/205, 34%; range 15 hours to 3 months).
FIGURE 2.
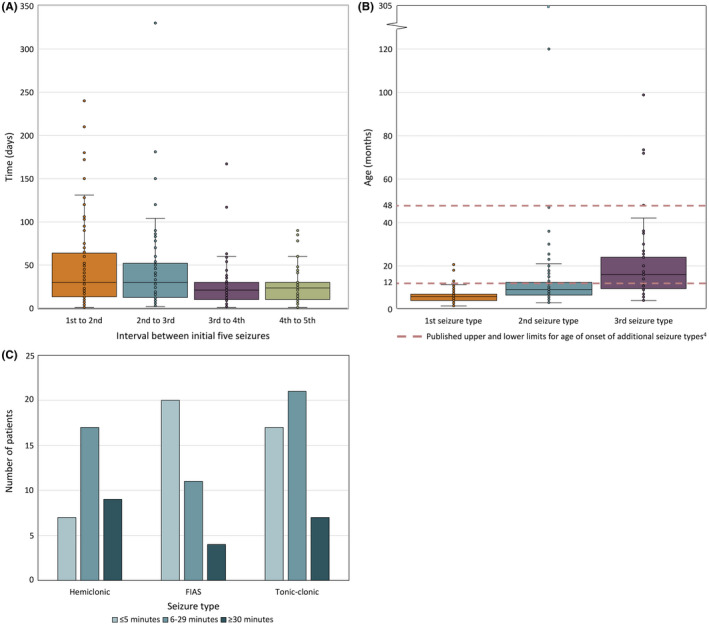
(A) Days between each of the first five seizures. Boxplots show individual data points (dots), median (solid line), interquartile range (boxes), and 1.5× the interquartile range (whiskers). (B) Age at onset of the first three seizure types. Boxplots show individual data points (dots), median (solid line), interquartile range (boxes), and 1.5× the interquartile range (whiskers). Dashed lines denote published upper and lower limits for age of onset of additional seizure types. 4 (C) Average duration of initial hemiclonic, focal impaired awareness seizures, and tonic‐clonic seizures in 33, 35, and 45 patients with available data, respectively
3.4. Evolution of additional seizure types
Hemiclonic seizures are considered the hallmark seizure type of Dravet syndrome, yet only 147 of 201 (73%) of our cohort had hemiclonic seizures (Table 1, Figure 1B). For the 84 of 147 individuals (57%) in whom lateralization was known, 13 of 84 (16%) had hemiclonic seizures involving only one side, whereas the remaining patients had hemiclonic seizures involving either side independently. The most frequent seizure type was tonic‐clonic, occurring in 191 of 200 patients (96%), followed by myoclonic seizures (143/200, 72%), FIAS (142/196, 72%), absence (116/200, 58%), tonic (50/190, 26%), and atonic seizures (43/194, 22%, Figure 1B).
Status epilepticus is frequent in Dravet syndrome. We found that, over time, 186 of 200 patients (93%) had status epilepticus; 178 of 199 (89%) had convulsive status epilepticus; and 43 of 178 (24%) had non‐convulsive status epilepticus.
Multiple seizure types are usual in Dravet syndrome; 202 of 205 patients (99%) developed multiple seizure types. Myoclonic, FIAS, and absence seizures occurred earlier in their disease course (Figure 1B). Of the cohort, 64 of 205 (31%) had four seizure types and 56 of 205 (27%) had five seizure types (range 1–7, Table 1). Tonic and atonic seizures typically occurred in patients with at least three other seizure types.
In Dravet syndrome, the onset of additional seizure types is classically described between 1 and 4 years of age. 4 In our cohort, the median age at onset of the second seizure type was 9.1 months (range 3 months to 25.4 years, data available for 130 of 202 patients [64%], Figure 2B) where 88 of 130 patients (68%) developed a second seizure type before 12 months. The median age at onset of the third seizure type was 15.5 months (range 4 months to 8.2 years, data available for 86 of 179 patients [48%]).
At the onset of tonic‐clonic seizures, hemiclonic seizures, and FIAS, we reviewed the duration of each seizure type. FIAS lasted ≤5 minutes in 20 of 35 patients (57%). In contrast, convulsive seizures were longer, lasting between 6 and 29 minutes in 21 of 45 patients (47%) having tonic‐clonic seizures and 17 of 33 individuals (52%) having hemiclonic seizures (Figure 2C).
