

Abstract
Purpose:
Molecular profiling has been used to select patients for targeted therapy and determine prognosis. Noninvasive strategies are critical to hepatocellular carcinoma (HCC) given the challenge of obtaining liver tissue biopsies.
Experimental Design:
We analyzed blood samples from 206 HCC patients using comprehensive genomic testing (Guardant Health, CA) of circulating tumor DNA (ctDNA).
Results:
153/206 (74.3%) were men; median age, 62 years (range, 18–91 years). 181/206 patients had ≥1 alteration. The total number of alterations was 680 (non-unique); median number of alterations/patient was 3 (range, 1–13); median mutant allele frequency (% cfDNA), 0.49% (range 0.06 – 55.03%). TP53 was the common altered gene (>120 alterations (non-unique)) followed by EGFR, MET, ARID1A, MYC, NF1, BRAF, and ERBB2 (20–38 alterations (non-unique)/gene). Of the patients with alterations, 56.9% (103/181) had ≥1 actionable alterations, most commonly in MYC, EGFR, ERBB2, BRAF, CCNE1, MET, PIK3CA, ARID1A, CDK6, and KRAS. In these genes, amplifications occurred more frequently than mutations. Hepatitis B (HBV)-positive patients, were more likely to have ERBB2 alterations, 35.7% (5/14) versus 8.8% HBV- negative (p=0.04).
Conclusions:
This study represents the first large-scale analysis of blood-derived ctDNA in HCC in USA. The genomic distinction based on HCC risk factors and the high percentage of potentially actionable genomic alterations suggests potential clinical utility for this technology.
Keywords: cell-free DNA, hepatocellular carcinoma, molecular profiling
INTRODUCTION
Recent technological advances in molecular diagnostics have allowed for the study of solid malignancies through non-invasive blood sampling. Importantly, intact circulating tumor cells (CTCs) and cell-free DNA (cfDNA) [leukocyte-derived and tumor-derived (circulating tumor DNA; ctDNA)] can now be interrogated through advanced sequencing methods in order to identify somatic mutations that may be druggable targets for future therapies1, 2. Although cfDNA and ctDNA are similar in that they both derive from cell lysis and apoptosis3, ctDNA is the fraction of cfDNA, which can range from <0.1% to >10%4, specifically derived from primary or metastatic tumors5. Therefore, profiling the mutational landscape of ctDNA from solid tumors may represent a particularly attractive method for identifying tumor-associated somatic mutations. Applications that can be envisioned to be of clinical utility for hepatocellular carcinoma (HCC) include detection of genomic changes, mutational analysis, prognostication, oncogenic pathway determination, prediction/monitoring of treatment response, drug resistance alterations, and identification of mechanisms of malignant/metastatic transformation based on underlying risk factor and genetic predisposition.
CtDNA has been found to be inversely correlated with HCC prognosis6 and shorter overall survival7. Although several studies have investigated the global alterations in cfDNA of HCC8–13, the small number of patients and heterogeneity of demographics and geographic locations posed a challenge to the application of this methodology in the US12, 13.
Therefore, the purpose of the current study was to examine a large multi-institutional cohort of HCC tumors with a CLIA-certified cfDNA technology that is currently used in practice and to identify the mutational landscape that may be contributing to HCC tumorigenesis for the purpose of selecting patients for targeted therapy trials. This investigation represents the largest reported series of HCC patients analyzed to date for the genomic portraits derived from blood-derived cfDNA.
METHODS
Patients:
During the period extending from November 2014 to July 2016, 219 patients with advanced HCC underwent blood-derived cfDNA testing by Guardant Health (Redwood City, CA). The diagnosis of HCC was established either by histopathological examination or by characteristics imaging findings, following clinical guidelines by the American Association for the Study of Liver Disease (AASLD)14. The majority of the patients were from the following 4 institutions: MD Anderson Cancer Center (MDACC; 37 patients), University of California San Diego, Moores Cancer Center (UCSD; 36 patients), Washington University School of Medicine-Siteman Cancer Center (18 patients), and University of California San Francisco-Helen Diller Family Comprehensive Cancer Center (UCSF) (11 patients). The remaining patients were from other US institutions. From the 219 patients, 12 had serial testing. For patients who had more than one test, only the first test result was considered. The analysis and consent of patients in this cohort followed guidelines of various institutional review boards and clinical protocols, as follows: MDACC: NCT01772771- Molecular Testing for the MDACC Personalized Cancer Therapy Program; UCSD: NCT02478931 (PREDICT)-Study of Personalized Cancer Therapy to Determine Response and Toxicity; Washington University School of Medicine-Siteman Cancer Center: WU IRB protocol 201606097, A retrospective analysis of circulating cell free DNA in patients with gastrointestinal malignancies; UCSF, UCSF IRB# 12–09576 Hepatobiliary Tissue Bank and Registry. The latter two institutional IRB approvals were for non-therapeutic/non-interventional use and hence are not registered in clinicaltrials.gov. The study was conducted in concordance with the Declaration of Helsinki.
Demographic information (i.e., gender, age) and date of blood collection were available for all patients. Additional patient information was requested from the institutions that contributed more than 10 patients for cfDNA testing (UCSD, MDACC, Siteman Cancer Center, and UCSF). The information requested was as follows: (i) confirmation of HCC diagnosis; (ii) risk factor information, such as hepatitis A, B, and C status, non-alcoholic steatohepatitis (NASH), alcoholic liver disease, and metabolic syndrome; (iii) relevant clinicopathologic information including Child-Pugh score, Cancer of the Liver Italian Program (CLIP), Barcelona Clinic Liver Cancer (BCLC), Tumor-Node-Metastasis (TNM) Staging, and treatment status at time of ctDNA testing. Metabolic syndrome as established as a clinical diagnosis as per guidelines by the 2001 National Cholesterol Education Program ATP III15. The diagnosis of NASH was established by pathologic diagnosis of NASH in patients with available clinical data.
Comprehensive genomic testing in plasma:
cfDNA was extracted from whole blood collected in 10 mL Streck tubes. Samples were shipped to a Clinical Laboratory Improvement Act (CLIA)-certified, College of American Pathologists-accredited laboratory (Guardant Health, Redwood City, California). After double ultracentrifugation, 5 ng – 30 ng of cfDNA was isolated for digital sequencing as previously described12, 13, 16.
Cell-free DNA fragments, both leukocyte- and tumor-derived, were simultaneously sequenced. The variant allele fraction (VAF) was calculated as the proportion of cfDNA harboring the variant in a background of wild-type cell-free DNA. The analytical sensitivity allowed detection of 1–2 mutant fragments in a 10 ml blood sample (0.1% limit of detection) with analytic specificity > 99.9999%. Twelve CNAs were reported as the absolute gene copy number in plasma. Since most cfDNA is leukocyte-derived, the gene copy number is generally 2.0. Tumor-derived DNA shed into the bloodstream raises this value but, due to the relative proportions of tumor-derived versus leukocyte-derived cfDNA, it is typically a minor contributor. Gene copy number in plasma is thus a function of both copy number in tissues and the degree to which tumor DNA is shed into circulation. Plasma copy number of 2.5–4.0 is reported as ++ amplification and >4.0 as +++, representing the 50th-90th and >90th percentile, respectively of all CNA calls in the Guardant360 database12, 13, 16.
