

SUMMARY
Grain filling and grain development are essential biological processes in the plant’s life cycle, eventually contributing to the final seed yield and quality in all cereal crops. Studies of how the different wheat (Triticum aestivum L.) grain components contribute to the overall development of the seed are very scarce. We performed a proteomics and metabolomics analysis in four different developing components of the wheat grain (seed coat, embryo, endosperm, and cavity fluid) to characterize molecular processes during early and late grain development. In‐gel shotgun proteomics analysis at 12, 15, 20, and 26 days after anthesis (DAA) revealed 15 484 identified and quantified proteins, out of which 410 differentially expressed proteins were identified in the seed coat, 815 in the embryo, 372 in the endosperm, and 492 in the cavity fluid. The abundance of selected protein candidates revealed spatially and temporally resolved protein functions associated with development and grain filling. Multiple wheat protein isoforms involved in starch synthesis such as sucrose synthases, starch phosphorylase, granule‐bound and soluble starch synthase, pyruvate phosphate dikinase, 14‐3‐3 proteins as well as sugar precursors undergo a major tissue‐dependent change in abundance during wheat grain development suggesting an intimate interplay of starch biosynthesis control. Different isoforms of the protein disulfide isomerase family as well as glutamine levels, both involved in the glutenin macropolymer pattern, showed distinct spatial and temporal abundance, revealing their specific role as indicators of wheat gluten quality. Proteins binned into the functional category of cell growth/division and protein synthesis/degradation were more abundant in the early stages (12 and 15 DAA). At the metabolome level all tissues and especially the cavity fluid showed highly distinct metabolite profiles. The tissue‐specific data are integrated with biochemical networks to generate a comprehensive map of molecular processes during grain filling and developmental processes.
Keywords: wheat, Triticum aestivum, proteomics, metabolomics, grain filling, seed development, seed coat, embryo, endosperm, cavity fluid
Significance Statement
We have applied a multiomics approach to analyze the grain filling process in wheat (Triticum aestivum L.). The spatial distribution of proteins and metabolites provides an integrated view of the biochemical networks during the developmental processes of the seed. This will provide the seed biologist with a comprehensive molecular map to improve the breeding process.
INTRODUCTION
Wheat (Triticum aestivum L.) is one of the three most important cereal crops. It is widely cultivated due to its value as a staple food and as a primary starch, protein source and due to its unique suitability for bread production. The total global annual wheat production is more than 600 million tons, which accounts for more than 20% of calories consumed worldwide (Brenchley et al., 2012; Pfeifer et al., 2014; Shewry, 2009). The constantly increasing world population and extreme weather conditions due to climate change impose a severe threat to agriculture and its productivity. Hence it is imperative to improve our understanding of the essential plant processes such as grain development, contributing to final grain quality and yield.
Grain development plays an essential role in the life cycle of angiosperms. Grain stores genetic information and nutrients that guarantee the reproduction of the next generation. Wheat grain development is a complex process that involves three successive stages: cellularization (i.e., cell division and differentiation), grain filling, and maturation/desiccation (Nadaud et al., 2010). This process involves coordinated interaction with three main seed components: seed coat, endosperm, and embryo (Chaudhury et al., 2001). The first stage comprises cellularization and differentiation, leading to the development of both the embryo and endosperm—this stage lasts until up to 10 days after anthesis (DAA). The grain filling stage is characterized by the onset of synthesis and accumulation of storage molecules such as starch and gluten proteins. This stage lasts up to 20 days (i.e., from 11 to 30 DAA); this period is further divided into three stages: the Medium milk (11–16 DAA), Soft dough (17–21 DAA), and Hard dough (22–30 DAA) stages. Farmers use these stages to describe grain development in the field (www.wheatbp.com). ‘Medium milk’ is the beginning of the grain filling stage. In this stage, the endosperm meristem cells continue to divide and form storage compartments. At 14 DAA, larger A‐type starch particles appear, and at the same time, liposomes and protein bodies also develop. As the grain enters the ‘Soft dough’ stage, endosperm cell division stops. At the same time, protein and starch granules start to accumulate, and embryo growth is driven by starch digestion of the endosperm and surrounding tissue. In the final stage of grain filling, ‘Hard dough’ activities are still dominated by protein accumulation and starch synthesis (McIntosh et al., 2007). The maturation/desiccation stage is characterized by dehydration of the grain, which gradually enters a quiescent dormancy state. The duration of each stage varies, largely depending upon the genetic background and environmental conditions (Jin et al., 2013).
In brief, wheat grain is constituted by three distinct components: embryo, endosperm, and seed coat. Each component has a different gene expression pattern during the grain filling process. The filial organs (embryo and endosperm) develop within maternal tissue (seed coat). Mature wheat grain consists of 2–3% embryo, 13–17% outer layers (seed coat), and 80–85% endosperm (all components converted to dry matter) (Barron et al., 2007; Shi et al., 2019). The seed coat provides a protective layer for the developing zygotes. It contains chlorophyll, has photosynthetic activity, and provides nutrients for grain development (Yu et al., 2015). In the early stage of grain growth, the seed coat is an assimilation reservoir (Radchuk et al., 2009) which is gradually degenerated. The green tissue layer and crease region remain viable until the grain dehydration stage (Tran et al., 2014). Photosynthesis in the seed coat entirely depends on the CO2 produced by respiration in the endosperm and embryo. The seed coat of barley (Hordeum vulgare) grain re‐fixes 79% of the CO2 produced by respiration in the endosperm and embryo and the carbon conversion efficiency of barley grain reaches 95%. It is speculated that this also occurs in wheat grain (Rolletschek et al., 2015). Within 2–10 DAA, the seed coat begins to accumulate starch granules. The number of starch granules and protein bodies gradually increases throughout development, and the particles become larger and larger (Christiaens et al., 2012; Loussert et al., 2008). At 10–20 DAA, the seed coat nutrients begin to decompose, as they are partly used for their physiological processes and partly transported to the endosperm and embryos for their growth (Christiaens et al., 2012; Zhou et al., 2009).
The wheat endosperm is the essential nutrient storage organ, accounting for approximately 83% of the whole grain (Barron et al., 2007). It is specifically used for the accumulation of starch and protein. The spatial heterogeneity of starch and protein directly affects the quality of wheat flour. The embryo is located on the dorsal side of the wheat caryopsis’ proximal end (Shi et al., 2019). As an important reproductive organ in wheat grain, the embryo absorbs about 15% of the assimilation through the peel and is used to produce and accumulate a mixture of starch, lipids, and proteins (Neuberger et al., 2008; Rolletschek et al., 2015). During grain filling, the amino acids produced by proteolysis in the leaves are transported via the phloem into the grain, where they serve as substrates for protein synthesis (Fisher and Gifford, 1986). In barley grain, free amino acids need to be unloaded into the endosperm cavity (cavity fluid) and enter the endosperm through the transfer cells or aleurone layer (Thiel et al., 2009).
In recent years, genetics has helped decipher the gene regulatory network that plays a vital role in seed development (Santos‐Mendoza et al., 2008). DNA microarray and RNA sequencing (RNA‐seq) techniques are advantageous for large‐scale genome‐wide studies (Girke et al., 2000; Xu et al., 2012b; Xue et al., 2012). However, it has been noted that mRNA levels are not always correlated with protein abundance (Gygi et al., 1999; Weckwerth et al., 2020), and genomics tools cannot always provide precise information about protein levels and regulation, limiting our understanding of the metabolic regulatory networks (Anderson and Anderson, 1998). Therefore, to unravel the developmental patterns of proteins and metabolic regulation and their role in the grain filling process, it is essential to record and analyze these biochemical (proteins and metabolites) dynamics at their interface. In this context, proteomics and metabolomics have become the central element of plant systems biology research (Weckwerth, 2011; Weckwerth et al., 2020). The ultimate aim is to detect, identify, quantify, and interpret the abundance of proteins and metabolites of a given system. Here, an integrated approach comprising different analytical platforms such as liquid chromatography coupled to mass spectrometry (LC‐MS) and gas chromatography coupled to mass spectrometry (GC‐MS) enables high‐throughput profiling of proteins and metabolites (Morgenthal et al., 2005; Weckwerth et al., 2004c; Wienkoop et al., 2008).
Although considerable work has been performed in the investigation of proteomics and metabolomics in wheat grain, these studies are mainly focused on selected wheat grain components (such as embryo and endosperm), several time points, and abiotic stress (Das et al., 2017; Dupont, 2008; Dupont et al., 2011; Francki et al., 2016; Ge et al., 2012; Gu et al., 2015; Guo et al., 2012b; Han et al., 2017; Yang et al., 2016; Yang et al., 2017; Zhang et al., 2015). Wheat also has a much larger genome and a more complex proteome (approximately 104 091 different proteins) (Clavijo et al., 2017) than model plants such as Arabidopsis and rice (Oryza sativa). Thus, the important regulatory mechanisms for wheat grain development still require further study. To the best of our knowledge, a systematic proteomics and metabolomics analysis of the entire development process in wheat grain has not been reported previously. Therefore, we analyzed the dynamic changes in protein and metabolite levels in wheat grain at four sequential developmental stages, i.e., 12, 15, 20, and 26 DAA, in four different components, i.e., seed coat, embryo, endosperm, and cavity fluid. Our results revealed a global pattern of proteins and metabolites corresponding to grain development, which serves as a valuable resource and provides new insights into the potential metabolic networks that control grain yield and quality.
RESULTS
Morphology and physiology during grain filling
Whole wheat grains were sampled from 12 to 35 DAA, and the characteristics of the developing grain were recorded at each time point (Figure 1(a,b)). The moisture content gradually decreased during this period (Figure 1(a)), while the dry weight gradually increased (Figure 1(b)). The fresh weight was highest at 29 DAA and then declined (Figure 1(b)), indicating that the developing grain entered the desiccation stage at 29 DAA. The cavity fluid also disappeared after 26 DAA. Overall, up to 12 DAA developing grain mainly undergoes active cell division and differentiation, followed by grain filling up to 29 DAA, and then enters the desiccation stage. Therefore, the harvesting time points for this experiment were set at 12, 15, 20, and 26 DAA, a primary phase of the grain filling process. During this phase, grain grew gradually in length and width (Figure 1(c)), and the color of the grain switched from white to green (Figure S1). The fresh weight of the seed coat, embryo, endosperm, and cavity fluid varies significantly between different developmental stages (Figure 1(d)). The fresh weight of the seed coat and cavity fluid peaked at 15 DAA and then decreased significantly. The embryo and endosperm grew gradually from 12 to 26 DAA. It is noteworthy that the period from 15 to 20 DAA is important for endosperm growth. The texture of the cavity fluid becomes sticky at 26 DAA. All the recorded observations are reported in Table S1.
Figure 1.
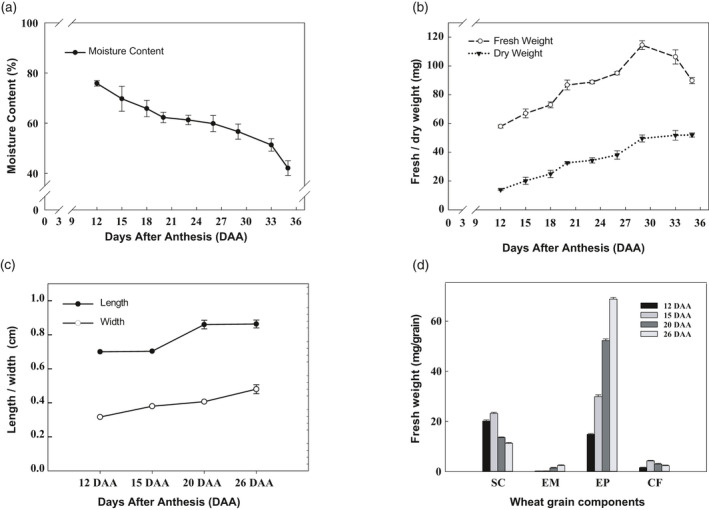
Development of wheat grain cultivar Yangmai 16. (a) Changes in the total moisture content of developing grain. Values are expressed as the percentage of dry grain weight. (b) Changes in fresh and dry weight of developing grain. At least 70 grains were analyzed at each stage. (c) Changes in grain length and width between 12 and 16 DAA. (d) Dynamic changes in the fresh weight of four grain components in different grain filling stages (70 seeds/stage). Error bars represent the standard deviation of three replicates. DAA, days after anthesis; SC, seed coat; EM, embryo; EP, endosperm; CF, cavity fluid.
Comparative proteome characterization of developing seed coat, embryo, endosperm, and cavity fluid during grain filling
From all the detected peptides, 3806 proteins were detected in the seed coat, 4560 proteins in the embryo, 4144 proteins in the endosperm, and 2974 proteins in the cavity fluid during the grain filling process (Tables S2 and S3). A Venn diagram analysis was conducted to broadly survey the identified proteins with altered abundance in each component, which determines the dynamics of the proteome in different developmental stages (12, 15, 20, and 26 DAA) (Figure 2(a)). In endosperm, in total 4144 proteins were identified, of which 2966 proteins were expressed at all four time points (Figure 2(a)). Interestingly, five and four unique proteins were identified at 15 DAA and 26 DAA, respectively. Aspartic proteinase nepenthesin I was identified at 15 DAA. This protein is a unique member of a novel subfamily of aspartic proteinases. Short‐chain dehydrogenase/reductase, functioning as a molecular link between nutrient signaling and plant hormone biosynthesis, was identified at 26 DAA (Table S4).