3.5. Developmental course
Development in Dravet syndrome is described as normal in the first year of life followed by developmental slowing or regression. 4 In our cohort, 194 of 198 (98%) had normal development prior to seizure onset (Table 1). The remaining 4 of 198 patients (2%) had a history of developmental delay prior to seizure onset; all had truncation mutations, consistent with loss‐of‐function, and severe intellectual disability with seizure onset from 2–6.5 months.
Although the median age at developmental slowing or plateauing was 20 months (range 3 months to 5 years; data available for 104/202 patients [52%]), we found that 28 of 104 (27%) showed slowing in the first year of life, which is earlier than usually accepted (Figure 3). At the mild end, there were 2 of 104 patients (2%), who did not show definite slowing until 5 years of age. Lack of independent walking occurred in 2 of 187 patients (1%) and 6 of 190 patients (3%) were nonverbal. Eight patients had borderline intellect and one patient had emerging learning difficulties at the time of his death at age 5 years.
FIGURE 3.
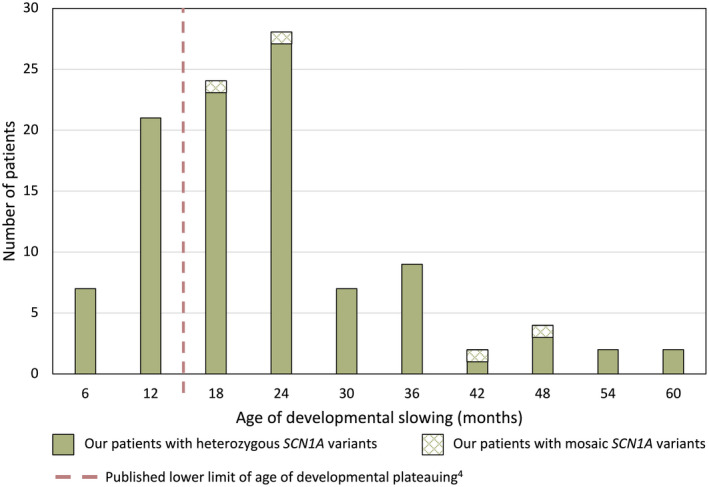
Age at onset of developmental plateauing in 106 patients with SCN1A‐Dravet syndrome
3.6. EEG findings
One hundred and thirty‐nine of 205 patients (68%) had EEG results available. The median age at first EEG showing epileptiform abnormalities was 26 months (range 4 months to 18 years). The first epileptiform EEG findings were generalized spike‐wave (GSW) in 61 of 139 patients (44%), focal or multifocal epileptiform abnormalities in 59 of 139 (42%), and GSW with focal or multifocal epileptiform abnormalities in 19 of 139 (14%, Table 1).
3.7. MRI findings
MRI brain reports, available for 189 of 205 patients (92%), were normal in 130 of 189 patients (69%). In the 59 with abnormalities, only 5 had hippocampal sclerosis, with the remainder having nonspecific findings (Table S1).
3.8. Mortality
Twenty‐five of 205 patients (12%) died at a median of 6.5 years (range 11 months to 39 years, Figure 4). Causes of death included sudden unexpected death in epilepsy (SUDEP) or probable SUDEP in 13 of 25 (52%), 20 cerebral edema in 5 of 25 (20%), 21 accidental drowning in 4 of 25 (16%), status epilepticus in 2 of 25 (8%) and one patient died of asphyxia due to aspiration of gastric contents, which may have occurred during a seizure.
FIGURE 4.
