

Abstract
The implementation of high‐throughput sequencing (HTS) technologies in research and diagnostic laboratories has linked many new genes to rare bleeding, thrombotic, and platelet disorders (BTPD), and revealed multiple genetic variants linked to those disorders, many of them being of uncertain pathogenicity when considering the accepted evidence (variant consequence, frequency in control datasets, number of reported patients, prediction models, and functional assays). The sequencing effort has also resulted in resources for gathering disease‐causing variants associated with specific genes, but for BTPD, such well‐curated databases exist only for a few genes. On the other hand, submissions by individuals or diagnostic laboratories to the variant database ClinVar are hampered by the lack of a submission process tailored to capture the specific features of hemostatic diseases. As we move toward the implementation of HTS in the diagnosis of BTPD, the Scientific and Standardization Committee for Genetics in Thrombosis and Haemostasis has developed and tested a REDCap‐based interface, aimed at the community, to submit curated genetic variants for diagnostic‐grade BTPD genes. Here, we describe the use of the interface and the initial submission of 821 variants from 30 different centers covering 14 countries. This open‐access variant resource will be shared with the community to improve variant classification and regular bulk data transfer to ClinVar.
Keywords: blood, genes, hemorrhage, mutation, platelets, thrombosis
1. INTRODUCTION
High‐throughput sequencing (HTS) technologies have entered the field of bleeding, thrombotic, and platelet disorders (BTPD) 1 , 2 , 3 , 4 and, following guidelines for sequence variant interpretation, clinical laboratories perform genetic testing for diagnostics and improved clinical management. Accurate genetic diagnosis of BTPD is essential for differential diagnosis; for guiding treatment in the case of disorders sharing the same laboratory platelet phenotype, such as platelet‐type von Willebrand disease and von Willebrand disease type 2B; 5 or for determining prognosis and informing patient management when a phenotype‐‐genotype correlation exists, such as in Hermansky‐Pudlak syndrome 6 or MYH9‐related disorders. 7 The identification of a genetic variant might have a strong impact on a patient’s life, for example when a variant is found in RUNX1, ETV6, or ANKRD26 genes associated with increased leukemic risk, and may represent an unsolicited finding. 8
An important first step in setting up an accredited diagnostic HTS panel test for BTPD is to define the set of diagnostic‐grade (or TIER1) genes; this task was taken up by the Scientific and Standardization Committee (SSC) for Genetics in Thrombosis and Haemostasis (GinTH), which resulted in an initial list of 91 genes. 9 This list is re‐assessed by the SSC‐GinTH at the yearly International Society on Thrombosis and Haemostasis (ISTH) meeting and currently contains 93 TIER1 genes (www.isth.org/page/GinTh_GeneLists). This webpage also contains a list of TIER2 genes that currently lack sufficient evidence to be considered proven diagnostic‐grade genes, mostly because they were typically discovered in single or small pedigrees and still require confirmation studies in independent pedigrees and functional assays or a mouse model.