Over the course of the study, the panel composition expanded from 54 to 68 to 70 genes. The initial 54-gene panel consisted of complete exon coverage or critical exon coverage in 54-cancer related genes, and amplifications in 3 genes. In the 68-gene panel, 8 genes were retired from the single nucleotide variation (SNV) gene set, while coverage of gene amplifications expanded from 3 to 16 genes, and detection of fusions in 4 genes, and insertions or deletion of bases (indels) in 1 gene. The 70-gene panel includes all NCCN somatic genomic targets, including complete or critical exon coverage in 30 and 40 genes, respectively; amplifications in 18 genes, fusions in 6 genes, and indels in 3 genes [Supplementary Table 1]. Seven patients were tested on the 54-gene panel was used, eighty-six on the 68-gene version of the original, and one-hundred-forty-two on the 70-gene panel version [Supplementary Table 2].
Actionable genes and variants defined
To determine whether an alteration was actionable or not, we referenced the actionable gene list set in place by the Institute for Personalized Cancer Therapy-Precision Oncology Decision Support (PODS) team at MD Anderson Cancer Center17. By these standards, a gene is considered actionable if there is supporting evidence that such gene is a driver of tumorigenesis, wherein actionability of the gene can refer to either sensitivity and/or resistance to a drug(s), and sensitivity and/or resistance can be inclusive of all alterations types or specific to alteration classes, i.e., applicable to amplifications, but not mutations. Furthermore, there must be a clinically available agent targeting such gene, and for a specific agent, there must be at least pre-clinical evidence that supports its role in targeting the specific gene. While the PODS team aims at maintaining the actionable gene list as comprehensive and up to date as possible, we relied heavily on the availability of a drug in the context of clinical trial at MDACC. However, we acknowledge that drug availability and trials varies greatly by institution. Finally, the actionability of a gene can be in the context of tumor type(s). For example, in colorectal cancer, the use of the EGFR monoclonal antibodies cetuximab and panitumumab is contraindicated in tumors with activating KRAS mutations, specifically those in codons G12 and G13 (FDA label)18–20. Hence, KRAS is deemed actionable for treatment with MEK inhibitors in all tumor types. However, in the case of colorectal cancer, it is also deemed actionable for resistance to cetuximab and panitumumab21. At the moment, there are no HCC-specific actionable gene.
Based on the aforementioned criteria, the following genes were considered actionable based on their sensitivity towards respective targeted agents: AKT1, ALK, ARAF, ARID1A, ATM, BRAF, BRCA1, BRCA2, CCND1, CCND2, CDK4, CDK6, CDKN2A, CDKN2B, EGFR, ERBB2, FGFR1, FGFR2, FGFR3, HRAS, IDH1, IDH2, JAK2, JAK3, KIT, MAP2K1, MAP2K2, MET, MPL, MYC, NF1, NOTCH1, NRAS, NTRK1, PDGFRA, PIK3CA, PTEN, PTPN11, RET, ROS1, SMO, STK11, and TSC1.
The following genes were deemed actionable based on context-specific criteria: CCNE1 (sensitivity to CDK2 inhibitors; resistance to CDK4/6 inhibitors); ESR1 (presence is sensitizing to hormone therapy, mutations cause resistance to anti-hormone therapy); KRAS (sensitivity to MEK inhibitors; resistance to cetuximab and panitumumab in colorectal and erlotinib, and gefitinib in NSCLC; NRAS (sensitivity to MEK inhibitors, resistance to cetuximab and panitumumab); RAF1 (activating alterations cause sensitivity to MEK inhibitors and resistance to RAF inhibitors; inactivating alterations cause resistance to dasatinib). Only AR and RB1 were deemed actionable solely because of their resistance to anti-hormone therapy and to CDK4/6 inhibitors, respectively.
At the time of analysis, several genes included in the Guardant panel were denoted as non-actionable including: TP53, CTNNB1, APC, GNAS, NFE2L2, MLH1, RIT1, SMAD4, HNF1A, CDH1, GATA3, VHL, FBXW7, and RhoA. To note, some investigators, including co-authors on this manuscript, have considered TP53 actionable for Wee-1 or VEGF/VEGFR inhibitors 22–24. Since this remains a matter of debate, TP53 was not considered actionable in this paper. Likewise, since the initial analysis, MLH1 has also become actionable based on FDA-approval of nivolumab and pembrolizumab in 2017. Finally, variants found in actionable genes but which lacked any supporting, functional evidence were designated as “variants of unknown functional significance.” A comprehensive list of all genes considered and their actionability is included in Supplementary Table S1b.
Statistical analysis
The distribution of each continuous variable was summarized by its mean, standard deviation, and range. The distribution of each categorical variable was summarized in terms of its frequencies and percentages. Continuous variables were compared between groups by Wilcoxon rank sum test, and for categorical data the comparison was conducted by Fisher Exact test. Heat map with dendogram was generated to explore and visualize the gene mutation/amplification relationship along with the corresponding various risk factors. The relationship between gene types with regard to mutation and amplification as well as synonymous and targetable status was evaluated with Spearman’s rank correlation and displayed in the correlation matrix in which the non-significant correlations are marked with “blank” in the graph [Figure 6]. All computations were carried out in SAS version 9.3 and R version 3.13.
Figure 6:

Correlation between molecular landscape and metabolic syndrome and/or history of hepatitis.
a) Correlation with metabolic syndrome: A significant number of patients with metabolic syndrome, 30% (3/10) had alterations detected in PDGFRA (p=0.0113), while patients without metabolic syndrome (n=30) had no alterations in this gene.
b) Correlation with hepatitis B status: A significant number of patients with hepatitis B 35.7% (5/9) had alterations in ERBB2 (p=0.0365), while HBV-negative patients had none.
c) Correlation with hepatitis C status: 16.7% (3/18) of patients with hepatitis C (HCV) had alterations in BRCA1 (p=0.0472), while HCV-negative patients had none.
d) Correlation with Child-Pugh score: 23.1% (3/13) of patients with Child-Pugh score B had alterations in PIK3CA (p=0.0341), versus 0% (0/27) Score A or C (0/1) (p=0.03).
RESULTS
Patient Characteristics:
A cohort of 219 patients was enrolled in the study. The median age was 62.3 years (range: 18–91; 160 males and 59 females). Irrespective of gene actionability, the median number of alterations per patient was 3 (range: 1–13); the median allele frequency (AF) of altered cfDNA (%cfDNA) was 0.49% (range: 0.06–55.03).