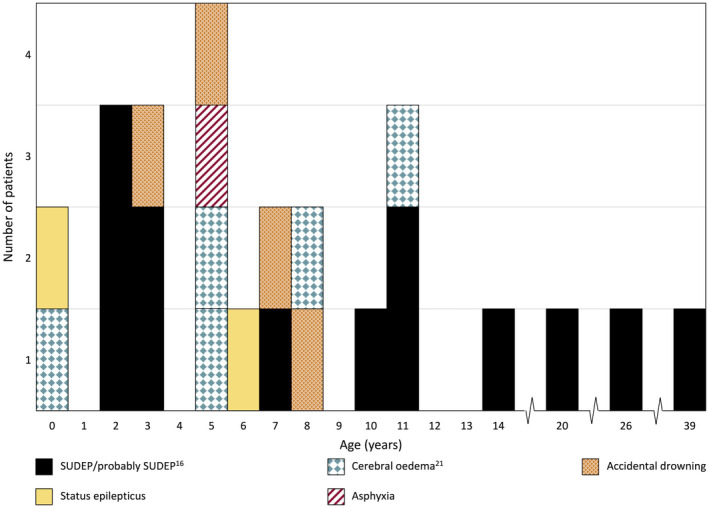
Age and cause of death in 25 patients from our cohort with SCN1A‐Dravet syndrome
4. DISCUSSION
The recent revolution in next generation sequencing has led to a remarkable increase in genetic diagnoses in individuals with DEEs. The prototypic DEE, Dravet syndrome, is associated with SCN1A pathogenic variants in 92% of our cohort of 228 patients with Dravet syndrome, whereas 8 patients had pathogenic variants in other genes (GABRA1 in 3 patients and one each of GABRG2, SCN1B, STXBP1, HCN1, and KCNA2) and 11 did not have a pathogenic variant identified. Understanding how Dravet syndrome presents is essential for early, accurate diagnosis of patients, which, together with an understanding of the natural history, is critical for evaluating the impact of novel therapies on disease course. Never has this been more pertinent than with the imminent promise of precision medicine trials, following recent success in murine models of Dravet syndrome. 5 As molecular studies are increasingly performed at seizure onset in infancy, interpreting their significance depends on the patient's phenotype. For an SCN1A pathogenic variant, the distinction between a benign self‐limited outcome such as GEFS+, 7 and a more severe outcome due to a loss‐of‐function mutation in Dravet syndrome or a gain‐of‐function mutation in early infantile SCN1A‐DEE is crucial. 8 , 9
Epilepsy syndromes are defined initially by recognition of seizure types that co‐occur, developmental course, EEG, and imaging features. 1 With increasing recognition of an epilepsy syndrome, the phenotypic spectrum is often expanded with relatively small series of cases. As more patients are identified, the limits of the diagnostic criteria may be drawn into question. Here, we carefully dissect phenotypic features in patients with SCN1A‐positive Dravet syndrome. By drawing on a large, well‐phenotyped cohort, we distil the key elements underpinning a diagnosis of Dravet syndrome and provide a diagnostic algorithm (Figure 5). This will allow clinicians to make an earlier diagnosis in patients who are not presenting with the heretofore considered classical phenotype.
FIGURE 5.
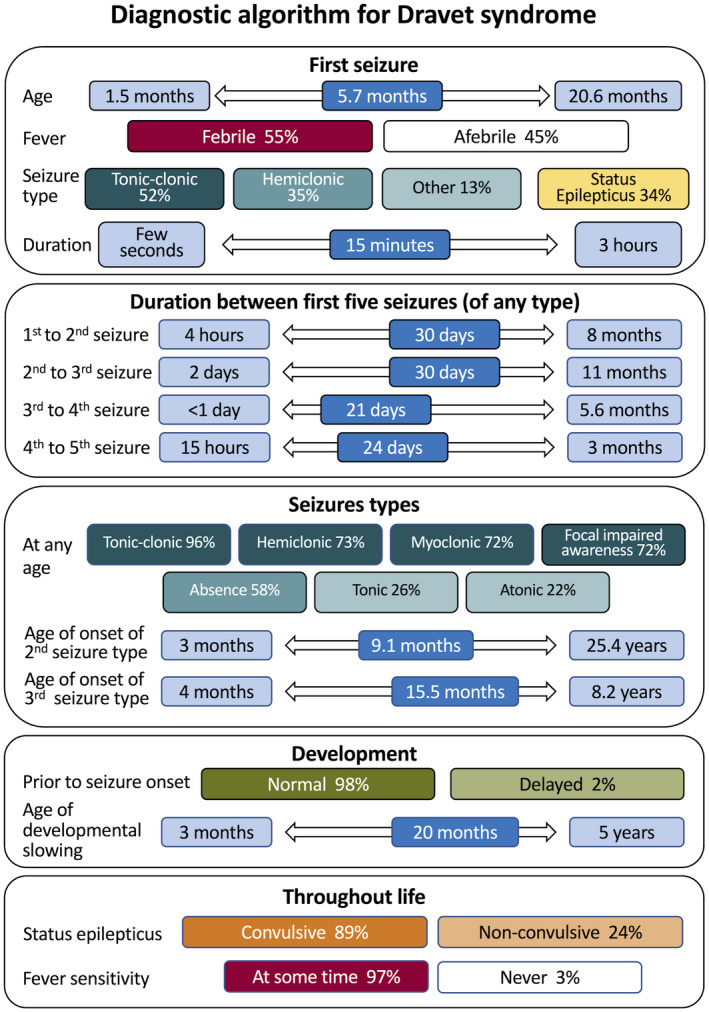
Diagnostic algorithm for SCN1A‐Dravet syndrome
Our findings crystallize the defining features of Dravet syndrome and dispel some of the rigid boundaries previously used. First, some authors state that age at onset must be in the first year of life. 22 In our cohort, five patients had seizure onset beyond the first year of life (13–20 months), confirming previous reports of nine patients (1/6, 2/26, 6/333) with onset from 13 to 18 months (Figure 1A). 15 , 16 , 17 Thus we extend the age at seizure onset in heterozygous SCN1A‐Dravet syndrome to 19 months. Of interest, mosaicism can be associated with later onset at 20 months (22% allele frequency) or under 1 year (13%–40% allele frequency). 18 , 19 Another of our patients with 30% mosaicism had onset at 10 months but a more severe seizure phenotype, with 600 convulsive seizures per year and moderate intellectual disability. Considering early seizure onset, our 10 patients with onset before age 3 months did not have the profound impairment or movement disorder that are characteristic of early infantile SCN1A‐DEE. 8
