

Abstract
Immuno‐oncology has been at the forefront of cancer treatment in recent decades. In particular immune checkpoint and chimeric antigen receptor (CAR)‐T cell therapy have achieved spectacular results. Over the years, CAR‐T cell development has followed a steady evolutionary path, focusing on increasing T cell potency and sustainability, which has given rise to different CAR generations. However, there was less focus on the mode of interaction between the CAR‐T cell and the cancer cell; more specifically on the targeting moiety used in the CAR and its specific properties. Recently, the importance of optimizing this domain has been recognized and the possibilities have been exploited. Over the last 10 years—in addition to the classical scFv‐based CARs—single domain CARs, natural receptor‐ligand CARs, universal CARs and CARs targeting more than one antigen have emerged. In addition, the specific parameters of the targeting domain and their influence on T cell activation are being examined. In this review, we concisely present the history of CAR‐T cell therapy, and then expand on various developments in the CAR ectodomain. We discuss different formats, each with their own advantages and disadvantages, as well as the developments in affinity tuning, avidity effects, epitope location, and influence of the extracellular spacer.
Keywords: adoptive T cell therapy, cancer, chimeric antigen receptor, immunotherapy, T cell receptor, tumor‐infiltrating lymphocyte
1. INTRODUCTION: EXPLOITING THE T CELL'S POTENTIAL IN TUMOR CELL KILLING
The principle of cancer immunotherapy is to harness the immune system to selectively attack cancer cells and build an immunological memory to avoid relapse when cancer cells reoccur. This concept is over a century old, however was long received with skepticism. 1 Only in the last decades, immunotherapy became trending owing to increased knowledge in cancer immunology and linked herewith the birth of immuno‐oncology. 2 , 3
It is well‐established that cancer cells are genetically altered cells that carry tumor‐associated antigens (TAAs). These TAAs serve as an identification card for immune cells to discriminate cancer cells from the healthy cells they arise from. In particular, CD8+ cytotoxic T lymphocytes (CTLs) have been proposed as important effectors of the anticancer immune response. 4 Activated CTLs use their T cell receptor (TCR) to scan for TAA‐derived peptides bound to major histocompatibility complex class I (MHC‐I) molecules on cancer cells. 5 , 6 Functional recognition of peptide/MHC‐I complexes by the TCR triggers CTL‐mediated cancer cell killing. Important effector molecules during this process are Fas ligand (FasL), perforin and granzymes, and cytokines such as interferon‐γ (IFN‐γ) and tumor necrosis factor‐α (TNF‐α). 4 CTLs are key in killing cancer cells, however, they do not act alone. Activation of CD8+ T cells to become potent cancer killing CTLs requires antigen presentation by professional antigen‐presenting cells such as dendritic cells (DCs) and is supported by CD4+ T helper 1 (TH1) cells. Moreover, IFN‐γ produced by both TH1 cells and CTLs further stimulates macrophages to perform antitumor activities, showing that the anticancer immune response is a multifaceted process, involving intricate communication between cells of the innate and adaptive immune system (Figure 1). 7 , 8
Figure 1.
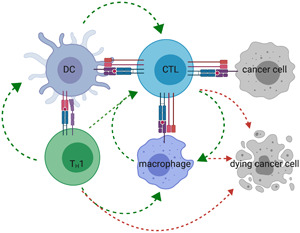
The anticancer immune response. DCs that acquired TAAs present these in MHC‐II molecules to CD4+ T cells and cross‐present these in MHC‐I molecules to CD8+ T cells. When presentation of TAAs is performed in the context of costimulation, provided by ligand–receptor interactions like B7.1/2‐CD28, and cytokines like interleukin‐12 (IL‐12), CD4+ T cells are activated to TH1 cells. These TH1 cells license DCs to become more potent in CD8+ T cell activation as well as they directly support CD8+ T cell proliferation and survival through cytokine secretion. Activated CD8+ T cells (CTLs) kill their target using FasL, release of cytokines, perforin, and granzymes. Moreover, IFN‐γ released by TH1 cells and CTLs triggers classical macrophage polarization, as such amplifies anticancer responses, as these macrophages support CTL activation and exert tumoricidal activities. Green arrows indicate communication between immune cells, while red arrows indicate immune cells that can exert tumoricidal activities. Of these, CTLs are key in cancer cell killing. CTL, cytotoxic T lymphocyte; MHC, major histocompatibility complex; TAA, tumor‐associated antigen [Color figure can be viewed at wileyonlinelibrary.com]
Clinically manifest cancer cells have found ways to escape immune‐mediated destruction. 9 Nonetheless, pre‐existing immunity and the presence of tumor‐infiltrating lymphocytes (TILs) in human cancers are well‐known determinants of prognosis. 10 This knowledge has instigated research into strategies to reinforce anticancer immunity and as such permanently deal with cancer cells. One of these strategies is adoptive T cell therapy, which focuses on expanding cancer‐specific T cells with cytolytic capacity ex vivo before returning the T cells to the patient. 11 Adoptive T cell therapy is an umbrella term that covers therapies ranging from adoptive transfer of ex vivo expanded TILs to adoptive transfer of lymphocytes genetically engineered to carry a cancer‐specific TCR or CAR (Figure 2).
Figure 2.
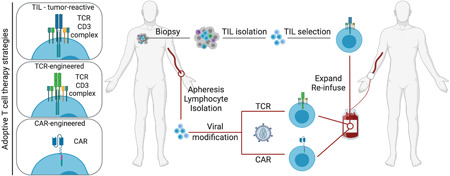
Adoptive T cell therapy strategies. Three different adoptive T cell therapy modalities can be distinguished; (1) re‐infusion of expanded tumor‐reactive TILs that have been isolated from a tumor biopsy, and re‐infusion of blood lymphocytes that are genetically engineered to express a tumor‐specific (2) TCR or (3) CAR. CAR, chimeric antigen receptor; TCR, T cell receptor; TIL, tumor‐infiltrating lymphocyte [Color figure can be viewed at wileyonlinelibrary.com]
Therapy with TILs dates to the late 1980s and was first performed in melanoma patients, whose tumors are often immunogenic and therefore infiltrated with tumor‐reactive CTLs. 12 While promising in melanoma, TIL therapy has not been generally adopted as a cancer therapy, as obtaining sufficient TIL numbers for each individual patient is challenging (30–50 billion TILs are required) and time‐consuming (typically taking 5–6 weeks). Moreover, TIL therapy in other cancer types, such as breast, colon, ovary, prostate, and pancreatic cancer, did not show the same activity as in melanoma. This is likely because only a fraction of TILs harvested from these cancer types is tumor‐reactive. 13 , 14 Meanwhile, increasing knowledge of TAAs and TCRs, together with advancements in strategies to engineer TCRs and transfer them to peripheral blood T cells, enabled adoptive T cell therapy with T cells genetically engineered to express a tumor‐specific TCR, a strategy that was first attempted for the treatment of melanoma patients. 15 However, TCR‐engineered T cells are restricted by MHC presentation, which poses two limitations. First, TCRs recognizing TAA‐derived peptides can only be applied in populations expressing the TAA and correct MHC molecule. Second, cancer cells can hide from CTLs by preventing TAA presentation in MHC‐I molecules using mechanisms such as downregulation of TAA expression and/or processing, and the loss of MHC components (e.g., loss of β2‐microglobulin, deletion of the MHCα‐chain alleles). 16 To overcome these limitations, scientists have looked into ways of circumventing the MHC‐dependency of T cell activation, which has led to a further extension of the adoptive cell therapy domain with CARs. 17
2. INTRODUCING CAR‐T CELLS TO THE FIELD OF ADOPTIVE CELL TRANSFER
The first steps toward the design of CAR molecules were taken in the late 1980s by the group of Zelig Eshhar, who developed a functional chimeric molecule consisting of antibody‐derived variable domains that are able to bind antigens, coupled to TCRα‐ or β‐derived constant domains. When transfected into T cells, heterodimers were formed that enabled antigen‐specific IL‐2 production and MHC‐independent cytolytic activity. 18 The same group refined this technology by designing a single gene, encoding an antibody‐derived single‐chain variable fragment (scFv) coupled to the CD3ζ chain of the TCR complex, resulting in a chimeric protein that was capable of similar T cell activation. 17 This scFv‐CD3ζ chimeric design would later be referred to as a first generation CAR (Figure 3). The in vitro functionality of these first generation CARs was demonstrated in patient‐derived T cells in 1999. 19 In the same study, it was found that the levels of secreted IL‐2 and IFN‐γ were significantly elevated when target cells were CD80+, highlighting the synergistic potential of these early CAR‐T cells with the CD28/CD80 costimulation pathway. The same group previously showed the possibility of incorporating scFv‐CD28 chimeric molecules in T cells, and in that way activating the costimulatory pathway in an antigen‐dependent way. 20 The combination of this information led to the design of second generation CAR molecules in 2002, when a single molecule was designed, incorporating a CD28 costimulatory molecule on the intracellular side of the scFv‐CD3ζ chimera. 21 This design elevated CAR‐T cells from cytotoxic cells to living drugs, as in addition to antigen‐specific T cell cytotoxicity (artificially provided “Signal 1” by the CD3ζ domain), the CD28 domain allows for T cell proliferation (“Signal 2”) when in contact with a target cell (Figure 3). These second generation CAR‐T cells therefore bypass the predestination of T cell anergy or apoptosis, even in the absence of costimulatory ligands. 22 , 23 Later on, clinical data confirmed these preclinical insights, as first generation CAR‐T cells were insufficiently persistent to obtain a sustained CAR‐T cell response, 24 , 25 while CD28‐based second generation CAR‐T cells showed increased proliferation and persistence in patients. 26 Over the years, other costimulatory molecules have been incorporated in the CAR molecule, replacing CD28. These include 4‐1BB (CD137, TNFRSF9), CD27 (TNFRSF7), OX40 (CD134, TNFRSF4), and ICOS (CD278), of which the exact functions have been reviewed elsewhere. 23 , 27 So far, second generation CAR molecules incorporating either CD28 or 4‐1BB as a costimulatory moiety are by far the best examined, and different T cell activation kinetics are attributed to each format. Generally, CD28‐based CAR costimulation is more aggressive and less sustained than 4‐1BB‐driven costimulation, which evolves more gradually and over a more extended period of time. CD28‐based CAR‐T cells will therefore result in more rapid tumor elimination, but this is matched by 4‐1BB‐based CAR‐T cells over time. 28 , 29 The different and complementary kinetics of both costimulatory molecules led to the idea of combining both. In 2007, Stephan et al. 30 showed that CAR‐T cells in which both the CD28 and the 4‐1BB costimulation pathways are triggered outperform the same CAR‐T cells in which either one of them is activated. As a consequence of these results, third generation CAR molecules, incorporating two costimulatory domains on the cytoplasmic side of the receptor, were designed (Figure 3). 31 , 32 , 33
Figure 3.
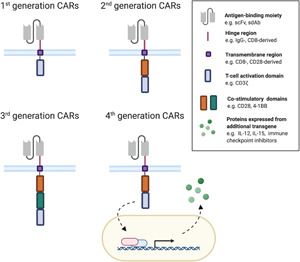
Different CAR generations. The difference between different generation CAR molecules lies in the intracellular domain. First‐generation CARs incorporate a CD3ζ‐derived T cell activation domain. Second‐generation CARs carry the same activation domain and an additional costimulatory domain on the intracellular side. In third generation CARs, multiple costimulatory domains are incorporated in the receptor. In fourth generation CARs, usually only a single costimulatory domain is incorporated, however, the modified cells are further engineered with transgenes that encode T cell‐stimulating molecules such as cytokines or other factors enhancing CAR‐T cell activity—for example, tumor stroma destroying enzymes (heparanase) or immune checkpoint inhibiting proteins [Color figure can be viewed at wileyonlinelibrary.com]
Since CAR‐T cells found their way to the clinic, the CAR‐T cell generation protocols have been optimized. Although the general recipe is currently well defined (Figure 2), there is variation possible at many points throughout the process, rendering consensus guidelines unavailable at the moment. In short, peripheral blood mononuclear cells (PBMCs) are isolated from patient blood via a leukapheresis procedure. Within the bulk of these PBMCs, lymphocytes are isolated, 34 and T cells are stimulated to proliferate. These activated T cells are then genetically modified, usually via gamma‐retroviral or lentiviral transduction systems, although transposon‐based techniques are also emerging, as are CRISPR/Cas‐based genome editing systems. After culture, the transduced cells are cryopreserved and as such readily available to be re‐infused intravenously to the patient after a lymphodepleting pretreatment. In this basic protocol, different parameters can be optimized, evoking a wide subfield of CAR‐T cell therapy research, as reviewed by Stock et al. 35 Variables influencing CAR‐T cell efficacy include the ratio of CD4+/CD8+ CAR‐T cells infused to the patient, as well as their specific subtypes and T cell differentiation and/or exhaustion status upon transduction. Other parameters under investigation are the added value of isolating specific cells from PBMCs or polarization before manipulation, the manner of T cell activation, the genetic manipulation method, and the CAR‐T cell expansion protocol details. 35 These variabilities are beyond the genetic construction of the CAR module, beyond attempts to generate allogeneic CAR‐T cells, and beyond newer ideas of in vivo T cell modification.
In 2013, CAR‐T cell therapy—together with immune checkpoint therapy—was proclaimed as breakthrough of the year by the journal “Science”. 36 Deservingly, as second generation CAR‐T cell products directed against the B cell antigen CD19 induce complete remission in 60%–93% of patients with chemo‐refractory B cell acute lymphoblastic leukemia (ALL) and achieve 50%–93% overall response rates among patients with B cell non‐Hodgkin lymphomas (NHLs). 37 , 38 , 39 , 40 , 41 , 42 These impressive outcomes have resulted in market authorization of four CAR‐T cell products since 2017, that is, Novartis' tisagenlecleucel (CTL‐019, Kymriah®), Kite Pharma's axicabtagene ciloleucel (axi‐cel, Yescarta®) and brexucabtagene autoleucel (KTE‐X19, Tecartus®), 43 and Juno Therapeutics' lisocabtagene maraleucel (liso‐cel, Breyanzi®). 43 These are the reason that other pharmaceutical companies are investing in developing new CAR‐T cell products, exploring new targets on hematological as well as on solid tumors.
Currently, the best clinically validated CAR‐T cells are still second generation CAR‐T cells, of which CD28‐based Yescarta® 44 and Tecartus® and 4‐1BB‐based Breyanzi®, Kymriah®, 45 bb2121 (NCT03651128) and LCAR‐B38M/JNJ‐68284528 (NCT04181827) are leading examples. Earlier‐phase clinical trials validating third generation CARs are ongoing and show somewhat contradictory results. Morgan et al. 46 described an alarming toxicity of third generation CAR‐T cells, whereas others have described the clinical use of third generation CAR‐T cells as safe 47 and showing more favorable expansion profiles compared to second generation CAR‐T cells. 48 A recent meta‐analysis confirms the uncertainty about the added value of third generation CAR‐T cells compared to the previous generation. 49 Indeed, the “living drug” nature of both second and third generation CAR‐T cell products is associated with significant toxicity, resulting mainly from a hyperactivated immune system and a correlated cytokine storm. Two forms of cytokine‐mediated toxicity are characteristic of CAR‐T cell therapy: cytokine release syndrome (CRS) and neurotoxicity. Without going into much detail, symptoms of CRS range from fever to life‐threatening events such as multiorgan failure or disseminated coagulation. Neurotoxicity may co‐occur with or occur after CRS, and can evoke multiple symptoms, such as confusion and seizures, potentially with a lethal outcome. At the basis of this form of toxicity lies immune activation‐mediated disruption of the blood–brain barrier. A detailed review on the underlying mechanisms of both toxicities is written by Abreu et al. 50 Besides these cytokine‐mediated adverse events, targeted therapy is always associated with the risk of on‐target/off‐tumor toxicity. This is especially limiting in solid tumors, as antigen expression is typically not limited to cancerous tissue. In hematological malignancies, off‐target antigen expression is mainly restricted to non‐vital tissue and is therefore more manageable. 51
An alternative and/or additional initiative to improve the efficacy of second or third generation CARs, is the inclusion of an extra transgene under a constitutive or inducible promotor, to create fourth generation CARs (Figure 3). Examples of these additional assets are cytokines such as interleukin‐12 (IL‐12), 52 , 53 IL‐15, 54 and IL‐18 55 ; costimulatory ligands (e.g., CD40L) 56 , 57 ; enzymes (e.g., heparanase) 58 ; or secreted immune checkpoint inhibitors (e.g., PD‐1 blocking scFv). 59 Another form of fourth generation CARs incorporates a suicide gene, which enhances the safety and controllability of CAR‐T cell therapy—as reviewed by Yu et al. 60 Recently, reports on fifth generation CAR‐T cells have emerged, albeit that the definition of this fifth generation CAR‐T cells is not unequivocally. Some have described them as single molecules encoding an IL‐2Rβ domain and a STAT3/5‐binding‐tyrosine motif in addition to a costimulatory molecule and a CD3ζ T cell activation domain. 61 In this way, activation, costimulation, and cytokine production are encoded by a single molecule. Others describe fifth generation CAR‐T cells as off‐the‐shelf products, when T cells from healthy donors are modified to lose HLA and TCR genes. 62 These newer CAR‐T cell variations are still in the early stages of development.
As described in previous paragraphs, CAR design has seen many developments over the past 30 years, with CD19‐specific second generation CARs putting CAR‐T cell therapy on the map. However, since its first development in 1993, the antigen‐binding domain of the classical CAR molecule has not deviated from an scFv. Recently, some groups have been focusing on optimizing this extracellular domain and its parameters. Several studies show that fine‐tuning this underinvestigated CAR domain is more important than anticipated. Here, we provide an overview of these studies and our insights on future CAR design.
3. DIFFERENT ANTIGEN‐BINDING MOIETIES IN CARs
The ability of CAR‐T cells to recognize TAAs is key for their therapy success, implying that the antigen‐binding domain has an important role in shaping the ensuing CAR‐T cell response. To date, scFvs, single‐domain antibodies (sdAbs), natural receptors and ligands, and scaffold proteins are among the antigen‐binding moieties that have been studied (Figure 4). In the next section and as summarized in Table 1, we outline these different domains, their properties, specific advantages, and disadvantages, as well as their progress toward the clinic.
Figure 4.