Figure 2.
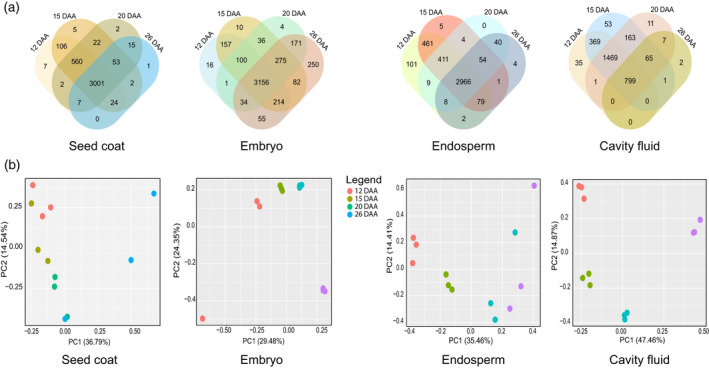
(a) Venn diagrams representing total proteome identification from different components (seed coat, embryo, endosperm, and cavity fluid) of developing grain (12, 15, 20, and 26 days after anthesis [DAA]) of wheat cultivar Yangmai 16. (b) Principal component analysis (PCA) of the protein profiles of wheat grain components (seed coat, embryo, endosperm, and cavity fluid). Orange, green, dark green, and violet colors represent 12, 15, 20, and 26 DAA. Three independent biological replicates were used for proteomic analysis.
Principal component analysis (PCA) of protein normalized spectral abundance factor (NSAF) scores was performed using the R (package ggplot2). PCA demonstrated that the proteome regulation pattern of the four grain filling stages differed significantly in every grain component. Figure 2(b) shows that all grain components were clearly separated as a sequential process determined by the first principal component (PC1), which accounted for 36.79%, 29.48%, 35.46%, and 47.46% of variability in the seed coat, embryo, endosperm, and cavity fluid, respectively. The loadings of PC1 determine unique sets of proteins in each developmental stage (12, 15, 20, and 26 DAA). Positive loadings of PC1 represent proteins with higher abundances in the late grain filling stage (20 and 26 DAA), whereas negative loadings represent proteins with higher levels during early grain filling stages (12 and 15 DAA) (Table S5). The highest negative PCA loadings of seed coat include proteins involved in signal transduction, members of the protein disulfide isomerase (PDI) family, and proteins involved in sugar synthesis. In contrast, the negative loadings of endosperm include proteins involved in DNA replication and tRNA pseudouridine. The negative PCA loadings in the embryo include glucan hydrolases and vacuolar processing enzyme, responsible for vacuolar protein maturation and activation. The highest PCA loadings of cavity fluid revealed proteins involved in the nuclear export of proteins, rRNA, snRNA, and some mRNA. Other negative loadings revealed proteins involved in the synthesis of histidyl‐tRNA and protein ubiquitination. The highest positive PCA loadings include proteins which are involved in photosynthesis in the seed coat, oleosin and peroxidase in embryo, protein storage and lipid metabolism in the endosperm, and disease resistance (endochitinase and PBSP domain protein) and sugar hydrolysis in cavity fluid (Table S5).
Differentially expressed proteins at different developmental stages in wheat grain components
A fold change (FC) cutoff value of 1.5 and P < 0.05 were used to identify differentially expressed proteins (DEPs). Comparisons were made between grain developmental stages, i.e., 15 vs. 12 DAA, 20 vs. 15 DAA, and 26 vs. 20 DAA, in each grain component (Table S6). Comparing 15 vs. 12 DAA, a total of 66 DEPs were identified in the seed coat, 309 in embryo, 134 in endosperm, and 170 in cavity fluid. Comparing 20 vs. 15 DAA, a total of 123 DEPs were identified in the seed coat, 195 in embryo, 188 in endosperm, and 123 in cavity fluid. Comparing 26 vs. 20 DAA, 229 DEPs were identified in the seed coat, 417 in embryo, 59 in endosperm, and 121 in cavity fluid (for detailed information about these DEPs refer to Table S6). This analysis aimed to determine the overall trend of enrichment in the specific functional categories during the grain filling process, as shown in Figure 3. Functional categorization of the identified proteins was performed according to Ghatak and co‐workers (Ghatak et al., 2021). Functional distributions of the total proteome in the four developmental stages (12, 15, 20, and 26 DAA) of the different grain components (seed coat, embryo, endosperm, and cavity fluid) are depicted in Figures S2–S5 via heatmap biclustering using the total NSAF score summed up for different functional categories (Chaturvedi et al., 2013; Ghatak et al., 2021) (Table S7). In this analysis, DEPs were categorized according to the MapMan plant functional ontology (see Experimental Procedures). In total, 410 unique DEPS were identified in the seed coat, 815 in embryo, 372 in endosperm, and 492 in cavity fluid (Figure 3, Table S8).
Figure 3.
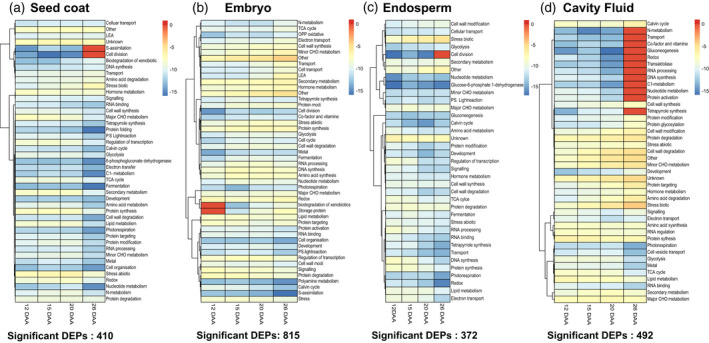
Heatmaps. (a) Differentially expressed proteins (DEPs) in the seed coat. (b) DEPs in the embryo. (c) DEPs in the endosperm. (d) DEPs in the cavity fluid.
For the four dissected wheat grain components, K‐means cluster analysis of proteins in different grain filling stages was performed by the COVAIN toolbox (Sun and Weckwerth, 2012), revealing spatially coordinated proteome changes during four different stages of grain development (Figures S6 and S7). K numbers were different according to the different grain components. The value of k was 35 in seed coat and cavity fluid, 48 in embryo, and 49 in endosperm (Table S9). For the discussion we selected specific clusters which revealed the temporal dynamics of the proteome (see below).
Comparative metabolite profiling in wheat grain components during grain filling
Metabolomics analysis of the different developing wheat grain components (seed coat, embryo, endosperm, and cavity fluid) was conducted using GC‐time‐of‐flight (TOF)‐MS. In total, 34 metabolites showed different levels at various developmental stages (12, 15, 20, and 26 DAA). The identified metabolites were grouped into carbohydrates, amino acids, and organic acids. PCA showed a clear separation between all developmental stages for each wheat grain component (Figure S8, Table S10).
Distinct metabolite patterns for early (12 and 15 DAA) and late (20 and 26 DAA) grain filling stages were determined (Figure S9, Table S10). The identified metabolites demonstrated very similar levels between 12 and 15 DAA, which could be due to the short time interval (3 days) between the two respective stages. Between 20 and 26 DAA, which coincide with the accumulation of starch reserves (Dhatt et al., 2019), a clear difference was observed in the levels of the identified metabolites (Figure S8, Table S10). Selected metabolites and their levels are depicted in Figure 4.
Figure 4.
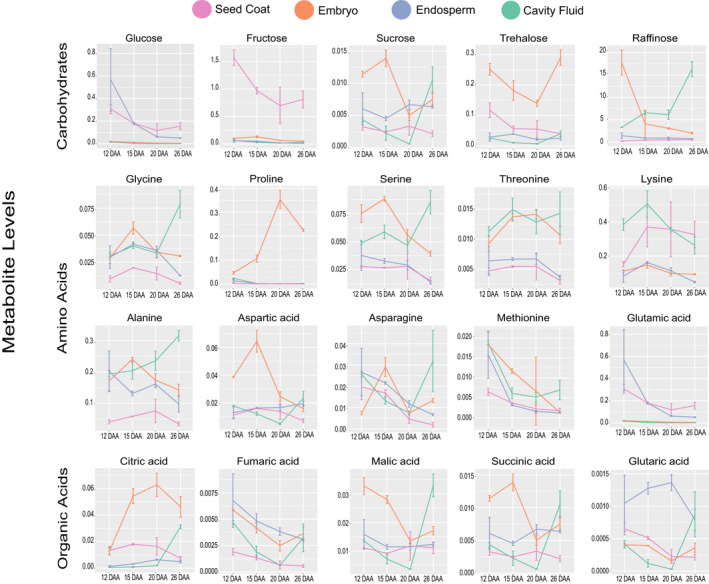
Abundance levels of selected metabolites. The means of the abundance levels from four different components (seed coat, embryo, endosperm, and cavity fluid) during different grain developmental stages (12, 15, 20, and 26 DAA) are shown. Data are presented as the mean ± standard deviation from three biological replicates for each grain component and developmental stage. DAA, days after anthesis.
Asparagine and aspartic acid levels changed consistently except in the endosperm. Asparagine accumulated to maximum levels at 12 DAA in the endosperm, while aspartic acid showed the highest levels at 26 DAA (Figure 4). Fructose and glucose levels were highest in the early grain filling stage (12 and 15 DAA) in all the four grain components analyzed. Isoleucine, methionine, threonine, valine, and lysine accumulated mostly in all components at 15 and 20 DAA (Figure 4). Galactose and maltose showed the same trend in the endosperm, embryo, and cavity fluid, with maximum accumulation at 12 and 26 DAA, respectively (Figure S9). Some amino acids showed similar dynamic changes in the seed coat, endosperm, and embryo (but not the cavity fluid). For example, putrescine and fumaric acid showed the highest levels at 12 and 15 DAA—similarly, leucine and lactic acid showed the highest levels at 20 and 26 DAA (Figure S9, Table S10).
In the seed coat, all primary metabolites showed low levels in the late grain filling stage (20 and 26 DAA). In contrast, the levels of some photosynthesis‐related metabolites and carbohydrate storage metabolites were higher. Most metabolites (but not tyrosine, proline, leucine, maltose, and trehalose) in the embryo showed the highest level at 15 DAA, indicating that primary metabolism was active in early grain filling stages. These metabolites were highly accumulated at 26 DAA. The embryo accumulated proline in late grain filling stages (20 and 26 DAA), probably to combat various stresses. In the cavity fluid, in contrast to other components, most metabolites showed the highest levels at 26 DAA, except methionine, galactose, glucose, and fructose, whose levels were highest at 12 DAA. Hence, it can be concluded that in the early grain filling process the cavity fluid supplies sugars to the endosperm (Figure 4, Table S10).
DISCUSSION
Previously, when studying the transportation and accumulation of nutrients in wheat plants, typically whole wheat grain was considered (Barneix, 2007). All the physiological and developmental processes in plants undergo a necessary integration process, which depends on the transport of hormones, metabolites, and other signals of the vascular system (van Bel et al., 2013). These integration processes stop at the seed coat, which wraps the endosperm and embryo (Radchuk and Borisjuk, 2014). These three grain components follow their own developmental program to form an interactive system (Sreenivasulu et al., 2006). Grain filling substrates are transported in the aqueous solution of the cavity fluid to the developing grain. There are indications that it is first transported into the endosperm cavity and thus released into the endosperm (Radchuk and Borisjuk, 2014). Accordingly, to understand the spatial and temporal dynamics of the grain filling process and the developing grain, it is necessary to divide them into four parts, i.e., seed coat, endosperm, embryo, and cavity fluid, for analysis. To reveal spatially coordinated proteome changes, K‐means cluster analysis of proteins in different grain filling stages was performed using the COVAIN toolbox (Sun and Weckwerth, 2012) (Table S9). In the following, we discuss selected clusters, revealing the temporary proteome and metabolome dynamics of the four grain compartments (Figures 5 and 6).
Figure 5.
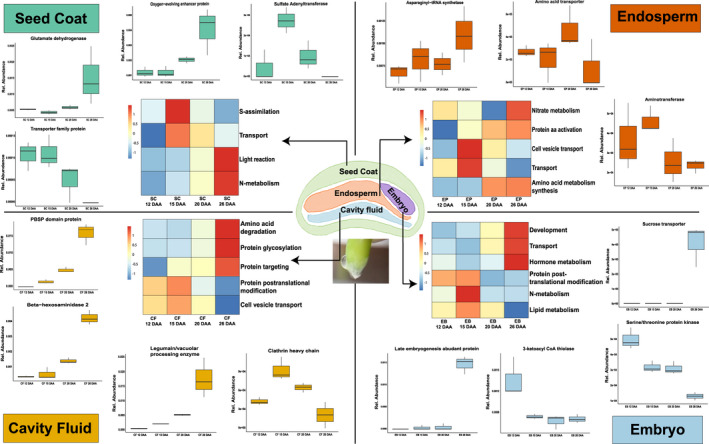
Selected functional categorization of four different components during different grain filling stages using bicluster plots. NSAF values were averaged over three biological replicates. SC, seed coat; EB, embryo; EP, endosperm; CF, cavity fluid.