Second, and somewhat surprisingly, only 55% of our patients had a fever at seizure onset, contrasting with previous studies (57%–74% 22 , 23 , 24 ) and showing that the expectation of fever‐related seizures at onset is not met in almost half the patients with Dravet syndrome. In infants with afebrile seizure onset, other factors may trigger the first seizure, such as vaccination‐related onset, which we reported previously in one‐third of patients with Dravet syndrome. 25 In our cohort, 46% of patients had vaccination‐related seizure onset, 35% of whom were afebrile. Although ongoing seizures are often triggered by fever in Dravet syndrome, 4 , 25 3% of our cohort never had a seizure with fever.
Third, the most common type of seizure at onset was a tonic‐clonic seizure (52%), with hemiclonic seizures the second most frequent seizure type (35%). This contrasts with the classical description of an infant with Dravet syndrome presenting with a hemiclonic seizure. Sixteen percent of those who had hemiclonic seizures always had seizures on the same side. Furthermore, only 73% of our patients had hemiclonic seizures over the course of their disease, highlighting that hemiclonic seizures are not an essential feature of Dravet syndrome. 23 , 24 , 26
Fourth, when Dravet syndrome begins with nonconvulsive seizures, convulsive seizures are mooted to follow within days or weeks. 4 Of the 18 patients who had nonconvulsive seizures at onset, 7 had onset of convulsive seizures within 1 month. However, 10 of 18 patients did not have onset of convulsive seizures until 3–9 months after their initial nonconvulsive seizures (age at onset of convulsive seizures unknown for one patient).
Fifth, contrary to the teaching that infants with Dravet syndrome present with status epilepticus, we found that 66% of our cohort had seizures lasting less than 30 minutes at onset, mirroring the 75% and 66% of patients in Italian and French studies. 22 , 27 The median duration was 15 minutes overall, and 27% of patients presented with seizures lasting 5 minutes or less. Some authors suggest that Dravet syndrome is unlikely in patients with onset of status epilepticus after 18 months 28 ; however, 18 of our patients had their first episode of status epilepticus after age 18 months.
Sixth, the time between the first three seizures may be many months, thereby hampering consideration of a Dravet syndrome diagnosis, for which seizures are usually expected to occur more frequently. 4 The median time between the first and second seizure, and the second and third seizure was brief (30 days); however, the range was broad from 4 hours to 11 months. A diagnosis of Dravet syndrome in a patient whose first three seizures occurred at 6 and 14 and 22 months would often not be contemplated. The interval between the subsequent seizures tightened up considerably, with all patients having their fourth and fifth seizures within 6 months.
Seventh, additional seizure types in Dravet syndrome are usually stipulated as beginning between 1 and 4 years of age. 4 Surprisingly, we found an earlier median age at onset of a second seizure type at 9.1 months and the third seizure type at 15.5 months. This highlights the need for clinicians to ask carefully about specific seizure types that may have escaped recognition by the family. Equally though, the second seizure type may not emerge until as late as the third decade.
Although the usual diagnostic criteria for Dravet syndrome state that development is normal in the first year of life, 4 we found that 27% of our cohort showed developmental slowing by 12 months. Three patients were never normal, and a fourth patient had developmental delay from 3 months with seizure onset at 5 months. Conversely, slowing did not emerge until 5 years of age in two patients. Developmental criteria should therefore be less rigid than is currently accepted.