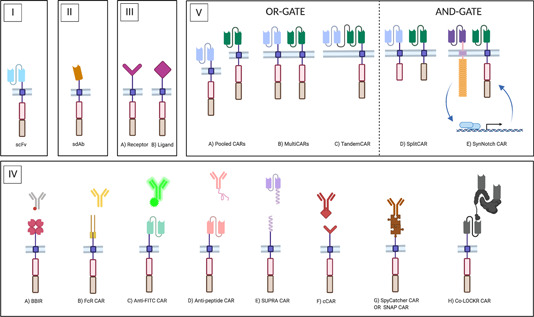
Different antigen‐binding moieties in CARs. CAR endodomains are schematically represented by a costimulatory domain (red box) and a T cell activation domain (brown box). The CAR ectodomain comprises the hinge region and the antigen‐binding moiety, which differs according to the configuration. (I) Classical CAR molecule. Antigen binding is provided by a monoclonal antibody (mAb)‐derived scFv. (II) NanoCAR. Antigen binding is provided by a sdAb, either a VHH (derived from camelid heavy‐chain‐only antibodies, HCAbs), or a VH or VL domain from the human repertoire. (III) Receptor‐ligand CARs. Either the receptor (III.A) or the ligand (III.B) portion of a naturally associating receptor‐ligand pair is incorporated as the targeting moiety. (IV) uCARs. In the uCAR configuration, the targeting domain is disconnected from the CAR module. The in vivo re‐association that must lead to CAR‐T cell engagement can take place by different types of binding: (IV.A) In the biotin‐binding immune receptor (BBIR), the CAR encodes an avidin motif which can associate with high affinity with biotinylated targeting molecules; (IV.B) FcR‐based CARs encode a FcγRIII ectodomain that can associate with the Fc‐portion of IgG‐type mAbs, thus resulting in an engineered form of antibody‐dependent cellular cytotoxicity (ADCC); (IV.C) Classical CARs with an scFv that has specificity for a FITC‐tag or a peptide tag (IV.D) on targeting molecules; (IV.E) SUPRA CARs have an extracellular leucine zipper that zips in with a complementary zipper on targeting molecules. The zipper affinity is tunable; (IV.F) Convertible CARs (cCARs) are based on a natural receptor‐ligand interaction. The CAR incorporates a variant of the NKG2D receptor that can associate with a ligand‐derivative, which is conjugated to the tracer molecule; (IV.G) SpyTag/SpyCatcher CARs and SNAP CARs rely on the formation of a covalent bond between the CAR and the adaptor molecule, via a chemical and an enzymatic reaction, respectively; (IV.H) The colocalization‐dependent protein (co‐LOCKR) CAR system consists of an anti‐tag CAR, a tagged adaptor module and a second untagged adaptor molecule that influences the conformational availability of the tag to the CAR, thereby generation AND‐, OR‐, and NOT‐gate possibilities; (V) CARs targeting more than one antigen. Left: OR‐gate CARs are designed to reduce the risk of antigen escape and to tackle heterogeneous tumors, as they only require one targeted antigen to be expressed. These include pooled CAR‐T cell populations (Pooled CARs, V.A), multiple CARs expressed by a single cell population (MultiCARs, V.B), and TandemCARs in which a single CAR molecule encodes multiple scFvs (V.C). Right: AND‐gate CARs provide increased safety, as they are active only against tumors expressing both targeted TAAs and spare healthy tissues with a single TAA expression pattern. These include (V.D) SplitCARs in which the costimulatory domain is split from the T cell activation domain, in a MultiCAR set‐up and (V.E) SynNotch CARs, in which expression of a classical CAR molecule is under control of TAAbinding of a synthetic Notch receptor that recognizes a different TAA. For simplicity reasons, the CAR moieties in (V.) are depicted with an scFv as the targeting moiety, but variations are possible. SUPRA, split, universal, and programmable [Color figure can be viewed at wileyonlinelibrary.com]
Table 1.
Pros and contras of different antigen‐binding moieties in a CAR
| CAR ectodomain type | CAR ectodomain subtype | Advantages | Disadvantages | Clinically tested |
|---|---|---|---|---|
| scFv CARs |
|
|
Yes | |
| NanoCARs |
|
|
Yes | |
| Receptor‐ligand CARs |
|
|
||
| Receptor‐based CARs |
|
Yes | ||
| Ligand‐based CARs |
|
|
Yes | |
| uCARs |
|
|
||
| Biotin‐based |
|
|
No | |
| SUPRA CAR |
|
|
No | |
| Anti‐FITC CAR |
|
|
No | |
| FcγRIII CAR |
|
|
Yes | |
| Peptide‐specific CAR |
|
|
Yes | |
| Convertible CAR |
|
|
No | |
| SpyTag/SpyCatcher CAR |
|
|
No | |
| SNAP CAR |
|
|
No | |
| Co‐LOCKR CAR |
|
|
No | |
| CARs targeting more than 1 antigen | ||||
| Pooled CARs |
|
|
Yes | |
| MultiCARs |
|
|
Yes | |
| TandemCARs |
|
|
Yes | |
| SplitCARs |
|
|
No | |
| SynNotch CARs |
|
|
No | |
| Others |
|
|||
| Protein scaffolds |
|
|
No | |
| dcCARs |
|
|
No | |
| TRuCs |
|
|
No |
3.1. Single‐chain variable fragment
The CAR concept originates from a combination of antibodies with TCRs. Logically, therefore, the antigen‐binding portion of classical CARs is a monoclonal antibody (mAb)‐derived fragment, more specifically an scFv. An scFv is the reconstructed antigen‐binding part of a conventional mAb, traditionally of the IgG type. An IgG is a heterotetramer consisting of two identical disulfide‐bonded heavy and two identical light chains, each consisting of constant domains and a variable domain. Antigen‐binding occurs through the combined variable domains of the heavy chain (VH) and light chain (VL), together forming the Fv‐domain (Figure 5A). Within each variable domain, three complementarity determining regions (CDRs) are hypervariable and are the main antigen‐interaction regions, forming the paratope that binds the antigen epitope. 63
Figure 5.
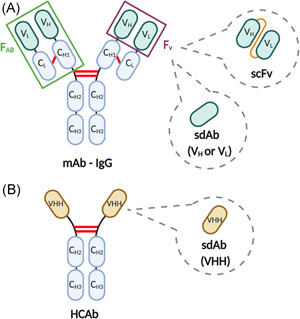
Comparison of a monoclonal antibody (mAb), a heavy‐chain‐only antibody (HCAb) and their derivatives. (A) IgG‐type mAbs are made up of two identical heavy and two identical light chains. Heavy chains consist of a variable domain (VH) and three constant domains (CH1, CH2, and CH3). Light chains consist of a variable (VL) and a single constant (CL) domain. The fragment responsible for antigen‐binding (FAB) is comprised of VH‐CH1‐VL‐CL domains. More specifically, the VH/VL‐combination is responsible for antigen‐binding and is termed the variable domain FV. This is the smallest mAb‐derived fragment capable of binding the antigen, but it needs to be stabilized with a peptide linker (orange) to create an scFv. Single variable domains (VH or VL) are categorized as sdAbs, but hydrophilic mutations in the VH–VL interface are required for individual domain solubilization. (B) HCAbs are homodimers, built out of two heavy chains, each with a variable fragment (VHH) and two constant regions (CH2 and CH3). Evolutionary mutations in the VHH domain allow the VHH to behave as a stable monomer, rendering the HCAb‐derived sdAb stable and soluble by nature. Interchain disulfide bridges that stabilize the mAb and HCAb are indicated in red [Color figure can be viewed at wileyonlinelibrary.com]
In a mAb, VH–VL interactions are weak (micromolar affinity) and mediated by hydrophobic interactions between residues in the framework regions, away from the paratope. Therefore, the heterodimeric Fv fragment is held together by additional hydrophobic interactions between adjacent constant domains of the heavy and light chain, and especially by a cysteine bridge between the latter (Figure 5A). Hence, an isolated Fv domain is only functional when the VH and VL domains are artificially connected through a covalent bond, most commonly through a genetically encoded peptide, resulting in so‐called scFvs. 63
In most CAR‐T success stories so far, a classical scFv‐based receptor is incorporated, with the scFv most often derived from a validated mAb (Figure 4.I). 64 , 65 , 66 For example, Kymriah®, Yescarta®, Tecartus®, and Breyanzi® all incorporate an anti‐CD19 scFv derived from mAb FMC63. 64 , 67 , 68 Meanwhile, Bluebird Bio/Celgene's BCMA‐specific bb2121 (Idecabtagene vicleucel) CAR—currently finalizing Phase III clinical trials (NCT03651128) and pending for FDA approval 69 —, encodes a c11d5.3‐derived scFv. 70 However, drawbacks to this CAR‐T cell design are well exemplified during the development of bb2121, as different BCMA‐specific mAb‐derived scFv molecules behaved unpredictably different in CAR‐T cells. 71 This highlights the discrepancy between targeting domains that function well in mAb format but fail to lead to adequate T cell activation. Moreover, as the scFvs used in CARs are usually derived from (clinically) validated mAbs, 64 , 65 , 66 the availability of candidate binding domains is limited. This renders the design of classical CAR molecules largely dependent on serendipity.
Another important limitation lies in the molecular structure of scFvs. As explained, scFvs are natural dimers of variable domains that associate via hydrophobic interactions in the framework regions, in which an artificial stabilization bond has been introduced. In some cases, hydrophobic framework regions favor receptor aggregation on the T cell surface, leading to antigen‐independent T cell activation, called “tonic signaling,” and eventually resulting in premature T cell exhaustion and loss of therapy efficacy. 72 Moreover, scFvs are known to be sensitive to domain swapping—a phenomenon in which two variable domains of different scFvs associate with each other, which can also lead to CAR oligomerization. 73 Fujiwara et al. 74 for example compared several scFv‐based CARs against the same target side‐by‐side. Differences were observed in aggregation and ability to bind the target antigen with one out of four CARs showing low membrane expression, although transduction rates were similar. This could be reversed by CDR grafting on framework regions of a highly‐expressed scFv, however, only when there was sufficient similarity between the frameworks. All in all, the intrinsic stability of scFvs plays an important role in CAR expression and functionality. 74
Finally, clinical efficacy of CAR‐T cell therapy is largely dependent on in vivo CAR‐T cell persistence. As scFvs incorporated in CAR modules are often of murine origin, concerns are raised about their immunogenicity. Indeed, several groups reported an immune response against the CAR transgene, 75 of which some pinpointed this response as a reaction to the mouse‐derived scFv, 70 , 76 , 77 , 78 as well as to epitopes arising from retroviral transduction. 76 In 2013, Maus et al. 79 reported a serious anaphylactic event after CAR‐T cell infusion, highlighting the danger of anti‐CAR‐T cell immune responses. These data lead to the implementation of humanization strategies in CAR design, either by specific framework mutations 80 or by grafting the murine CDRs onto a human scFv framework. 81 Humanized CD19‐specific CARs have proven to be effective in 64% of patients that relapsed after murine CD19‐CAR‐T cell treatment. 81 Alternatively, fully human scFvs can be derived from human B cell‐derived scFv libraries, limiting the immunogenic risk, and allowing for multiple binding candidates to be screened in a CAR context. 82 Sommermeyer and Smith reported on fully human CD19‐ and BCMA‐specific scFv‐based CARs, respectively, that perform well in a preclinical setting. 82 , 83 However, these libraries are not widely available and the risk of an immune response against the scFv idiotype or against unnatural peptide junctions in the CAR is not avoided with either of these strategies. Of note in view of immunogenicity, lymphodepletion regimens before CAR‐T cell infusion have been used in clinical practice as a means to eliminate immunosuppressive cells. 84 In addition, by eliminating lymphoid‐derived cells, the body's ability to generate a response against the infused cells is also delayed and/or avoided, therefore increasing the depth of response to CAR‐T cell therapy and extending the persistence of infused cells. 78 The optimal preconditioning regimen has not been pinpointed in detail, but clinical experience shows that a combination of cyclophosphamide and fludarabine (Cy/Flu) often evokes the desired effects. 78 , 85 Cy/Flu preconditioning is also the regimen used in the CAR‐T cell products that are currently the most advanced in development, such as Yescarta®, Kymriah®, Tecartus®, Breyanzi®, bb2121, and JNJ‐68284528 (NCT04181827). 44 , 86 , 87 For example, in the case of Kymriah®, the effect of an increase in cellular or humoral immunity toward the transgenic cell product on the 3‐month response rate was shown to be negligible 88 and neither pre‐existing or treatment‐induced anti‐scFv Abs had a negative effect on CAR‐T cell treatment outcome. 89 These clinical studies were performed with preconditioning chemotherapy, which raises the question of whether immunogenicity is still of great importance with established clinical practice nowadays. Indeed, blood persistence of Kymriah® has been shown months to years after infusion. 38 , 45 , 90
Owing to the availability of well‐characterized mAbs against a broad range of relevant target antigens, as well as to the initial idea behind the CAR concept, most CARs that are being developed are (murine) scFv‐based. Despite their proven clinical value, alternative targeting moieties are finding their way to the clinic, indicating the awareness of scFvs' unpredictable and suboptimal behavior, and their dependency on trial‐and‐error research.
3.2. NanoCARs
VHHs, also called nanobodies, are the variable domains of heavy‐chain‐only antibodies (HCAbs), found in camelids and sharks. These HCAbs are heavy‐chain homodimers that lack a light chain, which reduces the size of the antigen‐binding part to one domain instead of two and the paratope to a total of three CDRs instead of six (Figure 5B). Evolutionary mutations in the VHH framework regions allow that these molecules are soluble protein domains that are stable in aqueous solutions, while enlargements and stabilizations in the CDR regions permit antigen binding with affinities like those of antibodies. 91 In addition, “camelization” of the hydrophobic regions in VH or VL domains from human conventional antibodies generate antigen‐binding proteins with features akin to nanobodies (Figure 5A). 92 Together, these molecules are referred to as sdAbs. Building on the results from Long et al. 72 that show that scFv framework regions may cause CAR aggregation and tonic signaling, different groups are now looking into the possibilities of sdAb‐based CARs, here referred to as NanoCARs, as an alternative (Figure 4.II).
As the concept of NanoCAR‐T cells is relatively young, most reports on this CAR format are limited to preclinical data. Nevertheless, diverse target antigens and application possibilities are being investigated. As such, NanoCAR‐T cells against markers of hematological malignancies (CD20, CD33, and CD38), 93 , 94 as well as against solid tumor TAAs (PSMA, MUC1, GPC2, and HER2) 95 , 96 , 97 , 98 , 99 , 100 and tumor stroma‐associated targets (PD‐L1, EIIIB fibronectin splice variant and VEGFR2) 96 , 101 , 102 have been developed.
Beyond this straightforward sdAb‐incorporation strategy, sdAb‐based uCARs 103 and tandemCARs 104 —both introduced below—are also under investigation. Lam et al. 70 recently reported on the incorporation of a human sdAb—isolated from transgenic rats that carry the human VH gene repertoire—in a NanoCAR. Furthermore, NanoCAR‐T cells targeting CD13 were developed after immunization of a llama with tumor cells, followed by in vitro panning and in vivo NanoCAR selection. 105 Another study uses tumor stroma‐specific NanoCARs that locally secrete VHH‐type immune modulators targeting CD47 or PD‐L1. This highlights the versatility of NanoCARs, owing to the small size and single domain nature of sdAbs, which makes them suited building blocks for combined approaches. 102
Most clinically advanced CAR‐T cell products incorporate an scFv antigen‐binding domain, but Nanjing Legend Biotech/Janssen's LCAR B38M (JNJ 68284528), currently in Phase III clinical trials (NCT04181827) and awarded with a PRIME designation by the EMA in 2019, 106 forms an exception. Antigen binding is provided by a single CAR molecule that encodes two linker‐separated sdAbs, both BCMA‐specific, but targeting a different epitope. 107 Besides this, two NanoCAR‐T cell products have entered clinical evaluation: a CD19/CD20‐bispecific NanoCAR (NCT03881761) and a BCMA‐specific NanoCAR (NCT03664661), both in Phase I trials.
While NanoCARs counter the aggregation problem associated with scFv‐based CARs, this design does not fence off concerns about potential immunogenicity and unpredictability in behavior. An advantage that sdAbs have is that the use of immune libraries is more obvious, because of their monomeric structure. 91 , 108 This allows for multiple candidate binders to be identified and compared side‐by‐side, thereby reducing concerns about unpredictable behavior. Also, the immunogenicity profile of soluble camelid‐derived VHHs is more advantageous than that of murine scFvs, since the sequence of the VHH framework regions closely resembles the one of the human VH3 family. 109 Still, immunity problems are possible due to the nonhuman origin of these molecules. Humanization protocols that would theoretically minimize these problems are available, 110 however, reports on the success of this approach in the NanoCAR‐T domain are lacking. Human VH‐based NanoCARs, such as those of Lam 70 and Li, 100 are theoretically less susceptible to this problem. Similar to the evolution seen with scFv‐based CARs, the trend of humanization and/or incorporation of fully human targeting domains exists with NanoCARs. Here too, the future will have to show how important these concerns are in clinical practice in combination with the preconditioning regimens used.