After further evaluation, it was determined that 13 of these patients had a diagnosis other than HCC, namely, fibrolamellar HCC, cholangiocarcinoma or mixed HCC/cholangiocarcinoma. This determination was based on Liver Imaging Reporting and Data System (LI-RADS) and tumor markers for HCC, while fibrolamellar and mixed HCC/cholangiocarcinoma were mostly confirmed by biopsy. These patients were excluded from further analysis. In addition, for the patients who had more than one cfDNA test ordered (n=12 patients), only the first order was considered for further analysis. After exclusion of these unique cases, as well as the non-HCC cases, the initial data set was reduced from 219 to 206 patients, and from 777 to 680 alterations [Figure 1a–d].
Figure 1:
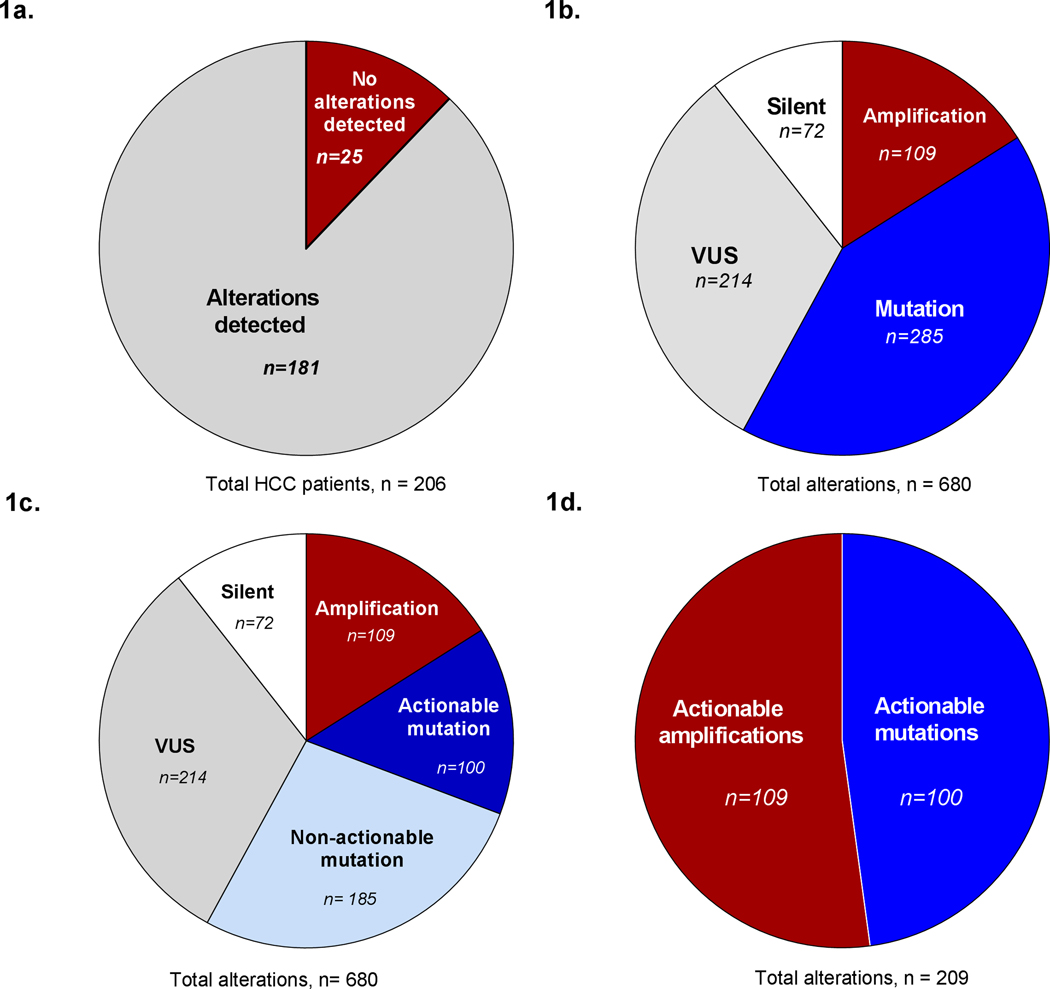
Overview of Findings
a) Number of HCC confirmed cases with alterations detected through cfDNA.
b) Total number of alterations identified among the 181 patients with alterations, and the proportion of alteration types identified.
c) Action ability among mutations identified.
d) Total number of actionable alterations, after removing silent, VUS, and non-actionable mutations.
Overall distribution of alteration types
The majority of patients (87.8%=181/206) had at least one alteration; only 12.2% (25/206) had no alterations [Figure 1a]. A total of 680 alterations (non-unique) were detected among the former patient population, of which 10.5% (72/680) were synonymous/silent in nature, and 16% were amplifications (109/680). The remaining 73.3% of alterations (499/680) consisted of indels, missense, nonsense, and splice variants. A fraction of these alterations (214/499), were classified as “variants of unknown functional significance” (VUS) due to lack of supporting evidence for their functionality. The remaining variants (285/499) were broadly categorized as “mutations”, and made up 42% (285/680) of the total alterations identified [Figure 1b].
Further analysis of the 42% of alterations that made up the “mutation” category revealed that 53% of these alterations occurred within actionable genes (actionable mutations) while the remaining 47% occurred within genes not considered actionable (non-actionable mutations) at the time of analysis [Figure 1c]. On the other hand, all of the reported amplifications occurred within actionable genes. After accounting for synonymous alterations, variants of unknown significance (VUS), and mutation in non-actionable genes, the total number of actionable alterations was 209 out of the initial 680 (non-unique) alterations identified [Figure 1d]. ). Of the patients with alterations, 56.9% (103/181) had ≥1 actionable alterations.
Most frequently mutated genes identified
We assessed the overall molecular landscape of the 181 HCC patients with detected alterations, both in terms of number of alterations reported per gene and number of patients per gene. Additionally, to assess the true utility of cfDNA findings, in terms of their potential for guiding clinical decisions, genes were parsed into “actionable” and “non-actionable genes”, as defined above (see Methods section for details). Inclusive of all alteration types (i.e., synonymous, VUS), the most frequently altered genes (≥20 events/gene) were as follows: within actionable genes--EGFR, MET, ARID1A, MYC, NF1, BRAF, and ERBB2; among non-actionable genes--TP53, followed by CTNNB1 and APC [Figure 2a]. Overall, TP53 was the most commonly altered gene, with >120 alterations. By comparison, EGFR had 38 alterations. Of note, the gene panel evolved over the course of this study, hence the number of patients evaluated for each of the genes varied depending on the panel available at that time. Moreover, detection of copy number variations was limited to amplifications in a subset of genes (see Material and Methods and Supplementary Table S1a and S2 for details).
