

Abstract
Acute kidney injury (AKI) is a devastating condition with high morbidity and mortality. AKI is characterized by tubular injury, inflammation, and vascular impairment. However, the role of interstitial fibroblasts in the pathogenesis of AKI is largely unknown. Here, we show that fibroblasts were activated, as defined by vimentin expression, at 1 h after AKI triggered by ischemia‐reperfusion injury (IRI). They rapidly entered the cell cycle with Ki‐67‐positive staining, which started at 1 h and peaked at 12 h after IRI, whereas tubular cell proliferation peaked at 3 d. The trigger for such an early activation of fibroblasts was identified as sonic hedgehog (Shh), which was rapidly induced in renal tubules and could target interstitial fibroblasts. Tubule‐specific knockout of Shh in mice inhibited fibroblast activation and aggravated kidney injury and functional decline after IRI. Likewise, pharmacologic inhibition of Shh signaling with cyclopamine also hindered fibroblast activation and exacerbated kidney damage. These studies uncover that tubule‐derived Shh triggers the early activation of fibroblasts, which is required for kidney repair and regeneration. Our findings for the first time illustrate a previously unrecognized importance of interstitial fibroblasts in conferring renal protection in AKI.—Zhou, D., Fu, H., Liu, S., Zhang L., Xiao, L., Bastacky, S. I., Liu, Y. Early activation of fibroblasts is required for kidney repair and regeneration after injury. FASEB J. 33, 12576–12587 (2019). www.fasebj.org
Keywords: acute kidney injury, sonic hedgehog, Wnt‐β‐catenin, injury repair
ABBREVIATIONS
- α‐SMA
α‐smooth muscle actin
- β‐Gal
β‐galactosidase
- AKI
acute kidney injury
- CKD
chronic kidney disease
- CPN
cyclopamine
- HGF
hepatocyte growth factor
- Gli1
glioma‐associated oncogene homolog 1
- Hhip
hedgehog‐interacting protein
- IRI
ischemia‐reperfusion injury
- NGAL
neutrophil gelatinase‐associated lipocalin
- NRK‐49F
normal rat kidney interstitial fibroblast
- PDGFR‐β
platelet‐derived growth factor receptor‐β
- qPCR
quantitative PCR
- Shh
sonic hedgehog
- TBM
tubular basement membrane
- Wnt
Wingless‐related integration site
- X‐Gal
5‐bromo‐4‐chloro‐3‐indolyl‐β ‐D‐galactopyranoside
Acute kidney injury (AKI) is a devastating condition with high morbidity and mortality, and it is responsible for ~2 million deaths each year worldwide (1–3). Over the past decades, great efforts have been made in delineating the courses, mediators, and pathomechanisms of AKI, particularly ischemic AKI, in animal models and patients (4–13). It has become clear that renal tubular epithelial cells are the primary targets of injury, and they lose functional integrity or die by apoptosis and necrosis. Such tubular injury is typically accompanied by, and may be initiated or propagated by, renal vascular endothelial dysfunction and the infiltration of proinflammatory cells, such as neutrophils, lymphocytes, or macrophages (14, 15). However, the potential role and contribution of interstitial fibroblasts in AKI has been largely, if not totally, overlooked.
Fibroblasts in normal adult kidneys are situated in the interstitial space between the capillaries and the epithelia throughout the renal parenchyma (16, 17). Morphologically, these cells are spindle‐shaped and possess multiple cellular processes, which connect them to the tubular and capillary basement membranes (16). Functionally, they maintain tissue homeostasis and contribute to proper cell communication (17, 18). Because vascular pericytes possess similar features and cell markers except the location, the general term “fibroblast” is used herein, which may include interstitial fibroblasts and pericytes. It is widely accepted that interstitial fibroblasts or pericytes are major effector cells responsible for synthesis and deposition of extracellular matrix, and therefore their activation plays a key role in renal fibrogenesis after injury (19–21). Although sporadic reports also suggest a transient myofibroblastic differentiation of interstitial fibroblasts in animal models of AKI (22, 23), little is known about the functional implications of such an activation in tissue injury, repair, and regeneration. We recently showed that fibroblast‐specific β‐catenin signaling is able to control the outcome of AKI (24). These findings suggest that the importance of fibroblasts in dictating kidney responses in the setting of AKI is much greater than one previously envisioned.
In the present study, we systematically studied the dynamics and trigger of fibroblast activation in AKI induced by ischemia‐reperfusion injury (IRI). We uncover that fibroblast activation is an extremely early event and fibroblast proliferation and expansion clearly precedes tubular cell proliferation and repopulation. Functionally, blockade of fibroblast activation by genetic or pharmacologic approaches aggravates kidney damage and functional decline after AKI. Our studies indicate that early activation of fibroblasts is crucial and required for kidney repair and regeneration.
MATERIALS AND METHODS
Animal models
Male C57BL/6J mice weighing about 20‐25 g were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). Renal IRI was performed by using an established protocol as previously described by Zhou et al. (25). Briefly, bilateral renal pedicles were clamped for 30 min using microaneurysm clamps to generate ischemic injury. During the ischemic period, body temperature was maintained between 35 and ~37.5°C by using a temperature‐controlled heating system. The mice were euthanized at 0, 1, 4, 12 h and 1, 3, and 7 d, respectively. For the pharmaceutics inhibition experiment, mice received a daily intraperitoneal injection of cyclopamine (CPN) (MilliporeSigma, Burlington, MA, USA) at 5 mg per kg body weight prior to surgery for 2 d as well as on the same day of surgery. Mice were euthanized at 1 d after IRI, and kidney tissues were collected for various analyses.
In separate experiments, homozygous sonic hedgehog (Shh)‐floxed mice were obtained from The Jackson Laboratory (004293). Transgenic mice that expressed Cre recombinase under the control of kidney‐specific Ksp‐cadherin promoter (ksp‐Cre) were reported elsewhere. By mating Shh‐floxed mice with Ksp‐Cre‐transgenic mice, conditional knockout mice (Ksp‐Shh‐/‐) in which the Shh gene was specifically disrupted in renal tubular epithelial cells (genotype Shhfl/fl and Cre+/‐)were created. These mice were crossbred with homozygous Shh‐floxed mice (genotype Shhfl/fl) to generate offspring with 50% Ksp‐Shh‐/‐ mice and 50% control mice (Shh‐floxed mice) within the same litters. A routine PCR protocol was used for genotyping of tail DNA samples with the following primer pairs: Cre transgene, 5′‐AGGTGTAGAGAAGGCACTTAGC‐3′ and 5′‐CTAATCGCCATCTTCCAGCAGG‐3′, which generated a 411‐bp fragment; and Shh genotyping, 5′‐ATGCTGGCTCGCCTGGCTGTGGAA‐3′ and 5′‐GAAGAGATCAAGGCAAGCTCTGGC‐3′, which yielded a 483‐bp band for the floxed alleles. All animals were born normally at the expected Mendelian frequency, and they were normal in size and did not display any gross physical or behavioral abnormalities.
The glioma‐associated oncogene homolog 1 (Gli1)lacZ mutant mouse with a 129S1/SvImJ background, which harbors a gene‐encoding β‐galactosidase (LacZ) knockin mutation that also abolishes the endogenous Gli1 gene, was obtained from The Jackson Laboratory (008211). Heterozygous mice were mated, and the offspring were genotyped by PCR according to the protocol specified by The Jackson Laboratory. Mutant mice (Gli1‐/‐) and their wild‐type (Gli1+/+) littermates from the colony at the age of 8 wk underwent IRI. Kidney tissues were collected for various analyses at 1 d after IRI. All animal experiments were approved by the Institutional Animal Care and Use Committee at the University of Pittsburgh.