3.3. Natural ligand‐receptor CARs
Both previously mentioned CAR formats rely on patient‐foreign antibodies. A different approach is the incorporation of molecules that have a natural affinity for a certain target. These CARs typically rely on receptor–ligand interactions, of which either the receptor or the ligand portion is incorporated in the CAR, giving rise to receptor‐based and ligand‐based CARs, respectively (Figure 4.III). The main advantage of this CAR format is the limited immunogenicity due to the unpolished character of the extracellular moiety. On the other hand, as receptors and ligands rarely have a single partner and expression of this partner is not usually tumor‐restricted, receptor‐ligand CARs may be associated with an increased risk of on‐target/off‐tumor toxicity. Because antigen binding is based on natural interactions, on‐target/off‐tumor toxicity can to some extent be evaluated in preclinical studies via murine homologs of the CAR–antigen interaction. 111 , 112
An important class of receptor‐based CARs is the one that contains an extracellular domain derived from NK cell receptors that typically recognize stress‐associated ligands. 113 A major advantage of these is the universal behavior of this CAR‐type, in that sense that the targeted antigens can be expressed by multiple cancer types, including antigen‐escape variants after other forms of targeted therapy. 114 The most advanced of these receptor‐based CARs are the NKG2D CAR‐T cells, which incorporate the NKG2D ectodomain, that has a variety of ligands. First‐generation NKG2D CAR‐T cells have been proven to be effective in for example multiple myeloma, 115 ovarian cancer, 116 , 117 and lymphoma, 117 while second generation NKG2D CARs are effective against amongst others Ewing's Sarcoma 118 and gastric cancer. 114 Of note, in these first generation NKG2D CARs, costimulation is provided by the natural association of the NKG2D receptor with the DAP10 adaptor protein in T cells. 119 Furthermore, it is established that NKG2D CAR‐T cells not only target tumor cells expressing signs of stress, but also immunosuppressive cells and vasculature in the tumor microenvironment, and thereby tackle solid tumors on different levels. 116 , 120 Indeed, Spear et al. 117 highlight that even tumors in which not all tumor cells express NKG2D ligands can be controlled by NKGD2 CAR‐T cells, indicating an interplay with the tumor environment. Belgian biotech company Celyad has multiple NKG2D CAR‐T cell products in its pipeline, both as allogeneic and autologous cell therapies and against a number of indications. These are mainly in preclinical stages or in Phase I/II clinical evaluation (NCT04167696, NCT03466320, NCT03018405, and NCT03692429). Other examples of NK receptor‐based CARs include NKp30‐ and DNAM‐I‐based CARs, 121 , 122 but concerns about the bulkiness of incorporating the complete DNAM‐I receptor in the CAR molecule have been raised.
Besides these NK receptor‐based CARs, CARs incorporating the extracellular portion of the T cell costimulatory receptor CD27 are under evaluation. CD27 naturally binds CD70, a cell surface marker that is upregulated in various cancer types. 123 CD27‐based CARs were designed by either coupling full‐length CD27 or the extracellular part of the CD27 receptor (trCD27) to different combinations of costimulatory domains and the CD3ζ T cell activation domain. 111 , 124 The trCD27‐4‐1BB‐CD3ζ CAR‐T cells proved most successful and have been brought to clinical trials for the treatment of CD70+ melanoma as well as pancreatic, renal cell, breast, and ovarian cancer (NCT02830724).
The ligand‐based CAR domain is diverse in comparison to the receptor‐based CAR domain, with various classes of ligands under evaluation. A first class incorporates a cytokine or a cytokine‐derived receptor‐binding peptide, as is the case in IL‐11Rα‐specific CAR‐T cells. Huang et al. 125 identified IL‐11Rα as an important target for metastatic osteosarcoma, and developed a second generation CAR in which the targeting is foreseen by an IL‐11‐derived peptide. Similarly, IL‐13Rα2‐expressing glioblastoma can be targeted by first generation CARs incorporating an IL‐13 mutein that spares IL‐13Rα1+ cells but has a specific binding affinity for the IL‐13Rα2 receptor. 126 Repeated intracranial injection of these CAR‐T cells in glioblastoma patients indicated the safety of this approach, however, the CAR‐T cells showed limited efficacy and persistence. 127 After adding a 4‐1BB costimulation domain and limiting Fc‐receptor interactions by mutating the IgG4‐Fc linker, 128 a Phase I clinical trial was initiated for the treatment of glioblastoma (NCT02208362). A case report from this clinical trial demonstrated a clear antitumor response lasting for 7.5 months after intracranial CAR‐T cell administration. 129 Other cytokine‐based CARs that have been developed incorporate full‐length CD116L that targets CD116 on juvenile myelomonocytic leukemic cells 130 or APRIL, which has a natural affinity for BCMA and TACI, both important surface antigens on multiple myeloma cells. 131 However, the third generation APRIL‐based CAR has proven insufficiently effective in clinical trials which led to an early termination of its clinical evaluation (NCT03287804). In a more recent study, the trimeric form of APRIL is incorporated into second generation CAR molecules, holding true to the naturally secreted form of APRIL. 132 This approach is still in preclinical development.
Other ligand‐based CARs are including hormones in the CAR ectodomain, to create chimeric endocrine receptors (CERs). Follicle‐stimulating hormone receptor (FSHR) is often upregulated in subtypes of ovarian cancer and second generation CAR molecules incorporating full‐length FSH as the antigen‐binding moiety has proven effective in PDX models. 112
Growth factor‐based CARs represent yet another class of ligand‐based CARs and are exemplified by the T1E CAR. T1E is a TGFα/EGF chimeric molecule that has an affinity for homo‐ and heterodimers incorporating EGFR and for HER2/HER3 heterodimers, all upregulated and redundantly important in several solid cancers. Second‐generation CARs with a T1E ectodomain have proven functional in preclinical models of breast, head and neck, 133 and ovarian cancer, 134 and have been brought to clinical trials for the treatment of advanced/recurrent head and neck squamous cell carcinoma (NCT01818323).
Apart from these different classes of ligand‐based CARs, there are other ligands under evaluation for their targeting capacity in CAR‐T cells. For example, the chlorotoxin (CLTX)‐based CAR‐T cell product, recently developed by Wang et al. 135 CLTX has a natural affinity for membrane‐bound matrix metalloproteinase‐2 on glioblastoma cells, and second generation CTLX CAR‐T cells showed preclinical effectiveness against glioblastoma after intracranial CAR‐T cell injection. Another example is the CAR that incorporates an LFA‐I domain with natural affinity for both human and mouse ICAM‐1. This third generation CAR proved efficacious in systemic solid tumor models. 136
3.4. CARs targeting more than one antigen
Disease relapse due to antigen downregulation or loss is a notorious problem in CAR‐T cell therapy and by extension in all targeted therapies. In addition, not all tumors exhibit a sufficiently homogenous antigen pattern and many TAAs are not necessarily tumor restricted. These issues can be circumvented by strategies in which multiple antigens are targeted. Targeting more than one antigen is possible in several ways. In the strategy of pooled CAR‐T cells, multiple CAR‐T cell products are administered to the same patient (Figure 4.V). 137 , 138 However, it has been shown that two different CAR‐T cell populations can expand disproportionally, resulting in one population outnumbering the other. 139 , 140 Alternatively, MultiCAR‐T cells express more than one CAR molecule. Such dual CARs have been developed by either cotransduction with multiple CAR‐encoding vectors or with a single bicistronic plasmid carrying multiple CARs (Figure 4.V). 137 , 141 Others have reported triple CAR‐T cells expressed from a tricistronic vector. 142 Of note, these approaches require the modification of T cells with large transgenes, which impacts viral vector packaging and transduction efficiency. In that regard, TandemCARs have been developed, which—in contrast to the previously mentioned bi‐ or tri‐specific CAR‐T cell products—, require a modified extracellular CAR domain. TandemCARs encode multiple antigen‐binding domains in a single CAR molecule (Figure 4.V). This has been described with multiple scFvs, 140 , 143 , 144 , 145 , 146 , 147 but a general risk with these TandemCARs, is the potential cross‐pairing between the VL and VH chains of different scFvs. Therefore, TandemCARs incorporating combined scFv/muteins, 148 scFv/natural ligands, 146 multiple VHHs, 104 and combined protein scaffolds (see below) 149 have been reported. It should be noted that Fry et al. 150 recently reported on dual antigen loss in patients as a consequence of sequential CAR‐T cell therapies against different targets.
Other approaches in line with these include SplitCARs and SynNotch CARs. SplitCAR‐T cells, like dual CAR‐T cells, express two different CAR molecules on the same T cell, but these are both incomplete: one receptor encodes the T cell activation domain, while the other encodes the costimulatory domain (Figure 4.V). Therefore, unlike dual CARs, SplitCARs require the expression of both antigens to be activated (AND‐gate), while dual CARs only require the expression of either one of the antigens (OR‐gate). The AND‐gate ensures an extra safety mechanism in which healthy tissues that only express one antigen are spared from CAR‐T cell‐mediated killing. This has been described with scFvs targeting MUC1 and HER2, 151 although the complementary signaling achieved was not as effective as with second generation CARs. The approach was therefore further exploited by Lanitis et al., 152 who did achieve similar efficacy and enhanced specificity with an scFv‐based SplitCAR, compared to a second generation CAR. However, as SplitCARs incorporate an antigen‐specific CD3ζ chimera (i.e., a first generation CAR molecule), some (unsustained) T cell activity, resulting toxicity and T cell anergy is observed toward cells expressing this antigen. To counter this problem, an scFv leading to suboptimal signal transduction was incorporated in the first generation module by Kloss et al., 153 while the chimeric costimulatory receptor was armed with an optimal scFv for activation. This approach resulted in a strict realization of the envisaged AND‐gate, where single‐antigen expressing cells were spared. More recently, SplitCARs encoding a CD13‐specific CD3ζ NanoCAR and a TIM3‐specific CD28/4‐1BB scFv‐based CAR have been reported. 105 The downside of the SplitCAR technology lies in the fact that signal 1 and signal 2 rely on different antigen expression patterns and CAR expression levels, while optimally an equivalent signaling ratio is required. This problem is circumvented by Split SUPRA CARs (with one CD3ζ ZipCAR and a CD28/4‐1BB ZipCAR, see Section 3.5), in which zipper affinity and ZipFv dosage can be varied to equilibrate both signaling pathways. 154
An alternative AND‐gate strategy is created by SynNotch CARs, where a synthetic Notch receptor with an affinity for one antigen drives the expression of a single CAR with an affinity for a second antigen (Figure 4.V). This has been described with CD19‐specific scFv‐based or anti‐GFP nanobody‐based synNotch receptors enabling mesothelin‐specific or CD19‐specific second generation CAR expression. 155 ScFv‐based synNotch receptors targeting EpCAM or B7‐H3 that result in ROR1‐specific second generation CAR expression have also been described to induce AND‐gate killing. 156 Of note, to obtain a functional AND‐gate with synNotch CAR‐T cells and thereby avoiding toxicity, physical distance between healthy cells expressing solely the CAR target and cells expressing both targeted antigens is required.
3.5. Universal CARs
The requirement of consistent antigen expression and the scalability are the main limitations of the previously mentioned CAR‐T cell approaches. Since each of these CARs is designed to target one (or multiple) predefined antigen(s), heterogenous tumors might not be fully susceptible to the treatment. 157 Furthermore, there is always a risk of antigen escape, even if the therapy is successful initially. 157 , 158 , 159 In addition, these CAR types imply that for each antigen of interest, a different CAR is designed and optimized, leading to immense costs. These limitations have prompted scientists to develop universal CARs (uCARs), in which the targeted antigen is not predefined in the CAR module, because the targeting domain is split from the CAR molecule. These uCAR‐T cells are typically modified with a receptor that contains the classical CAR transmembrane and intracellular modules, however, has an extracellular domain that can recognize a distinct protein or motif other than a TAA. Additionally, a switching module is administered to the patients, which is a bispecific fusion molecule with one binding domain against the TAA and one epitope that is specifically recognized by the uCAR ectodomain (Figure 4.IV). This makes that the target antigen can be easily switched, and as such the T cells can be redirected without the requirement of alternatively modified T cells. The flexibility in dosage of the antigen‐binding portion also contributes to the controllability of this form of therapy. 160 Furthermore, this system elegantly allows for a termination of the T cell therapy in case of unpredicted toxicity, by the administration of a nontumor targeting moiety that outcompetes binding of the adaptor molecule to the uCAR.
The first variant of uCARs, the biotin‐binding immune receptor (BBIR) CAR, is based on the binding of biotin to avidin and was conceptualized by Ubranska et al. 160 This CAR construct is composed of an extracellular multimeric avidin motif which serves as a ligand‐binding domain. The “switch on” molecule is a biotinylated antigen‐binding protein such as a mAb or an scFv (Figure 4.IV). 160 Infusion of avidin‐based uCARs, along with biotinylated mAbs resulted in efficient tumor recognition and eradication of tumor cells in mice. A few years later, a new CAR construct using the affinity‐enhanced monomeric streptavidin 2 (mSA2) biotin‐binding domain was engineered and demonstrated cancer cell lysis and IFN‐γ production in a mAb dose‐dependent manner. 161 However, concerns remain about the use of chicken avidin in humans which might cause an immune response. In addition, the presence of natural anti‐biotin mAbs as well as free biotin in the sera of patients might limit the use of this CAR‐T approach. 162
Circumventing the need for biotinylation, the first steps toward Fc‐binding uCAR‐T cells were taken in 2006, when Clémenceau et al. 163 transduced T cells with FcγRIIIa ectodomain—FcεRIγ transmembrane/endodomain chimeras. These CAR‐T cells are based on the interaction between FcγRIII and the Fc part of IgGs, such as the clinically approved mAb rituximab, thereby relying on engineered antibody‐dependent cellular cytotoxicity (ADCC) toward the mAb‐targeted antigen. 163 The same chimeras later proved efficacious in CAR‐NK cells in a xenograft model of breast cancer, when combined with the HER2‐specific mAb trastuzumab. 164 Ochi et al. 165 made the bridge to a more classic first generation CAR by introducing a FcγRIII‐CD3ζ chimera in T cells, which was effective in a preclinical model of lymphoma in combination with rituximab. Second generation FcyRIII CAR‐T cells proved to be universally functional when combined with trastuzumab, rituximab, or anti‐GD2 mAb in vitro and with the latter two in vivo (Figure 4.IV). 166 Unum Therapeutics brought CD28‐based ACTR707 167 and 4‐1BB‐based ACTR087 168 second generation FcγRIII uCARs to clinical trials in combination with rituximab (NCT02776813 and NCT03189836), trastuzumab (NCT03680560), and a BCMA‐specific mAb (NCT03266692), but of these, the trials combining mAbs other than rituximab have been terminated due to commercial reasons. Remarkably also, both ACTR707 and ACTR087 trials have been temporarily put on hold by the FDA for safety reasons. 169 , 170 Of note, all of these CARs incorporate the FcyRIIIa variant V158, which has a superior Fc‐binding capacity compared to the more common F158 variant. The main advantage of these CARs is the availability of various ADCC‐mediating clinical‐grade mAbs. However, these uCARs are potentially activated by any circulating IgG antibody and could therefore induce severe inflammation at unpredicted locations. Recently, Rataj et al. 171 showed that glycoengineering of the administered mAbs might reduce this risk by elevating the specificity for the therapeutic mAb.
Another uCAR module relies on the targeting of the fluorescent dye fluorescein isothiocyanate (FITC) (Figure 4.IV). An scFv targeting FITC has been incorporated in both second generation 172 , 173 and third generation 174 CAR formats, which have been combined with FITC‐labeled mAbs trastuzumab and cetuximab, 174 folate (natural ligand of folate receptor, which is overexpressed on multiple cancer types) 172 and CD22‐ and CD19‐mAb‐derived FAB fragments. 173 As the position and stoichiometry of the FITC‐conjugation to the different TAA‐targeting FABs proved crucial to the potency of uCAR‐T cell activity, site‐specific protein conjugation strategies need to be optimized, which hampers the fast adaptability of these uCARs. 173 In addition, despite a fully human anti‐FITC scFv in CAR‐T cell constructs, 173 the immunogenic potential of FITC itself remains a concern for clinical application. 174
An alternative approach is to equip the TAA tracing molecule with a peptide tag that can be recognized by peptide‐specific CARs. Examples include the yeast transcription factor GCN4‐derived Peptide Neo‐Epitope (PNE) and E5B9, a peptide derived from the human La/SS‐B nuclear protein (Figure 4.IV). PNE‐tagged FAB fragments targeting CD20 and CD19 in combination with anti‐PNE scFv‐based second generation CAR‐T cells were shown to be functional by the same group who designed FITC‐conjugated FAB‐based uCARs. 175 Again, the authors highlight the importance of the peptide incorporation site and valency on the targeting moiety, as well as the spacer length (see below)—and the case‐by‐case variety thereof. PNE‐based uCAR‐T cells also proved efficacious in PDX models of pancreatic cancer. 176 Comparing FITC‐ and PNE‐based anti‐HER2 uCARs, they conclude that both approaches are equivalent if an equal cell‐to‐cell geometry can be obtained. 177 Alternatively, E5B9 peptide‐specific second generation CARs have been developed and combined with monospecific and bispecific E5B9‐tagged CD33‐ and CD123‐specific scFvs. 178 Furthermore, these CARs were combined with a PSCA‐specific scFv, and demonstrated efficacy in prostate cancer models. 179 By extension, this uCAR module was also combined with EGFR‐specific nanobodies with an E5B9 tag, 103 which, as soluble proteins, have an advantage over scFvs in terms of stability, production yield, immunogenicity, biodistribution profile, and theranostic potential. 91 , 180 La/SS‐B is an auto‐antigen in particular autoimmune diseases, but no specific antibodies are present in the sera of healthy individuals. 181 Both PNE‐ and E5B5‐based uCARs (UniCARs) are currently under clinical evaluation (NCT04450069 and NCT04633148, respectively).