In terms of which seizure types occur at which age, we found some interesting differences from previous series. Myoclonic seizures before age 12 months are reportedly rare, 4 , 29 yet they were seen in 44 patients (22%). Onset of myoclonic seizures before 2 years is said to be a key clinical feature of Dravet syndrome, 30 but this was observed in only 18 patients, or 9% of our cohort. The median age at onset of focal impaired awareness seizures was earlier in our cohort (18 months) compared to others (33–35 months), 4 , 29 , 30 and although absence seizures are usually seen in children older than 2 years of age, 30 35 patients (18%) had onset before 2 years. We found that 26% of patients had tonic seizures with a median onset of 3 years (range 3.7 months to 25.4 years); previous reports suggest that tonic seizures occur in older patients with Dravet syndrome. 27 , 31 , 32 We acknowledge that diagnosis of tonic seizures is challenging without ictal EEG studies, particularly as tonic seizures often occur in sleep and may be very subtle. In addition, 22% of patients had atonic seizures with median onset at 2 years and the latest onset at 7 years.
In 1998 and 2003, German and Japanese authors reported a form of Dravet syndrome in which only convulsive seizures (tonic‐clonic and hemiclonic seizures) occurred using the syndromic names of Severe Idiopathic Generalized Epilepsy of Infancy with Generalized Tonic‐Clonic Seizures and Intractable Childhood Epilepsy with Generalized Tonic‐Clonic Seizures, respectively. 33 , 34 These patients follow a neurodevelopmental course similar to that of patients with classical Dravet syndrome, including status epilepticus, fever sensitivity, and drug‐resistant seizures. Only 8 of 205 (4%) of our patients had convulsive seizure types alone, akin to this specific Dravet phenotype.
Dravet syndrome carries a well‐established mortality of 17% by 20 years of age. 20 Our overall figure was 12% (25 patients) but this likely reflects the young median age of our cohort of 8.5 years. Definite or probable SUDEP occurred in 52% of our deaths, including three adults up to 39 years of age.
Our study has a number of limitations. The diagnosis of Dravet syndrome was based on an understanding of the gestalt of this complex disease by three independent experts. Some of the data were retrospective, which is the case for many patients assessed in studies of DEEs. Although we strived to obtain objective data from the time of presentation (eg, hospital emergency department notes, temperature recordings, seizure durations), we sometimes relied on parental history, which is subject to recall bias. Of interest, 69% of patients with formal documentation of temperature had a fever at seizure onset, whereas only 55% of patients were reported as febrile at seizure onset when data from parental report was included. Limited EEG data were available for review, as 68% patients had early EEG reports available, which reflects the evolution of the seizure disorder that often seems mild at onset.
These data offer an opportunity to understand the key features underpinning a diagnosis of Dravet syndrome due to a SCN1A pathogenic variant. The most frequent features were:
SCN1A pathogenic variant in all (inclusion criteria)
Hemiclonic or tonic‐clonic seizures (all patients)
Seizure onset <12 months (98%)
Normal development prior to seizure onset (98% patients with adequate data)
Developmental impairment prior to 5 years (98% patients with adequate data)
Fever‐sensitive seizures (97% patients with adequate data)
Tonic‐clonic seizures (96% patients with adequate data)
Intellectual disability (93% patients with adequate data)
Status epilepticus (93% patients with adequate data)
No single clinical feature was common to all patients, except the eventual occurrence of convulsive seizures (ie, hemiclonic or tonic‐clonic seizures), making stringent rules regarding the diagnosis of Dravet syndrome challenging to define. We determined that two of the following three features will capture all patients in our cohort with SCN1A‐Dravet syndrome: hemiclonic and/or tonic‐clonic seizures, fever‐sensitive seizures, and status epilepticus. It is noteworthy that only 115 of 205 patients (56%) presented with at least two of these three features at onset; yet by 12 months of age, 73% (149/205) and by 2 years of age, 79% (162/205) had developed two of these three features.