Figure 6.
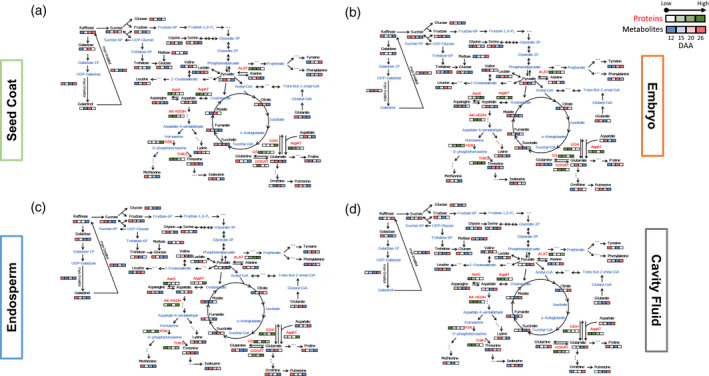
Integration of metabolite and protein dynamics in a biochemical pathway map. (a) Seed coat. (b) Embryo. (c) Endosperm. (d) Cavity fluid. Metabolites are written in black letters; enzymes are written in red letters. Levels of metabolites and proteins were averaged over three biological replicates after normalization. Four consecutive squares from left to right indicates four developmental stages (12, 15, 20, and 26 days after anthesis). Values of metabolite levels from minimal to maximal are colored from blue to red and protein concentrations are colored from white to green. Metabolites identified by GC‐MS are written in black; metabolites not identified by GC‐MS are written in blue. ALAT, alanine aminotransferase; AK‐HSDH, bifunctional aspartokinase/homoserine dehydrogenase I; AsnS, asparagine synthetase; AspAT, aspartate aminotransferase; GDH, glutamate dehydrogenase; GOGAT, glutamate synthase; GS, glutamine synthetase; HSK, homoserine kinase; THRS, threonine synthase.
Seed coat
In the seed coat, cluster 32 (Table S9) showed the proteins with the highest level at 12 DAA during early grain filling. The proteins included trafficking protein particle complex subunit 9, which may play a role in vesicular transport from the endoplasmic reticulum (ER) to the Golgi apparatus. Proteins involved in cell elongation were also found (Elongation factor‐like protein and Transcription elongation factor SPT6). At 15 DAA, proteins which are involved in protein trafficking (trafficking protein particle complex subunit 2 and Clathrin‐binding protein) were highly expressed. Additionally, the abundance of non‐specific lipid‐transfer protein was also highest at 15 DAA. Sulfate adenylyltransferase and a transporter family protein, which are involved in transport and sulfate assimilation, respectively, were upregulated at 15 DAA. The seed coat turns green at 20 DAA. The levels of proteins such as the calcium‐binding protein Caleosin and the lipid A export ATP‐binding/permease protein MsbA were increased at this stage, indicating high lipid metabolism. The fresh weight of the seed coat decreased at 26 DAA. Late embryogenesis abundant (LEA) protein and PBSP domain protein were found at high levels. Proteins involved in light reactions and nitrate metabolism were upregulated at 26 DAA. Among them, oxygen‐evolving enhancer protein 2 and glutamate dehydrogenase (GDH) were activated at 26 DAA (Figure 5). Most primary metabolites showed low levels in the seed coat, but with a few exceptions. Phenylalanine, tyrosine, lactate, raffinose, and myo‐inositol showed high levels at 26 DAA. Raffinose and myo‐inositol peaked at 26 DAA, showing carbohydrate storage was activated in the seed coat. Furthermore, myo‐inositol is important for phosphate storage and transport of plant hormones.
Endosperm
In the endosperm at 12 DAA, the levels of proteins involved in energy metabolism and protein trafficking and elongation factors were highest. Legumain/vacuolar processing enzyme was highly expressed at 15 DAA, illustrating high protein processing activity at this stage (Table S9). The levels of many proteins involved in ubiquitin/proteasome processes were also increased at 15 DAA. Proteins involved in cell vesicle transport and amino acid metabolism/synthesis were upregulated at 15 DAA. The protein transportation is still active at 20 DAA (Vesicle transport v‐SNARE 11‐like). Moreover, glutathione S‐transferase 1 levels were increased at 20 DAA, indicating a defense reaction against exogenous toxins. In the late grain filling stage (at 26 DAA), proteins involved in protein storage (Vicilin) and amino acid metabolism (aminomethyltransferase) were highly abundant. Proteins involved in nitrate metabolism and protein amino acid activation showed upregulation at 26 DAA. Asparaginyl‐tRNA synthetase, which is involved in translation, was upregulated at 26 DAA. Amino acid transporter and amino acid transferase were upregulated at 20 and 26 DAA, respectively (Figure 5). Only a few metabolites were highly accumulated at 26 DAA: aspartic acid, glutamic acid, leucine, citric acid, and maltose. Aspartic acid and glutamic acid are important substrates for nitrogen (N) supply, indicating that N metabolism is active at 26 DAA.
Embryo
The embryo has a highly distinct proteome compared to the other tissues. In the early grain filling stage (12 DAA), the levels of proteins which are involved in energy and lipid metabolism were increased. The proteins involved in sugar hydrolysis and DNA replication were present at high levels at 15 DAA. Proteins involved in post‐translational protein modification and cell vesicle transport were upregulated at 15 DAA. Clathrin heavy chain was significantly upregulated at 15 DAA (Table S9). The levels of proteins related to ubiquitin, lipid storage, and protein transport were increased at 20 DAA. Vicilin, which is involved in protein storage, and the LEA protein were present at high levels at 26 DAA. Proteins involved in amino acid metabolism and degradation, protein glycosylation, and protein targeting were upregulated at 26 DAA. Among them, PBSP domain protein, beta‐hexosaminidase 2, and legumain/vacuolar processing enzyme were upregulated at 26 DAA (Figure 5). Most of the metabolites showed the highest levels at 15 DAA, except tyrosine, proline, leucine, maltose, and trehalose. These metabolites were highly accumulated at 26 DAA. Primary metabolism was activated in early grain filling, and carbohydrates were accumulated as maltose and trehalose at 26 DAA. The embryo stored proline in the late grain filling stage, most probably as a protective mechanism to various kinds of stresses.
Cavity fluid
Cavity fluid is the sap in the grain cavity, which is surrounded by endosperm and the vascular bundle. Cavity fluid sampling is prone to contamination by surrounding cells and tissues. Accordingly, all our data on cavity fluid need to be handled with care and contamination cannot be excluded. However, proteins and metabolites showed significant differences in comparison to the endosperm and also the seed coat and embryo. Therefore, we discuss these data but remind the reader about these difficulties in sampling.
In cavity fluid, proteins whose levels were increased during the early grain filling stage were involved in disease resistance (endochitinase and PBSP domain protein) and sugar hydrolysis, while the proteins whose levels were decreased during grain filling were involved in the nuclear export of proteins, rRNA, snRNA, and some mRNA. Other proteins whose levels were decreased during grain filling were involved in histidyl‐tRNA synthesis and protein ubiquitination. No glutamine‐related enzymes were detected in cavity fluid. At 12 DAA, several reductases and transferases related to amino acid metabolism were present at high levels, such as gamma‐glutamyl phosphate reductase and homoserine kinase (HSK). The levels of detected enzymes involved in transformations of amino acids decreased during grain filling, indicating that the cavity fluid contained enzymes at high concentrations. Among all the detected metabolites, methionine, galactose, glucose, and fructose peaked at 12 DAA (during early grain filling), indicating that the cavity fluid supplies sugars to the endosperm. At 15 DAA, the levels of proteins involved in ubiquitin and signal transduction increased. Furthermore, many enzymes like isocitrate dehydrogenase related to the tricarboxylic acid cycle (TCA) were also found to be highly abundant. Peroxidase and FAD‐binding domain‐containing protein were identified at 20 DAA. Cavity fluid turned less voluminous and stickier at 26 DAA. Proteins which are involved in disease resistance such as endochitinase and PBSP domain protein were identified at high levels at this stage (Figure 5, Table S9). Proteins involved in development, transport, and hormone metabolism were upregulated. Unlike other grain compartments, most metabolites in the cavity fluid showed the highest levels at 26 DAA, highlighting the importance of this stage and the active transport of all necessary metabolic components by the cavity fluid during the last phase of grain filling.
Regulation of proteins during the grain filling process
Protein disulfide isomerase family
Different isoforms of the PDI family proteins participate in the formation of intermolecular or intramolecular disulfide bonds and the assembly of glutenin macropolymers (GMPs) through intramolecular disulfide bonds between wheat grain proteins (Wilkinson and Gilbert, 2004). Therefore, they play an important role in the maturation of secreted plasma membranes and storage proteins (Houston et al., 2005). In wheat, nine PDI and PDIL genes were cloned, and their transcript levels during endosperm development indicate that they are related to storage protein synthesis and deposition, which is closely related to gluten and bread quality (d'Aloisio et al., 2010). Analysis based on iTRAQ showed that two PDI proteins (B9A8E3 and Q9FE55) are highly expressed during early grain development (7–14 DAA), which helps the folding and maturation of storage proteins and increases gluten quality (Ma et al., 2014a). In our study, several different isoforms of PDI family proteins showed tissue‐dependent expression patterns. The highest abundance was observed at 12 DAA in all four components. In endosperm and cavity fluid, levels remained relatively high from 12 to 20 DAA. But in the embryo, its abundance at 12 DAA was more than two times that of other stages (Table S2, S7). Accordingly, our analysis demonstrates the complex expression pattern of PDIs during development and paves the way for breeding strategies to manipulate GMP content in the grain.
LEA proteins
LEA proteins accumulate at later stages, especially under stress conditions such as drought and low temperature (He et al., 2011). Seven LEA proteins were present from 20 to 26 DAA in the embryo. The LEA proteins have an important role in protecting grain from serious dehydration during the late developmental stages. In a recent study, the levels of eight LEA proteins increased significantly at 28 DAA (Ma et al., 2014a). Compared to other components, the seed coat and embryo contained much more isoforms. LEA protein 2 was present in all four grain components (Table S2, S7). It showed decreased abundance during grain filling except in the embryo, where its levels peaked at 26 DAA.
Serpin family
Several other proteins related to stress/defense pathways were identified, including nine, five, eight, and one serpin isoforms in the endosperm, seed coat, cavity fluid, and embryo, respectively (Tables S2 and S7). Serpin expression increased in all tissues from 12 to 26 DAA except for the embryo. The serpin family proteins irreversible inhibit endogenous and exogenous proteases. These proteases play an important role in plant growth, development, the adversity response, and defense against insects and pathogens (Roberts and Hejgaard, 2008). Serpins are present in wheat grain, and their content is as high as a few percent of the total protein content. They may be present in the seeds as a defense barrier that protects stored proteins from digestion (Vensel et al., 2005). Previous studies have shown that serpin is upregulated under osmotic conditions such as salt and cold stress (Lampl et al., 2010).
Transporters, maturation and transport of proteins
Amino acid selective channel proteins, which are involved in transmembrane amino acid transporter activity, showed the highest abundance at 12 DAA in the seed coat and embryo. In endosperm, their levels gradually increased during grain filling, with a peak during late grain filling (Tables S2 and S7).
The general vesicular transport factor p115 plays an important role in intracisternal transport in the Golgi stack. It was highly accumulated at 12–20 DAA in the seed coat and cavity fluid. In the endosperm, its levels remained high (peaking at 20 DAA). In the embryo, it was highly accumulated at 12 DAA. The situation was the same for the protein transport protein Sec23, which promotes the formation of transport vesicles from the ER. Legumain/vacuolar processing enzyme has been identified as the proteinase responsible for the maturation and activation of vacuolar proteins in plants. It was significantly accumulated at 26 DAA in cavity fluid. The signal recognition particle (SRP) protein SRP72, which has a crucial role in targeting secretory proteins to the rough ER membrane, showed high abundance at 12–20 DAA in the seed coat, embryo, and cavity fluid. However, in the endosperm, it was highly accumulated at 20–26 DAA. The vacuolar sorting receptor is involved in the sorting and packaging of soluble vacuolar proteins into transport vesicles. It was highly accumulated at 20–26 DAA in three grain organs, while in cavity fluid, it had high abundance at 12–20 DAA.
Alternative energy pathways
During late wheat grain development, due to insufficient energy for glycolysis and the TCA cycle, the alcohol fermentation pathway can provide supplementary energy under anaerobic conditions. In addition, the expression of formate dehydrogenase increased at 15 DAA and further increased at the late filling stage and the drying period (Tables S2 and S7). This is consistent with the transition from cell growth and differentiation to starch synthesis. The shift from central carbon (C) metabolism to alcohol fermentation may be important for starch synthesis and accumulation during grain development (Guo et al., 2012a). Alcohol fermentation is a branch of the pyruvate glycolysis pathway and is a two‐step reaction. The focus is on oxidizing NADH to NAD+ and then generating ATP without consuming oxygen. This is essential for pre‐adapting to hypoxia and seed development (Cao et al., 2016).