Figure 2:
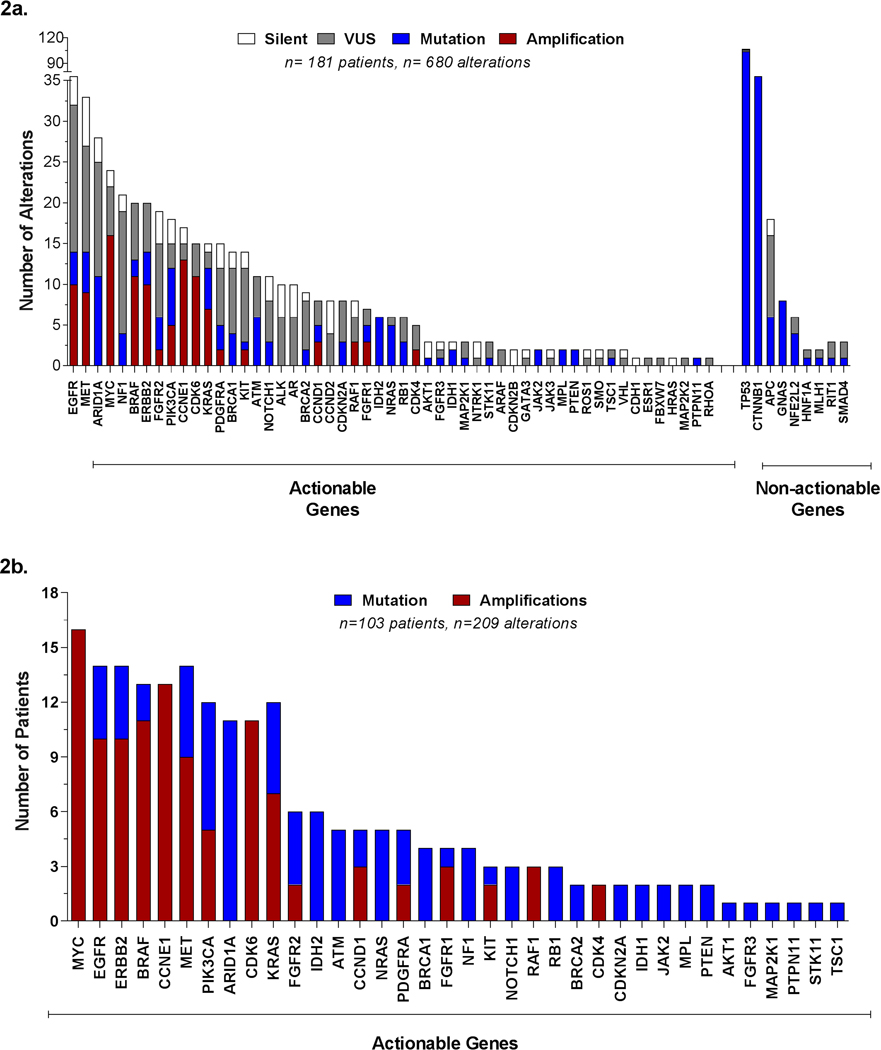
Most frequently mutated genes identified in cfDNA among HCC patients.
a) Distribution of all alterations identified, actionable and non-actionable genes.
b) Proportion of patients with alterations in actionable genes only.
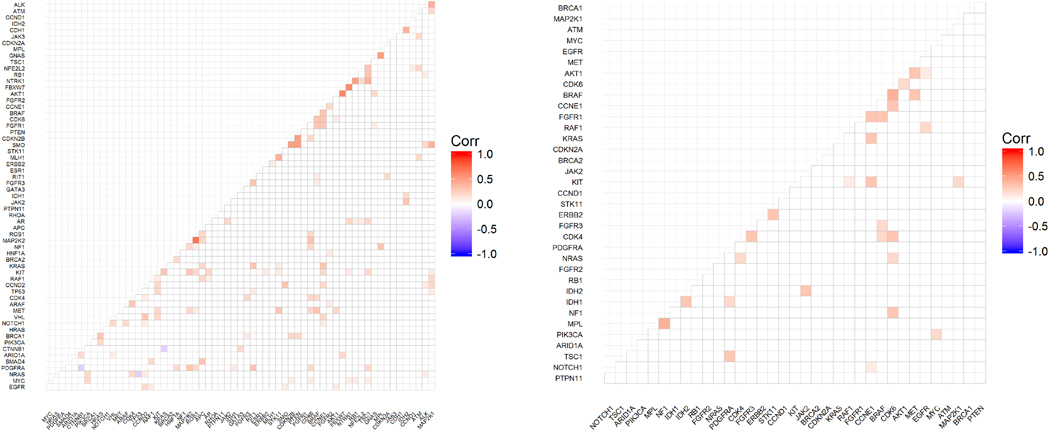
c) Gene correlation matrix among all patients with alterations detected. All patients with alterations detected, irrespective of the actionability of the alteration (i.e. variant in non-actionable genes, VUS, and/or synonymous) were considered for this matrix. In all from 181 patients and 62 genes were evaluated with Spearman’s rank correlation and displayed in the correlation matrix above. Non-significant correlations are marked with “blank” in the graph. Values < 0 indicate no agreement, 0 – 0.20 slight agreement, 0.21 – 0.40 fair agreement, 0.41 – 0.60 as moderate agreement, 0.61 – 0.80 as substantial agreement, and 0.81 – 1 as almost perfect agreement.
d) Gene correlation matrix among HCC patients with alterations in actionable genes (synonymous and VUS alteration excluded). Exclusion of non-actionable genes, VUS, and synonymous alterations reduced the patient population size considered for this matrix from 181 to 103 patients and from 62 genes to only 36 actionable genes. The correlation between mutated genes was evaluated with Spearman’s rank correlation and displayed in the correlation matrix above. Non-significant correlations are marked with “blank” in the graph. Values < 0 indicate no agreement, 0 – 0.20 slight agreement, 0.21 – 0.40 fair agreement, 0.41 – 0.60 as moderate agreement, 0.61 – 0.80 as substantial agreement, and 0.81 – 1 as almost perfect agreement.
Exclusion of non-actionable alterations, i.e., VUS and synonymous alterations, further reduced the total number of alterations from 680 alterations to 209 alterations. Moreover, the patient population was also reduced from 181 to 103 patients, as many patients only had alterations detected within non-actionable genes, or all alterations were synonymous or VUS. Among the 103 patients with actionable alterations, the most commonly altered genes (≥10 patients/gene) were MYC, EGFR, ERBB2, BRAF, CCNE1, MET, PIK3CA, ARID1A, CDK6, and KRAS. Likewise, amplifications were more common than mutations among the top genes [Figure 2b].
Gene correlation matrices to visualize co-expression and mutual exclusivity of genes with alterations detected
Two correlation matrices were generated, independent of the availability of risk factor information: the first matrix considered all confirmed HCC patients with alterations detected, and included all alterations types (synonymous, VUS, mutations, amplifications) in both actionable and non-actionable genes. This amounted to 181 patients and 62 genes analyzed [Figure 2c]. The premise of this matrix was to allow an overall appreciation of all alterations identified. For more hypothesis-generating analysis, the second matrix, which focused only on mutations and amplifications within actionable genes (see S1b), and excluded synonymous and VUS alterations [Figure 2d] was generated. Application of these exclusion criteria reduced the initial correlation matrix from 181 to 103 patients, and from 62 to 36 actionable genes. Concordance estimates and 95% intervals (Cohen’s kappa) for each pair of genes were generated to better understand any association in terms of co-expression or mutual exclusivity of genes analyzed. In this analysis, any value <0 indicated no agreement, 0 – 0.20 slight agreement, 0.21–0.40 as fair agreement, 0.41–0.60 as moderate agreement, 0.61–0.80 as substantial agreement, and 0.81–1 as almost perfect agreement [Figure 2c–d]. Firm conclusions were not possible with these matrices, given the small sample size, but hypothesis-generating observations could be deduced.