Human studies
Human serum samples were obtained from the Department of Nephrology of the First Affiliated Hospital of Nanjing University of Chinese Medicine. Some serum samples were also obtained from healthy volunteers. The studies were approved by the Ethics Committee at the First Affiliated Hospital of Nanjing University of Chinese Medicine (2012NL‐063‐01‐08). All the participants signed written consent forms prior to inclusion. A cohort of 9 cases of patients with AKI caused by percutaneous coronary intervention, chemotherapy, contrast medium, and hemosiderosis was used for measurement of Shh levels. The diagnosis and demographic data of the patients were presented in Table 1 . Human kidney specimens were obtained from diagnostic renal biopsies performed at the Presbyterian Hospital of the University of Pittsburgh Medical Center. Nontumor kidney tissue from the patients who had renal cell carcinoma and underwent nephrectomy was used as normal controls. Paraffin‐embedded human kidney biopsy sections (2.5‐μm thickness) were prepared using a routine procedure. All studies involving human kidney sections were approved by the Institutional Review Board at the University of Pittsburgh.
Table 1.
Demographic characteristics of the participants included in the study
| Cohort | ||
|---|---|---|
| Characteristic | Healthy person (n = 9) | Patient with AKI (n =9) |
| Gender [n (%)] | ||
| Male | 7 (77.8) | 6 (66.7) |
| Female | 2 (22.2) | 3 (33.3) |
| Age at entry (yr) | ||
| Means ± sem | 47.6 ± 3.7 | 57.6 ± 6.2 |
| Range | 35‐68 | 19‐76 |
Cell culture and treatment
The normal rat kidney interstitial fibroblast (NRK‐49F) cell line was obtained from the American Type Culture Collection (Manassas, VA, USA). Cells were maintained as previously described by Zhou et al. (25). Serum‐starved NRK‐49F was treated with recombinant human Shh protein (StemRD, Burlingame, CA, USA) at different concentrations for various periods of time as indicated. Cells were then collected and subjected to various analyses.
Determination of serum creatinine
Serum was collected from mice at 0, 1, 4, and 12 h and 1, 3, and 7 d after IRI, respectively. Serum creatinine level was determined by use of a QuantiChrom Creatinine Assay Kit according to the protocols specified by the manufacturer (BioAssay Systems, Hayward, CA, USA). The level of serum creatinine was expressed as milligrams per 100 ml.
Shh ELISA
The Human Shh ELISA Kit was purchased from Abcam (ab100639; Abcam, Cambridge, MA, USA). This assay employed an antibody specific for human Shh coated on a 96‐well plate. Standards and samples were pipetted into the wells, and Shh present in a sample was bound to the immobilized antibody. The wells were washed, and biotinylated anti‐human Shh antibody was added. After the washing away of unbound biotinylated antibody, horseradish peroxidase‐conjugated streptavidin was pipetted to the wells. The wells were again washed and a 3,3‘,5,5‘‐tetramethylbenzidine substrate solution was added and color developed in proportion to the amount of Shh bound. The stop solution changed the color from blue to yellow, and the intensity of the color was measured at 450 nm.
RT‐PCR and real‐time PCR
Total RNA was extracted using Trizol RNA isolation system (Thermo Fisher Scientific, Waltham, MA, USA). First‐strand cDNA synthesis was carried out by using a reverse transcription system kit according to the instructions of the manufacturer (Promega, Madison, WI, USA). Real‐time quantitative PCR (qPCR) was performed on an ABI Prism 7000 sequence‐detection system (Thermo Fisher Scientific) as previously described by Zhou et al. (25). The majority of the primer pairs used in this study were reported in our previous publications (24–28). Two sets of the new primer sequences were listed as follows: Patch1, forward 5′‐CAGCCGAGACAAGCCCATCGAC‐3′ and reverse 5′‐ATGTTGGCCTGGGAGGCAGC‐3′; and hedgehog‐interacting protein (Hhip) forward 5′‐CAACCTGCCCAGCCACTGACC‐3′ and reverse 5 ‐GACACCTCCATGACGGCACGC‐3′. PCR was run by using standard conditions. The mRNA levels of various genes were calculated after normalizing with β‐actin.
Western blot analysis
Kidney tissues were lysed with RIPA buffer containing 1% nonyl phenoxypolyethoxylethanol (NP‐40), 0.1% SDS, 100 μg/ml PMSF, 1% protease inhibitor cocktail, and 1% phosphatase I and II inhibitor cocktail (MilliporeSigma) in PBS on the ice. The supernatants were collected after centrifugation at 13, 000 g at 4°C for 15 min. Protein expression was analyzed by Western blot analysis as previously described by Zhou et al. (25). The primary antibodies used were as follows: anti‐Shh (sc‐9024), anti‐platelet‐derived growth factor receptor‐β (PDGFR‐β) (sc‐432; Santa Cruz Biotechnology, Dallas, TX, USA), anti‐a‐smooth muscle actin (a‐SMA) (A2547), anti‐β‐catenin (610154; BD Biosciences, San Jose, CA, USA), anti‐hepatocyte growth factor (HGF) (AF2207; R&D Systems, Minneapolis, MN, USA), anti‐α‐tubulin (T9026; MilliporeSigma), and anti‐actin (MAB1501; Thermo Fisher Scientific).
Histology and immunohistochemical staining
Paraffin‐embedded mouse kidney sections (3‐μm thickness) were prepared by a routine procedure. The sections were stained with periodic acid Schiff reagents by standard protocol. Immunohistochemical staining was performed according to the established protocol as previously described by Zhou et al. (29). The antibodies against vimentin (5741s; Cell Signaling Technology, Danvers, MA, USA), Shh (sc‐9024; Santa Cruz Biotechnology), a‐SMA (A2547; MilliporeSigma), β‐catenin (ab15180), Ki‐67 (ab66155; Abcam), and matrix metallopeptidase 7 (104658; GeneTex, Irvine, CA, USA) were used.
Coimmunofluorescence staining and confocal microscopy
Kidney cryosections were fixed with 3.7% paraformaldehyde for 15 min at room temperature and immersed in 0.2% Triton X‐100 for 10 min. After blocking with 10% donkey serum in PBS for 1 h, slides were coimmunostained with the following antibodies: anti‐Ki‐67 (ab66155; Abcam), anti‐laminin (L8271; MilliporeSigma), anti‐vimentin (5741s; Cell Signaling Technology), anti‐CD31 (550274; BD Biosciences), and anti‐F4/80 (MCA497GA; Bio‐Rad Laboratories, Hercules, CA, USA). To visualize the primary antibodies, slides were stained with cyanine 2‐ or cyanine 3‐conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Stained slides were viewed under a Leica TCS‐SL confocal microscope equipped with a digital camera (Leica Microsystems, Buffalo Grove, IL, USA).
5‐Bromo‐4‐chloro‐3‐indolyl‐β‐D‐galactopyranoside staining
For detecting functional β‐Gal activity, optimal cutting temperature‐embedded kidneys from the Gli1‐LacZ knockin reporter mice were cryosectioned into 7‐μm sections and fixed with paraformaldehyde solution and then stained with standard 5‐bromo‐4‐chloro‐3‐indolyl‐β‐d‐galactopyranoside (X‐Gal) for 2 d at 37°C. The specimens were washed 3 times with PBS. Tissue sections were viewed using light microscopy. The X‐Gal staining protocol was provided by Dr. Ben Humphreys (Washington University, St. Louis, MO, USA).
Statistical analyses
All data were expressed as means ± sem. Statistical analysis of the data was performed using SigmaStat software (Systat Software, San Jose, CA, USA). Comparison between groups was made using 1‐way ANOVA, followed by the Student's Newman‐Keuls test. A value of P < 0.05 was considered significant.