Differential accumulation patterns of proteins during the grain filling process
Starch biosynthesis during grain filling and development
In cereals, grain yield is largely determined by starch accumulation during the grain filling period. There are three sucrolytic enzymes in higher plants: sucrose synthase (SuSy), acid invertase, and neutral invertase. In most rapidly filling sinks such as seeds, SuSy has the highest activity (Sung et al., 1989). SuSy acts as the first enzyme in the conversion of sucrose to starch, and thus, SuSy activity is an excellent predictor of sink strength (Sung et al., 1989). Sink strength is the ability of a sink to attract or import carbohydrates. Accordingly, SuSy may be valuable to assess relative sink strength among rice breeding lines (Counce and Gravois, 2006). In the present study, key proteins related to starch biosynthesis, including SuSy, starch synthase (SS), starch phosphorylase, granule‐bound SSI, and soluble SS, were shown to be significantly up‐ or downregulated (Table S11). The levels of all identified proteins increased gradually in endosperm from 12 to 26 DAA, and the accumulation of starch biosynthesis proteins in endosperm was most pronounced (Table S11). A similar pattern was observed in the embryo (gradual increase from 12 to 26 DAA). There was a gradual decrease in the abundance of starch biosynthesis proteins in the seed coat during the grain filling period. As discussed below, decreased expression of 14‐3‐3 isoforms in the cavity fluid and the embryo could result in an increase of starch synthesis during the grain filling period. Similarly, in the endosperm, pyruvate phosphate dikinase (PPDK) has an essential role in starch synthesis and energy supply during grain development, i.e., the upregulation of PPDK could support starch biosynthesis. The endosperm is the primary storage site of starch. Its function is to support and nurture the developing embryo. It has a central role in regulating embryo development (An et al., 2020), so the accumulation of starch biosynthesis proteins in endosperm tissue is not surprising.
14‐3‐3 proteins, pyruvate phosphate dikinase, and starch synthesis during grain development
In this study, five, seven, eight, and seven isoforms of 14‐3‐3 proteins were significantly accumulated in the endosperm, seed coat, cavity fluid, and embryo, respectively. In the embryo, two isoforms were upregulated at 20 DAA and five isoforms were downregulated at 26 DAA. Out of eight isoforms in the cavity fluid, the levels of seven isoforms decreased during 15–26 DAA (three isoforms decreased gradually from 12 to 26 DAA, two isoforms only at 15 DAA, and two isoforms at 20 DAA). The highest downregulation of 14‐3‐3 proteins was observed at 15 and 20 DAA (Table S11). Proteins of the 14‐3‐3 family play regulatory roles in various cellular physiological processes, such as cell signal transduction, cell cycle regulation, N and C assimilation, and defense mechanisms (Ferl, 1996; Fulgosi et al., 2002). It has been proposed that 14‐3‐3 proteins may decrease starch biosynthesis by inhibiting SS in a large protein complex, suggesting its possible role in grain development (Fulgosi et al., 2002; Sehnke et al., 2001). In the present study, three isoforms of granule‐bound SSI were identified. The levels of all isoforms increased gradually from 12 to 26 DAA (Table S11). This upregulation can be correlated with the expression of 14‐3‐3 proteins, which was reduced dramatically after 15 DAA, when starch synthesis was upregulated. Zhang and co‐workers observed low expression of 14‐3‐3 proteins in the rice grain with high starch content compared to grain with low starch content (Zhang et al., 2014). Consistent with this hypothesis, we found here that the decreased expression of 14‐3‐3 proteins is inversely related to starch synthesis and accumulation. A proteomics study on maize (Zea mays) grain reported significant accumulation for three isoforms of 14‐3‐3 proteins at the early stage (3–10 DAA), and then downregulation until grain maturity (Yu et al., 2016). A similar pattern has also been observed in castor (Ricinus communis) grain (Houston et al., 2009).
In endosperm, one 14‐3‐3 protein isoform (A0A1D6CLX4) was significantly downregulated at 20 DAA, whereas proteins related to starch synthesis, including three isoforms of granule‐bound SS and soluble SS, increased in their levels gradually from 12 to 26 DAA. Interestingly, we found three isoforms of PPDK, all of them upregulated from 12 to 26 DAA (Table S11). Several proteomic studies reported that multiple isoforms of PPDK accumulate at high levels in the developing grain of cereals such as rice (Xu et al., 2008b) and wheat (Ma et al., 2014b), indicating that PPDK has an essential role in starch synthesis and energy supply during grain development (Wang et al., 2020). PPDK catalyzes the reversible conversion of pyruvate, ATP, and Pi to phosphoenolpyruvate (PEP), AMP, and PPi (Yu et al., 2016). PPDK is highly efficient in remobilizing carbon skeletons that must be metabolized via pyruvate, hence clearly demonstrating the gluconeogenic function of converting pyruvate to PEP and consuming ATP (Eastmond et al., 2015). The formation of pyruvate may be beneficial to synthesis at the grain filling stage and the PPi could be used for the initial AGPase step in starch biosynthesis. Interestingly, the levels of all three isoforms of PPDK in the seed coat and cavity fluid decreased gradually at 20–26 DAA; these isoforms were not detected in the embryo (Table S11). Therefore, these results indicate that PPDK in the endosperm and 14‐3‐3 in the cavity fluid may play important roles in starch synthesis and energy supply during wheat grain filling and development.
Cell division and cell wall modification during grain filling
Effective grain filling involves a transition from cellularization (cell growth) to starch synthesis and accumulation. At the early stage of grain filling (6–13 DAA), grain demonstrates a rapid increase in cell number and size. In the mid‐stage of grain development (14–24 DAA), there is a small increase in cell number and size, which eventually decrease completely. In contrast, storage materials (mainly starch) are synthesized and accumulate rapidly (Laudencia‐Chingcuanco et al., 2007). In the endosperm, the levels of proteins related to cell division decreased from 20 to 26 DAA (stronger decrease at 26 DAA), consistent with the simultaneous starch accumulation in the endosperm. A small increase in the levels of cell division proteins from 12 to 20 DAA was followed by a sudden downregulation at 26 DAA. The levels of cell division proteins in the embryo decreased gradually from 12 to 26 DAA (Table S11). Similarly, many cell wall degradation proteins were expressed in all grain components, demonstrating upregulation from 12 to 20 DAA and then downregulation at 26 DAA (Table S11).
Interestingly, in this study, we identified seven expansin protein isoforms that significantly accumulated only in the cavity fluid. Expansins could be specifically involved in grain enlargement and seed growth control (Mehdi et al., 2020). The functional mechanism of expansin involves disrupting hydrogen bonds between cellulose microfibrils and cross‐linking glycans in the cell wall, permitting turgor‐driven cell wall extension (Mcqueenmason and Cosgrove, 1994; Mcqueenmason and Cosgrove, 1995). The obtained results are consistent with the cellularization phase (cell growth). A small increase in expansin protein levels was observed from 12 to 20 DAA and then a sudden downregulation was observed at 26 DAA (Table S11). It is important to mention that a few expansin protein isoforms were also found in the endosperm and embryo. However, none of them showed any significant up‐ or downregulation during grain filling (Table S11).
Protein degradation and synthesis during grain filling
Our analysis showed that many proteins involved in protein degradation were significantly expressed in all four components, peaking from 12 to 15 DAA in the endosperm and embryo and from 12 to 20 DAA in the seed coat and cavity fluid. A significant decrease was observed at 26 DAA (Table S11). Some of these proteins are key components of the ubiquitin/26S proteasome pathway and play important roles in protein degradation and in cellular and developmental events (Moon et al., 2004; Vierstra, 2009). Many proteins related to protein synthesis, e.g., proteolysis‐related proteins, showed similar expression patterns in all tissues except for the embryo, in which the expression of proteins involved in protein synthesis increased gradually from 12 to 26 DAA. Active protein turnover (proteolysis and synthesis) was found during the early development of rice and maize grain (Xu et al., 2008b; Yu et al., 2016). Overall, these expression patterns of proteins involved in protein biosynthesis in the endosperm, seed coat, and cavity fluid suggest that protein turnover and rearrangements are also important and effective in the early stage of wheat grain cell division and enlargement.
It is observed that the embryo remains quite small until 15–20 DAA. Apparently, from 15 to 30 DAA, growth of the embryo is quite rapid and it is during this period that differentiation of the axial organs (coleoptile, shoot and root primordia, and coleorhiza) is initiated (Smart and Obrien, 1983). Therefore, the expression of proteins involved in protein synthesis in the embryo increases until 26 DAA to support its rapid growth and development from 15 to 30 DAA.
Integration of metabolites and proteins into biochemical pathways during the grain filling process
We obtained dynamic metabolomic and proteomic profiles of wheat grain components (seed coat, embryo, endosperm, and cavity fluid) during different developmental stages (12, 15, 20, and 26 DAA). For a more intuitive display of the dynamic changes in the levels of the identified metabolites (blue‐white‐red color code) and related enzymes detected from shotgun proteomics (white‐green color code) during the grain filling period, the metabolic pathways in four grain components are presented in Figure 6.
In the seed coat, most primary metabolites were present at low levels in the late grain filling stage, with a few exceptions. Phenylalanine, tyrosine, lactate, raffinose, and myo‐inositol were present at high levels at 26 DAA. Raffinose and myo‐inositol levels peaked at 26 DAA, indicating carbohydrate storage was activated in the seed coat. Furthermore, myo‐inositol is important for phosphate storage and transport of plant hormones. Myo‐inositol is linked to the decline in chlorophyll loss for photosynthetic maintenance, protecting against oxidative damage under drought stress (Li et al., 2020). The highly accumulated raffinose and myo‐inositol at 26 DAA serve either as an energy and carbon storage reserve for future biological processes or as protectants against stresses (Peterbauer and Richter, 2001). Among the detected enzymes in the seed coat, only GDH and HSK, which are related to N metabolism (Diab and Limami, 2016) and amino acid interconversion, showed increased expression during the grain filling period. These observations suggest that the seed coat serves as the ‘source’ for other grain components in the late grain filling period. The situation was similar in the endosperm. After the free amino acids are transported to the destination (developing endosperm), the conversion between amino acids is essential to provide suitable substrates for protein synthesis. Asparagine and glutamine were present at high levels at 12 DAA, which was consistent with the observed expression levels of asparagine synthetase (AsnS) and glutamine synthetase (Figure 6, Table S12). AsnS catalyzes the formation of asparagine and glutamate from glutamine and aspartate (Gaufichon et al., 2016). This observation indicates that primary N metabolism is activated in the early grain filling stage. Aspartic acid and glutamic acid which are important substrates for N supply (Xu et al., 2012a), were highly accumulated at 26 DAA (Figure 6). This is probably because aspartic acid and glutamic acid serve as the carbon skeletons in amino acid metabolism during late grain filling (Lam et al., 1996; de la Torre et al., 2014). The levels of glutamate dehydrogenase, which produces glutamic acid from glutamine, increased from 15 DAA and then kept increasing during grain filling, which was in line with the glutamic acid content. At 26 DAA, the levels of most of the other amino acids decreased (Figure 6), possibly because the amino acids are incorporated into storage proteins (Wang et al., 2016).
In the embryo, most primary metabolites were present at high levels during early grain filling, while carbohydrates such as maltose and trehalose were highly accumulated at 26 DAA (Figure 6, Table S12). The non‐reducing sugar trehalose is involved in embryo development (Eastmond et al., 2002), reflecting the status of sucrose metabolism and trehalose‐6‐phosphate (Dhatt et al., 2019). This result suggests that sugar storage is activated in the embryo during the grain filling period. In contrast to other components, AsnS and threonine synthase showed increasing expression levels, indicating that amino acids originating from aspartate (asparagine and threonine) are in high demand in the embryo during the grain filling process.
Cavity fluid only exists during the grain filling process, and starts to disappear after 26 DAA. It is the metabolic supply route for grain development and filling. In contrast to other components, in the cavity fluid, most metabolites showed the highest levels at 26 DAA, with several exceptions, including galactose, glucose, and fructose. These hexoses serve as the major carbon sources to metabolic pathways such as glycolysis, the oxidative pentose phosphate pathway, and the TCA cycle (Gibson, 2005; Jeon et al., 2010). Detected enzymes related to amino acid transformations were all downregulated during grain filling. During early grain filling, the cavity fluid supplies sugars as the carbon skeleton and aminotransferases to the neighboring endosperm. During late grain filling, the cavity fluid becomes a source of amino acids.