Identifying patients with alterations in actionable genes and available risk factor data
To understand if molecular findings correlated with any of the risk factors commonly observed among patients diagnosed with HCC, we requested additional patient information from institutions having >10 HCC participants, as detailed under Methods. These included: University of California San Diego Moores Cancer Center: 36 patients, 48 orders; MD Anderson Cancer Center: 37 patients, 37 orders; Siteman Cancer Center: 18 patients, 19 orders; and University of California San Francisco Comprehensive Cancer Center: 11 patients, 11 orders) for a total of 103 patients.
The 13 non-HCC cases previously excluded were identified from within this population. Hence, the final number of patients from participating institutions was 89 patients, of which 82 patients had alterations detected while 7 had none [Figure 3]. We again parsed this subset of alterations into actionable or non-actionable groups; the latter included synonymous alterations, VUS, and non-actionable genes. We identified a subset of patients (n=34) who only had alterations detected in non-actionable genes or the alteration type was non-actionable, in the case of synonymous or VUS. Ultimately, this reduced the patient population with alterations from 82 to 48 patients contributing, for a total of 98 alterations (non-unique) in 31 actionable genes. This small subset of patients was statistically evaluated for their correlation with risk factors contributing to HCC, and consisted of 14 women, 34 men, mostly Caucasian patients (n=30) followed by Hispanic (n = 8), Asian (n = 5), Black (n = 2), Native American (n = 1) and other (n = 2). Nineteen patients were reported to have alcoholic liver disease, 8 had confirmed non-alcoholic steatohepatitits (NASH) and 10 had metabolic syndrome. Staging information as classified by Child-Pugh Score (CPS), Barcelona Clinic Liver Cancer staging system (BCLC), CLIP, and/or TNM staging was requested. There were 27 patients with Child-Pugh Score of A: (5–6, well compensated), 13 with stage B: (7–9, significant functional compromise), and 1 with C: (10–15, decompensated). Six patients had an unknown BCLC classification, while 1 was classified as stage A, 4 as stage B and 37 as stage C. Hepatitis A was reported in 12 patients; hepatitis B in 14 patients; and hepatitis C in 17 patients. There were only a handful of cases with both hepatitis A & B (n=4), hepatitis B & C (n=6) or hepatitis A & C (n=7). [Supplementary Table 3, Supplementary Figure 1a–d].
Figure 3:
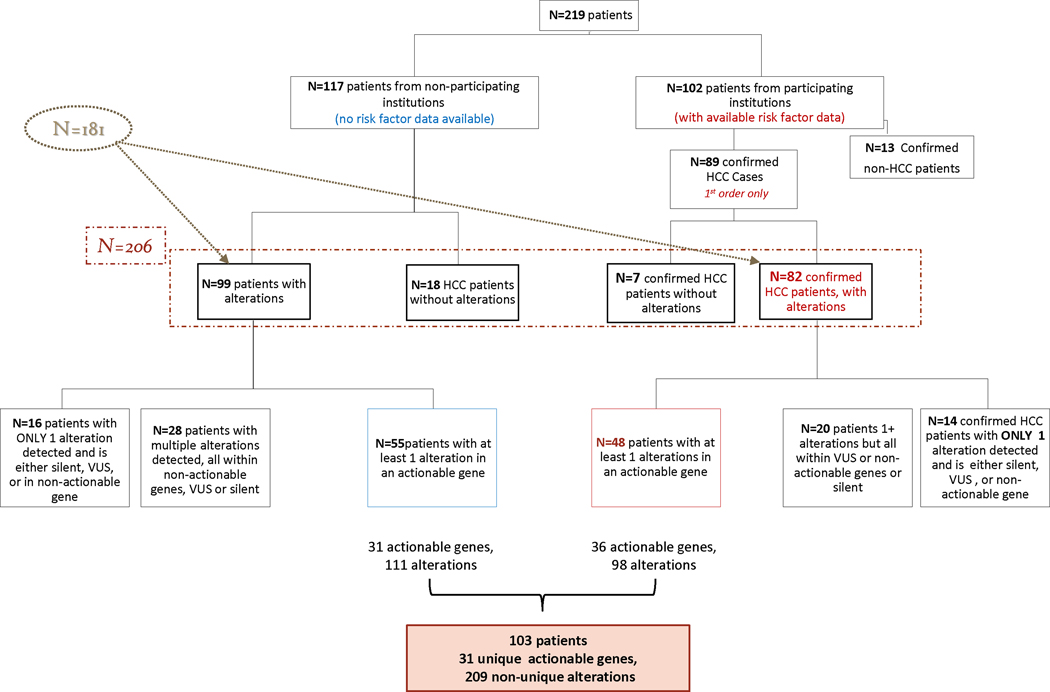
Stratification of patients for analysis over a period of approximately 2 years. 219 patients from various institutions underwent biopsy-free, next generation sequencing. We collected additional risk factor information for 102 patients, irrespective of alterations detected or not. Two correlation matrices were generated, independent of risk factor information. The first considered all patients, and all alteration types (Fig 3a, n=181 patients), and the second considered only patients with at least 1 alteration in an actionable gene and excluded VUS and synonymous alterations (Fig 3b, n=103 patients)
Although this smaller subset (only patients with available risk factor information and only actionable genes) included only 48/206 patients, it maintained the overall molecular landscape of the larger subset (see Figure 2a–b). Whereas in the overall population the top 10 most frequently altered actionable genes were MYC, EGFR, ERBB2, BRAF, CCNE1,MET, PIK3CA, ARID1A, CDK6, and KRAS, in this subset of patients (n= 48) the top 10 most frequently altered actionable genes were: EGFR, ERBB2, MET, CCNE1,MYC, BRAF, CCND1, CDK6, FGFR2, and ARID1A [Figure 4a–b]. Hence, this subgroup should be considered a suitable and representative model of the molecular profile of circulating cell-free DNA in patients with hepatocellular carcinoma.
Figure 4:

Representative molecular landscape among patients with available risk factor data.
a) Distribution of all alterations identified, actionable genes only, by alteration.
b) Distribution of all alterations identified, actionable genes only, by patient.