RESULTS
Fibroblast activation is an early cellular event after AKI
To study the potential role of interstitial fibroblasts in AKI, we systemically investigated the dynamics of fibroblasts activation in the kidney after IRI, a classic model of AKI. Kidney sections were obtained from mice at different time points (0, 1, 4, and 12 h and 1, 3, and 7 d) after IRI and subjected to immunostaining for vimentin, an intermediate cytoskeleton protein that is present in activated fibroblasts (18, 21). As shown in Fig. 1A, fibroblast activation, as reflected by vimentin expression, was an extremely early event that occurred as early as 1 h after IRI. At that time, there was no appreciable tubular damage and renal inflammation yet (Fig. 1B). The numbers of vimentin‐positive cells did not further increase significantly until 3 d after IRI, suggesting that the majority of residential fibroblasts were activated at 1 h after injury (Fig. 1B). However, vimentin‐positive cells were dramatically increased at 3 d after IRI, presumably because of the expansion of the activated fibroblast population after severe AKI.
Figure 1.
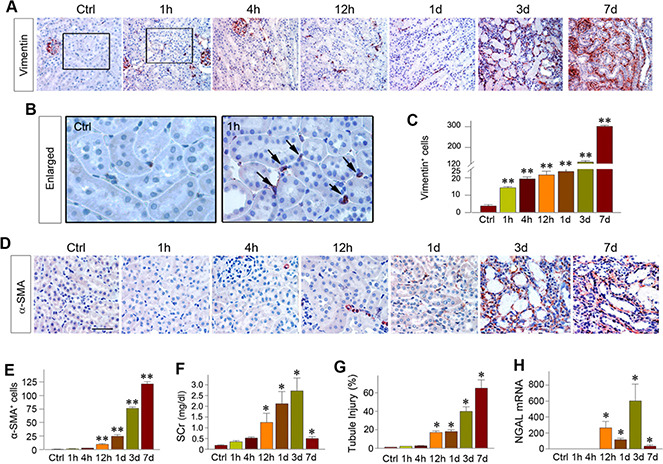
Activation of interstitial fibroblasts is an early event after AKI. A, B) Immunohistochemical staining showed the vimentin‐positive fibroblasts at different time points (1, 4, and 12 h, and 1, 3 and 7 d) after IRI (A). Enlarged boxes are presented (B) that indicate vimentin expression at 1 h after IRI compared with the sham‐treated controls. Black arrows indicate vimentin‐positive cells. C) Bar graph showed the numbers of vimentin‐positive cells in each high‐power field at different time points after IRI. **P < 0.01 vs. sham‐treated (n =3). D) Representative micrographs show renal a‐SMA‐positive cell population at different time points after IRI. Kidney sections were immunostained with antibody against a‐SMA. Scale bar, 50 μm. E) Bar presentation of the numbers of the a‐SMA‐positive myofibroblasts at different time points after IRI. Shown are the cell numbers per high‐power field. **P < 0.01 vs. sham‐treated (n = 3). F) Serum creatinine levels at different time points after IRI. *P < 0.05 vs. sham‐treated controls (n = 4). G) Tubular injury index (percentage of injured tubules) at different time points after IRI. Tubular injury was defined by loss of brush borders, cell flatting, and cell death or detachment. *P < 0.05 vs. sham‐treated controls (n = 4). H) Realtime qPCR showed renal expression of NGAL mRNA at different time points after IRI. *P < 0.05 vs. sham‐treated controls (n = 3‐4). Ctrl, control; SCr, serum creatinine.
We also examined the expression of a‐SMA, the molecular signature of myofibroblasts. As shown in Fig. 1D, a‐SMA‐positive myofibroblasts were not significantly increased in the kidney until 12 h after IRI. Interestingly, kidney injury and dysfunction, as illustrated by serum creatinine level and tubular injury index, were not observed until 12 h after IRI (Fig. 1F, G ). Similarly, real‐time qPCR analyses revealed that neutrophil gelatinase‐associated lipocalin (NGAL), one of the biomarkers linked to tubules injury (30, 31), was not induced until 12 h after injury (Fig. It was consistent with the pattern of kidney injury molecule‐1 expression after injury in the current model (unpublished results). These data indicate that fibroblast activation, reflected by vimentin expression, is an extremely early cellular event in AKI when compared with the timeline of tubular and inflammatory responses after IRI.
Fibroblast proliferation precedes tubular repair and regeneration
We next examined the dynamics and patterns of cell proliferation in AKI. As shown in Fig. 2A, interstitial cells, presumably fibroblasts or pericytes, started to enter the cell cycle as early as 1 h and peaked at 12 h after IRI as shown by immunohistochemical staining for Ki‐67. Particularly in the corticomedullary junctional region in which tubular damage was most severe in this model, abundant proliferative fibroblasts were lined along the tubular basement membrane (TBM) at 12 and 24 h after IRI (Fig. 2B). To confirm the identity of these Ki‐67‐positive interstitial cells, we carried out double immunofluorescence staining for Ki‐67 and cell type‐specific markers. As shown in Fig. 2C, the vast majority of the Ki‐67‐positive proliferating cells coexpressed vimentin at 12 h after IRI, suggesting that they are activated fibroblasts. A few Ki‐67‐positive cells also costained with a‐SMA, the marker of myofibroblasts (Fig. 2C ). However, little costaining of Ki‐67 with macrophage‐specific marker F4/80 or endothelial cell‐specific marker CD31 was observed (Fig. 2C ). Taken together, these results suggest that the majority of the Ki‐67‐positive proliferative cells in the early phase of AKI (<24 h) are vimentin‐positive activated fibroblasts.
Figure 2.
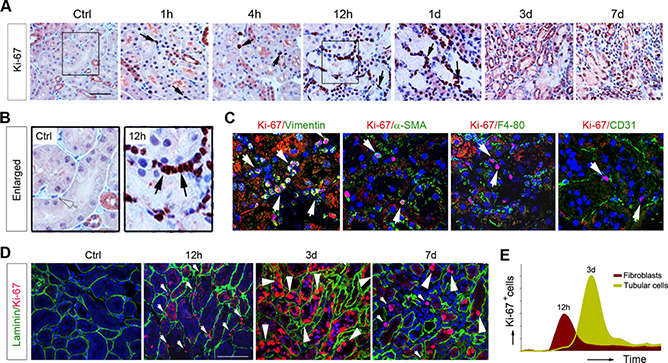
Fibroblast proliferation precedes tubular repair and regeneration after AKI. A, B) Immunohistochemical staining for Ki‐67 showed that fibroblast proliferation is an early event preceding tubular regeneration. A) Representative micrographs of renal Ki‐67 staining at different time points after IRI are shown. Scale bar, 50 μm. B) Enlarged boxes are presented. Arrows indicate Ki‐67‐positive cells in renal interstitium. C) Double immunostaining showed renal Ki‐67 and cell type‐specific markers, such as vimentin, α‐SMA, F4/80, and CD31, at 12 h after IRI. White arrows indicate Ki‐67‐positive cells. D) Coimmunostaining with antibodies against Ki‐67 and laminin demonstrated tissue compartment‐specific cell proliferation at different time points after IRI. Small arrowheads indicate interstitial cells, whereas large arrowheads indicate tubular epithelial cells. E) Graphic presentation showed different dynamics of fibroblast and tubular cell proliferation after AKI. Ctrl, control.
To further delineate the temporal and spatial patterns of cell proliferation in AKI, we then carried out double immunostaining for Ki‐67 and laminin, a major TBM protein that outlines the tubular and interstitial compartments (32). As shown in Fig. 2D, at 12 h after IRI, the Ki‐67‐positive proliferating cells were largely localized in the interstitial compartment. However, at 3 d after injury, the majority of the Ki‐67‐positive cells were found in the tubular compartment (Fig. 2D, E ). Therefore, it seems that there are 2 waves of cell proliferation, with fibroblast proliferation peaking at 12 h and tubular cell proliferation peaking at 3 d after IRI (Fig. 2E).
Shh triggers fibroblast activation in AKI
To explore how fibroblasts become activated rapidly after injury, we sought to identify the trigger responsible for fibroblast activation after IRI. To this end, we studied the potential involvement of Shh because earlier studies implicate it as a tubule‐derived growth factor for interstitial fibroblasts in chronic kidney disease (CKD) (25, 33, 34). As shown in Fig. 3A, B, Shh protein was rapidly induced in the renal tubular epithelium as early as 1 h after IRI, and its induction was sustained throughout the experiments. Similarly, real‐time qPCR revealed that Shh mRNA was also rapidly induced in the kidney at 1 h after IRI (Fig. 3C). Various downstream targets of Shh signaling, such as Gli1, Patch1, and hedgehog interacting protein, were quickly induced as early as 1 h after IRI as well (Fig. 3D‐F).