The implementation of HTS testing for BTPD diagnostics has led to an explosion of genetic information, which required improved variant curation and data sharing. The American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) guidelines proposed a variant classification system using five levels: benign, likely benign, variant of uncertain significance (VUS), likely pathogenic, and pathogenic. Variants in the pathogenic and likely pathogenic categories have clinical significance and VUS might subsequently develop clinical significance if they become re‐classified into one of these categories. 10 Data sharing of genotype and phenotype information is essential to provide curated evidence for genetic variants, especially for VUS, which cannot be conclusively assigned to either a benign or pathologic category on the basis of information often obtained from a case report. Although several gene‐specific variant databases have arisen over time for BTPD, such as those from the European Association for Haemophilia and Allied Disorders (EAHAD) for F5, F7, F8, F9, F10 and VWF deficiencies (https://dbs.eahad.org) and the Glanzmann Thrombasthenia Database (https://glanzmann.mcw.edu), such databases do not exist for most BTPD genes. In contrast, variant information for all genes is available from the commercial Human Gene Mutation Database (HGMD), although this database doesn’t use the ACMG/AMP criteria for variant classification and is populated with false‐positive pathogenic variants. 2 The free‐access version of HGMD database does not include variants published in the last 3 years. Since 2013, the open‐access ClinVar (Clinically Relevant Variations) archival database aggregates information about genomic variation and the relationship to human disease. However, ClinVar contains numerous variants that have been submitted by separate labs applying the variant classification criteria differently, resulting in heterogeneous pathogenicity assignments, some being classified as both benign and pathogenic. It is the task of the ClinGen Variant Curation Expert Panels to define gene‐specific modification to the general ACMG/AMP variant curation rules and re‐evaluate ClinVar variants based on the modified rules, as currently done for ITGA2B and ITGB3 variants causing Glanzmann thrombasthenia. 11 ClinGen is a National Institutes of Health (NIH)‐funded research consortium that aims to build a central resource that defines the clinical relevance of genes and variants for use in clinical diagnostics. The ClinGen clinical domain working group for hemostasis and thrombosis (https://clinicalgenome.org/working‐groups/clinical‐domain/hemostasis‐thrombosis/) will oversee gene and variant curation activities for BTPD. Because these curation processes are expected to take several years for all BTPD genes, it is critical in the meantime to capture all variants identified by HTS activities in our community. Variant data sharing is recognized as a real necessity, especially for rare disorders; however, clinical laboratories might not have the time or interest to deposit their rare causal variants to public repositories. The submission of variants to the ClinVar database is laborious and many clinical laboratories using HTS do not submit identified variants. In addition, for established gene‐disease associations, causal variants identified in patients are not routinely published, and without submission to a public database, this information, essential for evidence‐based variant interpretation, is not available for clinical diagnostics. Finally, some general laboratories lack the appropriate knowledge in BPTD and submit variants to ClinVar without sufficient or even no phenotype information.
To aid variant sharing, the SSC‐GinTH has developed “GoldVariants,” an open‐access and user‐friendly tool to capture variants. It has a REDCap‐based interface and has been designed for a rapid and easy submission of BTPD variants, which will then be deposited in ClinVar half‐yearly. Here we describe the development and testing of GoldVariants by 30 expert centers, resulting in the deposition of 821 variants in ClinVar.
2. METHODS
2.1. GoldVariants project and resource
The ISTH GoldVariants project was an initiative of the SSC GinTH and was supported by other SSCs (Table S1 in supporting information). One aim in this project was the development of the GoldVariants resource. Experts with publications related to HTS for diagnostics of BTPDs were contacted and the responders are the centers involved in this study. These experts participated in at least one of the two SSC‐GinTH workshops (ISTH 2019 or virtual workshop in September 2020) organized to develop and test the GoldVariants resource. The resource, built around REDCap, is maintained by ISTH and will be open access for the scientific community. Transfer to ClinVar will be coordinated by GinTH.
2.2. GoldVariants workflow
The GoldVariants resource is an intermediate platform for the deposition of genetic variants to ClinVar. The workflow is presented in Figure S1 in supporting information. Briefly, variants are submitted by the community and aggregated in a single file. Twice a year the file is “frozen,” versioned, and deposited in ClinVar and on the ISTH website.
3. RESULTS
3.1. The ISTH GoldVariants resource: A REDCap–based interface to submit rare variants for BTPD
The aim was to deliver an ISTH community‐based REDCap solution for the safe sharing of genotype and phenotype data among clinicians, scientists, and the public to support international collaboration and improve variant classification for BTPD. The GoldVariants submission interface can be accessed via https://is.gd/GinTH_VariantCapture (or indirectly via the TIER1 gene list webpage www.isth.org/page/GinTh_GeneLists). Genetic variants can be submitted in two ways: (1) uploading a single variant via the interface by filling in a form, or (2) uploading a batch of variants by completing a worksheet (containing examples) that can be submitted via email. Table 1 provides an overview of the mandatory and optional information requested in the five sections: submitter, gene, variant, clinical significance, and phenotype‐related patient information. These fields are in line with the requirements for variant deposition in ClinVar. The only compulsory information regarding the phenotype is the “disorder name,” while other clinical or laboratory data can be entered under the optional box “clinical features.” As stated above, sufficient phenotype information is critical for variant interpretation.