Another variant is the Split, Universal, and Programmable CAR or SUPRA CAR that consists of two components: a leucine zipper‐containing uCAR (zipCAR) and a separate switch molecule with a cognate leucine zipper linked to an antigen‐specific scFv (zipFv) (Figure 4.IV). Cho et al. 154 developed the SUPRA CAR system, capable of targeting TAAs HER2, AXL, and mesothelin without re‐engineering the T cells. Unique to this approach is the possibility of tuning the binding affinity of the zipper components, which influences the CAR‐T cell's cytokine profile and killing capacity more than the scFvs affinity for the targeted antigen does. Moreover, the flexibility in zipper affinity also allows for controllable zipFv competition, replacement, and CAR‐T cell therapy termination. Another advantage of SUPRA CARs is the ability to designate a safety protein. Cells expressing the safety protein and the target TAA will be bound by two zipFvs on the surface, and this zipper will close. As such, the zipCAR can no longer be engaged with these cells. In addition, zipper components can be derived from human transcription factors, thereby lowering the potential immunogenicity of these domains. SUPRA CAR‐T cells were able to demonstrate preclinical efficacy in both solid and hematological cancer models, with blood cytokine levels controlled by the dose of the zipFv and by the zipper affinity. 154
Convertible CARs (cCARs) combine the advantages of uCARs with those of natural receptor‐based CARs (Section 3.3). The antigen‐binding part of the cCAR consists of a modified NKG2D receptor that has lost its affinity for its natural ligands. Instead, it exclusively binds U2S3, a mutant of NKG2D's natural ligand ULBP2 that no longer binds the wild‐type receptor. An antibody‐U2S3 fusion, termed a MicAbody, then functions as a natural ligand‐based adaptor molecule for universal CAR targeting (Figure 4.IV). 182 , 183
Two more recent uCAR developments rely on a spontaneous covalent bond formation between the TAA‐specific switching molecule and the uCAR. In the SpyTag/SpyCatcher system, an isopeptide bond is formed between two parts of a Streptococcus pyogenes‐derived protein. These peptides are site‐specifically conjugated or genetically engineered at the C‐terminal end of the switching molecule (SpyTag) and the N‐terminal end of the uCAR (SpyCatcher) (Figure 4.IV). Minutolo et al. 184 introduced second generation SpyCatcher uCAR‐T cells in combination with SpyTagged mAbs and Designed Ankyrin Repeat Proteins (DARPins, introduced below), and Liu et al. 185 expanded this with scFv‐SpyTag adaptor molecules. Formation of the covalent bond allows for arming the uCAR‐T cells with their adaptor module before infusion. However, continued administration of the switch molecule is needed for a prolonged therapeutic effect. 184 Since the adaptor system is of bacterial origin, immunogenicity remains a concern when clinical translation is envisaged. The SNAP CAR technology was described by Jason Lohmueller and makes use of a self‐labeling enzyme. SNAP uCARs encode a modified O‐6‐methylguanine‐DNA methyltransferase enzyme (SNAPtag) at the extracellular side of a second generation CAR. This human enzyme is capable of covalently attaching itself to benzylguanine groups in vivo. In turn, benzylguanine groups can be randomly conjugated on lysine residues of adaptor proteins, such as mAbs or FAB fragments (Figure 4.IV). 186 Compared to the SpyCatcher system, SNAP uCARs are predicted to be less immunogenic, due to the human origin of the encoded enzyme. However, as adaptor conjugation does not occur site‐specifically, more optimization might be required. Different from other uCARs, both systems rely on the formation of a covalent bond, maximizing the resemblance to classical CARs, but reducing the adaptability of the system. Termination of CAR‐T cell activity is indeed dependent on in vivo degradation and is elusive to competition. 184
The last uCAR system that we will discuss combines the universal platform with AND‐ and OR‐gate strategies, as described in Section 3.4. The colocalization‐dependent protein (Co‐LOCKR) system consists of three components: (i) a uCAR with an extracellular Bcl2 domain with an affinity for a peptide named Bim; (ii) a Bim‐coupled CAGE protein that binds a TAA; and (iii) a KEY protein that binds a second TAA, and has an influence on the conformation of the CAGE (Figure 4.IV). Only when both the CAGE and the KEY bind their respective antigen, the Bim peptide will be accessible to the uCAR, and T cell activation can take place. By adding a second KEY module, an X‐AND‐Y OR X‐AND‐Z gate is established. Additionally, a Decoy protein (which binds an antigen only present on healthy cells) can ensure a NOT‐gate by trapping the KEY, which in turn leaves the CAGE in an inactive conformation. Lajoie et al. 187 established the system with TAA‐specific DARPins, in combination with a second generation CAR. Although conceptually innovative, the Co‐LOCKR system is still in the early stages of development, and questions about the pharmacokinetic profile of the different components exist. Furthermore, as these multiple components render this uCAR format complex and bulky, immunogenicity will also be a future concern. 187
Although the concept of uCARs is very elegant in theory, there are some drawbacks. In addition to the immunogenicity of various uCAR moieties, switching between targeting modules is not self‐evident. Indeed, different parameters of cell–cell interaction will be required for different antigens. If a single uCAR is deployed against different targets, not every interaction will lead to optimal T cell activation, and therefore the CAR‐T cell therapy will not be universally flexible. Furthermore, each type of antigen‐binding moiety will come with its own specific interaction and blood‐retention profile. 180 For example, although clinical‐grade antibodies are widely available, they possess a slow biodistribution profile which impairs the envisaged action‐reaction capacity of the uCAR technology. In that respect, nanobodies or scFvs might be more suited due to a more favorable pharmacokinetic profile. 180
3.6. Others
Other CAR designs incorporate for example protein scaffolds with a designed specificity. DARPins are a class of genetically engineered antibody‐mimetic proteins and are comprised of two or three repeat units of 33 amino acids each, flanked by N‐ and C‐terminal hydrophilic caps. Repeat proteins are common in nature and they are smaller in size, less prone to aggregation, more thermodynamically stable, and considered less immunogenic compared to murine scFvs. 188 CARs utilizing HER2‐targeting DARPins were already developed preclinically and were as effective as scFv‐CARs in inhibiting tumor growth in vivo. 189 Balakrishnan et al. 190 directly compared the use of scFv‐CARs and DARPin‐based CARs using EGFR as a target and demonstrated improved antitumor effects of DARPin‐based CARs compared to scFv‐based CARs in EGFR+ cancer models. Moreover, DARPins specific for EGFR, EpCAM, and HER2 were combined in one CAR construct and linking these three DARPins in tandem resulted in synergistic antitumor effects in cancer cells that expressed more than one antigen. 190
Another class of scaffold protein‐based CAR‐T cells ensures tumor targeting via the incorporation of antigen‐specific adnectins. These are proteins derived from the 10th Type III domain of human fibronectin (10Fn3), which is a small and stable protein scaffold that contains three mutable loops, responsible for target binding. Because these molecules are human fibronectin‐derived, these CAR formats are expected to be low‐immunogenic. Due to their monomeric nature, the risk of tonic signaling is also avoided. Adnectin‐based CARs targeting EGFR have been developed preclinically. 191 Recently, CARs with combined HER2‐specific DARPin/adnectin and EGFR‐specific affibody/adnectin targeting domains were reported. 149 Affibodies form yet another class of protein scaffolds, which are helical targeting moieties derived from Protein A (Staphylococcus aureus), with a designed specificity.
An entirely different development in the CAR field, is the Dual Chain CAR (dcCAR), which incorporates a complete mAb heavy chain/light chain heterodimer on the extracellular side of the CAR. This renders the dcCAR bulkier than a classical CAR molecule but circumvents the artificial scFv format and the therewith associated instability, antigen‐independent clustering and tonic signaling. These CARs do not require the extensive design of an scFv, which is often unstable and binds suboptimally to the target antigen compared to the parent mAb. The avoidance of artificial linkers also leads to less potentially immunogenic epitopes. 192
Yet another development is the TCR fusion protein (TRuC). TRuCs revert to the original idea of the combination of T cell cytotoxicity and mAb‐based antigen recognition, by making scFv‐TCR chimeras. These molecules are actually not CARs, but TCRs in which the CD3 subunit is linked to an scFv. In terms of activation biology, these chimeras have shown a more natural pattern than the artificial CARs. Apart from CD19‐ and BCMA‐specific scFv‐based TRuCs, IL‐13Rα2‐specific TRuCs incorporating a VHH have been functionally developed. TRuCs have demonstrated their efficacy preclinically, however are still in the early stages of development. 193
4. THE AFFINITY OF THE EXTRACELLULAR DOMAIN AS A MAJOR DETERMINANT OF CAR‐T CELL EFFICACY AND SAFETY
4.1. The influence of affinity on the efficacy of CAR‐T cells
Interactions between TCRs or CARs and their targets differ in multiple aspects such as target and receptor density, spatial interactions, and binding affinity. 194 The most conventional way of describing the affinity of a molecular interaction is via the equilibrium dissociation constant (KD). Natural affinities of TCRs for peptide‐MHC (pMHC) complexes are in the micromolar affinity range (KD = 10−4–10−6 M). 136 , 194 , 195 This relatively weak affinity allows serial engagement and triggering of multiple TCRs, which is thought to be necessary for a fully functional T cell response and tumor eradication. 195 , 196 Contrarily, mAbs and scFvs naturally interact with their target epitopes with (sub)nanomolar affinity. 194 Thus, as opposed to TCRs, CARs that rely on scFvs to bind target antigens exhibit strong affinities (nanomolar or high picomolar range: KD = 10−7–10−10 M) for surface‐expressed antigens. 194
A consensus regarding CAR affinity is that there is a minimum threshold that is required for sufficient antigen recognition and the induction of adequate signaling and CAR‐T cell activation. 197 , 198 , 199 , 200 Hudecek et al. 199 demonstrated increased antitumor efficacy of anti‐ROR1 CAR‐T cells designed with a 5.6 × 10−10 M affinity scFv compared to 6.5 × 10−8 M affinity scFv. 198 , 199 Accordingly, Lynn et al. 198 showed that anti‐folate receptor beta (FRβ) CAR‐T cells employing strong‐affinity scFvs (KD = 2.48 × 10−9 M) outperformed weaker affinity scFvs (KD = 5.43 × 10−8 M) with significantly enhanced in vivo anti‐leukemic activity. 198 However, Chmielewski et al. 201 generated a panel of five anti‐HER2 scFvs with affinities ranging from 3.2 × 10−7 to 1.5 × 10−11 M by site‐directed mutagenesis of the anti‐HER2 C6.5 scFv. When incorporated in CAR‐T cells, an affinity ceiling of 10−8 M was identified. 201 CAR‐T cells with stronger affinities responded to HER2‐expressing target cells independently of antigen density while CAR‐T cells with weaker affinity were only activated in response to HER2‐overexpressing tumor cells and not cells expressing HER2 at physiological levels. 201
4.2. Tuning CAR affinity to enhance tumor cell selectivity
Especially in solid tumors, TAAs are often shared with (vital) healthy tissue. Overexpression is observed in cancer cells, but on‐target/off‐tumor toxicity remains a limiting factor for TAA‐targeted therapy, such as CAR‐T cells. 200 Indeed, this is illustrated by a case in which strong‐affinity trastuzumab‐derived CAR‐T cells targeting HER2 induced fatal pulmonary toxicity. 46 , 195 This was attributed to CAR‐T cell reactivity against low HER2‐expressing lung epithelial cells. 46 , 195 In another example, CAR‐T cells bearing a high‐affinity scFv variant targeting GD2 ganglioside were generated by inducing a mutation predicted to improve stability and affinity. 202 However, it was observed that while the changes clearly improved the in vitro and in vivo function of the CAR, they were associated with lethal on‐target/off‐tumor toxicity in a GD2+ human neuroblastoma xenograft model. 202 These studies highlight the challenge of finding the right balance between optimal CAR‐T cell function and safety. 200
Building on Chmielewski's results, 201 tuning CAR affinity to enhance CAR‐T cell selectivity toward antigen‐overexpressing tumor cells has therefore been explored to reduce on‐target/off‐tumor toxicity. 195 , 200 Caruso et al. 196 generated CARs with scFvs derived from two anti‐EGFR mAbs with different affinities: nimotuzumab and cetuximab. They demonstrated that the weaker affinity CAR, derived from nimotuzumab, exhibited reduced T cell activation as the density of EGFR decreased, enabling these CAR‐T cells to selectively kill malignant cells overexpressing EGFR and spare nonmalignant cells. Contrarily, activation of T cells bearing the stronger affinity cetuximab‐based CAR was associated with equally efficient activation, independent of EGFR density. 196 Similar results were provided by Han et al., 191 who compared the scFv‐based CAR derived from cetuximab (KD = 3.9 × 10−10 M) with four CARs incorporating adnectins targeting EGFR with different affinities. The weakest affinity adnectin E3 (KD = 9.92 × 10−9 M) had a similar affinity for EGFR as the nimotuzumab‐based CAR of Caruso et al. 191 , 196 Accordingly, they observed that while the E3‐CAR‐T cells displayed a higher selectivity against EGFR‐overexpressing cancer cells, the cetuximab‐based CAR‐T cells did not show this selectivity. Importantly, the authors raise the concern that weaker affinity adnectins may pose disadvantages in terms of persistence and proliferation, as E3‐CAR‐T cells infiltrated the tumors at lower percentages as compared to cetuximab‐based CAR‐T cells. Furthermore, they highlight the risk of off‐target activation induced by weaker affinity adnectins, since these may bear reduced specificity. 191
Arcangeli et al. 203 generated and tested two scFv CAR mutants (generated by single‐residue substitution) targeting CD123 with an affinity approximately 10‐ and 100‐fold weaker than the parent mAb. They observed that weakening CAR affinity to 10−7 M did not impair CAR‐T cell lytic activation but enhanced the safety profile of the anti‐CD123 CAR‐T cells. 203 Furthermore, Liu et al. 204 explored affinity‐tuning of CAR‐T cells targeting HER2 and EGFR. Four anti‐HER2 and four anti‐EGFR scFvs were generated by mutating the variable domains of mAbs 4D5‐8 and C10, respectively. 204 Anti‐EGFR CARs with weaker affinity (high nanomolar) were associated with optimal functionality and safety profiles. 204 In case of the anti‐HER2 CARs, an scFv with micromolar affinity was shown to effectively eliminate tumors in vivo with reduced toxicity against normal tissues expressing physiologic levels of HER2. 204 Park et al. 136 evaluated a panel of anti‐ICAM‐1 CARs designed with affinity variants of LFA‐1, the natural ligand of ICAM‐1. Directed evolution allowed to identify variants with affinities spanning six orders of magnitude (1 × 10−9–1 × 10−3 M). Micromolar affinity CAR‐T cells were shown to outperform nanomolar CAR‐T cells and rapidly eliminated tumors while avoiding systemic toxicity in a solid tumor mouse model. 136 Drent et al. 205 presented a rational approach for tumor‐selective targeting of the TAA CD38. They used the light‐chain exchange technology to rapidly generate hundreds of new antibodies with a large range of affinities for CD38 and to methodically identify the optimal candidate for tumor‐selective cytotoxicity. Combining the heavy chains of two high‐affinity CD38 antibodies with 176 germline light chains, they generated approximately 124 new antibodies with 10 to >1000‐fold weaker affinities for CD38. 205 Anti‐CD38 CAR‐T cells of approximately 1000‐fold reduced affinity (KD = 1.915 × 10−6 M) showed optimal functionality against CD38‐overexpressing multiple myeloma cells but spared low CD38‐expressing healthy hematopoietic cells in vitro and in vivo. 205 All in all, these studies highlight the potential of tuning affinity to enhance tumor cell selectivity. Moreover, in a clinical study, a novel anti‐CD19 scFv CAR (named CAT) that has a weaker affinity (KD = 1.438 × 10−8 M) than FMC63 (KD = 3.28 × 10−10 M) was evaluated and 12 out of 14 patients achieved molecular remission. FMC63 is the strong‐affinity binder used in many clinical studies and in the approved CAR‐T cell products Kymriah®, Yescarta®, Tecartus®, and Breyanzi®. 68 , 206 Interestingly, prolonged persistence of CAT CAR‐T cells and enhanced CAR‐T cell expansion was observed in these patients compared to published data for Kymriah®, while Kymriah® only differs from CAT in the scFv and the promoter. 206 These results were especially remarkable as most patients had a lower tumor burden, which is usually associated with decreased CAR‐T cell expansion. Furthermore, in most cases, expansion was not associated with significant increases in pro‐inflammatory cytokines, which suggests that expansion of weaker affinity CAR‐T cells may depend less on cytokine secretion. 206
While affinity‐tuning of CARs seems promising, optimal affinities differ strongly when targeting different antigens. Nonetheless, the results of these studies suggest that improving CAR affinity beyond the activation threshold is not required for optimal CAR‐T cell function, and weaker affinity CARs have been associated with improved functionality and safety profiles for certain antigens. 136 , 195 Therefore, generating CARs with antigen‐binding domains with different affinities and selecting the CAR that associates with maximal antitumor efficacy and minimal off‐tumor toxicity may improve future CAR‐T cell design. 195 Of note in this context is that antigen‐downregulation remains one of the most frequently reported mechanisms of tumor escape upon CAR‐T cell therapy, and current studies regarding affinity modulation have not yet reported on whether employing weaker affinity CARs facilitates these antigen‐escape mechanisms. 196 , 200
4.3. Beyond affinity: The influence of CAR expression levels
Besides CAR affinity, both TAA and CAR expression levels are determinants for the avidity of the CAR/TAA interaction, and may therefore influence CAR‐T cell behavior. 195 The level of surface CAR expression is directly influenced by the transgene delivery method and the promoter used to drive CAR expression. 64 , 207 , 208 , 209 Both viral and nonviral genetic engineering strategies have been used for the generation of CAR‐T cells, resulting in lasting or temporary expression of the CAR transgene. 64 Low CAR expression has been associated with impaired effector functions and limited antitumor efficacy. 203 , 210 Indeed, anti‐CD19 CAR‐T cells with high CAR‐expression levels, but not those with low CAR‐expression levels demonstrated robust antitumor functionality. 211 Contrarily, it was shown that—in addition to CAR structure—high CAR density can result in clustering of CAR molecules at the T cell surface leading to antigen‐independent tonic signaling. 64 , 72 , 195 , 208 , 209 High expression may occur when the CAR transgene is driven by a strong constitutive promoter. This may lead to chronic T cell activation, increased T cell differentiation, exhaustion, and activation‐induced cell death (AICD), resulting in inferior in vivo antitumor efficacy and CAR‐T cell persistence. 64 , 72 , 195 , 207 , 208 , 209 Mitigating CAR clustering is possible by using alternative promoters to reduce CAR expression levels, although this has also been associated with reduced efficacy. 64 , 207 Targeted gene delivery of a CD19‐specific CAR to the TCRα constant locus using CRISPR/Cas9 technology resulted in constant CAR expression and more potent CAR‐T cells in which tonic CAR signaling was prevented, and delayed CAR‐T cell differentiation and exhaustion was observed. 212 Altogether, fine‐tuning CAR expression to sufficient levels for optimal T cell signaling, while keeping it under the threshold for tonic signaling is important to obtain optimal CAR‐T cells. 195
In conclusion, CAR affinity, CAR expression levels, and target antigen density can all influence CAR‐T cell functionality. 195 , 210 While target antigen density cannot be modified (with few exceptions 213 ), CAR affinity and CAR surface expression can to some extent be controlled. 195 Notably, disparities in results exist between studies 201 , 204 , 205 presumably as a consequence of the impact of other factors such as antigen density on target cells, epitope location on the antigen (as discussed below), the range of affinities/avidities tested and/or the incorporated intracellular domains. 201 , 204 Still, the above mentioned studies underline the importance of these features in future CAR‐T cell design.