Correlation of risk factors with alterations identified
Significant differences were noted in the molecular profile of patients based on their underlying hepatic reserve and risk factors for HCC. Among patients with metabolic syndrome, 30% (3/10) had alterations detected in PDGFRA (p=0.0113), while patients without metabolic syndrome (n=30) had no alterations in this gene. In terms of hepatitis status, 35.7% (5/9) of patients with hepatitis B had alterations in ERBB2 (p=0.0365), and 16.7% (3/18) of patients with hepatitis C had alterations in BRCA1 (p=0.0472). Hepatitis B and Hepatitis C negative patients, did not have any alterations in these respective genes. In addition, 23.1% (3/13) of patients with Child-Pugh score B had alterations in PIK3CA (p=0.0341) versus 0% (0/27) Score A (0/27) or C (0/1) (p=0.03). Heat map with dendogram was generated to better visualize and study the gene mutation/amplification relationships along with the corresponding various risk factors. The relationships between gene types, with regard to mutation and amplification as well as synonymous and targetable status, was evaluated with Spearman’s rank correlation and displayed in the correlation matrix in which the non-significant correlations are marked with “blank” in the graph. All computations were carried out in SAS version 9.3 and R version 3.13. [Figure 5–6; Supplementary figure 2a–c].
Figure 5:

Correlation between molecular landscape and risk factors contributing to HCC. Heat map dendrogram was generated to explore and visualize the gene mutation/amplification relationship along with the corresponding various risk factors. The relationship between gene types with regard to mutation and amplification, synonymous and targetable status was evaluated with Spearman’s rank correlation and displayed in the correlation matrix in which the non-significant correlations are marked with “blank” in the graph.
DISCUSSION
In the vast majority of patients, HCC comprises a two-disease state; HCC tumors and concomitant liver disease. Therefore, HCC staging/prognostication systems take into account tumor parameters in addition to hepatic reserve grade, manifested by Child-Pugh classification. While mounting evidence in the literature suggests different molecular pathogenesis pathways based on the degree of liver fibrosis and underlying HCC risk factors, this has not been translated into defining molecular subclasses in HCC. The challenge to defining molecular nosology is largely due to lack of access to tissue sampling in HCC, given the risk of bleeding due to concomitant coagulopathy and thrombocytopenia in patients with advanced fibrosis and portal hypertension. This bleeding risk led to the development and wide acceptance of guidelines to diagnose HCC without tissue sampling25. Therefore, despite recent advances in our understanding of HCC molecular profiling based on tissue sampling26, firm conclusions and biomarker integration into HCC staging were not achieved due to major challenges in prospective validation studies that are powered to study risk factor-specific HCC. Liquid biopsies could bridge this gap, given the practicality of obtaining them in this patient population, and are expected to help advance the field of molecular prognostication and patient selection in targeted therapy trials. The current study represents a step towards achieving this major goal given our important observations related to specific common risk factors in HCC. This study also represents the largest US cohort ever reported. Hence, our correlative findings of molecular alterations, as they relate to specific risk factors and demographics, highlight the importance of cfDNA methodology in HCC and warrant future validation and exploration on its utility.
Chronic infection with hepatitis B with its incorporation into the human genome is a common HCC risk factor, particularly in Asia. The pathogenesis can be due to oncogenic viral proteins or insertional mutagenesis. The mutational spectrum in HCC arising from hepatitis B infection is known to be enriched with TERT, CCNE1, and MLL4 due to insertional events, while hepatitis C infection is typically chronic and leads to chronic inflammatory cascade events in the microenvironment and mutations.
Interestingly, our study showed statistically significant associations between certain alterations and specific risk factors, such as metabolic syndrome and PDGFRA (p=0.0113), HBV and ERBB2 (p=0.0365), HCV and BRCA1 (p=0.0472), and Child-Pugh score B and PIK3CA (p=0.0341). This relationship between gene types with regard to alteration type, risk factor, and targetable status was evaluated with Spearman’s rank correlation and displayed in the correlation matrix. However, given the small number of patients in each category, we consider these findings as hypothesis-generating associations that should be studied in future validation studies However, given the small number of patients in each category, we consider these findings as hypothesis-generating associations that should be studied in future validation studies. For example, while ERBB2 dysregulation is not known to be a significant driver or hallmark of HCC, upregulation, correlation with HCC poor outcome, and cell line data suggests that HBV interacts with ERBB2 and ERBB3 resulting in protein stabilization [20,21]. Similarly, dysregulation of BRAF, BRCA1, MET, CDK6, ARID1A, CCNE1, EGFR, FGFR1, MYC, and NOTCH1 correlated with a history of HCV in our study [Supplementary Figure 4a-d]. In the case of BRCA1, while there is an exhaustive amount of literature supporting the role of BRCA1 in breast and ovarian tumor types, less is known about the role of BRCA1 in HCC, much less in regards to its role in HCV affected HCC patients. Nonetheless, one whole genome-sequencing analysis of 88-matched HCC tumor/normal tissue samples, reported that BRCA1 alterations were identified in 1.1% of HCC patients 27. By comparison, our study using cfDNA assay identified BRCA1 alterations in 4 of 212 patients (1.8%) by ctDNA. Although this is not direct evidence for BRCA1 in HCV in HCC, overall these findings are consistent with NGS results in solid tumor testing. Furthermore, increasing evidence supports the role of common molecular signatures across distinct tumor types 28–30. Hence, there is the potential that BRCA1 and/or other genes that have specifically been implicated in other tumor types, may also be relevant to HCC.
Notably, alterations in TERT promoter and in CTNNB1 gene were reported among the most frequent alterations in HCC31, and in particular viral hepatitis-related HCC32. Similarly, our data indicated the high prevalence of CTNNB1 mutations but did not confirm the hepatitis status correlation, most likely because of the relatively small sample size of patients with available risk factors for analysis 33–38.
One of the major strengths of our study is the large number of patients representing numerous US institutions driven by samples submitted prospectively to aid in therapeutic decision making using a CLIA-certified assay approved for clinical practice. Moreover, almost half of the patients had risk factors. Another major advantage to our study is the critical importance to assessing ctDNA in HCC patients, where, obtaining initial and repeat tissue sampling may be challenging as previously mentioned. However, the study has some limitations. First, firm conclusions of the correlation with risk factors were not possible given the small number of patients with available risk factors from five institutions. However, the pattern of the alterations was similar to the overall cohort. Second, the cfDNA panel is not as comprehensive as some tissue molecular profiling panels. However, the panel used is clinically driven in that it is designed to identify somatic alterations in 73 genes, primarily focused on genomic alterations that are associated with 1) FDA-approved targeted therapies, 2) targeted therapies in late stage clinical trials, 3) known predictive or prognostic value, or 3) informative of the presence of cfDNA. It is also important to note that the Guardant360 assay reports out only somatic alterations. Mutant allele fractions (MAF) are reported quantitatively for somatic single nucleotide variants (SNVs) of clinical significance and distinguished from heterozygous and homozygous germline variants by their low concentrations, which are filtered out by the Guardant360 molecular tumor board39. Another study limitation was that there was no direct comparison with tumor tissue, in large part because of the difficulty in doing biopsies in HCC; therefore, our work reflects current clinical practice, in which the imaging diagnosis of HCC in the appropriate setting without tissue diagnosis is widely adopted in both academic and community centers. However, surgical resection may be a source of tissue in patients - this procedure is more frequent in South East Asia where HBV patients with HCC may not have cirrhosis. Finally, we acknowledge the definition of actionability is dynamic and institution-dependent, and data on enrollment on targeted trials based on cfDNA results was not available. Prospective studies using ctDNA profiling to guide therapy decisions are warranted.