TABLE 1.
Information required for the variant submission interface
| Items | Mandatory information | Optional information |
|---|---|---|
| 1. Submitter |
|
|
| 2. Gene |
|
|
| 3. Variant |
Single nucleotide variant/indel
|
|
|
Structural variant
|
||
| 4. Clinical significance |
|
|
| 5. Patient information |
|
|
Abbreviations: ACMG, American College of Medical Genetics and Genomics; AMP, Association for Molecular Pathology; VUS, variant of uncertain significance.
3.2. GoldVariants test phase with the submission of 821 genetic variants
So far, 821 (611 unique) variants have been submitted to the GoldVariants resource since it opened after July 2020. Submissions originate from 30 different expert centers in 14 countries across the world (North and South America, Europe, Asia and Oceania; Table S2 in supporting information). Submissions ranged from a single variant, via the online portal, to 249 variants, via the batch upload. The majority of the variants were single nucleotide variants or small insertion‐deletions (indels), with the remaining variants submitted being large deletions (Figure 1A). A total of 18 deletions have been submitted in 12 different genes with 3 deletions in both VWF and RUNX1 as most common. These ranged from single exon deletions to large deletions encompassing multiple genes that are relatively simple to detect using HTS data, compared to complex structural variants or those not detectable using HTS methods (e.g., F8 intron 1 and 22 inversions). 1 Out of the 821 variants, 81 were identified as homozygous; 32 as hemizygous; and 702 were heterozygous, of which 57 were in compound heterozygosity.
FIGURE 1.
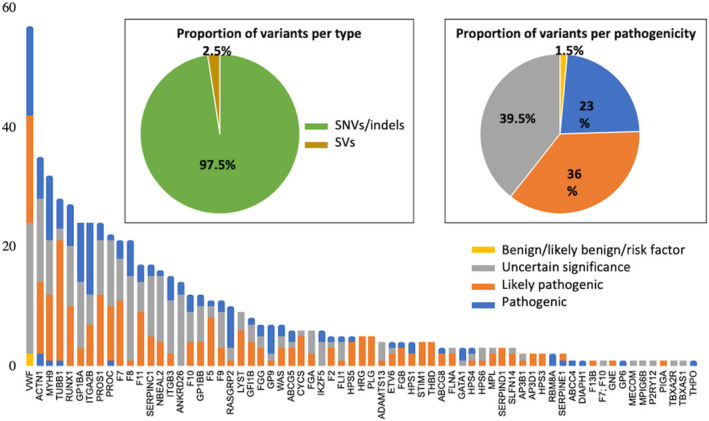
Summary of the variant types, pathogenicity, and genes. A, Proportion of unique variants per type single nucleotide variants (SNVs)/indels or structural variants (SVs). B, Number of unique variants, per gene and per pathogenicity and (insert) proportion of unique variants that are benign/likely benign/risk factor, variant of uncertain significance, likely pathogenic, or pathogenic
The majority of the 611 unique variants have been classified by the submitter as pathogenic (23%), likely pathogenic (36%), or VUS (39.5%), while 1.5% have been defined as risk factors or benign (Figure 1B, insert). Only 6 variants were submitted as likely benign and benign. We especially encourage the submission of variants listed in ClinVar as VUS or (likely) pathogenic where additional evidence obtained through co‐segregation or functional studies suggests reclassification as (likely) benign. Variants were detected in 68 out of the 93 diagnostic‐grade genes (Version ISTH2020.1, 9 ), with the number of variants per gene varying from nearly 60 for VWF to a single variant detected in 14 different genes (Figure 1B).