5. THE LOCATION OF THE EPITOPE RECOGNIZED BY THE CAR AND THE SPACER LENGTH INFLUENCE CAR‐T CELL EFFICACY
5.1. Impact of the epitope location on the targeted antigen on CAR‐T cell efficacy
Formation of a stable immune synapse between effector T cells and target cells upon engagement of the TCR is essential for the induction of specific target cell killing. 214 As opposed to the strict structural dimensions between the TCR and the pMHC complex that dictate the spatial interaction between T cell and target cell, CAR‐T cells are not subject to this scripted relationship. 214 , 215 The distance between CAR‐T and target cells is dependent on multiple variables including the structural dimensions of the targeted antigen, the epitope location, and the length and flexibility of the spacer region. 214 , 215 , 216 In this context, it was shown that CAR‐T cells were more potent in vitro when targeting membrane‐proximal epitopes on carcinoembryonic antigen (CEA) and CD22 as compared to membrane‐distal epitopes. 217 , 218 Generation of a truncated version of CEA and CD22 in which the membrane distal epitope was translocated to a more membrane‐proximal position induced more efficient CAR‐T cell function, which highlighted the impact of the epitope location. 217 , 218 These findings were later confirmed by another group that demonstrated that CAR‐T cells targeting a membrane‐proximal CD22 epitope induced superior survival in mice compared to CAR‐T cells targeting a membrane distal epitope on CD22. 219 More recently, a similar outcome was shown for CAR‐T cells targeting glypican 3 (GPC3) in hepatocellular carcinoma. 220 Furthermore, CAR‐T cells targeting the membrane‐proximal epitope showed significantly higher polyfunctionality, which refers to the production of two chemokines and/or cytokines by a single cell and is associated with long‐term immune responses in a clinical setting. 220 These studies highlight the importance of placing CAR‐T and target cells in close proximity to each other, which is in line with the kinetic segregation model. In this model of T cell activation, it is hypothesized that T cell and target cell should be placed in a sufficiently close proximity to form an immune synapse that excludes large inhibitory phosphatases (e.g., CD45 and CD148) 197 , 221 which could otherwise prevent adequate T cell signaling upon entering the immune synapse. 104 , 197 , 218
5.2. Tuning spacer length depending on epitope location and accessibility
While antigen structure is predefined and location of the targeted epitope is determined by the incorporated antigen‐binding domain, spacer length can be tuned for each CAR to adjust the intercellular distance between T cell and target cell for optimal T cell activation. 216 A spacer region is incorporated in each CAR to provide flexibility and appropriate length to facilitate access to the target antigen. 200 Since the spacer should ensure a suitable intercellular distance between the CAR‐T and target cells, as proposed by the kinetic segregation model, the optimal spacer length is thought to depend on both the accessibility as well as location of the targeted epitope (membrane‐proximal or distal).
CARs have been designed with spacers of different sizes and origins. Spacers derived from human IgGs (mostly IgG1 and IgG4), CD8α, or CD28 are commonly used. 197 , 222 Long spacers are generally composed of a human IgG hinge region and the CH2 and CH3 domains (IgG‐Fc), while intermediate spacers often refer to spacers composed of a human IgG hinge region and a CH3 domain. Short spacers have been derived from human IgG hinge regions or the extracellular domains of CD8α or CD28. 197 Multiple groups have shown that CARs designed with relatively shorter spacers (intermediate/short/absent) targeting CD19, IL13Rα2 and membrane distal epitopes within ROR1 and CEA were associated with optimal T cell activation. 197 , 199 , 215 , 221 , 223 Contrarily, when targeting more inaccessible membrane‐proximal epitopes within ROR1, MUC1, NCAM, and 5T4, introduction of a longer/flexible spacer was needed to overcome steric hindrance and ensure optimal antigen targeting. 197 , 215 , 221 , 224
5.3. Intrinsic spacer characteristics
Besides the influence of spacer length on intercellular distance, intrinsic features of the spacer regions can affect CAR‐T cell behavior. In this regard, an often described phenomenon is that CARs designed with IgG‐Fc‐derived spacers retain their ability to bind FcγR‐bearing monocytic and NK cells via their CH2 domain. 197 , 215 , 222 , 225 , 226 , 227 This interaction can induce activation and cytokine secretion by the innate immune cells as well as antigen‐independent/unspecific activation of CAR‐T cells resulting in toxicity, limited CAR‐T cell persistence, and increased AICD. 197 , 215 , 227 Moreover, trapping of CAR‐T cells in the lung by residing FcγR‐bearing cells has been described. 93 , 197 Nonetheless, it was shown that deleting the CH2 and/or CH3 regions as well as mutating essential amino acid residues in the CH2 region that participate in binding to the FcγR abrogate this unwanted interaction, which is important to design CARs where a long spacer is required for optimal antigen binding. 215 , 222 , 225 , 227
IgG‐derived spacer‐related issues can also be avoided by incorporating spacer sequences derived from CD8α or CD28. 222 These spacer sequences have been extensively studied in anti‐CD19 CAR‐T cell clinical trials that demonstrated potent antitumor efficacy, as Yescarta® incorporates a CD28‐derived and Kymriah® a CD8α‐derived spacer. 64 , 197 However, the impact of the structure of these spacers on CAR‐T cell performance is less reported. 197 One study by Alabanza et al. 228 compares anti‐CD19 CARs incorporating either CD8α or CD28‐derived spacer and transmembrane domains and showed that while both CAR‐T cell products were equally efficient in eliminating tumors from mice, CD8α‐based CAR‐T cells showed less cytokine‐mediated toxicity and AICD. However, because the transmembrane regions were simultaneously exchanged with the spacer regions, the relative contribution of each domain could not be specified. 228
5.4. Cell‐to‐cell distance in receptor‐based CARs, CARs targeting multiple antigens and uCARs
The potential impact of cell‐to‐cell distance on CAR signaling has also been described in the particular context of natural receptor‐based CARs. As described before, Wu et al. 122 evaluated a CAR incorporating the full‐length DNAM‐1 protein, recognizing PVR and nectin‐2 ligands. While high cytotoxicity could be observed, in vitro cytokine secretion was low. The interaction between DNAM‐1 and its ligand was assumed to lead to the weak T cell signaling observed. DNAM‐1 has a double Ig‐fold structure and its ligands PVR and nectin‐2 exhibit a triple Ig‐fold structure. Therefore, the overall structure of this complex is rather bulky, potentially hampering CAR signaling due to an increased intercellular distance. 122
The intercellular distance between CAR‐T and target cells is also particularly difficult to optimize in tandemCARs, as here the spacer length should be optimized for both targeted antigens in a single molecule. 64 Indeed, impaired CAR‐T cell function has been observed for tandemCARs targeting CD19/CD20 and CD19/CD22, which was attributed to suboptimal targeting of one of the antigens. 139 , 140 , 144 However, optimizing scFv order, spacer length, and linker sequences is possible to resolve this issue, again highlighting the need for an adequate cell‐to‐cell distance. 64 , 139
In addition, the intercellular distance needs to be considered when designing uCARs that target antigen‐binding adaptor molecules. The development of a uCAR design that will induce optimal signaling for each target antigen and epitope is considered very complex. 195 It is doubtful that each adaptor molecule will lead to equally efficient signal transduction in the uCAR as the intercellular distance will not be adjusted for each epitope location. 195 As briefly mentioned before, this feature was addressed in a number of subsequently published studies evaluating uCAR‐T cells using adaptor molecules conjugated to FITC or to a PNE‐tag. 173 , 175 , 177 First, Ma et al. 173 site‐specifically labeled anti‐CD19 FAB fragments (from the FMC63 clone) with FITC at proximal, medial, or distal sites relative to the antigen‐binding region. When evaluating the lytic activity of these uCAR‐T cells, differences in cytotoxicity were observed depending on conjugation sites. Adaptors conjugated with FITC proximal to the antigen‐binding region were more potent than adaptors conjugated with FITC at distal sites. Although the epitope location targeted by mAb FMC63 is unknown, these findings suggest that proximal conjugation sites on the adaptor lead to a closer distance between anti‐FITC CAR‐T cells and CD19+ cells resulting in enhanced antitumor activity. Interestingly, a randomly conjugated anti‐CD19 FITC‐conjugated control adaptor showed inferior antitumor activity compared to the optimized proximal FITC‐labeled adaptor molecule, highlighting the need for a site‐specific labeling strategy. The same approach was followed using an anti‐CD22 FAB adaptor molecule (from the m971 clone). Intriguingly, here, they found that adaptor molecules with FITC conjugated at distal positions were more cytotoxic in comparison with proximal FITC conjugates, which is in contrast to the findings with the anti‐CD19 FAB adaptors. 173 This may be due to the epitope targeted by the m971 mAb, which is located membrane‐proximally on CD22. Altogether, differences in optimal conjugation sites likely depend on what is needed to approximate the natural immunological synapse distance of TCR/pMHC complexes (~130–150 Å), as well as on the accessibility of the adaptor molecules. 173 In a similar study, the same group evaluated uCAR‐T cells targeting anti‐CD19 (from the FMC63 clone) and anti‐CD20 (from ofatumumab) FAB adaptor molecules, N‐ or C‐terminally linked to a PNE‐tag. 175 Adaptors with a PNE‐tag proximal to the antigen‐binding site were generally more potent than adaptors with a PNE‐tag at the C‐terminus of the FAB. Improved uCAR‐T cell activity was further shown for anti‐CD19 FAB adaptors when the spacer sequence was shortened from a 45 amino acid CD8‐derived hinge to a 12 amino acid IgG4m hinge, derived from IgG4 bearing a serine‐to‐proline mutation to increase interchain disulfide formation. Interestingly, while similar results were obtained for CD19‐ and CD20‐targeting FABs when using a CD8‐based spacer, the shortened IgG4m spacer reverted the results for anti‐CD20 adaptors. Now, C‐terminal PNE adaptors were more potent than N‐terminal adaptors. Ofatumumab is known to bind a membrane‐proximal epitope on CD20, supposedly enabling close association between T cell and target cell. The authors suggest that the intercellular distance arising from N‐terminal‐tagged ofatumumab‐derived adaptors in combination with IgG4m‐based uCAR‐T cells may be too close for optimal synapse formation. It is also possible that the large neighboring loop of CD20 induces steric hindrance for binding of uCAR‐T cells with a shortened spacer sequence. 175 In a subsequent study the same authors extend this methodology to HER2‐expressing breast cancer cells. 177 They evaluate CD8‐ or IgG4m‐based uCAR‐T cells for optimal targeting of anti‐HER2 adaptor FABs (from the 4D5 clone) by introduction of FITC or PNE‐tags at defined sites. Again, they confirm that simultaneous optimization of both the spacer and the adaptor‐conjugation site is needed to obtain optimal intercellular distance and orientation for uCAR‐T cell activation. 177
In conclusion, cell‐to‐cell distance is an important influential factor on CAR‐T cell efficacy. So far, data are in line with the kinetic segregation model. As proposed by this model, optimal T cell signaling occurs when large inhibitory phosphatases are excluded from the immune synapse, which can be achieved by shortening the intercellular distance. In accordance, targeting of membrane‐proximal epitopes was often shown to be favorable. Nonetheless, target epitopes are sometimes differently located and steric hindrance can play a role. 218 , 219 , 220 , 221 , 225 These aspects can be tackled by optimizing spacer length which proved useful in multiple studies. While functional CAR‐T cells targeting membrane‐distal epitopes have successfully been designed using unedited short spacer regions, IgG‐Fc derived long spacers are sometimes essential and these should be edited to avoid off‐target CAR‐T cell activation. 199 , 215 , 221 , 222 , 224 , 227 Furthermore, tandemCAR and uCAR technologies are intriguing, but require additional attention in terms of intercellular distance optimization. 173 , 175 , 177
6. CAR‐T CELLS: THE MORE WE LEARN, THE LESS WE KNOW
Although there is and has been a great deal of research into the optimization of CAR‐T cell signaling, the behavior of different CAR‐T cells is still fairly unpredictable, and epitope location, spacer length, affinity, and avidity are all influential. Today, assessing the functionality of a CAR molecule remains largely trial‐and‐error, and the key mechanisms have not yet been revealed. For example, Smith et al. 82 designed several BCMA‐specific scFv‐based CARs with similar affinity and specificity. However, these CAR‐T cells behave differently in vitro and, correspondingly, in vivo. 82 Sommermeyer and colleagues observed different cytokine production profiles and proliferation patterns when altering the VH/VL order in the scFv. When comparing two structurally different scFv‐based CARs that target the same epitope with similar affinity, one of these outperformed the other in in vivo efficacy evaluation. 83 All CARs showed equal expression levels and were equally distributed on the T cell membrane, thus eliminating clustering as a potential reason for difference in potency. More recently, differences in CAR‐T cell activation kinetics and cytokine production were observed when replacing framework amino acids of an scFv to humanize the compound. 229 These data highlight the serendipity of CAR‐T cell design, even with emerging guidelines on which parameters are important.
7. OUTLOOK
CAR‐T cell therapy research has had enormous successes in recent years. Deservingly, much research and financing is spent on this therapy approach with the aim of expanding the patient population who can benefit from it. However, it seems that the initial CD19‐CAR‐T cell success stories were largely lucky shots and there is still much to learn about optimal CAR design and proper therapy tuning. A lot of research has already been done on the CAR endodomain, which has led to the development of different CAR generations. Over the years, this research has followed a clear line of improvements, with limited conflicts within the scientific community. However, research into the extracellular domain has been much more fragmented, and there are many different views on this. In general, it is believed that despite the scFv‐CAR success stories, there are many drawbacks to this format. Multiple alternatives exist and each come with their own advantages and disadvantages. Antibody‐based CARs have the advantage of being fairly predictable in terms of in vivo behavior. Within this format, NanoCARs have multiple advantages over scFv‐based CARs: a monomeric and stable nature, less immunogenicity concerns, and no predisposition for tonic signaling. Receptor‐ligand CARs further lower the risk of immunogenicity but come with increased concerns about unpredictability in terms of off‐tumor toxicity. In addition, these formats require individual (optimized) transgenes for each antigen of interest, with the risk of antigen‐escape variants that negate the principle of the therapy. In that sense, uCARs have a great advantage, since these have a tunable specificity. However, to have a sustained CAR‐T cell population, continuous triggering is required, which implies prolonged hospital time. In addition, due to immunogenicity concerns, repeated dosing of the tracing molecule might be hampered.
All in all, CAR‐T cell research that focuses on the CAR ectodomain is diverse with a plethora of candidates for CAR design being explored. Consensus about an optimal binding domain is currently not in sight. However, we believe that the successes achieved with the CD19‐CAR‐T cells should not be neglected, and mAb‐based CAR‐T cells should be further explored and exploited. As an alternative to the unstable scFvs, sdAb‐based CARs open doors. Within these, it is important to look at specific parameters that are important and optimize these in such a way that they lead to optimal T cell activation. For example, there is still a great lesson to be learned about affinity and spacer structures, which can be optimized on a case‐by‐case basis to arrive at clinically relevant CAR‐T cell products.
ACKNOWLEDGMENTS
Research support was provided by the Research Foundation Flanders (Fonds Wetenschappelijk Onderzoek [FWO]) through research project G028220N and personal mandates to Kim De Veirman (12I0917N), Heleen Hanssens (1S55619N) and Fien Meeus (1S68521N). All figures were created with BioRender.com.
Biographies
Heleen Hanssens holds an MSc in Bioengineering from VUB and focusses on medical biotechnology, applied immunology, and cell‐based cancer therapy during her PhD in Medical Sciences.
Fien Meeus obtained an MSc in Biomedical Sciences from VUB and focuses on the development of CAR‐T cell cancer therapy during her PhD in Medical Sciences.
Kim De Veirman is a post‐doctoral researcher at the Hematology and Immunology lab at the VUB in Belgium. She holds an MSc degree in Biomedical Sciences and a PhD in Medical Sciences at the VUB. Her research focuses on hematological cancers and immunotherapy.
Karine Breckpot is the head of the Laboratory for Molecular and Cellular Therapy at the Vrije Universiteit Brussel (VUB) in Belgium. She received an MSc in Biomedical Sciences and a PhD in Medical Sciences from the same university. Her research interests include cancer immunology and immunotherapy.
Nick Devoogdt is a principal investigator at the Medical Imaging department, Laboratory of In Vivo Cellular and Molecular Imaging, at the VUB in Belgium. He holds an MSc degree in Molecular Biology and a PhD degree in Applied Biological Sciences from the same university. His interests include antibody engineering, antibody‐based therapies, and molecular imaging.
Hanssens H, Meeus F, De Veirman K, Breckpot K, Devoogdt N. The antigen‐binding moiety in the driver's seat of CARs. Med Res Rev. 2022;42:306‐342. 10.1002/med.21818
Heleen Hanssens and Fien Meeus indicate shared co‐first authorship.
Kim De Veirman, Karine Breckpot, and Nick Devoogdt indicate shared co‐senior authorship.