Figure 3.
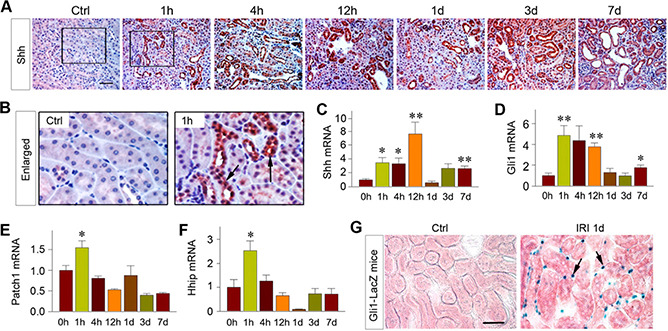
Tubule‐derived Shh triggers fibroblast activation in AKI. A, B) Shh was induced specifically in renal tubules as early as 1 h after IRI. A) Kidney sections at different time points after IRI were immunostained with anti‐Shh antibody. Scale bar, 50 μm. B) Boxed areas were enlarged and presented. Arrow indicates positive staining. C‐F) Shh signaling was rapidly activated in the kidney after AKI. Renal expression of Shh (C), Gli1 (D), Patch1 (E), and Hhip (F) mRNA at different time points after IRI was assessed by real‐time qPCR. **P < 0.01, *P < 0.05 vs. sham‐treated controls (n =3‐4). G) Shh targets interstitial fibroblasts in AKI. The Gli1‐LacZ reporter mice were subjected to IRI. At 1 d after IRI, kidney sections were subjected to X‐gal staining. Arrows indicate X‐gal‐positive interstitial cells in renal interstitium. Scale bar, 50 μm. Ctrl, control.
To further corroborate whether the tubule‐derived Shh targets interstitial fibroblasts, we carried out the studies of identifying the hedgehog‐responding cells in vivo by using Gli1‐LacZ reporter mice, which harbor a β‐Gal knockin mutation. Under the control of native Gli1 promoter, lacZ expression in these mice authentically recapitulates the endogenous Gli1 expression (35). As shown in Fig. 3G, interstitial fibroblasts were clearly β ‐Gal‐positive in the kidney after IRI, whereas tubular cells did not express β ‐Gal at all. Therefore, it appears that tubule‐derived Shh may target interstitial fibroblasts, leading to their activation and proliferation in AKI.
Fibroblast activation promotes tubular repair via Wingless‐related integration site‐β‐catenin and HGF
To elucidate how fibroblast activation by Shh mediates tubular repair and regeneration in AKI, we investigated whether Wingless‐related integration site (Wnt)‐β‐catenin signaling is involved in this process because earlier studies indicate a protective role of tubular β‐catenin in AKI (36). As shown in Fig. 4A‐D, β‐catenin induction was a relatively late event that peaked at 1 and 3 d after IRI, as shown by immunostaining and Western blot analysis. The β ‐catenin protein was predominantly localized in renal tubular epithelium (Fig. 4B), consistent with its role in promoting tubular cell survival and regeneration (36). Accordingly, 2 major direct downstream targets of β‐catenin, Snail1 (Fig. 4E) and fibronectin (Fig. 4F), were up‐regulated at 7 d after injury.
Figure 4.
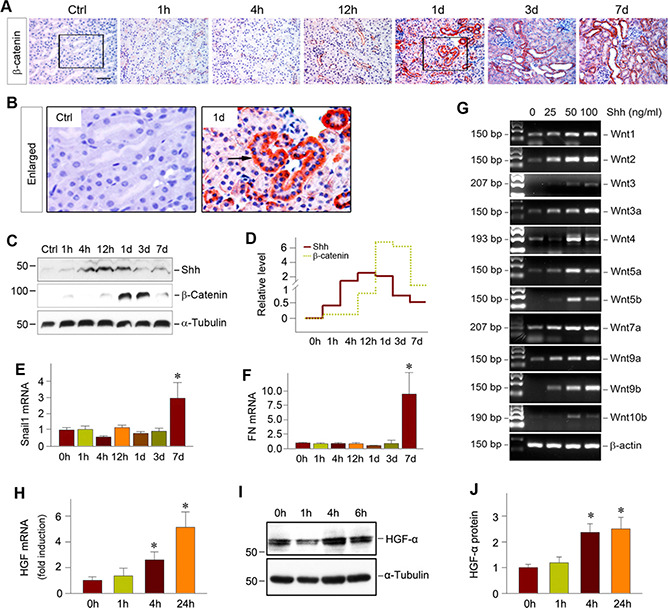
Shh induction precedes renal Wnt‐β‐catenin activation after AKI. A, B) Representative micrographs (A) show immunostaining of β‐catenin at different time points after IRI, as indicated. Scale bar, 50 μm. Enlarged boxes (B) show β‐catenin expression at 1 d after IRI. Arrow indicates positive staining. C, D) Western blot analyses demonstrated renal Shh and β‐catenin expression at different time points after IRI. Representative Western blot (C) and relative abundance of Shh and β‐catenin (D) are shown. E, F) Real‐time qPCR showed renal expression of Wnt‐β‐catenin targeted genes in the kidney after IRI. Relative abundance of renal Snail1 (E) and fibronectin (FN) (F) mRNA are presented. *P < 0.05 vs. sham‐treated controls (n =3‐4). G) Shh induces the expression of Wnt ligands in cultured interstitial fibroblasts. NRK‐49F cells were incubated with different doses of human recombinant Shh protein for 24 h, as indicated. The mRNA expression of various Wnt ligands was assessed by RT‐PCR. H‐J) Shh induces HGF expression in renal interstitial fibroblasts. NRK‐49F cells were incubated with Shh (50 ng/ml) for various periods of time as indicated. HGF mRNA (H) and protein levels (I, J) are shown. *P < 0.05 vs. sham‐treated controls (n = 3). Ctrl, control.
Because Shh induction was much earlier than Wnt‐β‐catenin activation in the kidney after IRI (Fig. 4C, D ), this prompted us to investigate whether there is a direct connection between them. As presented in Fig. 4G, incubation of NRK‐49F with Shh protein induced the mRNA expression of many Wnt ligands in a dose‐dependent fashion. Therefore, it is plausible that tubule‐derived Shh targets interstitial fibroblasts and induces Wnt ligands, which in turn activate β‐catenin in renal tubules after AKI.
We also explored the possibility of whether Shh elicits its beneficial action by regulating the expression of HGF, a pleiotropic factor that promotes injury repair and regeneration after AKI (37). As shown in Fig. 4H, Shh induced HGF mRNA expression in NRK‐49F cells.
Similarly, HGF protein was also up‐regulated in renal fibroblasts after incubation with Shh (Fig. 4I, J ).
Inhibition of Shh suppresses fibroblast activation and aggravates AKI
To define the role of fibroblast activation in AKI, we perturbed Shh signaling in vivo by pharmacologic inhibition using a specific small‐molecule inhibitor. As shown in Fig. 5A, mice were injected intraperitoneally with 5 mg per kg body weight of CPN for 3 d (d —2, —1, and 0), followed by IRI for 1 d. Interestingly, CPN at 5 mg/kg body weight did not exhibit any adverse effect (Fig. 5B). After CPN treatment, Gli1 induction after IRI was completely abolished (Fig. 5C) , suggesting the efficacy of suppressing Shh signaling in this model.
Figure 5.