3.3. Variant classification differences
A total of 17 single nucleotide variants (SNV) in 10 genes were submitted by more than one center and with different pathogenicity scores, illustrating the difficulty of the variant classification process even when using the same ACMG/AMP criteria (Table S3 in supporting information). This is also visible in the “ClinVar Interpretation” column of the table, providing the different pathogenicities for those variants in ClinVar. In addition, four SNVs (ANKRD26 c.‐126T>C, ITGA2B R1026W, PROS1 S501P, and VWF Y1584C) submitted by the same center received different classification scores reflecting the evolution of interpretation as evidence for pathogenicity accumulates over time (usually based on the occurrence of the same variant in unrelated patients with similar phenotypes or familial co‐segregation studies). Although the aim of this project is not to reclassify submitted variants, Table S3 shows the comparison with the current ClinVar variant interpretation and, if available, the information obtained from gene‐specific databases. By submitting these variants to ClinVar, ClinGen variant curators can classify these variants with similar scores. If more unrelated patients carry the same variant and share phenotypes, a VUS variant can become clinically significant. Providing sufficient clinical and laboratory phenotype information associated with each VUS variant is important for variant reclassification processes. Variant deposition into ClinVar through the GoldVariants resource will aid this curation work.
3.4. Accessing submitted variants and future updates
Variants will be deposited in ClinVar, but with indication of the submitter and their laboratory, as well as any associated publication describing the patients. This will help patient tracking in order to mitigate patients being represented more than once within the ClinVar database. Deposition will be performed twice a year, under the name “ISTH‐SSC Genomics in Thrombosis and Hemostasis” (Table S2). GoldVariants will also be accessible as a downloadable file from the TIER1 gene list ISTH webpage (www.isth.org/page/GinTh_GeneLists), which will also be updated twice a year.
4. DISCUSSION
The ISTH GoldVariants resource currently contains a total of 821 variants submitted by 30 different expert centers, using different gene screening methods for diagnosis of patients with BTPD. GoldVariants is now open for all submissions from the community and aims to deposit variants to ClinVar twice a year (July/December). Each submission center is responsible for the variant information and no additional variant curation steps will be performed after the initial variant submission to the GoldVariants resource or before their deposition to ClinVar. Each variant carries a link to the initial submitter to allow for contact in case additional information about that particular variant is required. One shortcoming of all current variant databases is the fact that, to preserve patient anonymity, no unique patient identifiers are assigned.
It remains important to submit variants that are already present in ClinVar or the GoldVariants resource if identified in another patient (or family). Multiple submissions of the same variant in different families with similar laboratory or clinical phenotypes will allow VUS variant reclassification to (likely) pathogenic where segregation with a specific trait is observed or (likely) benign when lack of segregation is demonstrated. The GoldVariants resource will be freely accessible for the community via the ISTH website (Figure 1). This resource will facilitate future variant curation work. Though ClinGen is not part of this project, the GoldVariants submission to ClinVar can support the variant curation work undertaken by the different ClinGen working groups for coagulation, platelet, and thrombotic disorders. To try and understand why the same variant identified in patients with similar phenotypes have different pathogenicity classifications by different laboratories, we will ask submitters to list which ACMG/AMP criteria were used; however, this is not mandatory for submission. A total of 56 variants (29 variants in 11 platelet genes and 27 variants in 10 coagulation genes) were submitted from at least two unrelated index patients, and such information allows for variant reclassification; this is of prime importance especially for VUS, to improve curation and appropriate counseling of patients with such variants in the future.
The next effort from this SSC‐GinTH initiative should be to capture variants in TIER2 genes that currently lack evidence to be considered as proven diagnostic‐grade. 9 Genotype‐‐phenotype information related to variants in these genes is one way to upgrade these genes to TIER1 status. Other support can come from specific functional assays or cellular and animal models. Variant curation is not only assisted by information available from pathogenic databases but also from knowledge about their population allele frequency. 2 The population size in databases that provide such information is also rapidly growing with the largest and most widely used database of variant allele frequency being gnomAD (V2 dataset in GRCh37 includes 125,748 exomes and 15,708 genomes and V3.1 dataset in GRCh38 includes 76,156 genomes). 12 The UK Biobank has released 200,000 exomes in October 2020 (interim analysis of these data in Van Hout et al. 13 ) and is expected to release variant information from 200,000 genomes by the end of 2021. In contrast to the gnomAD database, some phenotypic information about participants in the UK Biobank study will be available. These expanding population datasets will also significantly improve variant classification for rare diseases.