REFERENCES
- 1. Old LJ. Immunotherapy for cancer. Sci Am. 1996;275(3):136‐143. 10.1038/scientificamerican0996-136 [DOI] [PubMed] [Google Scholar]
- 2. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3(11):991‐998. 10.1038/ni1102-991 [DOI] [PubMed] [Google Scholar]
- 3. Sanmamed MF, Chen L. A paradigm shift in cancer immunotherapy: from enhancement to normalization. Cell. 2018;175(2):313‐326. 10.1016/j.cell.2018.09.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Farhood B, Najafi M, Mortezaee K. CD8+ cytotoxic T lymphocytes in cancer immunotherapy: a review. J Cell Physiol. 2019;234(6):8509‐8521. 10.1002/jcp.27782 [DOI] [PubMed] [Google Scholar]
- 5. Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer. 2014;14(2):135‐146. 10.1038/nrc3670 [DOI] [PubMed] [Google Scholar]
- 6. Smith CC, Selitsky SR, Chai S, Armistead PM, Vincent BG, Serody JS. Alternative tumour‐specific antigens. Nat Rev Cancer. 2019;19(8):465‐478. 10.1038/s41568-019-0162-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. De Vlaeminck Y, González‐Rascón A, Goyvaerts C, Breckpot K. Cancer‐associated myeloid regulatory cells. Front Immunol. 2016;7:113. 10.3389/fimmu.2016.00113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Awad RM, De Vlaeminck Y, Maebe J, Goyvaerts C, Breckpot K. Turn back the TIMe: targeting tumor infiltrating myeloid cells to revert cancer progression. Front Immunol. 2018;9:1977. 10.3389/fimmu.2018.01977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646‐674. 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 10. Galon J, Costes A, Sanchez‐Cabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960‐1964. 10.1126/science.1129139 [DOI] [PubMed] [Google Scholar]
- 11. Garber K. Pursuit of tumor‐infiltrating lymphocyte immunotherapy speeds up. Nat Biotechnol. 2019;37(9):969‐971. 10.1038/d41587-019-00023-6 [DOI] [PubMed] [Google Scholar]
- 12. Rosenberg SA, Packard BS, Aebersold PM, et al. Use of tumor‐infiltrating lymphocytes and interleukin‐2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med. 1988;319(25):1676‐1680. 10.1056/NEJM198812223192527 [DOI] [PubMed] [Google Scholar]
- 13. Tran E, Robbins PF, Rosenberg SA. 'Final common pathway' of human cancer immunotherapy: targeting random somatic mutations. Nat Immunol. 2017;18(3):255‐262. 10.1038/ni.3682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stevanović S, Draper LM, Langhan MM, et al. Complete regression of metastatic cervical cancer after treatment with human papillomavirus‐targeted tumor‐infiltrating T cells. J Clin Oncol. 2015;33(14):1543‐1550. 10.1200/JCO.2014.58.9093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314(5796):126‐129. 10.1126/science.1129003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Restifo NP, Kawakami Y, Marincola F, et al. Molecular mechanisms used by tumors to escape immune recognition: immunogenetherapy and the cell biology of major histocompatibility complex class I. J Immunother Emphasis Tumor Immunol. 1993;14(3):182‐190. 10.1097/00002371-199310000-00004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody‐binding domains and the gamma or zeta subunits of the immunoglobulin and T‐cell receptors. Proc Natl Acad Sci USA. 1993;90(2):720‐724. 10.1073/pnas.90.2.720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin‐T‐cell receptor chimeric molecules as functional receptors with antibody‐type specificity. Proc Natl Acad Sci USA. 1989;86(24):10024‐10028. 10.1073/pnas.86.24.10024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gong MC, Latouche JB, Krause A, Heston WD, Bander NH, Sadelain M. Cancer patient T cells genetically targeted to prostate‐specific membrane antigen specifically lyse prostate cancer cells and release cytokines in response to prostate‐specific membrane antigen. Neoplasia. 1999;1(2):123‐127. 10.1038/sj.neo.7900018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Krause A, Guo HF, Latouche JB, Tan C, Cheung NK, Sadelain M. Antigen‐dependent CD28 signaling selectively enhances survival and proliferation in genetically modified activated human primary T lymphocytes. J Exp Med. 1998;188(4):619‐626. 10.1084/jem.188.4.619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maher J, Brentjens RJ, Gunset G, Rivière I, Sadelain M. Human T‐lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta/CD28 receptor. Nat Biotechnol. 2002;20(1):70‐75. 10.1038/nbt0102-70 [DOI] [PubMed] [Google Scholar]
- 22. Sadelain M, Brentjens R, Rivière I. The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol. 2009;21(2):215‐223. 10.1016/j.coi.2009.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. van der Stegen SJ, Hamieh M, Sadelain M. The pharmacology of second‐generation chimeric antigen receptors. Nat Rev Drug Discov. 2015;14(7):499‐509. 10.1038/nrd4597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kershaw MH, Westwood JA, Parker LL, et al. A phase I study on adoptive immunotherapy using gene‐modified T cells for ovarian cancer. Clin Cancer Res. 2006;12(20 Pt 1):6106‐6115. 10.1158/1078-0432.CCR-06-1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Park JR, Digiusto DL, Slovak M, et al. Adoptive transfer of chimeric antigen receptor re‐directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther. 2007;15(4):825‐833. 10.1038/sj.mt.6300104 [DOI] [PubMed] [Google Scholar]
- 26. Savoldo B, Ramos CA, Liu E, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor‐modified T cells in lymphoma patients. J Clin Invest. 2011;121(5):1558‐8238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Weinkove R, George P, Dasyam N, McLellan AD. Selecting costimulatory domains for chimeric antigen receptors: functional and clinical considerations. Clin Transl Immunol. 2019;8(5):e1049. 10.1002/cti2.1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhao Z, Condomines M, van der Stegen SJC, et al. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell. 2015;28(4):415‐428. 10.1016/j.ccell.2015.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Abate‐Daga D, Davila ML. CAR models: next‐generation CAR modifications for enhanced T‐cell function. Mol Ther Oncolytics. 2016;3:16014. 10.1038/mto.2016.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stephan MT, Ponomarev V, Brentjens RJ, et al. T cell‐encoded CD80 and 4‐1BBL induce auto‐ and transcostimulation, resulting in potent tumor rejection. Nat Med. 2007;13(12):1440‐1449. 10.1038/nm1676 [DOI] [PubMed] [Google Scholar]
- 31. Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4‐1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl‐XL activation and CD8+ T cell‐mediated tumor eradication. Mol Ther. 2010;18(2):413‐420. 10.1038/mt.2009.210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tammana S, Huang X, Wong M, et al. 4‐1BB and CD28 signaling plays a synergistic role in redirecting umbilical cord blood T cells against B‐cell malignancies. Hum Gene Ther. 2010;21(1):75‐86. 10.1089/hum.2009.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Carpenito C, Milone MC, Hassan R, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci USA. 2009;106(9):3360‐3365. 10.1073/pnas.0813101106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fesnak A, Lin C, Siegel DL, Maus MV. CAR‐T cell therapies from the transfusion medicine perspective. Transfus Med Rev. 2016;30(3):139‐145. 10.1016/j.tmrv.2016.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stock S, Schmitt M, Sellner L. Optimizing manufacturing protocols of chimeric antigen receptor T cells for improved anticancer immunotherapy. Int J Mol Sci. 2019;20(24). 10.3390/ijms20246223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Couzin‐Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science. 2013;342(6165):1432‐1433. 10.1126/science.342.6165.1432 [DOI] [PubMed] [Google Scholar]
- 37. Curran KJ, Margossian SP, Kernan NA, et al. Toxicity and response after CD19‐specific CAR T‐cell therapy in pediatric/young adult relapsed/refractory B‐ALL. Blood. 2019;134(26):2361‐2368. 10.1182/blood.2019001641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B‐cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439‐448. 10.1056/NEJMoa1709866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lee DW, Kochenderfer JN, Stetler‐Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose‐escalation trial. Lancet. 2015;385(9967):517‐528. 10.1016/S0140-6736(14)61403-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schuster SJ, Svoboda J, Chong EA, et al. Chimeric antigen receptor T cells in refractory B‐cell lymphomas. N Engl J Med. 2017;377(26):2545‐2554. 10.1056/NEJMoa1708566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Locke FL, Ghobadi A, Jacobson CA, et al. Long‐term safety and activity of axicabtagene ciloleucel in refractory large B‐cell lymphoma (ZUMA‐1): a single‐arm, multicentre, phase 1‐2 trial. Lancet Oncol. 2019;20(1):31‐42. 10.1016/S1470-2045(18)30864-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cerrano M, Ruella M, Perales MA, et al. The advent of CAR T‐cell therapy for lymphoproliferative neoplasms: integrating research into clinical practice. Front Immunol. 2020;11:888. 10.3389/fimmu.2020.00888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.U.S. Food and Drug Administration. Approved Cellular and Gene Therapy Products. U.S. Food and Drug Administration. Accessed February 10, 2021.
- 44. Riedell PA, Bishop MR. Safety and efficacy of axicabtagene ciloleucel in refractory large B‐cell lymphomas. Ther Adv Hematol. 2020;11:2040620720902899. 10.1177/2040620720902899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ali S, Kjeken R, Niederlaender C, et al. The European medicines agency review of Kymriah (Tisagenlecleucel) for the treatment of acute lymphoblastic leukemia and diffuse large B‐cell lymphoma. Oncologist. 2020;25(2):e321‐e327. 10.1634/theoncologist.2019-0233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18(4):843‐851. 10.1038/mt.2010.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Enblad G, Karlsson H, Gammelgård G, et al. A phase I/IIa trial using CD19‐targeted third‐generation CAR T cells for lymphoma and leukemia. Clin Cancer Res. 2018;24(24):6185‐6194. 10.1158/1078-0432.CCR-18-0426 [DOI] [PubMed] [Google Scholar]
- 48. Ramos CA, Rouce R, Robertson CS, et al. In vivo fate and activity of second‐ versus third‐generation CD19‐specific CAR‐T cells in B cell non‐Hodgkin's lymphomas. Mol Ther. 2018;26(12):2727‐2737. 10.1016/j.ymthe.2018.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cao G, Lei L, Zhu X. Efficiency and safety of autologous chimeric antigen receptor T‐cells therapy used for patients with lymphoma: a systematic review and meta‐analysis. Med Baltim. 2019;98(42):e17506. 10.1097/MD.0000000000017506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Abreu TR, Fonseca NA, Gonçalves N, Moreira JN. Current challenges and emerging opportunities of CAR‐T cell therapies. J Control Release. 2020;319:246‐261. 10.1016/j.jconrel.2019.12.047 [DOI] [PubMed] [Google Scholar]
- 51. Bagley SJ, O'Rourke DM. Clinical investigation of CAR T cells for solid tumors: lessons learned and future directions. Pharmacol Ther. 2020;205:107419. 10.1016/j.pharmthera.2019.107419 [DOI] [PubMed] [Google Scholar]
- 52. Koneru M, Purdon TJ, Spriggs D, Koneru S, Brentjens RJ. IL‐12 secreting tumor‐targeted chimeric antigen receptor T cells eradicate ovarian tumors. Oncoimmunology. 2015;4(3):e994446. 10.4161/2162402X.2014.994446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. You F, Jiang L, Zhang B, et al. Phase 1 clinical trial demonstrated that MUC1 positive metastatic seminal vesicle cancer can be effectively eradicated by modified anti‐MUC1 chimeric antigen receptor transduced T cells. Sci China: Life Sci. 2016;59(4):386‐397. 10.1007/s11427-016-5024-7 [DOI] [PubMed] [Google Scholar]
- 54. Chen Y, Sun C, Landoni E, Metelitsa L, Dotti G, Savoldo B. Eradication of neuroblastoma by T cells redirected with an optimized GD2‐specific chimeric antigen receptor and interleukin‐15. Clin Cancer Res. 2019;25(9):2915‐2924. 10.1158/1078-0432.ccr-18-1811 [DOI] [PubMed] [Google Scholar]
- 55. Chmielewski M, Abken H. TRUCKs with IL‐18 payload: toward shaping the immune landscape for a more efficacious CAR T‐cell therapy of solid cancer. Adv Cell Gene Ther. 2018;1(1):e7. 10.1002/acg2.7 [DOI] [Google Scholar]
- 56. Curran KJ, Seinstra BA, Nikhamin Y, et al. Enhancing antitumor efficacy of chimeric antigen receptor T cells through constitutive CD40L expression. Mol Ther. 2015;23(4):769‐778. 10.1038/mt.2015.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kuhn NF, Purdon TJ, van Leeuwen DG, et al. CD40 ligand‐modified chimeric antigen receptor T cells enhance antitumor function by eliciting an endogenous antitumor response. Cancer Cell. 2019;35(3):473‐488. 10.1016/j.ccell.2019.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Caruana I, Savoldo B, Hoyos V, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR‐redirected T lymphocytes. Nat Med. 2015;21(5):524‐529. 10.1038/nm.3833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rafiq S, Yeku OO, Jackson HJ, et al. Targeted delivery of a PD‐1‐blocking scFv by CAR‐T cells enhances anti‐tumor efficacy in vivo. Nat Biotechnol. 2018;36(9):847‐856. 10.1038/nbt.4195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yu S, Yi M, Qin S, Wu K. Next generation chimeric antigen receptor T cells: safety strategies to overcome toxicity. Mol Cancer. 2019;18(1):125. 10.1186/s12943-019-1057-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kagoya Y, Tanaka S, Guo T, et al. A novel chimeric antigen receptor containing a JAK‐STAT signaling domain mediates superior antitumor effects. Nat Med. 2018;24(3):352‐359. 10.1038/nm.4478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhao L, Cao YJ. Engineered T cell therapy for cancer in the clinic. Front Immunol. 2019;10:2250. 10.3389/fimmu.2019.02250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Crivianu‐Gaita V, Thompson M. Aptamers, antibody scFv, and antibody Fab' fragments: an overview and comparison of three of the most versatile biosensor biorecognition elements. Biosens Bioelectron. 2016;85:32‐45. 10.1016/j.bios.2016.04.091 [DOI] [PubMed] [Google Scholar]
- 64. Guedan S, Calderon H, Posey AD, Maus MV. Engineering and design of chimeric antigen receptors. Mol Ther Methods Clin Dev. 2019;12:145‐156. 10.1016/j.omtm.2018.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Caruana I, Diaconu I, Dotti G. From monoclonal antibodies to chimeric antigen receptors for the treatment of human malignancies. Semin Oncol. 2014;41(5):661‐666. 10.1053/j.seminoncol.2014.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3(4):388‐398. 10.1158/2159-8290.CD-12-0548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. FDA okays second CAR‐T for Kite. Nat Biotechnol. 2020;38(9):1012. 10.1038/s41587-020-0676-z [DOI] [PubMed] [Google Scholar]
- 68. Chavez JC, Bachmeier C, Kharfan‐Dabaja MA. CAR T‐cell therapy for B‐cell lymphomas: clinical trial results of available products. Ther Adv Hematol. 2019;10:2040620719841581. 10.1177/2040620719841581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Center I‐OR. FDA Grants Priority Review to Idecabtagene Vicleucel for Advanced Multiple Myeloma. Immuno‐Oncology Resource Center. Accessed February 10, 2021
- 70. Lam N, Trinklein ND, Buelow B, Patterson GH, Ojha N, Kochenderfer JN. Anti‐BCMA chimeric antigen receptors with fully human heavy‐chain‐only antigen recognition domains. Nat Commun. 2020;11(1):283. 10.1038/s41467-019-14119-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Friedman KM, Garrett TE, Evans JW, et al. Effective targeting of multiple B‐cell maturation antigen‐expressing hematological malignances by anti‐B‐cell maturation antigen chimeric antigen receptor T cells. Hum Gene Ther. 2018;29(5):585‐601. 10.1089/hum.2018.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Long AH, Haso WM, Shern JF, et al. 4‐1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015;21(6):581‐590. 10.1038/nm.3838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Gil D, Schrum AG. Strategies to stabilize compact folding and minimize aggregation of antibody‐based fragments. Adv Biosci Biotechnol. 2013;4(4a):73‐84. 10.4236/abb.2013.44A011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Fujiwara K, Masutani M, Tachibana M, Okada N. Impact of scFv structure in chimeric antigen receptor on receptor expression efficiency and antigen recognition properties. Biochem Biophys Res Commun. 2020;527:350‐357. 10.1016/j.bbrc.2020.03.071 [DOI] [PubMed] [Google Scholar]
- 75. Jensen MC, Popplewell L, Cooper LJ, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19‐specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant. 2010;16(9):1245‐1256. 10.1016/j.bbmt.2010.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lamers CH, Willemsen R, van Elzakker P, et al. Immune responses to transgene and retroviral vector in patients treated with ex vivo‐engineered T cells. Blood. 2011;117(1):72‐82. 10.1182/blood-2010-07-294520 [DOI] [PubMed] [Google Scholar]
- 77. Turtle CJ, Hanafi LA, Berger C, et al. CD19 CAR‐T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016;126(6):2123‐2138. 10.1172/JCI85309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Turtle CJ, Hanafi LA, Berger C, et al. Immunotherapy of non‐Hodgkin's lymphoma with a defined ratio of CD8+ and CD4+ CD19‐specific chimeric antigen receptor‐modified T cells. Sci Transl Med. 2016;8(355):355ra116. 10.1126/scitranslmed.aaf8621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Maus MV, Haas AR, Beatty GL, et al. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol Res. 2013;1(1):26‐31. 10.1158/2326-6066.CIR-13-0006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Qian L, Li D, Ma L, et al. The novel anti‐CD19 chimeric antigen receptors with humanized scFv (single‐chain variable fragment) trigger leukemia cell killing. Cell Immunol. 2016;304–305:49‐54. 10.1016/j.cellimm.2016.03.003 [DOI] [PubMed] [Google Scholar]
- 81. Maeda T, Wakasawa T, Shima Y, Tsuboi I, Aizawa S, Tamai I. Efficacy of humanized CD19‐targeted chimeric antigen receptor (CAR)‐modified T cells in children and young adults with relapsed/refractory acute lymphoblastic leukemia. Blood. 2016;128(22):217. 10.1182/blood.V128.22.217.217 27207794 [DOI] [Google Scholar]
- 82. Smith EL, Staehr M, Masakayan R, et al. Development and evaluation of an optimal human single‐chain variable fragment‐derived BCMA‐targeted CAR T cell vector. Mol Ther. 2018;26(6):1447‐1456. 10.1016/j.ymthe.2018.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Sommermeyer D, Hill T, Shamah SM, et al. Fully human CD19‐specific chimeric antigen receptors for T‐cell therapy. Leukemia. 2017;31(10):2191‐2199. 10.1038/leu.2017.57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Klebanoff CA, Khong HT, Antony PA, Palmer DC, Restifo NP. Sinks, suppressors and antigen presenters: how lymphodepletion enhances T cell‐mediated tumor immunotherapy. Trends Immunol. 2005;26(2):111‐117. 10.1016/j.it.2004.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Lowe KL, Mackall CL, Norry E, Amado R, Jakobsen BK, Binder G. Fludarabine and neurotoxicity in engineered T‐cell therapy. Gene Ther. 2018;25(3):176‐191. 10.1038/s41434-018-0019-6 [DOI] [PubMed] [Google Scholar]
- 86. O'Leary MC, Lu X, Huang Y, et al. FDA approval summary: tisagenlecleucel for treatment of patients with relapsed or refractory B‐cell precursor acute lymphoblastic leukemia. Clin Cancer Res. 2019;25(4):1142‐1146. 10.1158/1078-0432.CCR-18-2035 [DOI] [PubMed] [Google Scholar]
- 87. Boyiadzis MM, Dhodapkar MV, Brentjens RJ, et al. Chimeric antigen receptor (CAR) T therapies for the treatment of hematologic malignancies: clinical perspective and significance. J Immunother Cancer. 2018;6(1):137. 10.1186/s40425-018-0460-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Awasthi R, Pacaud L, Waldron E, et al. Tisagenlecleucel cellular kinetics, dose, and immunogenicity in relation to clinical factors in relapsed/refractory DLBCL. Blood Adv. 2020;4(3):560‐572. 10.1182/bloodadvances.2019000525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Novartis. Kymriah: Full Prescribing Information. Novartis. Accessed July 23, 2020. https://www.novartis.us/sites/www.novartis.us/files/kymriah.pdf
- 90. Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507‐1517. 10.1056/NEJMoa1407222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lecocq Q, De Vlaeminck Y, Hanssens H, et al. Theranostics in immuno‐oncology using nanobody derivatives. Theranostics. 2019;9(25):7772‐7791. 10.7150/thno.34941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kim DY, Hussack G, Kandalaft H, Tanha J. Mutational approaches to improve the biophysical properties of human single‐domain antibodies. Biochim Biophys Acta. 2014;1844(11):1983‐2001. 10.1016/j.bbapap.2014.07.008 [DOI] [PubMed] [Google Scholar]
- 93. De Munter S, Van Parys A, Bral L, et al. Rapid and effective generation of nanobody based CARs using PCR and Gibson assembly. Int J Mol Sci. 2020;21(3). 10.3390/ijms21030883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. An N, Hou YN, Zhang QX, et al. Anti‐multiple myeloma activity of nanobody‐based anti‐CD38 chimeric antigen receptor T cells. Mol Pharm. 2018;15(10):4577‐4588. 10.1021/acs.molpharmaceut.8b00584 [DOI] [PubMed] [Google Scholar]
- 95. Hassani M, Hajari Taheri F, Sharifzadeh Z, et al. Construction of a chimeric antigen receptor bearing a nanobody against prostate a specific membrane antigen in prostate cancer. J Cell Biochem. 2019;120(6):10787‐10795. 10.1002/jcb.28370 [DOI] [PubMed] [Google Scholar]
- 96. Hajari Taheri F, Hassani M, Sharifzadeh Z, Behdani M, Arashkia A, Abolhassani M. T cell engineered with a novel nanobody‐based chimeric antigen receptor against VEGFR2 as a candidate for tumor immunotherapy. IUBMB Life. 2019;71(9):1259‐1267. 10.1002/iub.2019 [DOI] [PubMed] [Google Scholar]
- 97. Bakhtiari SH, Rahbarizadeh F, Hasannia S, Ahmadvand D, Iri‐Sofla FJ, Rasaee MJ. Anti‐MUC1 nanobody can redirect T‐body cytotoxic effector function. Hybridoma. 2009;28(2):85‐92. 10.1089/hyb.2008.0079 [DOI] [PubMed] [Google Scholar]
- 98. Khaleghi S, Rahbarizadeh F, Ahmadvand D, Rasaee MJ, Pognonec P. A caspase 8‐based suicide switch induces apoptosis in nanobody‐directed chimeric receptor expressing T cells. Int J Hematol. 2012;95(4):434‐444. 10.1007/s12185-012-1037-6 [DOI] [PubMed] [Google Scholar]
- 99. Jamnani FR, Rahbarizadeh F, Shokrgozar MA, et al. T cells expressing VHH‐directed oligoclonal chimeric HER2 antigen receptors: towards tumor‐directed oligoclonal T cell therapy. Biochim Biophys Acta. 2014;1840(1):378‐386. 10.1016/j.bbagen.2013.09.029 [DOI] [PubMed] [Google Scholar]
- 100. Li N, Fu H, Hewitt SM, Dimitrov DS, Ho M. Therapeutically targeting glypican‐2 via single‐domain antibody‐based chimeric antigen receptors and immunotoxins in neuroblastoma. Proc Natl Acad Sci USA. 2017;114(32):E6623‐E6631. 10.1073/pnas.1706055114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Xie YJ, Dougan M, Jailkhani N, et al. Nanobody‐based CAR T cells that target the tumor microenvironment inhibit the growth of solid tumors in immunocompetent mice. Proc Natl Acad Sci USA. 2019;116(16):7624‐7631. 10.1073/pnas.1817147116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Xie YJ, Dougan M, Ingram JR, et al. Improved antitumor efficacy of chimeric antigen receptor T cells that secrete single‐domain antibody fragments. Cancer Immunol Res. 2020;8(4):518‐529. 10.1158/2326-6066.CIR-19-0734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Albert S, Arndt C, Feldmann A, et al. A novel nanobody‐based target module for retargeting of T lymphocytes to EGFR‐expressing cancer cells via the modular UniCAR platform. Oncoimmunology. 2017;6(4):e1287246. 10.1080/2162402X.2017.1287246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. De Munter S, Ingels J, Goetgeluk G, et al. Nanobody based dual specific CARs. Int J Mol Sci. 2018;19(2). 10.3390/ijms19020403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. He X, Feng Z, Ma J, et al. Bispecific and split CAR T cells targeting CD13 and TIM3 eradicate acute myeloid leukemia. Blood. 2020;135(10):713‐723. 10.1182/blood.2019002779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Janssen Announces Investigational CAR‐T Therapy JNJ‐68284528 Granted PRIME Designation by the European Medicines Agency. 2019. Johnson & Johnson.