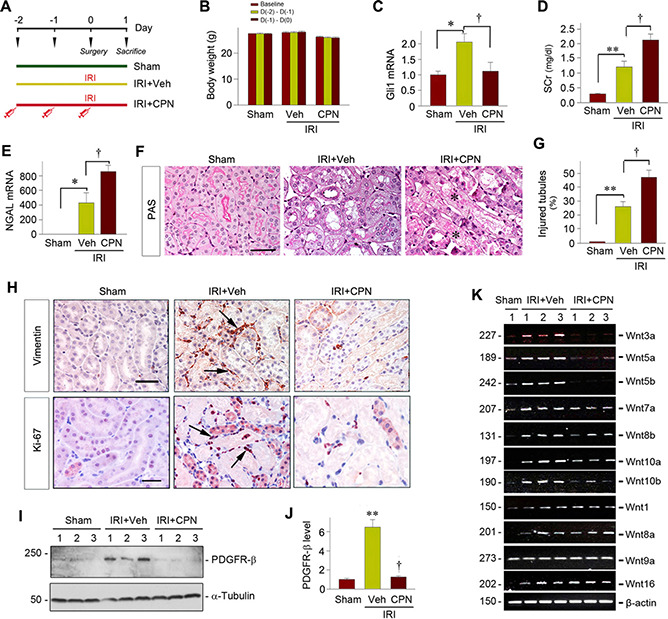
Blockade of Shh signaling inhibits fibroblast activation and aggravates AKI after IRI. A) Experiment design. CPN was administrated in mice 2 d prior to IRI, and mice were euthanized 1 d after IRI. B) Body weight changes after CPN administration. C) CPN abolished renal Gli1 mRNA induction at 1 d after IRI. *P < 0.05 vs. sham‐treated controls, +P < 0.05 vs. vehicle (n = 5, sham‐treated n = 3). D) Blockade of Shh signaling by CPN aggravated kidney dysfunction after IRI. Serum creatinine levels were assessed at 1 d after IRI in different groups as indicated. **P < 0.01, + P < 0.05 (n = 9, sham‐treated n =3). E) Real‐time qPCR revealed an increased NGAL mRNA expression in mice injected with CPN at 1 d after IRI compared with vehicle controls. *P< 0.05 vs. sham‐treated controls, +P < 0.05 vs. vehicles (n = 5, sham‐treated n = 3). F) Inhibition of Shh signaling by CPN worsened histologic injury after AKI. Representative micrographs show kidney histologic changes at 1 d after IRI. G) Bar presentation show the percentage of injured tubules in different groups as indicated. **P < 0.01, +P < 0.05 vs. control (n = 3‐5). H) Representative micrographs show that inhibition of Shh signaling blocked fibroblast activation and proliferation. Kidney sections were stained with antibodies against vimentin and Ki‐67. Arrows indicate positive staining. Scale bar, 50 μm. I, J) Western blot analysis showed renal PDGFR‐β levels in different groups at 1 d after IRI. Representative Western blot (I) and quantitative data (J) are shown. **P < 0.01 vs. sham‐treated controls, +P < 0.05 vs. vehicle (n =3‐5). K) Representative RT‐PCR showed renal mRNA levels of Wnt ligands in different groups as indicated at 1 d after IRI. PAS, periodic acid Schiff; SCr, serum creatinine; Veh, vehicle.
We then measured serum creatinine levels and assessed kidney damage after various treatments. As shown in Fig. 5D , IRI caused serum creatinine levels to rise at 1 d after IRI, and CPN treatment further deteriorated kidney dysfunction. Consistently, renal mRNA expression of NGAL at 1 d after IRI was also increased after injections of CPN compared with vehicle (Fig. 5E). Furthermore, IRI mice treated with CPN exhibited more severe morphologic injury, characterized by loss of brush border, tubular cells depletion, and casts formation in lumens (Fig. 5F, G ). Notably, our pilot experiment results indicated that a single dose of CPN injected at 1 d prior to IRI also aggravated, to a lesser extent, kidney injury, compared with 2 d protocol.
To confirm the implication of fibroblasts in this process, we then analyzed the extent of fibroblast activation after various treatments. As shown in Fig. 5H, blockade of Shh signaling by CPN reduced renal interstitial vimentin‐positive fibroblast accumulation after IRI. Similarly, CPN also inhibited fibroblast proliferation, as illustrated by immunostaining for Ki‐67 (Fig. 5H). Western blot analysis also revealed that CPN completely inhibited the induction of PDGFR‐β at 1 d after IRI (Fig. 5I, J). Furthermore, CPN repressed renal expression of Wnt ligands in vivo (Fig. 5K ). These data suggest that pharmacologic inhibition of Shh by CPN aggravates AKI primarily by inhibiting fibroblast activation.
We also examined the effect of Shh blockade on AKI at 2 d after IRI. We found that 1/5 mice died in the vehicle control group (20% mortality) and 5/10 mice died in the CPN group (50% mortality). These results suggest that blockade of Shh signaling aggravates AKI at 2 d after IRI as well.
Genetic ablation of Shh in renal tubules represses fibroblast activation and aggravates AKI
To validate the role of fibroblast activation in AKI, we next used a genetic approach by generating a conditional knockout mouse model in which Shh was specifically deleted in renal tubules. As shown in Fig. 6 A, mice with tubule‐specific ablation of Shh (designated as Ksp‐Shh‐/‐) were established by using a Cre‐locus of X‐over P1 (LoxP) system. Age‐ and gender‐matched Shh‐floxed mice from the same litters were used as controls. Mice with tubule‐specific deletion of Shh were phenotypically normal. There was no appreciable abnormality in kidney morphology of Ksp‐Shh‐/‐ mice under physiologic conditions. These mice were then subjected to IRI for 1 d. As expected, renal expression of Shh mRNA was lost in Ksp‐Shh‐/‐ mice compared with the controls (Fig. 6B). Immunostaining also revealed that Shh was expressed predominantly in renal tubules at 1 d after IRI in the controls but not in Ksp‐Shh‐/‐ mice (Fig. 6C). Consistently, loss of Shh in renal tubules also repressed the mRNA expression of its downstream genes, such as Gli1, Patch1, and tenascin C, at 1 d after IRI (Fig. 6D‐F).
Figure 6.