A diagnosis of Dravet syndrome is based on a complex gestalt of features, initially recognized by Charlotte Dravet in 1978. 35 Pediatric epileptologists identify patients with Dravet syndrome with a constellation of features, including those who may have features outside the classical criteria commonly taught. 4 , 30 In psychiatry, the Diagnostic and Statistical Manual of Mental Disorders, Fifth Revision (DSM‐5) emphasizes that diagnosis should allow for integration of diagnostic criteria with clinical judgement, where clinical experience results in a more nuanced understanding of a specific disease. 36 The DSM‐IV explicitly states that criteria need to serve as guidelines informed by clinical expertise rather than being applied in a “cookbook fashion.” 37 Indeed, a World Health Organization (WHO) survey of psychiatrists found maximal utility where diagnostic manuals allowed flexible guidance based on clinical judgment rather than fixed diagnostic criteria. 38 Here we interrogated a large cohort of patients with Dravet syndrome diagnosed by three clinical experts who understand the gestalt of this complex presentation. We provide data to delineate the broader limits of Dravet syndrome so that evidence‐based criteria can be developed and assist less‐expert clinicians in making an earlier Dravet syndrome diagnosis.
Our study highlights that epilepsy syndrome definitions need to be based on deep phenotyping of substantial numbers of patients with each specific genetic DEE. As the prototypic DEE, our work and that of others defining cohorts of patients with Dravet syndrome highlight the critical features that underpin a clinical diagnosis. It is notable that patients without all of the classical features of a syndrome need to be used to refine syndromic definitions to paint a true phenotypic picture of the disease. Never has this been more crucial and more relevant to each genetic DEE, as we face the need for early accurate diagnosis in preparation for precision medicine trials.
CONFLICT OF INTEREST
I.E. Scheffer has served on scientific advisory boards for UCB, Eisai, GlaxoSmithKline, BioMarin, Nutricia, Rogcon, Chiesi, Encoded Therapeutics, Xenon Pharmaceuticals, and Knopp Biosciences; has received speaker honoraria from GlaxoSmithKline, UCB, BioMarin, Biocodex, and Eisai; has received funding for travel from UCB, Biocodex, GlaxoSmithKline, Biomarin, and Eisai; has served as an investigator for Zogenix, Zynerba, Ultragenyx, GW Pharma, UCB, Eisai, Anavex Life Sciences, Ovid Therapeutics, Epigenyx, Encoded Therapeutics and Marinus; and has consulted for Zynerba Pharmaceuticals, Atheneum Partners, Ovid Therapeutics, Care Beyond Diagnosis, Epilepsy Consortium, and UCB. She may accrue future revenue on a pending patent WO2009/086591 (filed: 2008); has a patent for SCN1A testing held by Bionomics Inc and licensed to various diagnostic companies (WO/2006/133508), and has a patent for a molecular diagnostic/therapeutic target for benign familial infantile epilepsy (BFIE) [PRRT2] WO/2013/059884. W. Li and A.L. Schneider declare no potential conflict of interest. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
Table S1
ACKNOWLEDGMENTS
We thank the patients and their families for participating in our research. We thank Sophie Russ‐Hall for assistance with manuscript preparation. Funding was gratefully received from the National Health and Medical Research Council of Australia, Australian Medical Research Future Fund and the Australian Epilepsy Research Fund. Support for open access publication was provided by Fundación Síndrome de Dravet [grant number FSD‐OASPXI‐II].
Li W, Schneider AL, Scheffer IE. Defining Dravet syndrome: An essential pre‐requisite for precision medicine trials. Epilepsia. 2021;62:2205–2217. 10.1111/epi.17015
Wenhui Li and Amy L Schneider contributed equally to the manuscript
REFERENCES