- 107. Xu J, Chen LJ, Yang SS, et al. Exploratory trial of a biepitopic CAR T‐targeting B cell maturation antigen in relapsed/refractory multiple myeloma. Proc Natl Acad Sci USA. 2019;116(19):9543‐9551. 10.1073/pnas.1819745116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Muyldermans S. Nanobodies: natural single‐domain antibodies. Annu Rev Biochem. 2013;82:775‐797. 10.1146/annurev-biochem-063011-092449 [DOI] [PubMed] [Google Scholar]
- 109. Muyldermans S. Single domain camel antibodies: current status. J Biotechnol. 2001;74(4):277‐302. 10.1016/s1389-0352(01)00021-6 [DOI] [PubMed] [Google Scholar]
- 110. Vincke C, Loris R, Saerens D, Martinez‐Rodriguez S, Muyldermans S, Conrath K. General strategy to humanize a camelid single‐domain antibody and identification of a universal humanized nanobody scaffold. J Biol Chem. 2009;284(5):3273‐3284. 10.1074/jbc.M806889200 [DOI] [PubMed] [Google Scholar]
- 111. Wang QJ, Yu Z, Hanada KI, et al. Preclinical evaluation of chimeric antigen receptors targeting CD70‐expressing cancers. Clin Cancer Res. 2017;23(9):2267‐2276. 10.1158/1078-0432.CCR-16-1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Perales‐Puchalt A, Svoronos N, Rutkowski MR, et al. Follicle‐stimulating hormone receptor is expressed by most ovarian cancer subtypes and is a safe and effective immunotherapeutic target. Clin Cancer Res. 2017;23(2):441‐453. 10.1158/1078-0432.CCR-16-0492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol. 2013;31:413‐441. 10.1146/annurev-immunol-032712-095951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Tao K, He M, Tao F, et al. Development of NKG2D‐based chimeric antigen receptor‐T cells for gastric cancer treatment. Cancer Chemother Pharmacol. 2018;82(5):815‐827. 10.1007/s00280-018-3670-0 [DOI] [PubMed] [Google Scholar]
- 115. Barber A, Meehan KR, Sentman CL. Treatment of multiple myeloma with adoptively transferred chimeric NKG2D receptor‐expressing T cells. Gene Ther. 2011;18(5):509‐516. 10.1038/gt.2010.174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Barber A, Rynda A, Sentman CL. Chimeric NKG2D expressing T cells eliminate immunosuppression and activate immunity within the ovarian tumor microenvironment. J Immunol. 2009;183(11):6939‐6947. 10.4049/jimmunol.0902000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Spear P, Barber A, Rynda‐Apple A, Sentman CL. NKG2D CAR T‐cell therapy inhibits the growth of NKG2D ligand heterogeneous tumors. Immunol Cell Biol. 2013;91(6):435‐440. 10.1038/icb.2013.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Lehner M, Götz G, Proff J, et al. Redirecting T cells to Ewing's sarcoma family of tumors by a chimeric NKG2D receptor expressed by lentiviral transduction or mRNA transfection. PLOS One. 2012;7(2):e31210. 10.1371/journal.pone.0031210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Wu J, Song Y, Bakker AB, et al. An activating immunoreceptor complex formed by NKG2D and DAP10. Science. 1999;285(5428):730‐732. 10.1126/science.285.5428.730 [DOI] [PubMed] [Google Scholar]
- 120. Zhang T, Sentman CL. Mouse tumor vasculature expresses NKG2D ligands and can be targeted by chimeric NKG2D‐modified T cells. J Immunol. 2013;190(5):2455‐2463. 10.4049/jimmunol.1201314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Zhang T, Wu MR, Sentman CL. An NKp30‐based chimeric antigen receptor promotes T cell effector functions and antitumor efficacy in vivo. J Immunol. 2012;189(5):2290‐2299. 10.4049/jimmunol.1103495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Wu MR, Zhang T, Alcon A, Sentman CL. DNAM‐1‐based chimeric antigen receptors enhance T cell effector function and exhibit in vivo efficacy against melanoma. Cancer Immunol Immunother. 2015;64(4):409‐418. 10.1007/s00262-014-1648-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Jacobs J, Deschoolmeester V, Zwaenepoel K, et al. CD70: an emerging target in cancer immunotherapy. Pharmacol Ther. 2015;155:1‐10. 10.1016/j.pharmthera.2015.07.007 [DOI] [PubMed] [Google Scholar]
- 124. Shaffer DR, Savoldo B, Yi Z, et al. T cells redirected against CD70 for the immunotherapy of CD70‐positive malignancies. Blood. 2011;117(16):4304‐4314. 10.1182/blood-2010-04-278218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Huang G, Yu L, Cooper LJ, Hollomon M, Huls H, Kleinerman ES. Genetically modified T cells targeting interleukin‐11 receptor α‐chain kill human osteosarcoma cells and induce the regression of established osteosarcoma lung metastases. Cancer Res. 2012;72(1):271‐281. 10.1158/0008-5472.CAN-11-2778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Kahlon KS, Brown C, Cooper LJ, Raubitschek A, Forman SJ, Jensen MC. Specific recognition and killing of glioblastoma multiforme by interleukin 13‐zetakine redirected cytolytic T cells. Cancer Res. 2004;64(24):9160‐9166. 10.1158/0008-5472.CAN-04-0454 [DOI] [PubMed] [Google Scholar]
- 127. Brown CE, Badie B, Barish ME, et al. Bioactivity and safety of IL13Rα2‐redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res. 2015;21(18):4062‐4072. 10.1158/1078-0432.CCR-15-0428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Brown CE, Aguilar B, Starr R, et al. Optimization of IL13Rα2‐targeted chimeric antigen receptor T cells for improved anti‐tumor efficacy against glioblastoma. Mol Ther. 2018;26(1):31‐44. 10.1016/j.ymthe.2017.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Brown CE, Alizadeh D, Starr R, et al. Regression of glioblastoma after chimeric antigen receptor T‐cell therapy. N Engl J Med. 2016;375(26):2561‐2569. 10.1056/NEJMoa1610497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Nakazawa Y, Matsuda K, Kurata T, et al. Anti‐proliferative effects of T cells expressing a ligand‐based chimeric antigen receptor against CD116 on CD34(+) cells of juvenile myelomonocytic leukemia. J Hematol Oncol. 2016;9:27. 10.1186/s13045-016-0256-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Lee L, Draper B, Chaplin N, et al. An APRIL‐based chimeric antigen receptor for dual targeting of BCMA and TACI in multiple myeloma. Blood. 2018;131(7):746‐758. 10.1182/blood-2017-05-781351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Schmidts A, Ormhøj M, Choi BD, et al. Rational design of a trimeric APRIL‐based CAR‐binding domain enables efficient targeting of multiple myeloma. Blood Adv. 2019;3(21):3248‐3260. 10.1182/bloodadvances.2019000703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Davies DM, Foster J, van der Stegen SJ, et al. Flexible targeting of ErbB dimers that drive tumorigenesis by using genetically engineered T cells. Mol Med. 2012;18:565‐576. 10.2119/molmed.2011.00493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Parente‐Pereira AC, Whilding LM, Brewig N, et al. Synergistic chemoimmunotherapy of epithelial ovarian cancer using ErbB‐retargeted T cells combined with carboplatin. J Immunol. 2013;191(5):2437‐2445. 10.4049/jimmunol.1301119 [DOI] [PubMed] [Google Scholar]
- 135. Wang D, Starr R, Chang WC, et al. Chlorotoxin‐directed CAR T cells for specific and effective targeting of glioblastoma. Sci Transl Med. 2020;12(533). 10.1126/scitranslmed.aaw2672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Park S, Shevlin E, Vedvyas Y, et al. Micromolar affinity CAR T cells to ICAM‐1 achieves rapid tumor elimination while avoiding systemic toxicity. Sci Rep. 2017;7(1):14366. 10.1038/s41598-017-14749-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Ruella M, Barrett DM, Kenderian SS, et al. Dual CD19 and CD123 targeting prevents antigen‐loss relapses after CD19‐directed immunotherapies. J Clin Invest. 2016;126(10):3814‐3826. 10.1172/JCI87366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Wang N, Hu X, Cao W, et al. Efficacy and safety of CAR19/22 T‐cell cocktail therapy in patients with refractory/relapsed B‐cell malignancies. Blood. 2020;135(1):17‐27. 10.1182/blood.2019000017 [DOI] [PubMed] [Google Scholar]
- 139. Qin H, Ramakrishna S, Nguyen S, et al. Preclinical development of bivalent chimeric antigen receptors targeting both CD19 and CD22. Mol Ther Oncolytics. 2018;11:127‐137. 10.1016/j.omto.2018.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Zah E, Lin MY, Silva‐Benedict A, Jensen MC, Chen YY. T cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunol Res. 2016;4(6):498‐508. 10.1158/2326-6066.CIR-15-0231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Amrolia PJ, Wynn R, Hough RE, et al. Phase I study of AUTO3, a bicistronic chimeric antigen receptor (CAR) T‐cell therapy targeting CD19 and CD22, in pediatric patients with relapsed/refractory B‐cell acute lymphoblastic leukemia (r/r B‐ALL): Amelia study. Blood. 2019;134(suppl_1):2620. 10.1182/blood-2019-123424 [DOI] [Google Scholar]
- 142. Bielamowicz K, Fousek K, Byrd TT, et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro‐Oncology. 2018;20(4):506‐518. 10.1093/neuonc/nox182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Grada Z, Hegde M, Byrd T, et al. TanCAR: a novel bispecific chimeric antigen receptor for cancer immunotherapy. Mol Ther Nucleic Acids. 2013;2:e105. 10.1038/mtna.2013.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Schneider D, Xiong Y, Wu D, et al. A tandem CD19/CD20 CAR lentiviral vector drives on‐target and off‐target antigen modulation in leukemia cell lines. J Immunother Cancer. 2017;5:42. 10.1186/s40425-017-0246-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Martyniszyn A, Krahl AC, André MC, Hombach AA, Abken H. CD20‐CD19 bispecific CAR T cells for the treatment of B‐cell malignancies. Hum Gene Ther. 2017;28(12):1147‐1157. 10.1089/hum.2017.126 [DOI] [PubMed] [Google Scholar]
- 146. Zah E, Nam E, Bhuvan V, et al. Systematically optimized BCMA/CS1 bispecific CAR‐T cells robustly control heterogeneous multiple myeloma. Nat Commun. 2020;11(1):2283. 10.1038/s41467-020-16160-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Kang L, Zhang J, Li M, et al. Characterization of novel dual tandem CD19/BCMA chimeric antigen receptor T cells to potentially treat multiple myeloma. Biomark Res. 2020;8:14. 10.1186/s40364-020-00192-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Hegde M, Corder A, Chow KK, et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol Ther. 2013;21(11):2087‐2101. 10.1038/mt.2013.185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Ahn S, Li J, Sun C, et al. Cancer immunotherapy with T cells carrying bispecific receptors that mimic antibodies. Cancer Immunol Res. 2019;7(5):773‐783. 10.1158/2326-6066.CIR-18-0636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Fry TJ, Shah NN, Orentas RJ, et al. CD22‐targeted CAR T cells induce remission in B‐ALL that is naive or resistant to CD19‐targeted CAR immunotherapy. Nat Med. 2018;24(1):20‐28. 10.1038/nm.4441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Wilkie S, van Schalkwyk MC, Hobbs S, et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol. 2012;32(5):1059‐1070. 10.1007/s10875-012-9689-9 [DOI] [PubMed] [Google Scholar]
- 152. Lanitis E, Poussin M, Klattenhoff AW, et al. Chimeric antigen receptor T Cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol Res. 2013;1(1):43‐53. 10.1158/2326-6066.CIR-13-0008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31(1):71‐75. 10.1038/nbt.2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Cho JH, Collins JJ, Wong WW. Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell. 2018;173(6):1426‐1438. 10.1016/j.cell.2018.03.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Roybal KT, Rupp LJ, Morsut L, et al. Precision tumor recognition by T cells with combinatorial antigen‐sensing circuits. Cell. 2016;164(4):770‐779. 10.1016/j.cell.2016.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Srivastava S, Salter AI, Liggitt D, et al. Logic‐gated ROR1 chimeric antigen receptor expression rescues T cell‐mediated toxicity to normal tissues and enables selective tumor targeting. Cancer Cell. 2019;35(3):489‐503. 10.1016/j.ccell.2019.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. O'Rourke DM, Nasrallah MP, Desai A, et al. A single dose of peripherally infused EGFRvIII‐directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9(399). 10.1126/scitranslmed.aaa0984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Brudno JN, Maric I, Hartman SD, et al. T cells genetically modified to express an anti‐B‐cell maturation antigen chimeric antigen receptor cause remissions of poor‐prognosis relapsed multiple myeloma. J Clin Oncol. 2018;36(22):2267‐2280. 10.1200/JCO.2018.77.8084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Ruella M, Maus MV. Catch me if you can: leukemia escape after CD19‐directed T cell immunotherapies. Comput Struct Biotechnol J. 2016;14:357‐362. 10.1016/j.csbj.2016.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Urbanska K, Lanitis E, Poussin M, et al. A universal strategy for adoptive immunotherapy of cancer through use of a novel T‐cell antigen receptor. Cancer Res. 2012;72(7):1844‐1852. 10.1158/0008-5472.CAN-11-3890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Lohmueller JJ, Ham JD, Kvorjak M, Finn OJ. mSA2 affinity‐enhanced biotin‐binding CAR T cells for universal tumor targeting. Oncoimmunology. 2017;7(1):e1368604. 10.1080/2162402X.2017.1368604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Dale GL, Gaddy P, Pikul FJ. Antibodies against biotinylated proteins are present in normal human serum. J Lab Clin Med. 1994;123(3):365‐371. [PubMed] [Google Scholar]
- 163. Clémenceau B, Congy‐Jolivet N, Gallot G, et al. Antibody‐dependent cellular cytotoxicity (ADCC) is mediated by genetically modified antigen‐specific human T lymphocytes. Blood. 2006;107(12):4669‐4677. 10.1182/blood-2005-09-3775 [DOI] [PubMed] [Google Scholar]
- 164. Clémenceau B, Valsesia‐Wittmann S, Jallas AC, et al. In vitro and in vivo comparison of lymphocytes transduced with a human CD16 or with a chimeric antigen receptor reveals potential off‐target interactions due to the IgG2 CH2‐CH3 CAR‐spacer. J Immunol Res. 2015;2015:482089. 10.1155/2015/482089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Ochi F, Fujiwara H, Tanimoto K, et al. Gene‐modified human α/β‐T cells expressing a chimeric CD16‐CD3ζ receptor as adoptively transferable effector cells for anticancer monoclonal antibody therapy. Cancer Immunol Res. 2014;2(3):249‐262. 10.1158/2326-6066.CIR-13-0099-T [DOI] [PubMed] [Google Scholar]
- 166. Kudo K, Imai C, Lorenzini P, et al. T lymphocytes expressing a CD16 signaling receptor exert antibody‐dependent cancer cell killing. Cancer Res. 2014;74(1):93‐103. 10.1158/0008-5472.CAN-13-1365 [DOI] [PubMed] [Google Scholar]
- 167. Motz G, Whiteman K, Shin J, et al. Abstract B105: ACTR707: a novel T‐cell therapy for the treatment of relapsed or refractory CD20+ B cell lymphoma in combination with rituximab. Mol Cancer Ther. 2018;17(1 suppl):B105. 10.1158/1535-7163.targ-17-b105 [DOI] [Google Scholar]
- 168. Maeda T, Wakasawa T, Shima Y, Tsuboi I, Aizawa S, Tamai I. A phase 1 study of ACTR087 in combination with rituximab, in subjects with relapsed or refractory CD20‐positive B‐cell lymphoma. Blood. 2019;134(suppl_1):244. 10.1182/blood-2019-123810 [DOI] [Google Scholar]
- 169. FDA partially halts phase I study of ACTR707 in non‐Hodgkin lymphoma. Target Oncol. 2020. https://www.targetedonc.com/view/fda-partially-halts-phase-i-study-of-actr707-in-nonhodgkin-lymphoma [Google Scholar]