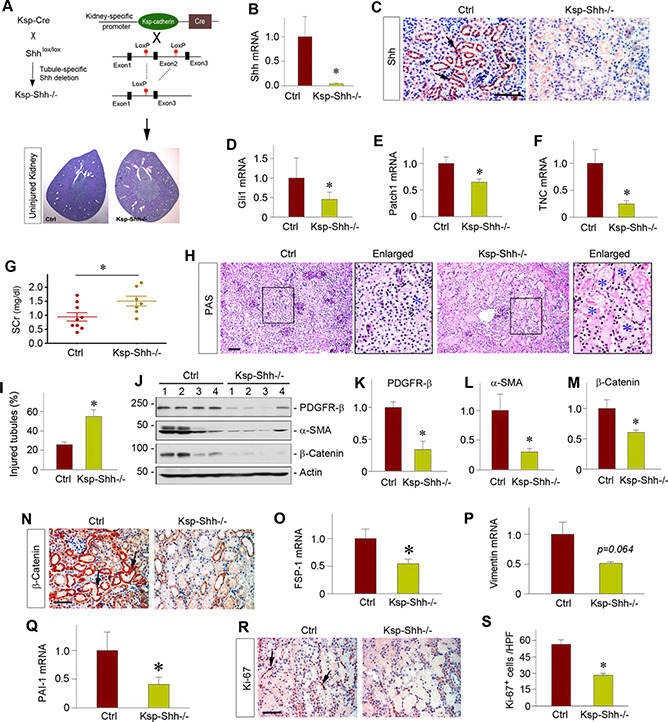
Tubule‐specific ablation of Shh inhibits fibroblast activation and aggravates AKI. A) Schematic diagram of the strategy used to generate tubule‐specific, Shh‐conditional knockout mice. Uninjured kidney histology was presented by periodic acid Schiff staining. B) Real‐time qPCR revealed a dramatic reduction of Shh mRNA in Ksp‐Shh‐/‐ kidney at 1 d after IRI compared with the controls. *P < 0.05 (n =5‐6). C) Representative micrographs show Shh protein expression at 1 d after IRI in the control and Ksp‐Shh‐/‐ mice. Arrows indicate positive staining. Scale bar, 50 μm. D‐F) Tubule‐specific ablation of Shh inhibited its downstream target genes after IRI. Renal expression of Gli1 (D), Patch1 (E), and tenascin C (TNC) (F) mRNA at 1 d after IRI was analyzed by real‐time qPCR in the control and Ksp‐Shh‐/‐ mice. *P < 0.05 (n =5‐6). G) Changes in serum creatinine at 1 d after IRI in the control and Ksp‐Shh‐/‐ mice. *P < 0.05 (n =7‐9). H) Histologic changes of the control and Ksp‐Shh‐/‐ kidneys at 1 d after IRI. Blue asterisks indicate injured tubules. I) Quantitative data show the percentage of injured tubules in the control and Ksp‐Shh‐/‐ mice. *P < 0.05 (n = 7). J‐M) Western blot analyses showed renal levels of PDGFR‐β, a‐SMA, and β‐catenin proteins at 1 d after IRI. Representative Western blot (J) and quantitative data on PDGFR‐β (K), a‐SMA (L), and β‐catenin (M) are presented. *P < 0.05 (n = 5). N) Representative micrographs show renal expression of β‐catenin protein in the control and Ksp‐Shh‐/‐ mice. Arrows indicate positive staining. O, P) Real‐time qPCR revealed substantial reductions of fibroblast‐specific protein 1 (FSP‐1), vimentin, and plasminogen activator inhibitor 1 mRNA levels in Ksp‐Shh‐/‐ kidney at 1 d after IRI compared with the controls. *P < 0.05 (n =5‐6). R) Representative micrographs show renal Ki‐67‐positive cells at 1 d after IRI in the control and Ksp‐Shh‐/‐ mice. Arrows indicate positive staining. Scale bar, 50 μm. S) Quantitative data show the Ki‐67‐positive cells at 1 d after IRI in the control and Ksp‐Shh‐/‐ mice. *P < 0.05 (n = 3). Ctrl, control; SCr, serum creatinine.
We next examined the impact of tubule‐specific ablation of Shh in kidney function after IRI. As shown in Fig. 6G, there was a significant increase in serum creatinine levels at 1 d after IRI in the Ksp‐Shh‐/_ mice compared with the controls. Similarly, more severe morphologic injury was found in the Ksp‐Shh‐/‐ kidney at 1 d after IRI (Fig. 6H, I), suggesting that ablation of tubular Shh exacerbates kidney injury and dysfunction after IRI.
We then assessed the status of fibroblasts activation in the kidneys after IRI. As shown in Fig. 6J‐L, renal expression of PDGFR‐β and a‐SMA proteins was repressed in Ksp‐Shh‐/‐ kidneys compared with the control group. Consistently, loss of tubular Shh also caused substantial reduction of renal expression of fibroblast‐specific protein 1 and vimentin mRNA in Ksp‐Shh‐/‐ mice (Fig. 6O, P ). Notably, tubule‐specific deletion of Shh also resulted in significant suppression of β‐catenin activation in renal tubules (Fig. 6J, M, N), which was accompanied by down‐regulation of Wnt‐β ‐catenin target plasminogen activator inhibitor 1 (Fig. 6Q ). Furthermore, selective deletion of Shh in renal tubules reduced Ki‐67‐positive interstitial cell populations after IRI in Ksp‐Shh‐/‐ mice (Fig. 6R, S ). Collectively, these results suggest that genetic ablation of Shh in a tubule‐specific fashion blunts fibroblast activation and aggravates AKI.
Shh induction and fibroblast activation in human AKI
To study the clinical relevance of fibroblast activation to human AKI, we investigated the expression of Shh and fibroblast activation in the early phase of kidney injury and repair in humans. The clinical features of patients with AKI were presented in Table 1. All of the patients with AKI recruited had no obvious kidney disease history and exhibited primary tubules damage in etiology. We first measured serum Shh level to assess its expression and secretion after acute injury. As shown in Fig. 7A , compared with healthy subjects, patients with AKI displayed an elevated level of Shh in the circulation. Immunohistochemical staining showed robust expression of Shh protein in the kidneys of patients with AKI, particularly in renal tubular epithelium, whereas Shh was not detectable in normal kidneys (Fig. 7B). Fibroblasts activation was prominent, as reflected by vimentin expression (Fig. 7B). However, a few fibroblasts expressed a‐SMA in that particular case, indicating that they were not myofibroblasts in the early stage of AKI. Interestingly, β‐catenin was also induced in the renal tubules of patients with AKI. These results suggest that tubule‐derived Shh induces fibroblast activation in humans as well, which presumably plays a protective role against AKI by activating tubular β ‐catenin.
Figure 7.
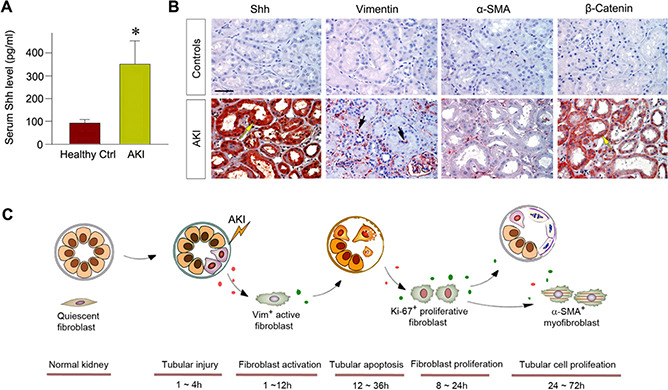
Interstitial fibroblasts are activated in human kidney biopsies of patients with AKI. A) Serum Shh levels in patients with AKI (n = 9) compared with the healthy subjects (n = 9). *P < 0.05 (n = 9). B) Representative micrographs showed protein expression of Shh, vimentin (Vim), a‐SMA, and β‐catenin in human kidney biopsy specimens from patients with AKI. Nontumor kidney tissue from the patients who had renal cell carcinoma and underwent nephrectomy was used as normal controls. Arrows indicate positive staining. Scale bar, 50 μm. C) Diagram depicts the renoprotective role of early activation of fibroblasts in AKI. After injury, kidney tubular cells express and secrete Shh, which targets and activates interstitial fibroblasts, highlighted by vimentin expression, as early as 1 h after IRI. Activated fibroblasts then undergo proliferation (Ki‐67‐positive) peaking at 12 h and secrete Wnt ligands and activate β‐catenin in tubular cells. This leads tubular cell proliferation and tubular repair and regeneration peaking at 3 d after IRI. Pharmacologic inhibition or genetic ablation of Shh blunts fibroblast activation and proliferation and aggravates kidney injury after AKI. Ctrl, control.
DISCUSSION
Kidneys possess substantial ability to repair and regenerate, leading to restoration of renal structure and function after AKI (38, 39). In this study, we present evidence that interstitial fibroblasts, the particular renal parenchymal cells that are long neglected in the AKI field, play an essential role in this process. Fibroblasts are rapidly activated in response to injury, produce and secrete trophic factors such as Wnt ligands and HGF, and promote tubular cell proliferation and regeneration. Blockade of fibroblast activation by genetic and pharmacologic approaches retards tubular repair and aggravates kidney dysfunction. It should be noted that the fibroblast referred to in this study is a relatively broad term that includes interstitial fibroblasts and perivascular pericytes because there is no specific marker to distinguish them with certainty. Our findings illustrate a previously unrecognized importance of interstitial fibroblasts in determining the outcomes of AKI. These studies also suggest a new paradigm in the biology of kidney repair and regeneration after injury.