Glycolysis, the TCA cycle, and the mitochondrial electron transport chain are essential for many cellular functions (Fernie et al., 2004). In the present study, we identified several organic acids, such as fumaric acid, malic acid, and pyruvic acid, all of which play important roles in the TCA cycle (Figure 6, Table S12). These organic acids are taken up by mitochondria, interconverted, and used to produce energy. In the present study, the levels of these organic acids were downregulated in the course of development in the seed coat, embryo, and endosperm (but not cavity fluid). At the proteome level, all TCA cycle‐related proteins were also downregulated in the course of development. In rice, the levels of proteins involved in the TCA cycle tend to peak in early stages, indicating the TCA cycle is very active during early grain developmental stages to provide the required energy (Xu et al., 2008a).
Interestingly, in the present study, the levels of amino acids such as alanine and glutamine declined in the seed coat, embryo, and endosperm (but not cavity fluid) at 26 DAA, suggesting their incorporation into storage proteins (Figure 6, Table S12). In a study by Ma and co‐workers, it was demonstrated that high‐molecular‐weight glutenin subunits mainly affect dough elasticity, while low‐molecular‐weight glutenin subunits influence dough viscosity. In the maturity period, the glutamine concentration plays a major role in the biosynthesis of glutenin proteins (Ma et al., 2005). In the present study, glutamine levels were reduced at 26 DAA in the seed coat, embryo, and endosperm, but not cavity fluid, implying its incorporation into glutenins in mature grain. Thus, these metabolites are important indicators for wheat gluten quality.
CONCLUSION
In the present study, we explored the dynamic changes in the levels of proteins and metabolites during early and late grain filling stages from 12 to 26 DAA in the wheat grain seed coat, embryo, endosperm, and cavity fluid. Our results indicate that the coordination of metabolism and cellular processes are associated with different developmental stages of the grain filling period in the wheat grain components. For example, cell growth/division are gradually downregulated from 12 to 26 DAA in all grain components. Simultaneously, starch biosynthesis‐ and proteolysis‐related proteins are significantly upregulated from the middle to the late stage. In addition, proteins such as PPDK and 14‐3‐3 proteins undergo major changes in the concentrations at specific developmental stages, suggesting their important roles in the wheat grain filling process. At the metabolite level, the seed coat, embryo, endosperm, and cavity fluid revealed different profiles. Most of the identified primary metabolites showed low levels in the seed coat at 26 DAA, with a few exceptions. Phenylalanine, tyrosine, lactate, raffinose, and myo‐inositol were present at high levels at 26 DAA. Interestingly, metabolites involved in the photosynthetic activity of the seed coat were more abundant at 26 DAA. Similarly, most of the metabolites showed increased levels in the later stages of development in the cavity fluid. Only methionine, galactose, glucose, and fructose peaked at 12 DAA. In early grain filling, cavity fluid supplies sugars and amino acids to the endosperm, but also proteins were detected. Future studies are needed to clarify the potential roles of the proteins in the cavity fluid during the grain filling process. This study provides a substantial amount of novel information on the post‐translational regulation in the grain filling process, facilitating forward and reverse genetic tools that could effectively contribute to improving crop resilience and grain quality.
EXPERIMENTAL PROCEDURES
Plant material and growth conditions
The elite Chinese bread wheat (T. aestivum L.) cultivar Yangmai 16, a medium gluten wheat variety with large spikes and high yield, was selected. This high‐quality wheat variety is widely cultivated in Jiangsu Province, China. The plants were grown under controlled conditions (12 h of light, 120 μmol m−2 sec−1, 23°C during day time, 20°C at night, 60% humidity). Plants were fertilized by slow‐release fertilizer and watered periodically. The main culm spikes were tagged upon anthesis, and the labeled spikes were sampled at 12, 15, 20, and 26 DAA. Grain was sampled from the four spikelets at the center of each spike. Samples from each stage consisted of at least 70 seeds. Further harvested grain was manually divided into seed coat, embryo, endosperm, and cavity fluid, which were immediately frozen in liquid N2 to stop all enzymatic activity and stored at −80°C until protein and metabolite extraction. Three biological replicates were analyzed at each developmental time point to minimize experimental errors.
Grain dissection and cavity fluid extraction
The seed coat and embryo were peeled from the endosperm as quickly as possible. The surface of peeled endosperms was usually smooth, with the aleurone layer still intact. As soon as each component was separated, they were placed in 2‐mL Eppendorf tubes and frozen immediately in liquid N2.
For the endosperm and cavity fluid, grain was held with the dorsal side upwards. The center wheat grain brush (black dot in Figure S1) was gently pierced with sharp tweezers and then the cavity fluid was squeezed out. Each grain’s cavity fluid was collected in 2‐mL Eppendorf tubes and immediately frozen in liquid N2, minimizing the chance for the chemical or enzymatic transformation of solutes. Compared with a previously described method (Ugalde and Jenner, 1990), this method is simpler, faster, and less contaminating and more fluid is collected.
Measurement of physiological parameters
Grain was collected at different developmental stages (12, 15, 20, and 26 DAA) from Yangmai 16, and the length and width of four grains in three replicates were recorded. Fresh weight, dry weight, and total moisture content were determined from each grain stage.
Protein extraction and pre‐fractionation
Frozen grain components were ground into fine powder in liquid N2 using a mortar and pestle, and proteins were extracted according to a previously described method (Ghatak et al., 2016; Jegadeesan et al., 2018). In brief, homogenized grain tissue was weighed (30 mg for seed coat, endosperm, and embryo and 30 µL of cavity fluid), suspended in 200 µL of protein extraction buffer (100 mm Tris‐HCl, pH 8.0, 5% SDS, 10% glycerol, 10 mm DTT, and 1% plant protease inhibitor cocktail [Sigma P9599]), and incubated at room temperature for 5 min, followed by incubation for 2.5 min at 95°C and centrifugation at 21 000 g for 5 min at room temperature. The supernatant was carefully transferred to a new tube. Two‐hundred microliters of 1.4 M sucrose was added to the supernatant and proteins were extracted twice with 200 µL Tris‐EDTA buffer‐equilibrated phenol followed by counter‐extraction with 400 µL of 0.7 M sucrose. Phenol phases were combined and subsequently mixed with 2.5 volumes of 0.1 M ammonium acetate in methanol for precipitation of proteins. After 16 h of incubation at −20°C, samples were centrifuged for 5 min at 5000 g . The pellet was washed twice with 0.1 M ammonium acetate in methanol and once with acetone and air‐dried at room temperature. The pellet was re‐dissolved in 6 M urea and 5% SDS, and the protein concentration was determined using the bicinchoninic acid (BCA) method. Proteins were pre‐fractionated by SDS‐PAGE. Total protein (40 μg per lane) was loaded onto the gel. Gels were fixed and stained with methanol/acetic acid/water/Coomassie Brilliant Blue R‐250 (40:10:50:0.001). Gels were destained in methanol/water (40:60).
Protein digestion and LC‐MS/MS
Gel pieces were destained, equilibrated, and digested with trypsin (using Trypsin Sequencing Grade from Roche [11418475001]) (Chaturvedi et al., 2015; Chaturvedi et al., 2013; Valledor and Weckwerth, 2014). Peptides were then desalted employing a Bond‐Elute C‐18 SPEC plate (Agilent Technologies, Santa Clara, CA, USA) and concentrated in a Speed Vac concentrator (SCANVAC Cool Safe 110‐4, Speed Vacuum concentrator, Labogene). Prior to MS measurements, the tryptic peptide pellets were dissolved in 4% (v/v) acetonitrile, 0.1% (v/v) formic acid. One microgram of each sample (three replicates for each component and each developmental stage) was loaded on a C18 reverse‐phase column (Thermo scientific, EASY‐Spray 500 mm, 2 µm particle size). The separation was achieved with a 90 min gradient from 98% solution A (0.1% formic acid) and 2% solution B (90% acetonitrile and 0.1% formic acid) at 0 min to 40% solution B at 90 min with a flow rate of 300 nL min−1. Nano‐electrospray ionization‐MS/MS measurements were performed on an Orbitrap Elite (Thermo Fisher Scientific, Bremen, Germany) with the following settings: full scan range 350–1800 m/z, resolution 120 000, max. 20 MS2 scans (activation type CID), repeat count 1, repeat duration 30 sec, exclusion list size 500, exclusion duration 30 sec, charge state screening enabled with the rejection of unassigned and +1 charge states, and minimum signal threshold 500.
Peptide and protein identification
Raw data were searched with the SEQUEST algorithm present in Proteome Discoverer version 1.3 (Thermo, Germany) as described previously (Chaturvedi et al., 2015; Chaturvedi et al., 2013). We used the following settings in Proteome Discoverer for data analysis: Peptide confidence, High, which is equivalent to a false discovery rate (FDR) of 1%, and Xcorr values of 2, 3, 4, 5, and 6 for peptides of charge 2, 3, 4, 5, and 6. The variable modifications were set to acetylation of the N‐terminus and methionine oxidation, with a mass tolerance of 10 ppm for the parent ion and 0.8 Da for the fragment ion. The number of missed and non‐specific cleavages permitted was two. There were no fixed modifications, as dynamic modifications were used (Paul et al., 2016).
For identification, the UniProt database containing the annotation of 136 865 wheat genes was used. Peptides were matched against these databases plus decoys, and a hit was considered significant when the peptide confidence was high. All the MS/MS spectra of the identified proteins and their meta‐information were further uploaded to the PRIDE repository. Submission details are as follows: project name, Spatial distribution of proteins and metabolites in developing wheat grain and their differential regulatory response during the grain filling process; project accession, PXD025761.
The identified proteins were quantified based on total ion count, followed by an NSAF normalization strategy (Paoletti et al., 2006):
The total number of spectra for the matching peptides from protein k (PSM) was divided by the protein length (L) and then divided by the sum of PSM/L for all N proteins.
GC‐TOF‐MS analysis for primary metabolites and data analysis
Metabolite extraction
Metabolite extraction was performed as described previously (Weckwerth et al., 2004b), with slight modifications. Grain tissue was weighed (30 mg for seed coat, endosperm, and embryo and 30 µL of cavity fluid), freeze‐dried, and grounded to a fine powder using a mortar and pestle. Next, 750 µL of pre‐chilled extraction solution (methanol/chloroform/water [MCW) = 2.5:1:0.5 v/v/v) was added to the tissue in safe‐lock Eppendorf tubes and thoroughly mixed at 4°C for 15 min to precipitate proteins and DNA/RNA and to disassociate metabolites from the membrane and cell wall components. Next, the tubes were centrifuged at 20 000 g for 4 min at 4°C, and the supernatant was transferred to a new Eppendorf tube. The remaining pellet was washed with pre‐cooled 250 µL MCW, vortexed thoroughly at 4°C for 15 min, and centrifuged at 20 000 g for 4 min at 4°C. The supernatant was pooled with the previously collected MCW, 300 mL water was added, and the emulsion was mixed and finally centrifuged for 2 min at 20 000 g at 25°C. The upper phase (methanol/water) and the lower phase (chloroform phase) were collected from each sample, separately transferred to new Eppendorf tubes, and lyophilized in a vacuum evaporator (SCANVAC Cool Safe 110‐4, Speed Vacuum concentrator, Labogene).
Metabolite profiling
Metabolite analysis was performed according to (Obermeyer et al., 2013; Weckwerth et al., 2004a). Lyophilized samples of the polar phase (upper phase, methanol/water) were dissolved in 20 µL methoxylamine hydrochloride in pyridine (40 mg mL−1) and incubated for 90 min at 30°C in a thermoshaker. Eighty microliters of N‐methyl‐N‐(trimethylsilyl) trifluoroacetamide (MSTFA) (Macherey Nagel, Germany) was spiked in all the samples, which were incubated at 37°C for 30 min in a thermoshaker. After the incubation, the samples were centrifuged for 2 min at 14 000 g and then transferred to GC‐microvials with microinserts and closed with crimp caps. Along with the samples, 60 µL retention index marker solution of even alkanes from C10 to C40 in hexane (Sigma‐Aldrich) at a concentration of 50 mg L−1 was also prepared with MSTFA spiked. For GC‐MS analyses of primary metabolites, a LECO Pegasus 4D GC × GC TOF‐MS instrument was used. Samples, alkanes, and blanks were injected with a split/splitless injector at a constant temperature of 230°C. Injection volume was 1 µL of the derivatized sample; the injection was performed at a split ratio of 1:5 and 1:100. GC separation was conducted on an HP‐5MS column (30 m × 0.25 mm × 0.25 mm, Agilent Technologies) using helium as carrier gas at a flow rate of 1 mL min−1. The temperature gradient started at 70°C isothermal for 1 min, followed by a heating ramp of 9°C min−1 to 330°C, where the temperature was held for 7 min. The transfer line temperature was 250°C, and the ion source temperature was set to 200°C. Mass spectra were acquired with an acquisition rate of 20 spectra sec−1 at an m/z range of 40–600 using a detector voltage of 1500 V and an electron impact ionization of 70 eV.