- 1. Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58:512–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Claes L, Del‐Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. De novo mutations in the sodium‐channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet. 2001;68:1327–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McTague A, Howell KB, Cross JH, Kurian MA, Scheffer IE. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 2016;15:304–16. [DOI] [PubMed] [Google Scholar]
- 4. Dravet C, Bureau M, Oguni H, Cokar O, Guerrini R, Wolf P. Dravet syndrome (previously severe myoclonic epilepsy in infancy). In: Bureau MPG, Dravet C, Delgado‐Escueta AV, Guerrini R, Tassinari CA, Thomas P, eds. Epileptic Syndromes in Infancy, Childhood and Adolescence, 6th edn. Arcueil, France: John Libbey Eurotext; 2019: 139–171. [Google Scholar]
- 5. Han Z, Chen C, Christiansen A, Ji S, Lin Q, Anumonwo C, et al. Antisense oligonucleotides increase Scn1a expression and reduce seizures and SUDEP incidence in a mouse model of Dravet syndrome. Sci Transl Med. 2020;26:12. [DOI] [PubMed] [Google Scholar]
- 6. de Lange IM, Gunning B, Sonsma ACM, van Gemert L, van Kempen M, Verbeek NE, et al. Influence of contraindicated medication use on cognitive outcome in Dravet syndrome and age at first afebrile seizure as a clinical predictor in SCN1A‐related seizure phenotypes. Epilepsia. 2018;59:1154–65. [DOI] [PubMed] [Google Scholar]
- 7. Zhang YH, Burgess R, Malone JP, Glubb GC, Helbig KL, Vadlamudi L, et al. Genetic epilepsy with febrile seizures plus: Refining the spectrum. Neurology. 2017;19(89):1210–9. [DOI] [PubMed] [Google Scholar]
- 8. Sadleir LG, Mountier EI, Gill D, Davis S, Joshi C, DeVile C, et al. Not all SCN1A epileptic encephalopathies are Dravet syndrome: early profound Thr226Met phenotype. Neurology. 2017;5(89):1035–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Berecki G, Bryson A, Terhag J, Maljevic S, Gazina EV, Hill SL, et al. SCN1A gain of function in early infantile encephalopathy. Ann Neurol. 2019;85:514–25. [DOI] [PubMed] [Google Scholar]
- 10. Dravet C, Roger J, Bereau M, Dalla Bernardina B. Myoclonic epilepsies in childhood. In: Akimoto H, Kazamatsuri H, Seino M, Ward A, eds. Advances in Epileptology: XIIIth Epilepsy International Symposium. New York, NY: Raven Press; 1982:135–140. [Google Scholar]
- 11. Reutens DC, Howell RA, Gebert KE, Berkovic SF. Validation of a questionnaire for clinical seizure diagnosis. Epilepsia. 1992;33(6):1065–71. [DOI] [PubMed] [Google Scholar]
- 12. Fisher RS, Cross JH, French JA, Higurashi N, Hirsch E, Jansen FE, et al. Operational classification of seizure types by the International League Against Epilepsy: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58:522–30. [DOI] [PubMed] [Google Scholar]
- 13. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wallace RH, Hodgson BL, Grinton BE, Gardiner RM, Robinson R, Rodriguez‐Casero V, et al. Sodium channel alpha1‐subunit mutations in severe myoclonic epilepsy of infancy and infantile spasms. Neurology. 2003;23(61):765–9. [DOI] [PubMed] [Google Scholar]
- 15. Catarino CB, Liu JY, Liagkouras I, Gibbons VS, Labrum RW, Ellis R, et al. Dravet syndrome as epileptic encephalopathy: evidence from long‐term course and neuropathology. Brain. 2011;134:2982–3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Depienne C, Trouillard O, Saint‐Martin C, Gourfinkel‐An I, Bouteiller D, Carpentier W, et al. Spectrum of SCN1A gene mutations associated with Dravet syndrome: analysis of 333 patients. J Med Genet. 2009;46:183–91. [DOI] [PubMed] [Google Scholar]
- 17. Kearney JA, Wiste AK, Stephani U, Trudeau MM, Siegel A, RamachandranNair R, et al. Recurrent de novo mutations of SCN1A in severe myoclonic epilepsy of infancy. Pediatr Neurol. 2006;34:116–20. [DOI] [PubMed] [Google Scholar]
- 18. de Lange IM, Koudijs MJ, van't Slot R, Gunning B, Sonsma ACM, van Gemert L, et al. Mosaicism of de novo pathogenic SCN1A variants in epilepsy is a frequent phenomenon that correlates with variable phenotypes. Epilepsia. 2018;59:690–703. [DOI] [PubMed] [Google Scholar]
- 19. Nakayama T, Ishii A, Yoshida T, Nasu H, Shimojima K, Yamamoto T, et al. Somatic mosaic deletions involving SCN1A cause Dravet syndrome. Am J Med Genet A. 2018;176:657–62. [DOI] [PubMed] [Google Scholar]