Fibroblast or pericyte activation is traditionally thought to play a central role in renal fibrosis, an eventual sequel of severe AKI and a common feature of CKD (28, 40). In this regard, the finding that fibroblast activation is an early event occurring at 1 h after IRI is quite surprising. At that time point, no appreciable tubular injury and inflammatory infiltration is evident (Fig. 1), consistent with the timeline established previously for tubular damage, endothelial impairment, and peritubular inflammation in this model (41–43). These results indicate that fibroblast activation, as defined by vimentin expression, is the earliest cellular event documented in AKI thus far. Notably, these vimentin‐positive cells are unlikely to result from surgery or stress because little staining was observed in sham‐treated kidneys at both 1 h (not shown) and 24 h (Fig. 1). Interestingly, the numbers of vimentin‐positive cells are not further increased significantly from 1 to 12 h, suggesting that the majority of the interstitial fibroblast population is already activated concurrently at 1 h right after IRI. It should be pointed out that vimentin is used to indicate fibroblast activation because this intermediate cytoskeleton protein is a hallmark of activated fibroblasts (19, 21). It is noticeable that a‐SMA‐positive myofibroblasts are not substantially increased until 12 h after IRI (Fig. 1). This finding may well explain why early activation of fibroblasts in AKI was not appreciated previously because most studies employ a‐SMA as the marker of fibroblast activation (22). The exact fate of vimentin‐positive cells is not known. However, we speculate that these cells will become myofibroblasts if injury is severe or sustained and become quiescent after renal repair or undergo apoptosis if injury is mild.
The present study also provides clear evidence supporting the existence of 2 waves of cell proliferation in the kidneys after AKI. The first wave, which starts at 1 h and peaks at 12 h, corresponds to fibroblast proliferation (Fig. 2), whereas the second wave, starting at 1 d and peaking at 3 d, matches with tubular cell proliferation and regeneration. Particularly in the corticomedullary region in which injury is most severe in this model, plentiful Ki‐67‐positive proliferating fibroblast cells are found along the denuded TBM at 12 h after IRI, a time point at which significant tubular cell proliferation is not evident yet (Fig. 2B). This observation unravels an intriguing conception that fibroblast activation and proliferation is an early and crucial episode after AKI, which could serve to inculcate survived tubular cells to proliferate for repairing the damaged tubules (Fig. 7). Along this line, fibroblast‐derived trophic factors, such as Wnt ligands and HGF, are well known for their ability to promote tubular cell proliferation and regeneration (36, 37).
The present study also identifies Shh, a hedgehog ligand that is specifically up‐regulated in the injured tubules (Fig. 3), as the trigger for the early activation of fibroblasts in AKI. Although other factors, such as TGF‐β and Wnt ligands, may also promote fibroblast activation (28, 29), kinetics analyses have pinpointed Shh as the likely inducer. Shh protein is up‐regulated in renal tubules at 1 h after IRI, and its downstream targets, such as Gli1 and Patch1, are also rapidly induced (Fig. 3). Most importantly, genetic ablation of Shh expression in renal tubules inhibits fibroblast activation and aggravates kidney injury and dysfunction (Fig. 5). Likewise, blockade of hedgehog signaling by small‐molecule inhibitor CPN also hinders fibroblast activation and worsens AKI (Fig. 6). It is interesting to point out that Shh may not directly regulate tubular cell proliferation and repair because it does not induce Gli1 expression in the Gli1‐LacZ reporter mice (Fig. 3G). Instead, Shh targets interstitial fibroblasts and induces the expression and secretion of a host of Wnt ligands (Fig. 4), which in turn activated β ‐catenin in injured tubular cells, leading to tubular cell survival and proliferation. In addition, Shh is able to induce HGF expression in interstitial fibroblasts, which promotes tubular cell survival, repair, and regeneration through paracrine fashion as well (37, 44). Therefore, there is a cascade of events to promote kidney repair and regeneration after AKI, in which tubular injury triggers Shh expression and secretion, followed by fibroblast activation leading to the release of Wnt ligands and HGF, resulting in tubular cell proliferation and regeneration (Fig. 7C). In this regard, Shh and Wnts or HGF are activated sequentially and work in concert to relay the signals and mediate the crosstalk between tubular epithelium and interstitial fibroblasts. These results are in harmony with the perception that the reactivation of developmental signals, such as Shh and Wnts or HGF, in AKI is renal protective in an attempt to promote injury repair and recovery, although a sustained and exaggerated activation of the same signaling may be detrimental, leading to CKD progression (25, 45).
In summary, we have shown that fibroblast activation is an extremely early event in AKI, which is triggered by the tubule‐derived Shh ligand. Such an early activation of fibroblasts is required for kidney repair and regeneration because blockade of fibroblast activation via genetic and pharmacologic approaches exacerbates kidney injury and dysfunction after AKI. These findings are clinically relevant to human AKI (Fig. 7). Our studies indicate that fibroblasts are a missing piece of the puzzle in comprehending the pathobiology of AKI.
AUTHOR CONTRIBUTIONS
D. Zhou and Y. Liu designed the research; D. Zhou, H. Fu, S. Liu, L. Zhang, and L. Xiao performed the research; D. Zhou, S. I. Bastacky, and Y. Liu analyzed the data; D. Zhou created the figures; D. Zhou and Y. Liu wrote the manuscript; and all authors approved the final version of the manuscript.
ACKNOWLEDGMENTS
The authors are grateful to the Center for Biologic Imaging at the University of Pittsburgh for the use ofcore facilities. This work was supported by the U.S. National Institutes of Health (NIH)/National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Grants DK064005 and DK106049. D.Z. was supported by NIH/NIDDK Grant 1K01DK116816. The authors declare no conflicts of interest.
Zhou, D. , Fu, H. , Liu, S., Zhang L. , Xiao, L. , Bastacky, S. I. , Liu, Y. Early activation of fibroblasts is required for kidney repair and regeneration after injury. FASEB J. 33, 12576–12587 (2019). www.fasebj.org
Contributor Information
Dong Zhou, Email: zhoudong@pitt.edu.
and Youhua Liu, Email: yhliu@pitt.edu.