Metabolite data analysis
Data were processed and analyses were conducted using ChromaTOF (LECO) software. Chromatograms of different samples were used to generate a reference peak list, and all other data files were processed against this reference list. Retention index markers were used to calculate retention indices of compounds and for chromatographic alignment. Deconvoluted mass spectra were matched against an in‐house mass spectral library, and this retention index was used for peak annotation. Peak annotations and peak integrations were checked manually before exporting peak areas for relative quantification to Microsoft Excel. Areas of different trimethylsilyl derivatives of single metabolites were summed, and from methoxyamine products, only one peak was selected for further analyses. All hydrophilic metabolite amounts are given in arbitrary units corresponding to the chromatograms’ peak areas (De La Harpe et al., 2020; Pazhamala et al., 2020). The heatmap was generated using the COVAIN toolbox in MATLAB (Sun and Weckwerth, 2012). Abundance levels for selected metabolites are presented as the mean ± standard deviation from three biological replicates for each grain component (seed coat, embryo, endosperm, and cavity fluid) and developmental stage (12, 15, 20, and 26 DAA).
Bioinformatics for functional annotation and statistics
Proteomics data were normalized using the NSAF approach, and metabolomics data were normalized with the internal standard and tissue’s fresh weight. The obtained data were subjected to multivariate (PCA, K‐means clustering) analysis, which was performed using the statistical toolbox COVAIN in MATLAB (Sun and Weckwerth, 2012) and R (package ggplot2). For K‐means cluster analysis, proteins were chosen only if they were present in all three biological replicates at least in one developmental stage. The Venn diagrams were produced using a Web tool (http://bioinformatics.psb.ugent.be/webtools/Venn/). Functional descriptions and annotations to wheat sequences were assigned according to a study by Ghatak and co‐workers (Ghatak et al., 2021).
To indicate the protein accumulation during the grain filling period among different components (endosperm, seed coat, cavity fluid, and embryo), we considered protein expression of the samples obtained at 12 DAA as a reference (Yu et al., 2016). The protein expression levels at each developmental stage relative to the levels at 12 DAA (15 DAA/REF, 20 DAA/REF, and 26 DAA/REF) were used to determine the abundance of a protein at each stage (Yu et al., 2016). DEPs between developmental stages for each grain component were identified considering two conditions: |log2FC| ≥ 1.5 and P < 0.05. DEPs are represented by heatmaps, prepared using the R (version 3.5, R Core Team 2019) (package ggplot2). All identified proteins were categorized into functional groups to allow a functional view of the proteome in the developmental stages of each grain component. Metabolic pathways were generated in‐house using MapMan classifications (Thimm et al., 2004).
CONFLICT OF INTEREST
The authors declare no competing financial interest.
AUTHOR CONTRIBUTIONS
SZ and WW conceived the idea of the study; PC, SZ, and AG performed the experiments; PC and SZ performed LC‐MS analysis; AG and SZ performed GC‐MS analysis; SZ, PC, PB, AG, MMB, RKV, DJ, and WW analyzed the data; PC, AG, SZ, DJ, and WW wrote the manuscript. All the authors read and agreed on the final version of the manuscript.
Supporting information
Figure S1. The color switch of the grain from white to green during the grain filling process.
Figure S2. (a) Total proteome of the seed coat (SC) during different developmental stages. (b) The proteome in selected functional categories.
Figure S3. (a) Total proteome of the embryo (EB) during different developmental stages. (b) The proteome in selected functional categories.
Figure S4. (a) Total proteome of the endosperm (EP) during different developmental stages. (b) The proteome in selected functional categories.
Figure S5. (a) Total proteome of the cavity fluid (CF) during different developmental stages. (b) The proteome in selected functional categories.
Figure S6. Selected clusters of highly expressed proteins in four grain components during early grain filling stages.
Figure S7. Selected clusters of highly expressed proteins in four grain components during late grain filling stages.
Figure S8. Principal component analysis (PCA) of the metabolite profiles from different grain components. (a) PCA of seed coat (SC). (b) PCA of embryo (EB). (c) PCA of endosperm (EP). (d) PCA of cavity fluid (CF).
Figure S9. Heatmap biclustering analysis to determine the levels of all 34 identified metabolites in different developmental stages of different grain components. (a) Seed coat (SC). (b) Embryo (EB). (c) Endosperm (EP). (d) Cavity fluid (CF).
Table S1. Physiological parameters (length and width of the grain, fresh weight, dry weight, and moisture content) were measured during wheat grain development. Additionally, fresh weight of the wheat grain components (seed coat, embryo, endosperm, and cavity fluid) was also recorded.
Table S2. Information of proteins identified from different developmental stages in the seed coat, embryo, endosperm, and cavity fluid. Data were normalized by the NSAF strategy.
Table S3. List of selected proteins that are present in all three biological replicates of at least one developmental stage of the seed coat, embryo, endosperm, and cavity fluid (max count = 3).
Table S4. Venn analyses of the identified proteins in the seed coat, embryo, endosperm, and cavity fluid during different developmental stages.
Table S5. PCA loadings of identified proteins in the seed coat, embryo, endosperm, and cavity fluid during different developmental stages. The top 100 proteins were selected in positive and negative loadings.
Table S6. Differentially expressed proteins (DEPs; proteins showing fold change ≥ 1.5 and P ≤ 0.05) between 15 vs. 12 DAA, 20 vs. 15 DAA, and 26 vs. 20 DAA in the seed coat, embryo, endosperm, and cavity fluid.
Table S7. Functional categories of proteins identified during different developmental stages in the seed coat, embryo, endosperm, embryo, and cavity fluid.
Table S8. Levels of unique DEPs during four developmental stages in the seed coat, embryo, endosperm, and cavity fluid.
Table S9. K‐means cluster analysis of the identified proteins during different developmental stages in the seed coat, embryo, endosperm, and cavity fluid.
Table S10. GC‐TOF‐MS data from different wheat grain components during four different developmental stages. Data were normalized to the internal standard and fresh weight.
Table S11. Protein accumulation pattern of the selected functional categories in seed coat, embryo, endosperm, and cavity fluid.
Table S12. Annotations and average abundance values of the identified metabolites and proteins used for pathway construction for the seed coat, embryo, endosperm, and cavity fluid during each developmental stage.
ACKNOWLEDGMENTS
Authors are thankful to the gardeners Andreas Schröfl and Thomas Joch for excellent plant cultivation in the glasshouse facility of the Molecular Systems Biology Lab (MOSYS), Department of Functional and Evolutionary Ecology, University of Vienna, Vienna, Austria. SZ is supported by a PhD scholarship provided by the China Scholarship Council (CSC) (grant number: 201706850037) and a completion grant of the Vienna Doctoral School Ecology and Evolution of the Faculty of Life Sciences, University of Vienna, Austria. AG is supported by the European Union Horizon 2020 research and innovation program (ADAPT) under grant agreement number GA 2020 862‐858.
Linked article: This paper is the subject of a Research Highlight article. To view this Research Highlight article visit https://doi.org/10.1111/tpj.15422
Contributor Information
Palak Chaturvedi, Email: palak.chaturvedi@univie.ac.at.
Wolfram Weckwerth, Email: wolfram.weckwerth@univie.ac.at.
DATA AVAILABILITY STATEMENT
The datasets presented in this study can be found in the online repositories of PRIDE (project accession: PXD025761). All details are provided in the Experimental Procedures section.
REFERENCES
- An, L. , Tao, Y. , Chen, H. , He, M.J. , Xiao, F. , Li, G.H. et al. (2020) Embryo‐endosperm interaction and its agronomic relevance to rice quality. Frontiers in Plant Science, 11, 587641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, N.L. & Anderson, N.G. (1998) Proteome and proteomics: new technologies, new concepts, and new words. Electrophoresis, 19, 1853–1861. [DOI] [PubMed] [Google Scholar]
- Barneix, A.J. (2007) Physiology and biochemistry of source‐regulated protein accumulation in the wheat grain. Journal of Plant Physiology, 164, 581–590. [DOI] [PubMed] [Google Scholar]
- Barron, C. , Surget, A. & Rouau, X. (2007) Relative amounts of tissues in mature wheat (Triticum aestivum L.) grain and their carbohydrate and phenolic acid composition. Journal of Cereal Science, 45, 88–96. [Google Scholar]
- Brenchley, R. , Spannagl, M. , Pfeifer, M. , Barker, G.L.A. , D'Amore, R. , Allen, A.M. et al. (2012) Analysis of the breadwheat genome using whole‐genome shotgun sequencing. Nature, 491, 705–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, H. , He, M. , Zhu, C. , Yuan, L. , Dong, L. , Bian, Y. et al. (2016) Distinct metabolic changes between wheat embryo and endosperm during grain development revealed by 2D‐DIGE‐based integrative proteome analysis. Proteomics, 16, 1515–1536. [DOI] [PubMed] [Google Scholar]
- Chaturvedi, P. , Doerfler, H. , Jegadeesan, S. , Ghatak, A. , Pressman, E. , Castillejo, M.A. et al. (2015) Heat‐treatment‐responsive proteins in different developmental stages of tomato pollen detected by Targeted Mass Accuracy Precursor Alignment (tMAPA). Journal of Proteome Research, 14, 4463–4471. [DOI] [PubMed] [Google Scholar]
- Chaturvedi, P. , Ischebeck, T. , Egelhofer, V. , Lichtscheidl, I. & Weckwerth, W. (2013) Cell‐specific analysis of the tomato pollen proteome from pollen mother cell to mature pollen provides evidence for developmental priming. Journal of Proteome Research, 12, 4892–4903. [DOI] [PubMed] [Google Scholar]
- Chaudhury, A.M. , Koltunow, A. , Payne, T. , Luo, M. , Tucker, M.R. , Dennis, E.S. et al. (2001) Control of early seed development. Annual Review of Cell and Developmental Biology, 17, 677–699. [DOI] [PubMed] [Google Scholar]
- Christiaens, A. , Van Labeke, M.C. , Pauwels, E. , Gobin, B. , De Keyser, E. & De Riek, J. (2012) Flowering quality of azalea (Rhododendron simsii.) following treatments with plant growth regulators. Xxviii International Horticultural Congress on Science and Horticulture for People (Ihc2010): International Symposium on Advances in Ornamentals, Landscape and Urban Horticulture, 937, 219–224. [Google Scholar]
- Clavijo, B.J. , Venturini, L. , Schudoma, C. , Accinelli, G.G. , Kaithakottil, G. , Wright, J. et al. (2017) An improved assembly and annotation of the allohexaploid wheat genome identifies complete families of agronomic genes and provides genomic evidence for chromosomal translocations. Genome Research, 27, 885–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counce, P.A. & Gravois, K.A. (2006) Sucrose synthase activity as a potential indicator of high rice grain yield. Crop Science, 46, 1501–1507. [Google Scholar]
- d'Aloisio, E. , Paolacci, A.R. , Dhanapal, A.P. , Tanzarella, O.A. , Porceddu, E. & Ciaffi, M. (2010) The Protein Disulfide Isomerase gene family in bread wheat ( T. aestivum L.). BMC Plant Biology, 10, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das, A. , Kim, D.W. , Khadka, P. , Rakwal, R. & Rohila, J.S. (2017) Unraveling key metabolomic alterations in wheat embryos derived from freshly harvested and water‐imbibed seeds of two wheat cultivars with contrasting dormancy status. Frontiers in Plant Science, 8, 1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Harpe, M. , Paris, M. , Hess, J. , Barfuss, M.H.J. , Serrano‐Serrano, M.L. , Ghatak, A. et al. (2020) Genomic footprints of repeated evolution ofCAMphotosynthesis in a Neotropical species radiation. Plant Cell and Environment, 43, 2987–3001. [DOI] [PubMed] [Google Scholar]
- de la Torre, F. , Canas, R.A. , Pascual, M.B. , Avila, C. & Canovas, F.M. (2014) Plastidic aspartate aminotransferases and the biosynthesis of essential amino acids in plants. Journal of Experimental Botany, 65, 5527–5534. [DOI] [PubMed] [Google Scholar]