- 170. FDA safety concerns halt ACTR087 B cell non‐Hodgkin lymphoma clinical trial. Lymphomahub. 2019. https://lymphomahub.com/medical-information/fda-safety-concerns-halt-actr087-b-cell-non-hodgkin-lymphoma-clinical-trial [Google Scholar]
- 171. Rataj F, Jacobi SJ, Stoiber S, et al. High‐affinity CD16‐polymorphism and Fc‐engineered antibodies enable activity of CD16‐chimeric antigen receptor‐modified T cells for cancer therapy. Br J Cancer. 2019;120(1):79‐87. 10.1038/s41416-018-0341-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Kim MS, Ma JS, Yun H, et al. Redirection of genetically engineered CAR‐T cells using bifunctional small molecules. J Am Chem Soc. 2015;137(8):2832‐2835. 10.1021/jacs.5b00106 [DOI] [PubMed] [Google Scholar]
- 173. Ma JS, Kim JY, Kazane SA, et al. Versatile strategy for controlling the specificity and activity of engineered T cells. Proc Natl Acad Sci USA. 2016;113(4):E450‐E458. 10.1073/pnas.1524193113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Tamada K, Geng D, Sakoda Y, et al. Redirecting gene‐modified T cells toward various cancer types using tagged antibodies. Clin Cancer Res. 2012;18(23):6436‐6445. 10.1158/1078-0432.CCR-12-1449 [DOI] [PubMed] [Google Scholar]
- 175. Rodgers DT, Mazagova M, Hampton EN, et al. Switch‐mediated activation and retargeting of CAR‐T cells for B‐cell malignancies. Proc Natl Acad Sci USA. 2016;113(4):E459‐E468. 10.1073/pnas.1524155113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Raj D, Yang MH, Rodgers D, et al. Switchable CAR‐T cells mediate remission in metastatic pancreatic ductal adenocarcinoma. Gut. 2019;68(6):1052‐1064. 10.1136/gutjnl-2018-316595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Cao Y, Rodgers DT, Du J, et al. Design of switchable chimeric antigen receptor T cells targeting breast cancer. Angew Chem Int Ed Engl. 2016;55(26):7520‐7524. 10.1002/anie.201601902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Cartellieri M, Feldmann A, Koristka S, et al. Switching CAR T cells on and off: a novel modular platform for retargeting of T cells to AML blasts. Blood Cancer J. 2016;6(8):e458. 10.1038/bcj.2016.61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Pishali Bejestani E, Cartellieri M, Bergmann R, et al. Characterization of a switchable chimeric antigen receptor platform in a pre‐clinical solid tumor model. Oncoimmunology. 2017;6(10):e1342909. 10.1080/2162402X.2017.1342909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. De Vos J, Devoogdt N, Lahoutte T, Muyldermans S. Camelid single‐domain antibody‐fragment engineering for (pre)clinical in vivo molecular imaging applications: adjusting the bullet to its target. Expert Opin Biol Ther. 2013;13(8):1149‐1160. 10.1517/14712598.2013.800478 [DOI] [PubMed] [Google Scholar]
- 181. Yiannaki EE, Tzioufas AG, Bachmann M, et al. The value of synthetic linear epitope analogues of La/SSB for the detection of autoantibodies to La/SSB; specificity, sensitivity and comparison of methods. Clin Exp Immunol. 1998;112(1):152‐158. 10.1046/j.1365-2249.1998.00558.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Herzig E, Kim KC, Packard TA, et al. Attacking latent HIV with convertibleCAR‐T cells, a highly adaptable killing platform. Cell. 2019;179(4):880‐894. 10.1016/j.cell.2019.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Landgraf KE, Williams SR, Steiger D, et al. convertibleCARs: a chimeric antigen receptor system for flexible control of activity and antigen targeting. Commun Biol. 2020;3(1):296. 10.1038/s42003-020-1021-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Minutolo NG, Sharma P, Poussin M, et al. Quantitative control of gene‐engineered T‐cell activity through the covalent attachment of targeting ligands to a universal immune receptor. J Am Chem Soc. 2020;142(14):6554‐6568. 10.1021/jacs.9b11622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Liu X, Wen J, Yi H, et al. Split chimeric antigen receptor‐modified T cells targeting glypican‐3 suppress hepatocellular carcinoma growth with reduced cytokine release. Ther Adv Med Oncol. 2020;12:1758835920910347. 10.1177/1758835920910347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Lohmueller J, Butchy AA, Tivon Y, et al. Post‐translational covalent assembly of CAR and synNotch receptors for programmable antigen targeting. bioRxiv. 2020. 10.1101/2020.01.17.909895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Lajoie MJ, Boyken SE, Salter AI, et al. Designed protein logic to target cells with precise combinations of surface antigens. Science. 2020;369(6511):1637‐1643. 10.1126/science.aba6527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Plückthun A. Designed ankyrin repeat proteins (DARPins): binding proteins for research, diagnostics, and therapy. Annu Rev Pharmacol Toxicol. 2015;55:489‐511. 10.1146/annurev-pharmtox-010611-134654 [DOI] [PubMed] [Google Scholar]
- 189. Siegler E, Li S, Kim YJ, Wang P. Designed ankyrin repeat proteins as Her2 targeting domains in chimeric antigen receptor‐engineered T cells. Hum Gene Ther. 2017;28(9):726‐736. 10.1089/hum.2017.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Balakrishnan A, Rajan A, Salter AI, et al. Multispecific targeting with synthetic ankyrin repeat motif chimeric antigen receptors. Clin Cancer Res. 2019;25(24):7506‐7516. 10.1158/1078-0432.CCR-19-1479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Han X, Cinay GE, Zhao Y, Guo Y, Zhang X, Wang P. Adnectin‐based design of chimeric antigen receptor for T cell engineering. Mol Ther. 2017;25(11):2466‐2476. 10.1016/j.ymthe.2017.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Faitschuk E, Nagy V, Hombach AA, Abken H. A dual chain chimeric antigen receptor (CAR) in the native antibody format for targeting immune cells towards cancer cells without the need of an scFv. Gene Ther. 2016;23(10):718‐726. 10.1038/gt.2016.48 [DOI] [PubMed] [Google Scholar]
- 193. Baeuerle PA, Ding J, Patel E, et al. Synthetic TRuC receptors engaging the complete T cell receptor for potent anti‐tumor response. Nat Commun. 2019;10:2041‐1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Johnson LA, June CH. Driving gene‐engineered T cell immunotherapy of cancer. Cell Res. 2017;27(1):38‐58. 10.1038/cr.2016.154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Watanabe K, Kuramitsu S, Posey AD, June CH. Expanding the therapeutic window for CAR T cell therapy in solid tumors: the knowns and unknowns of CAR T cell biology. Front Immunol. 2018;9:2486. 10.3389/fimmu.2018.02486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Caruso HG, Hurton LV, Najjar A, et al. Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Res. 2015;75(17):3505‐3518. 10.1158/0008-5472.CAN-15-0139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Stoiber S, Cadilha BL, Benmebarek MR, Lesch S, Endres S, Kobold S. Limitations in the design of chimeric antigen receptors for cancer therapy. Cells. 2019;8(5). 10.3390/cells8050472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Lynn RC, Feng Y, Schutsky K, et al. High‐affinity FRβ‐specific CAR T cells eradicate AML and normal myeloid lineage without HSC toxicity. Leukemia. 2016;30(6):1355‐1364. 10.1038/leu.2016.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Hudecek M, Lupo‐Stanghellini MT, Kosasih PL, et al. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1‐specific chimeric antigen receptor T cells. Clin Cancer Res. 2013;19(12):3153‐3164. 10.1158/1078-0432.CCR-13-0330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol. 2019;17:147‐167. 10.1038/s41571-019-0297-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Chmielewski M, Hombach A, Heuser C, Adams GP, Abken H. T cell activation by antibody‐like immunoreceptors: increase in affinity of the single‐chain fragment domain above threshold does not increase T cell activation against antigen‐positive target cells but decreases selectivity. J Immunol. 2004;173(12):7647‐7653. 10.4049/jimmunol.173.12.7647 [DOI] [PubMed] [Google Scholar]
- 202. Richman SA, Nunez‐Cruz S, Moghimi B, et al. High‐affinity GD2‐specific CAR T cells induce fatal encephalitis in a preclinical neuroblastoma model. Cancer Immunol Res. 2018;6(1):36‐46. 10.1158/2326-6066.CIR-17-0211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Arcangeli S, Rotiroti MC, Bardelli M, et al. Balance of anti‐CD123 chimeric antigen receptor binding affinity and density for the targeting of acute myeloid leukemia. Mol Ther. 2017;25(8):1933‐1945. 10.1016/j.ymthe.2017.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Liu X, Jiang S, Fang C, et al. Affinity‐tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res. 2015;75(17):3596‐3607. 10.1158/0008-5472.CAN-15-0159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Drent E, Themeli M, Poels R, et al. A rational strategy for reducing on‐target off‐tumor effects of CD38‐chimeric antigen receptors by affinity optimization. Mol Ther. 2017;25(8):1946‐1958. 10.1016/j.ymthe.2017.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Ghorashian S, Kramer AM, Onuoha S, et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low‐affinity CD19 CAR. Nat Med. 2019;25(9):1408‐1414. 10.1038/s41591-019-0549-5 [DOI] [PubMed] [Google Scholar]
- 207. Guedan S, Posey AD Jr, Shaw C, et al. Enhancing CAR T cell persistence through ICOS and 4‐1BB costimulation. JCI Insight. 2018;3(1). 10.1172/jci.insight.96976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Frigault MJ, Lee J, Basil MC, et al. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol Res. 2015;3(4):356‐367. 10.1158/2326-6066.CIR-14-0186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Gomes‐Silva D, Mukherjee M, Srinivasan M, et al. Tonic 4‐1BB costimulation in chimeric antigen receptors impedes T cell survival and is vector‐dependent. Cell Rep. 2017;21(1):17‐26. 10.1016/j.celrep.2017.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Walker AJ, Majzner RG, Zhang L, et al. Tumor antigen and receptor densities regulate efficacy of a chimeric antigen receptor targeting anaplastic lymphoma kinase. Mol Ther. 2017;25(9):2189‐2201. 10.1016/j.ymthe.2017.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Chang ZL, Silver PA, Chen YY. Identification and selective expansion of functionally superior T cells expressing chimeric antigen receptors. J Transl Med. 2015;13:161. 10.1186/s12967-015-0519-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Eyquem J, Mansilla‐Soto J, Giavridis T, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543(7643):113‐117. 10.1038/nature21405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Kailayangiri S, Altvater B, Wiebel M, Jamitzky S, Rossig C. Overcoming heterogeneity of antigen expression for effective CAR T cell targeting of cancers. Cancers. 2020;12(5). 10.3390/cancers12051075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Benmebarek MR, Karches CH, Cadilha BL, Lesch S, Endres S, Kobold S. Killing mechanisms of chimeric antigen receptor (CAR) T cells. Int J Mol Sci. 2019;20(6). 10.3390/ijms20061283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Hudecek M, Sommermeyer D, Kosasih PL, et al. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res. 2015;3(2):125‐135. 10.1158/2326-6066.CIR-14-0127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Künkele A, Johnson AJ, Rolczynski LS, et al. Functional tuning of CARs reveals signaling threshold above which CD8+ CTL antitumor potency is attenuated due to cell Fas‐FasL‐dependent AICD. Cancer Immunol Res. 2015;3(4):368‐379. 10.1158/2326-6066.CIR-14-0200 [DOI] [PubMed] [Google Scholar]
- 217. Hombach AA, Schildgen V, Heuser C, Finnern R, Gilham DE, Abken H. T cell activation by antibody‐like immunoreceptors: the position of the binding epitope within the target molecule determines the efficiency of activation of redirected T cells. J Immunol. 2007;178(7):4650‐4657. 10.4049/jimmunol.178.7.4650 [DOI] [PubMed] [Google Scholar]
- 218. James SE, Greenberg PD, Jensen MC, et al. Antigen sensitivity of CD22‐specific chimeric TCR is modulated by target epitope distance from the cell membrane. J Immunol. 2008;180(10):7028‐7038. 10.4049/jimmunol.180.10.7028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Haso W, Lee DW, Shah NN, et al. Anti‐CD22‐chimeric antigen receptors targeting B‐cell precursor acute lymphoblastic leukemia. Blood. 2013;121(7):1165‐1174. 10.1182/blood-2012-06-438002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Li D, Li N, Zhang YF, et al. Persistent polyfunctional chimeric antigen receptor T cells that target glypican 3 eliminate orthotopic hepatocellular carcinomas in mice. Gastroenterology. 2020;158(8):2250-2265. 10.1053/j.gastro.2020.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Guest RD, Hawkins RE, Kirillova N, et al. The role of extracellular spacer regions in the optimal design of chimeric immune receptors: evaluation of four different scFvs and antigens. J Immunother. 2005;28(3):203‐211. 10.1097/01.cji.0000161397.96582.59 [DOI] [PubMed] [Google Scholar]
- 222. Jonnalagadda M, Mardiros A, Urak R, et al. Chimeric antigen receptors with mutated IgG4 Fc spacer avoid Fc receptor binding and improve T cell persistence and antitumor efficacy. Mol Ther. 2015;23(4):757‐768. 10.1038/mt.2014.208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Krenciute G, Krebs S, Torres D, et al. Characterization and functional analysis of scFv‐based chimeric antigen receptors to redirect T cells to IL13Rα2‐positive glioma. Mol Ther. 2016;24(2):354‐363. 10.1038/mt.2015.199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Wilkie S, Picco G, Foster J, et al. Retargeting of human T cells to tumor‐associated MUC1: the evolution of a chimeric antigen receptor. J Immunol. 2008;180(7):4901‐4909. 10.4049/jimmunol.180.7.4901 [DOI] [PubMed] [Google Scholar]
- 225. Hombach A, Hombach AA, Abken H. Adoptive immunotherapy with genetically engineered T cells: modification of the IgG1 Fc 'spacer' domain in the extracellular moiety of chimeric antigen receptors avoids 'off‐target' activation and unintended initiation of an innate immune response. Gene Ther. 2010;17(10):1206‐1213. 10.1038/gt.2010.91 [DOI] [PubMed] [Google Scholar]
- 226. Almåsbak H, Walseng E, Kristian A, et al. Inclusion of an IgG1‐Fc spacer abrogates efficacy of CD19 CAR T cells in a xenograft mouse model. Gene Ther. 2015;22(5):391‐403. 10.1038/gt.2015.4 [DOI] [PubMed] [Google Scholar]
- 227. Watanabe N, Bajgain P, Sukumaran S, et al. Fine‐tuning the CAR spacer improves T‐cell potency. Oncoimmunology. 2016;5(12):e1253656. 10.1080/2162402X.2016.1253656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Alabanza L, Pegues M, Geldres C, et al. Function of novel anti‐CD19 chimeric antigen receptors with human variable regions is affected by hinge and transmembrane domains. Mol Ther. 2017;25(11):2452‐2465. 10.1016/j.ymthe.2017.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Perez‐Amill L, Suñe G, Antoñana‐Vildosola A, et al. Preclinical development of a humanized chimeric antigen receptor against B cell maturation antigen for multiple myeloma. Haematologica. 2020;106:173‐184. 10.3324/haematol.2019.228577 [DOI] [PMC free article] [PubMed] [Google Scholar]