Notwithstanding the fact that further validation with solid tumor testing results is necessary, we conclude that cfDNA testing is a valid methodology for HCC molecular profiling given the abundance of ctDNA observed in this, the largest collection of US-based blood samples. While this direct comparison was not possible within this patient population, as few patients had solid tumor testing results available, comparison to the findings reported recently by the Cancer Genome Atlas initiative40 and other prior studies34, 41, are greatly encouraging, considering the limitations of the current sample population and the limited comprehensiveness of the cfDNA gene panel. In both the TCGA results and this study, the most commonly altered genes were TP53 and CTNNB1, while amplifications were most commonly observed in MET and CCND1. Moreover, the interesting molecular alteration findings related to specific HCC risk factors, if validated, could also pave the way for developing a molecularly driven HCC classification, and could aid in stratification for clinical trials. Indeed, genomically driven trials, both ongoing and completed, have begun to allow enrollment based on cfDNA test results. In HCC specifically, Ikeda et al recently published their findings on the clinical utility of cfDNA testing in HCC, reporting 79% of tested patients having at least one clinically actionable alteration identified via cfDNA testing, with two patients showing good clinical response to matched targeted therapies42. Nonetheless, future validation studies with larger numbers of patients with available patient characteristic data are essential for directing HCC translational efforts. Such data is expected to carry critical importance in using mutational signatures for prognostication and for predicting response to targeted therapies. In addition, blood-derived ctDNA analysis is amenable to serial assessment while on therapy in order to investigate acquired mutations.
Supplementary Material
STATEMENT OF RELEVANCE:
Molecular subgrouping of hepatocellular carcinoma (HCC) is challenging due to lack of tissue sampling, and profiling the HCC mutational landscape by cfDNA may be particularly attractive given the challenge of obtaining liver biopsies. We report the largest USA cohort, using a CLIA-certified assay approved for clinical practice. Our results demonstrate significant trends between alterations and risk factors - metabolic syndrome and PDGFRA, HBV and ERBB2, HCV and BRCA1. In addition, we observed a high percentage of potentially actionable genomic alterations, suggesting clinical utility for this technology in HCC.
Acknowledgements:
Bili Project Foundation, Inc -UCSF Hepatobiliary Tissue Bank and Registry. This work was supported in part by the National Institutes of Health through grants CA170035 and CA190945 (to A.O.K.), CA106458 (to M.M.H.), and CA178744 (to J.S.M.), The Cancer Prevention and Research Institute of Texas RP1100584 (to N.S., F.M.B.), the Sheikh Khalifa Bin Zayed Al Nahyan Institute for Personalized Cancer Therapy 1U01 CA180964 ( to N.S.S., K.M.S, F.M.B), NCATS Grant UL1 TR000371 (Center for Clinical and Translational Sciences), and MD Anderson Cancer Center Support Grant (P30 CA016672).
Conflicts of Interest:
R.K. receives consultant fees from Actuate Therapeutics, XBiotech and Loxo, Speaker fees from Roche, has an ownership interest in CureMatch, and receives research funds from Genentech, Pfizer, Merck Serono, Sequenom, Guardant, Foundation Medicine, and Incyte.
F.M.B receives honoraria from Dialecta, serves in a consulting or advisory role for Genentech, Inflection Bio- sciences, Pieris Pharmaceuticals, Clearlight Diagnostics, Darwin Health; receives research funding from Novartis, AstraZeneca, Taiho Pharma-ceutical, Genentech, Calithera Biosciences, Debiopharm Group, Bayer, Aileron Therapeutics, PUMA Biotechnology, CytomX Therapeutics, Jounce Therapeutics, Zymeworks, Effective Pharmaceuticals, Curls.
R.K.K. receives institutional research support from Agios Inc., Adaptimmune, Astra Zeneca, Bristol Myers Squibb, Celgene, Eli Lilly and Co., Exelixis, Merck, Novartis, TARGET-Pharma, and Tekmira.
R.B.L is Chief Medical Officer at, and has an ownership interest (including patents) in Guardant Health, Inc.
K.C.B has an ownership interest (including patents) in Guardant Health.
R.L.B, K.C.B and A.T. receive support from Guardant Health, Inc. in the form of salaries.
The rest of the authors report no commercial associations (e.g., consultancies, stock ownership, equity interests or patent-licensing arrangements) that might pose a conflict of interest in connection with the submitted article.
Abbreviations:
- cfDNA
Cell-free DNA
- HCC
hepatocellular carcinoma
- TACE
transarterial chemoembolization
- MDACC
MD Anderson Cancer Center
- UCSD
University of California San Diego
- UCSF
University of California San Francisco
- NASH
non-alcoholic steatohepatitis
- CLIP
Cancer of the Liver Italian Program
- BCLC
Barcelona Clinic Liver Cancer
- TNM
Tumour-Node-Metastasis
- CLIA
Clinical Laboratory Improvement Act
- VAF
variant allele fraction
- SNV
single nucleotide variation
- PODS
Personalized Cancer Therapy-Precision Oncology Decision Support
- AF
allele frequency
- LI-RADS
Liver Imaging Reporting and Data System
- VUS
variants of unknown functional significance
- CPS
Child-Pugh Score
References
- 1.Haber DA, Velculescu VE. Blood-based analyses of cancer: circulating tumor cells and circulating tumor DNA. Cancer Discov 2014;4:650–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khemlina G, Ikeda S, Kurzrock R. The biology of Hepatocellular carcinoma: implications for genomic and immune therapies. Mol Cancer 2017;16:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stroun M, Lyautey J, Lederrey C, et al. About the possible origin and mechanism of circulating DNA apoptosis and active DNA release. Clin Chim Acta 2001;313:139–42. [DOI] [PubMed] [Google Scholar]
- 4.Diehl F, Schmidt K, Choti MA, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med 2008;14:985–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jen J, Wu L, Sidransky D. An overview on the isolation and analysis of circulating tumor DNA in plasma and serum. Ann N Y Acad Sci 2000;906:8–12. [DOI] [PubMed] [Google Scholar]
- 6.Ren N, Qin LX, Tu H, et al. The prognostic value of circulating plasma DNA level and its allelic imbalance on chromosome 8p in patients with hepatocellular carcinoma. J Cancer Res Clin Oncol 2006;132:399–407. [DOI] [PubMed] [Google Scholar]
- 7.Tokuhisa Y, Iizuka N, Sakaida I, et al. Circulating cell-free DNA as a predictive marker for distant metastasis of hepatitis C virus-related hepatocellular carcinoma. Br J Cancer 2007;97:1399–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tang JC, Feng YL, Guo T, et al. Circulating tumor DNA in hepatocellular carcinoma: trends and challenges. Cell Biosci 2016;6:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schwaederle M, Chattopadhyay R, Kato S, et al. Genomic Alterations in Circulating Tumor DNA from Diverse Cancer Patients Identified by Next-Generation Sequencing. Cancer Res 2017;77:5419–5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwaederle M, Husain H, Fanta PT, et al. Use of Liquid Biopsies in Clinical Oncology: Pilot Experience in 168 Patients. Clin Cancer Res 2016;22:5497–5505. [DOI] [PubMed] [Google Scholar]