- Dhatt, B.K. , Abshire, N. , Paul, P. , Hasanthika, K. , Sandhu, J. , Zhang, Q. et al. (2019) Metabolic dynamics of developing rice seeds under high night‐time temperature stress. Frontiers in Plant Science, 10, 1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diab, H. & Limami, A.M. (2016) Reconfiguration of N metabolism upon hypoxia stress and recovery: roles of Alanine Aminotransferase (AlaAT) and Glutamate Dehydrogenase (GDH). Plants, 5(2), 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont, F.M. (2008) Metabolic pathways of the wheat (Triticum aestivum) endosperm amyloplast revealed by proteomics. BMC Plant Biology, 8(1), 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont, F.M. , Vensel, W.H. , Tanaka, C.K. , Hurkman, W.J. & Altenbach, S.B. (2011) Deciphering the complexities of the wheat flour proteome using quantitative two‐dimensional electrophoresis, three proteases and tandem mass spectrometry. Proteome Science, 9(1), 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastmond, P.J. , Astley, H.M. , Parsley, K. , Aubry, S. , Williams, B.P. , Menard, G.N. et al. (2015) Arabidopsis uses two gluconeogenic gateways for organic acids to fuel seedling establishment. Nature communications, 6, 6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastmond, P.J. , van Dijken, A.J.H. , Spielman, M. , Kerr, A. , Tissier, A.F. , Dickinson, H.G. et al. (2002) Trehalose‐6‐phosphate synthase 1, which catalyses the first step in trehalose synthesis, is essential for Arabidopsis embryo maturation. Plant Journal, 29, 225–235. [DOI] [PubMed] [Google Scholar]
- Ferl, R.J. (1996) 14‐3‐3 proteins and signal transduction. Annual Review of Plant Physiology and Plant Molecular Biology, 47, 49–73. [DOI] [PubMed] [Google Scholar]
- Fernie, A.R. , Carrari, F. & Sweetlove, L.J. (2004) Respiratory metabolism: Glycolysis, the TCA cycle and mitochondrial electron transport. Current Opinion in Plant Biology, 7, 254–261. [DOI] [PubMed] [Google Scholar]
- Fisher, D.B. & Gifford, R.M. (1986) Accumulation and conversion of sugars by developing wheat grains. 6. Gradients along the transport pathway from the peduncle to the endosperm cavity during grain filling. Plant Physiology, 82, 1024–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francki, M.G. , Hayton, S. , Gummer, J.P.A. , Rawlinson, C. & Trengove, R.D. (2016) Metabolomic profiling and genomic analysis of wheat aneuploid lines to identify genes controlling biochemical pathways in mature grain. Plant Biotechnology Journal, 14, 649–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulgosi, H. , Soll, J. , Maraschin, S.D. , Korthout, H.A.A.J. , Wang, M. & Testerink, C. (2002) 14‐3‐3 proteins and plant development. Plant Molecular Biology, 50, 1019–1029. [DOI] [PubMed] [Google Scholar]
- Gaufichon, L. , Rothstein, S.J. & Suzuki, A. (2016) Asparagine metabolic pathways in arabidopsis. Plant and Cell Physiology, 57, 675–689. [DOI] [PubMed] [Google Scholar]
- Ge, P. , Ma, C. , Wang, S. , Gao, L. , Li, X. , Guo, G. et al. (2012) Comparative proteomic analysis of grain development in two spring wheat varieties under drought stress. Analytical and Bioanalytical Chemistry, 402, 1297–1313. [DOI] [PubMed] [Google Scholar]
- Ghatak, A. , Chaturvedi, P. , Bachmann, G. , Valledor, L. , Ramsak, Z. , Bazargani, M.M. et al. (2021) Physiological and proteomic signatures reveal mechanisms of superior drought resilience in pearl millet compared to wheat. Frontiers in Plant Science, 11, 600278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghatak, A. , Chaturvedi, P. , Nagler, M. , Roustan, V. , Lyon, D. , Bachmann, G. et al. (2016) Comprehensive tissue‐specific proteome analysis of drought stress responses in Pennisetum glaucum (L.) R. Br. (Pearl millet). Journal of Proteomics, 143, 122–135. [DOI] [PubMed] [Google Scholar]
- Gibson, S.I. (2005) Control of plant development and gene expression by sugar signaling. Current Opinion in Plant Biology, 8, 93–102. [DOI] [PubMed] [Google Scholar]
- Girke, T. , Todd, J. , Ruuska, S. , White, J. , Benning, C. & Ohlrogge, J. (2000) Microarray analysis of developing Arabidopsis seeds. Plant Physiology, 124, 1570–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu, A.Q. , Hao, P.C. , Lv, D.W. , Zhen, S.M. , Bian, Y.W. , Ma, C.Y. et al. (2015) Integrated proteome analysis of the wheat embryo and endosperm reveals central metabolic changes involved in the water deficit response during grain development. Journal of Agricultural and Food Chemistry, 63, 8478–8487. [DOI] [PubMed] [Google Scholar]
- Guo, G. , Lv, D. , Yan, X. , Subburaj, S. , Ge, P. , Li, X. et al. (2012a) Proteome characterization of developing grains in bread wheat cultivars (Triticum aestivum L.). BMC Plant Biology, 12, 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, G.F. , Lv, D.W. , Yan, X. , Subburaj, S. , Ge, P. , Li, X.H. et al. (2012b) Proteome characterization of developing grains in bread wheat cultivars (Triticum aestivum L.). BMC Plant Biology, 12(1), 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gygi, S.P. , Rochon, Y. , Franza, B.R. & Aebersold, R. (1999) Correlation between protein and mRNA abundance in yeast. Molecular and Cellular Biology, 19, 1720–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, C.X. , Zhen, S.M. , Zhu, G.R. , Bian, Y.W. & Yan, Y.M. (2017) Comparative metabolome analysis of wheat embryo and endosperm reveals the dynamic changes of metabolites during seed germination. Plant Physiology and Biochemistry, 115, 320–327. [DOI] [PubMed] [Google Scholar]
- He, D. , Han, C. , Yao, J. , Shen, S. & Yang, P. (2011) Constructing the metabolic and regulatory pathways in germinating rice seeds through proteomic approach. Proteomics, 11, 2693–2713. [DOI] [PubMed] [Google Scholar]
- Houston, N.L. , Fan, C. , Xiang, J.‐Q.‐Y. , Schulze, J.‐M. , Jung, R. & Boston, R.S. (2005) Phylogenetic analyses identify 10 classes of the protein disulfide isomerase family in plants, including single‐domain protein disulfide isomerase‐related proteins. Plant Physiology, 137, 762–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houston, N.L. , Hajduch, M. & Thelen, J.J. (2009) Quantitative proteomics of seed filling in castor: comparison with soybean and rapeseed reveals differences between photosynthetic and nonphotosynthetic seed metabolism. Plant Physiology, 151, 857–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jegadeesan, S. , Chaturvedi, P. , Ghatak, A. , Pressman, E. , Meir, S. , Faigenboim, A. et al. (2018) Proteomics of heat‐stress and ethylene‐mediated thermotolerance mechanisms in tomato pollen grains. Frontiers in Plant Science, 9, 1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon, J.S. , Ryoo, N. , Hahn, T.R. , Walia, H. & Nakamura, Y. (2010) Starch biosynthesis in cereal endosperm. Plant Physiology and Biochemistry, 48, 383–392. [DOI] [PubMed] [Google Scholar]
- Jin, X.N. , Fu, Z.Y. , Ding, D. , Li, W.H. , Liu, Z.H. & Tang, J.H. (2013) Proteomic identification of genes associated with maize grain‐filling rate. PLoS One, 8(3), e59353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam, H.M. , Coschigano, K.T. , Oliveira, I.C. , MeloOliveira, R. & Coruzzi, G.M. (1996) The molecular‐genetics of nitrogen assimilation into amino acids in higher plants. Annual Review of Plant Physiology and Plant Molecular Biology, 47, 569–593. [DOI] [PubMed] [Google Scholar]
- Lampl, N. , Budai‐Hadrian, O. , Davydov, O. , Joss, T.V. , Harrop, S.J. , Curmi, P.M.G. et al. (2010) Arabidopsis AtSerpin1, crystal structure and in Vivo interaction with its target protease RESPONSIVE TO DESICCATION‐21 (RD21) *. Journal of Biological Chemistry, 285, 13550–13560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laudencia‐Chingcuanco, D.L. , Stamova, B.S. , You, F.M. , Lazo, G.R. , Beckles, D.M. & Anderson, O.D. (2007) Transcriptional profiling of wheat caryopsis development using cDNA microarrays. Plant Molecular Biology, 63, 651–668. [DOI] [PubMed] [Google Scholar]
- Li, Z. , Fu, J. , Shi, D. & Peng, Y. (2020) Myo‐inositol enhances drought tolerance increeping bentgrass through alteration of osmotic adjustment, photosynthesis, and antioxidant defense. Crop Science, 60, 2149–2158. [Google Scholar]
- Loussert, C. , Popineau, Y. & Mangavel, C. (2008) Protein bodies ontogeny and localization of prolamin components in the developing endosperm of wheat caryopses. Journal of Cereal Science, 47, 445–456. [Google Scholar]
- Ma, C. , Zhou, J. , Chen, G. , Bian, Y. , Lv, D. , Li, X. et al. (2014a) iTRAQ‐based quantitative proteome and phosphoprotein characterization reveals the central metabolism changes involved in wheat grain development. BMC Genomics, 15, 1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, C.Y. , Zhou, J.W. , Chen, G.X. , Bian, Y.W. , Lv, D.W. , Li, X.H. et al. (2014b) iTRAQ‐based quantitative proteome and phosphoprotein characterization reveals the central metabolism changes involved in wheat grain development. Bmc Genomics, 15, 1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, W. , Appels, R. , Bekes, F. , Larroque, O. , Morell, M.K. & Gale, K.R. (2005) Genetic characterisation of dough rheological properties in a wheat doubled haploid population: additive genetic effects and epistatic interactions. Theoretical and Applied Genetics, 111, 410–422. [DOI] [PubMed] [Google Scholar]
- McIntosh, S. , Watson, L. , Bundock, P. , Crawford, A. , White, J. , Cordeiro, G. et al. (2007) SAGE of the developing wheat caryopsis. Plant Biotechnology Journal, 5, 69–83. [DOI] [PubMed] [Google Scholar]
- Mcqueenmason, S. & Cosgrove, D.J. (1994) Disruption of hydrogen‐bonding between plant‐cell wall polymers by proteins that induce wall extension. Proceedings of the National Academy of Sciences United States of America, 91, 6574–6578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcqueenmason, S.J. & Cosgrove, D.J. (1995) Expansin mode of action on cell‐walls ‐ Analysis of wall hydrolysis, stress‐relaxation, and binding. Plant Physiology, 107, 87–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehdi, C. , Virginie, L. , Audrey, G. , Axelle, B. , Colette, L. , Hélène, R. et al. (2020) Cell wall proteome of wheat grain endosperm and outer layers at two key stages of early development. International Journal of Molecular Sciences, 21, 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon, J. , Parry, G. & Estelle, M. (2004) The ubiquitin‐proteasome pathway and plant development. The Plant Cell, 16, 3181–3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenthal, K. , Wienkoop, S. , Scholz, M. , Selbig, J. & Weckwerth, W. (2005) Correlative GC‐TOF‐MS‐based metabolite profiling and LC‐MS‐based protein profiling reveal time‐related systemic regulation of metabolite–protein networks and improve pattern recognition for multiple biomarker selection. Metabolomics, 1, 109–121. [Google Scholar]
- Nadaud, I. , Girousse, C. , Debiton, C. , Chambon, C. , Bouzidi, M.F. , Martre, P. et al. (2010) Proteomic and morphological analysis of early stages of wheat grain development. Proteomics, 10, 2901–2910. [DOI] [PubMed] [Google Scholar]
- Neuberger, T. , Sreenivasulu, N. , Rokitta, M. , Rolletschek, H. , Gobel, C. , Rutten, T. et al. (2008) Quantitative imaging of oil storage in developing crop seeds. Plant Biotechnology Journal, 6, 31–45. [DOI] [PubMed] [Google Scholar]
- Obermeyer, G. , Fragner, L. , Lang, V. & Weckwerth, W. (2013) Dynamic adaption of metabolic pathways during germination and growth of lily pollen tubes after inhibition of the electron transport chain. Plant Physiology, 162, 1822–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti, A.C. , Parmely, T.J. , Tomomori‐Sato, C. , Sato, S. , Zhu, D.X. , Conaway, R.C. et al. (2006) Quantitative proteomic analysis of distinct mammalian Mediator complexes using normalized spectral abundance factors. Proceedings of the National Academy of Sciences United States of America, 103, 18928–18933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul, P. , Chaturvedi, P. , Selymesi, M. , Ghatak, A. , Mesihovic, A. , Scharf, K.D. et al. (2016) The membrane proteome of male gametophyte in Solanum lycopersicum. Journal of Proteomics, 131, 48–60. [DOI] [PubMed] [Google Scholar]
- Pazhamala, L.T. , Chaturvedi, P. , Bajaj, P. , Srikanth, S. , Ghatak, A. , Chitikineni, A. et al. (2020) Multiomics approach unravels fertility transition in a pigeonpea line for a two‐line hybrid system. Plant Genome‐Us, 13, e20028. [DOI] [PubMed] [Google Scholar]
- Peterbauer, T. & Richter, A. (2001) Biochemistry and physiology of raffinose family oligosaccharides and galactosyl cyclitols in seeds. Seed Science Research, 11, 185–197. [Google Scholar]
- Pfeifer, M. , Kugler, K.G. , Sandve, S.R. , Zhan, B.J. , Rudi, H. , Hvidsten, T.R. et al. (2014) Genome interplay in the grain transcriptome of hexaploid bread wheat. Science, 345(6194), 1250091. [DOI] [PubMed] [Google Scholar]
- Radchuk, V. & Borisjuk, L. (2014) Physical, metabolic and developmental functions of the seed coat. Frontiers in Plant Science, 5, 510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radchuk, V.V. , Borisjuk, L. , Sreenivasulu, N. , Merx, K. , Mock, H.P. , Rolletschek, H. et al. (2009) Spatiotemporal profiling of starch biosynthesis and degradation in the developing barley grain. Plant Physiology, 150, 190–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts, T.H. & Hejgaard, J. (2008) Serpins in plants and green algae. Functional & Integrative Genomics, 8, 1–27. [DOI] [PubMed] [Google Scholar]