- 20. Cooper MS, McIntosh A, Crompton DE, McMahon JM, Schneider A, Farrell K, et al. Mortality in Dravet syndrome. Epilepsy Res. 2016;128:43–7. [DOI] [PubMed] [Google Scholar]
- 21. Myers KA, McMahon JM, Mandelstam SA, Mackay MT, Kalnins RM, Leventer RJ, et al. Fatal cerebral edema with status epilepticus in children with Dravet syndrome: report of 5 cases. Pediatrics. 2017;139(4):e20161933. [DOI] [PubMed] [Google Scholar]
- 22. Cetica V, Chiari S, Mei D, Parrini E, Grisotto L, Marini C, et al. Clinical and genetic factors predicting Dravet syndrome in infants with SCN1A mutations. Neurology. 2017;14(88):1037–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brunklaus A, Ellis R, Reavey E, Forbes GH, Zuberi SM. Prognostic, clinical and demographic features in SCN1A mutation‐positive Dravet syndrome. Brain. 2012;135:2329–36. [DOI] [PubMed] [Google Scholar]
- 24. Ragona F, Brazzo D, De Giorgi I, Morbi M, Freri E, Teutonico F, et al. Dravet syndrome: early clinical manifestations and cognitive outcome in 37 Italian patients. Brain Dev. 2010;32:71–7. [DOI] [PubMed] [Google Scholar]
- 25. McIntosh AM, McMahon J, Dibbens LM, Iona X, Mulley JC, Scheffer IE, et al. Effects of vaccination on onset and outcome of Dravet syndrome: a retrospective study. Lancet Neurol. 2010;9:592–8. [DOI] [PubMed] [Google Scholar]
- 26. Xu X, Zhang Y, Sun H, Liu X, Yang X, Xiong H, et al. Early clinical features and diagnosis of Dravet syndrome in 138 Chinese patients with SCN1A mutations. Brain Dev. 2014;36:676–81. [DOI] [PubMed] [Google Scholar]
- 27. Nabbout R, Chemaly N, Chipaux M, Barcia G, Bouis C, Dubouch C, et al. Encephalopathy in children with Dravet syndrome is not a pure consequence of epilepsy. Orphanet J Rare Dis. 2013;13(8):176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Le Gal F, Lebon S, Ramelli GP, Datta AN, Mercati D, Maier O, et al. When is a child with status epilepticus likely to have Dravet syndrome? Epilepsy Res. 2014;108:740–7. [DOI] [PubMed] [Google Scholar]
- 29. Oguni H, Hayashi K, Awaya Y, Fukuyama Y, Osawa M. Severe myoclonic epilepsy in infants–a review based on the Tokyo Women's Medical University series of 84 cases. Brain Dev. 2001;23:736–48. [DOI] [PubMed] [Google Scholar]
- 30. Wirrell EC, Laux L, Donner E, Jette N, Knupp K, Meskis MA, et al. Optimizing the diagnosis and management of Dravet syndrome: recommendations From a North American Consensus Panel. Pediatr Neurol. 2017;68:18–34.e13. [DOI] [PubMed] [Google Scholar]
- 31. Losito E, Kuchenbuch M, Chemaly N, Laschet J, Chiron C, Kaminska A, et al. Age‐related "Sleep/nocturnal" tonic and tonic clonic seizure clusters are underdiagnosed in patients with Dravet Syndrome. Epilepsy Behav. 2017;74:33–40. [DOI] [PubMed] [Google Scholar]
- 32. Nabbout R, Desguerre I, Sabbagh S, Depienne C, Plouin P, Dulac O, et al. An unexpected EEG course in Dravet syndrome. Epilepsy Res. 2008;81:90–5. [DOI] [PubMed] [Google Scholar]
- 33. Doose H, Lunau H, Castiglione E, Waltz S. Severe idiopathic generalized epilepsy of infancy with generalized tonic‐clonic seizures. Neuropediatrics. 1998;29:229–38. [DOI] [PubMed] [Google Scholar]
- 34. Fujiwara T, Sugawara T, Mazaki‐Miyazaki E, Takahashi Y, Fukushima K, Watanabe M, et al. Mutations of sodium channel alpha subunit type 1 (SCN1A) in intractable childhood epilepsies with frequent generalized tonic‐clonic seizures. Brain. 2003;126:531–46. [DOI] [PubMed] [Google Scholar]
- 35. Dravet C. Les epilepsies Graves De L'enfant. Vie Med. 1978;59:543–8. [Google Scholar]
- 36. American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders (DSM‐5®). Washington, DC: American Psychiatric Publishing; 2013. [Google Scholar]
- 37. American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (Dsm‐IV). Washington, DC: American Psychiatric Publishing, Incorporated; 1994. [Google Scholar]
- 38. Reed GM, Mendonça Correia J, Esparza P, Saxena S, Maj M. The WPA‐WHO Global Survey of Psychiatrists' attitudes towards mental disorders classification. World Psychiatry. 2011;10:118–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1