- 11.Ono A, Fujimoto A, Yamamoto Y, et al. Circulating Tumor DNA Analysis for Liver Cancers and Its Usefulness as a Liquid Biopsy. Cell Mol Gastroenterol Hepatol 2015;1:516–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim ST, Lee WS, Lanman RB, et al. Prospective blinded study of somatic mutation detection in cell-free DNA utilizing a targeted 54-gene next generation sequencing panel in metastatic solid tumor patients. Oncotarget 2015;6:40360–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zill OA, Greene C, Sebisanovic D, et al. Cell-Free DNA Next-Generation Sequencing in Pancreatobiliary Carcinomas. Cancer Discov 2015;5:1040–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marrero JA, Kulik LM, Sirlin CB, et al. Diagnosis, Staging, and Management of Hepatocellular Carcinoma: 2018 Practice Guidance by the American Association for the Study of Liver Diseases. Hepatology 2018;68:723–750. [DOI] [PubMed] [Google Scholar]
- 15.Expert Panel on Detection E, Treatment of High Blood Cholesterol in A. Executive Summary of the Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). JAMA 2001;285:2486–2497. [DOI] [PubMed] [Google Scholar]
- 16.Lanman RB, Mortimer SA, Zill OA, et al. Analytical and Clinical Validation of a Digital Sequencing Panel for Quantitative, Highly Accurate Evaluation of Cell-Free Circulating Tumor DNA. Plos One 2015;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meric-Bernstam F, Johnson A, Holla V, et al. A decision support framework for genomically informed investigational cancer therapy. J Natl Cancer Inst 2015;107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heinemann V, Stintzing S, Kirchner T, et al. Clinical relevance of EGFR- and KRAS-status in colorectal cancer patients treated with monoclonal antibodies directed against the EGFR. Cancer Treat Rev 2009;35:262–71. [DOI] [PubMed] [Google Scholar]
- 19.Linardou H, Briasoulis E, Dahabreh IJ, et al. All about KRAS for clinical oncology practice: gene profile, clinical implications and laboratory recommendations for somatic mutational testing in colorectal cancer. Cancer Treat Rev 2011;37:221–33. [DOI] [PubMed] [Google Scholar]
- 20.Misale S, Yaeger R, Hobor S, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012;486:532–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Allegra CJ, Jessup JM, Somerfield MR, et al. American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol 2009;27:2091–6. [DOI] [PubMed] [Google Scholar]
- 22.Koehler K, Liebner D, Chen JL. TP53 mutational status is predictive of pazopanib response in advanced sarcomas. Ann Oncol 2016;27:539–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Said R, Hong DS, Warneke CL, et al. P53 mutations in advanced cancers: clinical characteristics, outcomes, and correlation between progression-free survival and bevacizumab-containing therapy. Oncotarget 2013;4:705–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwaederle M, Lazar V, Validire P, et al. VEGF-A Expression Correlates with TP53 Mutations in Non-Small Cell Lung Cancer: Implications for Antiangiogenesis Therapy. Cancer Res 2015;75:1187–90. [DOI] [PubMed] [Google Scholar]
- 25.Bruix J, Sherman M, Practice Guidelines Committee AAftSoLD. Management of hepatocellular carcinoma. Hepatology 2005;42:1208–36. [DOI] [PubMed] [Google Scholar]
- 26.Harding JJ, Nandakumar S, Armenia J, et al. Prospective Genotyping of Hepatocellular Carcinoma: Clinical Implications of Next-Generation Sequencing for Matching Patients to Targeted and Immune Therapies. Clinical Cancer Research 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoo C, Ryu M-H, Ryoo B- Y, et al. Efficacy, safety, and pharmacokinetics of imatinib dose escalation to 800 mg/day in patients with advanced gastrointestinal stromal tumors. Investigational New Drugs 2013;31:1367–1374. [DOI] [PubMed] [Google Scholar]
- 28.Damrauer JS, Hoadley KA, Chism DD, et al. Intrinsic subtypes of high-grade bladder cancer reflect the hallmarks of breast cancer biology. Proc Natl Acad Sci U S A 2014;111:3110–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choi W, Porten S, Kim S, et al. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell 2014;25:152–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoadley KA, Yau C, Wolf DM, et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 2014;158:929–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee SE, Chang SH, Kim WY, et al. Frequent somatic TERT promoter mutations and CTNNB1 mutations in hepatocellular carcinoma. Oncotarget 2016;7:69267–69275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pezzuto F, Izzo F, Buonaguro L, et al. Tumor specific mutations in TERT promoter and CTNNB1 gene in hepatitis B and hepatitis C related hepatocellular carcinoma. Oncotarget 2016;7:54253–54262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nault JC, De Reynies A, Villanueva A, et al. A hepatocellular carcinoma 5-gene score associated with survival of patients after liver resection. Gastroenterology 2013;145:176–87. [DOI] [PubMed] [Google Scholar]
- 34.Schulze K, Imbeaud S, Letouze E, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet 2015;47:505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nault JC, Bioulac-Sage P, Zucman-Rossi J. Hepatocellular benign tumors-from molecular classification to personalized clinical care. Gastroenterology 2013;144:888–902. [DOI] [PubMed] [Google Scholar]
- 36.Cleary SP, Jeck WR, Zhao X, et al. Identification of driver genes in hepatocellular carcinoma by exome sequencing. Hepatology 2013;58:1693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guichard C, Amaddeo G, Imbeaud S, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet 2012;44:694–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nault JC, Couchy G, Balabaud C, et al. Molecular Classification of Hepatocellular Adenoma Associates With Risk Factors, Bleeding, and Malignant Transformation. Gastroenterology 2017;152:880–894 e6. [DOI] [PubMed] [Google Scholar]
- 39.Lanman RB, Mortimer SA, Zill OA, et al. Analytical and Clinical Validation of a Digital Sequencing Panel for Quantitative, Highly Accurate Evaluation of Cell-Free Circulating Tumor DNA. PLoS One 2015;10:e0140712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cancer Genome Atlas Research Network. Electronic address wbe, Cancer Genome Atlas Research N. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017;169:1327–1341 e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Totoki Y, Tatsuno K, Covington KR, et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat Genet 2014;46:1267–73. [DOI] [PubMed] [Google Scholar]
- 42.Ikeda S, Tsigelny IF, Skjevik AA, et al. Next-Generation Sequencing of Circulating Tumor DNA Reveals Frequent Alterations in Advanced Hepatocellular Carcinoma. Oncologist 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.