- Rolletschek, H. , Grafahrend‐Belau, E. , Munz, E. , Radchuk, V. , Kartausch, R. , Tschiersch, H. et al. (2015) Metabolic architecture of the cereal grain and its relevance to maximize carbon use efficiency. Plant Physiology, 169, 1698–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos‐Mendoza, M. , Dubreucq, B. , Baud, S. , Parcy, F. , Caboche, M. & Lepiniec, L. (2008) Deciphering gene regulatory networks that control seed development and maturation in Arabidopsis. Plant Journal, 54, 608–620. [DOI] [PubMed] [Google Scholar]
- Sehnke, P.C. , Chung, H.J. , Wu, K. & Ferl, R.J. (2001) Regulation of starch accumulation by granule‐associated plant 14‐3‐3 proteins. Proceedings of the National Academy of Sciences United States of America, 98, 765–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shewry, P.R. (2009) Wheat. Journal of Experimental Botany, 60, 1537–1553. [DOI] [PubMed] [Google Scholar]
- Shi, Z.Q. , Wang, Y. , Wan, Y.F. , Hassall, K. , Jiang, D. , Shewry, P.R. et al. (2019) Gradients of gluten proteins and free amino acids along the longitudinal axis of the developing caryopsis of bread wheat. Journal of Agricultural and Food Chemistry, 67, 8706–8714. [DOI] [PubMed] [Google Scholar]
- Smart, M.G. & Obrien, T.P. (1983) The development of the wheat embryo in relation to the neighboring tissues. Protoplasma, 114, 1–13. [Google Scholar]
- Sreenivasulu, N. , Radchuk, V. , Strickert, M. , Miersch, O. , Weschke, W. & Wobus, U. (2006) Gene expression patterns reveal tissue‐specific signaling networks controlling programmed cell death and ABA‐ regulated maturation in developing barley seeds. The Plant Journal, 47, 310–327. [DOI] [PubMed] [Google Scholar]
- Sun, X. & Weckwerth, W. (2012) COVAIN: A toolbox for uni‐ and multivariate statistics, time‐series and correlation network analysis and inverse estimation of the differential Jacobian from metabolomics covariance data. Metabolomics, 8, 81–93. [Google Scholar]
- Sung, S.J. , Xu, D.P. & Black, C.C. (1989) Identification of actively filling sucrose sinks. Plant Physiology, 89, 1117–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel, J. , Muller, M. , Weschke, W. & Weber, H. (2009) Amino acid metabolism at the maternal‐filial boundary of young barley seeds: a microdissection‐based study. Planta, 230, 205–213. [DOI] [PubMed] [Google Scholar]
- Thimm, O. , Blasing, O. , Gibon, Y. , Nagel, A. , Meyer, S. , Kruger, P. et al. (2004) MAPMAN: a user‐driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. The Plant Journal, 37, 914–939. [DOI] [PubMed] [Google Scholar]
- Tran, V. , Weier, D. , Radchuk, R. , Thiel, J. & Radchuk, V. (2014) Caspase‐like activities accompany programmed cell death events in developing barley grains. PLoS One, 9(10), e109426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugalde, T.D. & Jenner, C.F. (1990) Substrate gradients and regional patterns of dry‐matter deposition within developing wheat endosperm. 2. Amino‐acids and protein. Australian Journal of Plant Physiology, 17, 395–406. [Google Scholar]
- Valledor, L. & Weckwerth, W. (2014) An improved detergent‐compatible gel‐fractionation LC‐LTQ‐Orbitrap‐MS workflow for plant and microbial proteomics. Methods in Molecular Biology, 1072, 347–358. [DOI] [PubMed] [Google Scholar]
- van Bel, A.J. , Helariutta, Y. , Thompson, G.A. , Ton, J. , Dinant, S. , Ding, B. et al. (2013) Phloem: The integrative avenue for resource distribution, signaling, and defense. Frontiers in Plant Science, 4, 471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vensel, W.H. , Tanaka, C.K. , Cai, N. , Wong, J.H. , Buchanan, B.B. & Hurkman, W.J. (2005) Developmental changes in the metabolic protein profiles of wheat endosperm. Proteomics, 5, 1594–1611. [DOI] [PubMed] [Google Scholar]
- Vierstra, R.D. (2009) The ubiquitin‐26S proteasome system at the nexus of plant biology. Nature Reviews Molecular Cell Biology, 10, 385–397. [DOI] [PubMed] [Google Scholar]
- Wang, H. , Ham, T.‐H. , Im, D.‐E. , Lar, S.M. , Jang, S.‐G. , Lee, J. et al. (2020) A new SNP in rice gene encoding Pyruvate Phosphate Dikinase (PPDK) associated with floury endosperm. Genes, 11, 465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Fu, J.L. , Li, M. , Fragner, L. , Weckwerth, W. & Yang, P.F. (2016) Metabolomic and proteomic profiles reveal the dynamics of primary metabolism during seed development of lotus (Nelumbo nucifera) . Frontiers in Plant Science, 7, 750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weckwerth, W. (2011) Green systems biology ‐ From single genomes, proteomes and metabolomes to ecosystems research and biotechnology. Journal of Proteomics, 75, 284–305. [DOI] [PubMed] [Google Scholar]
- Weckwerth, W. , Ghatak, A. , Bellaire, A. , Chaturvedi, P. & Varshney, R.K. (2020) PANOMICS meets germplasm. Plant Biotechnology Journal, 18, 1507–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weckwerth, W. , Loureiro, M.E. , Wenzel, K. & Fiehn, O. (2004a) Differential metabolic networks unravel the effects of silent plant phenotypes. Proceedings of the National Academy of Sciences United States of America, 101, 7809–7814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weckwerth, W. , Wenzel, K. & Fiehn, O. (2004b) Process for the integrated extraction identification, and quantification of metabolites, proteins and RNA to reveal their co‐regulation in biochemical networks. Proteomics, 4, 78–83. [DOI] [PubMed] [Google Scholar]
- Weckwerth, W. , Wenzel, K. & Fiehn, O. (2004c) Process for the integrated extraction, identification and quantification of metabolites, proteins and RNA to reveal their co‐regulation in biochemical networks. Proteomics, 4, 78–83. [DOI] [PubMed] [Google Scholar]
- Wienkoop, S. , Morgenthal, K. , Wolschin, F. , Scholz, M. , Selbig, J. & Weckwerth, W. (2008) Integration of metabolomic and proteomic phenotypes: Analysis of data covariance dissects starch and RFO metabolism from low and high temperature compensation response in Arabidopsis thaliana. Molecular & Cellular Proteomics: MCP, 7, 1725–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson, B. & Gilbert, H.F. (2004) Protein disulfide isomerase. Biochimica et Biophysica Acta (BBA) ‐ Proteins & Proteomics, 1699(1‐2), 35–44. [DOI] [PubMed] [Google Scholar]
- Xu, G.H. , Fan, X.R. & Miller, A.J. (2012a) Plant nitrogen assimilation and use efficiency. Annual Review of Plant Biology, 63(63), 153–182. [DOI] [PubMed] [Google Scholar]
- Xu, H. , Gao, Y. & Wang, J.B. (2012b) Transcriptomic analysis of rice (Oryza sativa) developing embryos using the RNA‐Seq technique. PLoS One, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, S.B. , Li, T. , Deng, Z.Y. , Chong, K. , Xue, Y. & Wang, T. (2008a) Dynamic proteomic analysis reveals a switch between central carbon metabolism and alcoholic fermentation in rice filling grains. Plant Physiology, 148, 908–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, S.B. , Li, T. , Deng, Z.Y. , Chong, K. , Xue, Y.B. & Wang, T. (2008b) Dynamic proteomic analysis reveals a switch between central carbon metabolism and alcoholic fermentation in rice filling grains. Plant Physiology, 148, 908–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue, L.J. , Zhang, J.J. & Xue, H.W. (2012) Genome‐wide analysis of the complex transcriptional networks of rice developing seeds. PLoS One, 7, e31081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, M.M. , Dong, J. , Zhao, W.C. & Gao, X. (2016) Characterization of proteins involved in early stage of wheat grain development by iTRAQ. Journal of Proteomics, 136, 157–166. [DOI] [PubMed] [Google Scholar]
- Yang, M.M. , Gao, X. , Dong, J. , Gandhi, N. , Cai, H.J. , von Wettstein, D.H. et al. (2017) Pattern of protein expression in developing wheat grains identified through proteomic analysis. Frontiers in Plant Science, 8, 962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, T. , Li, G. , Dong, S.T. , Liu, P. , Zhang, J.W. & Zhao, B. (2016) Proteomic analysis of maize grain development using iTRAQ reveals temporal programs of diverse metabolic processes. BMC Plant Biology, 16, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, X.R. , Chen, X.Y. , Zhou, L. , Zhang, J. , Yu, H. , Shao, S.S. et al. (2015) Structural development of wheat nutrient transfer tissues and their relationships with filial tissues development. Protoplasma, 252, 605–617. [DOI] [PubMed] [Google Scholar]
- Zhang, N. , Chen, F. , Hue, W. & Cui, D.Q. (2015) Proteomic analysis of middle and late stages of bread wheat (Triticum aestivum L.) grain development. Frontiers in Plant Science, 6, 735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z.X. , Zhao, H. , Tang, J. , Li, Z. , Li, Z. , Chen, D.M. et al. (2014) A proteomic study on molecular mechanism of poor grain‐filling of rice (Oryza sativa L.) inferior spikelets. PLoS One, 9, e89140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Z.Q. , Wang, L.K. , Li, J.W. , Song, X.F. & Yang, C.A. (2009) Study on programmed cell death and dynamic changes of starch accumulation in pericarp cells of Triticum aestivum L.*. Protoplasma, 236, 49–58. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The color switch of the grain from white to green during the grain filling process.
Figure S2. (a) Total proteome of the seed coat (SC) during different developmental stages. (b) The proteome in selected functional categories.
Figure S3. (a) Total proteome of the embryo (EB) during different developmental stages. (b) The proteome in selected functional categories.
Figure S4. (a) Total proteome of the endosperm (EP) during different developmental stages. (b) The proteome in selected functional categories.
Figure S5. (a) Total proteome of the cavity fluid (CF) during different developmental stages. (b) The proteome in selected functional categories.
Figure S6. Selected clusters of highly expressed proteins in four grain components during early grain filling stages.
Figure S7. Selected clusters of highly expressed proteins in four grain components during late grain filling stages.
Figure S8. Principal component analysis (PCA) of the metabolite profiles from different grain components. (a) PCA of seed coat (SC). (b) PCA of embryo (EB). (c) PCA of endosperm (EP). (d) PCA of cavity fluid (CF).
Figure S9. Heatmap biclustering analysis to determine the levels of all 34 identified metabolites in different developmental stages of different grain components. (a) Seed coat (SC). (b) Embryo (EB). (c) Endosperm (EP). (d) Cavity fluid (CF).
Table S1. Physiological parameters (length and width of the grain, fresh weight, dry weight, and moisture content) were measured during wheat grain development. Additionally, fresh weight of the wheat grain components (seed coat, embryo, endosperm, and cavity fluid) was also recorded.
Table S2. Information of proteins identified from different developmental stages in the seed coat, embryo, endosperm, and cavity fluid. Data were normalized by the NSAF strategy.
Table S3. List of selected proteins that are present in all three biological replicates of at least one developmental stage of the seed coat, embryo, endosperm, and cavity fluid (max count = 3).
Table S4. Venn analyses of the identified proteins in the seed coat, embryo, endosperm, and cavity fluid during different developmental stages.
Table S5. PCA loadings of identified proteins in the seed coat, embryo, endosperm, and cavity fluid during different developmental stages. The top 100 proteins were selected in positive and negative loadings.
Table S6. Differentially expressed proteins (DEPs; proteins showing fold change ≥ 1.5 and P ≤ 0.05) between 15 vs. 12 DAA, 20 vs. 15 DAA, and 26 vs. 20 DAA in the seed coat, embryo, endosperm, and cavity fluid.
Table S7. Functional categories of proteins identified during different developmental stages in the seed coat, embryo, endosperm, embryo, and cavity fluid.
Table S8. Levels of unique DEPs during four developmental stages in the seed coat, embryo, endosperm, and cavity fluid.
Table S9. K‐means cluster analysis of the identified proteins during different developmental stages in the seed coat, embryo, endosperm, and cavity fluid.
Table S10. GC‐TOF‐MS data from different wheat grain components during four different developmental stages. Data were normalized to the internal standard and fresh weight.
Table S11. Protein accumulation pattern of the selected functional categories in seed coat, embryo, endosperm, and cavity fluid.
Table S12. Annotations and average abundance values of the identified metabolites and proteins used for pathway construction for the seed coat, embryo, endosperm, and cavity fluid during each developmental stage.
Data Availability Statement
The datasets presented in this study can be found in the online repositories of PRIDE (project accession: PXD025761). All details are provided in the Experimental Procedures section.