CONFLICTS OF INTEREST
None of the authors have a conflict of interest related to this manuscript.
AUTHOR CONTRIBUTIONS
KM, KD, SB, and KF designed the GoldVariants submission platform. SB implemented the platform. KM, KD, MCMK, JMB, LB, EL, KG, NVM, MO, JPB, JR, HS, DAT, WHO, and KF have participated in the two SSC‐GinTH workshops to develop the GoldVariants resource and have submitted variants. KM analyzed the variants and KM and KF will coordinate their deposition in ClinVar. KD, KG, LB, DAT, and KF are SSC‐GinTH co‐chairs that were involved in this SSC project. All authors contributed to the paper writing and the critical review of the paper. All have approved the final version.
Supporting information
Fig S1
Tab S1
Tab S2
Tab S3
Megy K, Downes K, Morel‐Kopp M‐C, et al. GoldVariants, a resource for sharing rare genetic variants detected in bleeding, thrombotic, and platelet disorders: Communication from the ISTH SSC Subcommittee on Genomics in Thrombosis and Hemostasis. J Thromb Haemost. 2021;19:2612–2617. 10.1111/jth.15459
Manuscript handled by: Joost Meijers
Final decision: Joost Meijers, 09 July 2021
Funding Information
This work was supported by a research grant from the ISTH.
REFERENCES
- 1. Ver Donck F, Downes K, Freson K. J Strengths and limitations of high‐throughput sequencing for the diagnosis of inherited bleeding and platelet disorders. Thromb Haemost. 2020;18:1839‐1845. [DOI] [PubMed] [Google Scholar]
- 2. Freson K, Turro E. High‐throughput sequencing approaches for diagnosing hereditary bleeding and platelet disorders. J Thromb Haemost. 2017;15(7):1262‐1272. [DOI] [PubMed] [Google Scholar]
- 3. Bastida JM, Benito R, Lozano ML, et al. Molecular diagnosis of inherited coagulation and bleeding disorders. Semin Thromb Hemost. 2019;45:695‐707. [DOI] [PubMed] [Google Scholar]
- 4. Andres O, König EM, Althaus K, et al. Use of targeted high‐throughput sequencing for genetic classification of patients with bleeding diathesis and suspected platelet disorder. TH Open. 2018;2:e445‐e454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Othman M, Gresele P. Guidance on the diagnosis and management of platelet‐type von Willebrand disease: a communication from the Platelet Physiology Subcommittee of the ISTH. J Thromb Haemost. 2020;18:1855‐1858. [DOI] [PubMed] [Google Scholar]
- 6. Sánchez‐Guiu I, Torregrosa JM, Velasco F, et al. Hermansky‐Pudlak syndrome. Overview of clinical and molecular features and case report of a new HPS‐1 variant. Hamostaseologie. 2014;34:301‐309. [DOI] [PubMed] [Google Scholar]
- 7. Bury L, Megy K, Stephens JC, et al. Next‐generation sequencing for the diagnosis of MYH9‐RD: predicting pathogenic variants. Hum Mutat. 2020;41:277‐290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Downes K, Borry P, Ericson K, et al. Clinical management, ethics and informed consent related to multi‐gene panel‐based high throughput sequencing testing for platelet disorders: communication from the SSC of the ISTH. J Thromb Haemost. 2020;18:2751‐2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Megy K, Downes K, Simeoni I, et al. Curated disease‐causing genes for bleeding, thrombotic, and platelet disorders: communication from the SSC of the ISTH. J Thromb Haemost. 2019;17:1253‐2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ross JE, Zhang BM, Lee K, et al. Specifications of the variant curation guidelines for ITGA2B/ITGB3: ClinGen Platelet Disorder Variant Curation Panel. Blood Adv. 2021;5(2):414‐431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Collins RL, Brand H, Karczewski KJ, et al. A structural variation reference for medical and population genetics. Nature. 2020;581:444‐451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Van Hout CV, Tachmazidou I, Backman JD, et al. Exome sequencing and characterization of 49,960 individuals in the UK Biobank. Nature. 2020;586:749‐756. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Tab S1
Tab S2
Tab S3