REFERENCES
- 1. Kaddourah, A. , Basu, R. K. , Bagshaw, S. M. , and Goldstein, S. L. ; AWARE Investigators . (2017) Epidemiology of acute kidney injury in critically ill children and young adults. N. Engl. J. Med. 376, 11–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rewa, O. , and Bagshaw, S. M. (2014) Acute kidney injury‐epidemiology, outcomes and economics. Nat. Rev. Nephrol. 10, 193–207 [DOI] [PubMed] [Google Scholar]
- 3. Singbartl, K. , and Kellum, J. A. (2012) AKI in the ICU: definition, epidemiology, risk stratification, and outcomes. Kidney Int. 81, 819–825 [DOI] [PubMed] [Google Scholar]
- 4. Bonventre, J. V. , and Yang, L. (2011) Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Invest. 121, 4210–4221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang, L. , Humphreys, B. D. , and Bonventre, J. V. (2011) Pathophysiology of acute kidney injury to chronic kidney disease: maladaptive repair. Contrib. Nephrol. 174, 149–155 [DOI] [PubMed] [Google Scholar]
- 6. Sharfuddin, A. A. , and Molitoris, B. A. (2011) Pathophysiology of ischemic acute kidney injury. Nat. Rev. Nephrol. 7, 189–200 [DOI] [PubMed] [Google Scholar]
- 7. Kramann, R. , Tanaka, M. , and Humphreys, B. D. (2014) Fluorescence microangiography for quantitative assessment of peritubular capillary changes after AKI in mice. J. Am. Soc. Nephrol. 25, 1924–1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cianciolo Cosentino, C. , Skrypnyk, N.I. , Brilli, L.L. , Chiba, T. , Novitskaya, T. , Woods, C. , West, J. , Korotchenko, V.N. , McDermott, L. , Day, B.W. , Davidson, A. J. , Harris, R.C. , de Caestecker, M.P. , and Hukriede, N. A. (2013) Histone deacetylase inhibitor enhances recovery after AKI. J. Am.Soc.Nephrol. 24, 943–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen, J. , Chen, J. K. , and Harris, R. C. (2012) Deletion of the epidermal growth factor receptor in renal proximal tubule epithelial cells delays recovery from acute kidney injury. Kidney Int. 82, 45–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen, J. , Chen, J. K. , Conway, E. M. , and Harris, R. C. (2013) Survivin mediates renal proximal tubule recovery from AKI. J. Am. Soc. Nephrol. 24, 2023–2033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gao, G. , Zhang, B. , Ramesh, G. , Betterly, D. , Tadagavadi, R.K. , Wang, W. , and Reeves, W.B. (2013) TNF‐α mediates increased susceptibility to ischemic AKI in diabetes. Am. J. Physiol. Renal Physiol. 304, F515–F521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xiao, X. , Hu, Y. , Quirós, P. M. , Wei, Q. , López‐Otín, C. , and Dong, Z. (2014) OMA1 mediates OPA1 proteolysis and mitochondrial fragmentation in experimental models of ischemic kidney injury. Am. J. Physiol. Renal Physiol. 306, F1318–F1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ma, Z. , Wei, Q. , Dong, G. , Huo, Y. , and Dong, Z. (2014) DNA damage response in renal ischemia‐reperfusion and ATP‐depletion injury of renal tubular cells. Biochim. Biophys. Acta 1842, 1088–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kinsey, G. R. (2014) Macrophage dynamics in AKI to CKD progression. J. Am.Soc.Nephrol. 25, 209–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dhaun, N. , and Webb, D. J. (2013) The road from AKI to CKD: the role of endothelin. Kidney Int. 84, 637–638 [DOI] [PubMed] [Google Scholar]
- 16. Kaissling, B. , and Le Hir, M. (2008) The renal cortical interstitium: morphological and functional aspects. Histochem. Cell Biol. 130, 247–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schiessl, I. M. , Grill, A. , Fremter, K. , Steppan, D. , Hellmuth, M. K. , and Castrop, H. (2018) Renal interstitial platelet‐derived growth factor receptor‐β cells support proximal tubular regeneration. J. Am. Soc. Nephrol. 29, 1383–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kuzet, S. E. , and Gaggioli, C. (2016) Fibroblast activation in cancer: when seed fertilizes soil. Cell Tissue Res. 365, 607–619 [DOI] [PubMed] [Google Scholar]
- 19. Liu, Y. (2011) Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 7, 684–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. LeBleu, V. S. , Taduri, G. , O'Connell, J. , Teng, Y. , Cooke, V. G. , Woda, C. , Sugimoto, H. , and Kalluri, R. (2013) Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 19, 1047–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Grande, M. T. , and López‐Novoa, J. M. (2009) Fibroblast activation and myofibroblast generation in obstructive nephropathy. Nat. Rev. Nephrol. 5, 319–328 [DOI] [PubMed] [Google Scholar]
- 22. Fujigaki, Y. , Muranaka, Y. , Sun, D. , Goto, T. , Zhou, H. , Sakakima, M. , Fukasawa, H. , Yonemura, K. , Yamamoto, T. , and Hishida, A. (2005) Transient myofibroblast differentiation of interstitial fibroblastic cells relevant to tubular dilatation in uranyl acetate‐induced acute renal failure in rats. Virchows Arch. 446, 164–176 [DOI] [PubMed] [Google Scholar]
- 23. Sun, D. F. , Fujigaki, Y. , Fujimoto, T. , Goto, T. , Yonemura, K. , and Hishida, A. (2002) Mycophenolate mofetil inhibits regenerative repair in uranyl acetate‐induced acute renal failure by reduced interstitial cellular response. Am. J. Pathol. 161, 217–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhou, D. , Fu, H. , Xiao, L. , Mo, H. , Zhuo, H. , Tian, X. , Lin, L. , Xing, J. , and Liu, Y. (2018) Fibroblast‐specific β‐catenin signaling dictates the outcome of AKI. J. Am. Soc. Nephrol. 29, 1257–1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhou, D. , Li, Y. , Zhou, L. , Tan, R. J. , Xiao, L. , Liang, M. , Hou, F.F. , and Liu, Y. (2014) Sonic hedgehog is a novel tubule‐derived growth factor for interstitial fibroblasts after kidney injury. J Am. Soc. Nephrol. 25, 2187–2200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fu, H. , Tian, Y. , Zhou, L. , Zhou, D. , Tan, R. J. , Stolz, D. B. , and Liu, Y. (2017) Tenascin‐C is a major component of the fibrogenic niche in kidney fibrosis. J. Am. Soc. Nephrol. 28, 785–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xiao, L. , Xu, B. , Zhou, L. , Tan, R. J. , Zhou, D. , Fu, H. , Li, A. , Hou, F.F. , and Liu, Y. (2019) Wnt/β‐catenin regulates blood pressure and kidney injury in rats. Biochim. Biophys. Acta Mol. Basis Dis. 1865, 1313–1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xiao, L. , Zhou, D. , Tan, R. J. , Fu, H. , Zhou, L. , Hou, F. F. , and Liu, Y. (2016) Sustained activation of Wnt/β‐catenin signaling drives AKI to CKD progression. J. Am. Soc. Nephrol. 27, 1727–1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou, D. , Fu, H. , Zhang, L. , Zhang, K. , Min, Y. , Xiao, L. , Lin, L. , Bastacky, S. I. , and Liu, Y. (2017) Tubule‐derived Wnts are required for fibroblast activation and kidney fibrosis. J. Am.Soc.Nephrol. 28, 2322–2336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Maisel, A. S. , Wettersten, N. , van Veldhuisen, D. J. , Mueller, C. , Filippatos, G. , Nowak, R. , Hogan, C. , Kontos, M. C. , Cannon, C. M. , Müller, G. A. , Birkhahn, R. , Clopton, P. , Taub, P. , Vilke, G. M. , McDonald, K. , Mahon, N. , Nuñez, J. , Briguori, C. , Passino, C. , and Murray, P. T. (2016) Neutrophil gelatinase‐associated lipocalin for acute kidney injury during acute heart failure hospitalizations: the AKINESIS study. J. Am. Coll. Cardiol. 68, 1420–1431 [DOI] [PubMed] [Google Scholar]
- 31. Bonventre, J. V. (2014) Kidney injury molecule‐1: a translational journey. Trans. Am.Clin. Climatol. Assoc. 125, 293–299 [PMC free article] [PubMed] [Google Scholar]
- 32. Yang, L. , Besschetnova, T. Y. , Brooks, C. R. , Shah, J. V. , and Bonventre, J. V. (2010) Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 16, 535–543, 1p following 143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kramann, R. , Fleig, S.V. , Schneider, R.K. , Fabian, S.L. , DiRocco, D.P. , Maarouf, O. , Wongboonsin, J. , Ikeda, Y. , Heckl, D. , Chang, S. L. , Rennke, H. G. , Waikar, S. S. , and Humphreys, B. D. (2015) Pharmacological GLI2 inhibition prevents myofibroblast cell‐cycle progression and reduces kidney fibrosis. J. Clin. Invest. 125, 2935–2951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhou, D. , Tan, R. J. , and Liu, Y. (2016) Sonic hedgehog signaling in kidney fibrosis: a master communicator. Sci. China Life Sci. 59, 920–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bai, C. B. , Auerbach, W. , Lee, J. S. , Stephen, D. , and Joyner, A. L. (2002) Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development 129, 4753–4761 [DOI] [PubMed] [Google Scholar]
- 36. Zhou, D. , Li, Y. , Lin, L. , Zhou, L. , Igarashi, P. , and Liu, Y. (2012) Tubule‐specific ablation of endogenous β‐catenin aggravates acute kidney injury in mice. Kidney Int. 82, 537–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhou, D. , Tan, R. J. , Lin, L. , Zhou, L. , and Liu, Y. (2013) Activation of hepatocyte growth factor receptor, c‐met, in renal tubules is required for renoprotection after acute kidney injury. Kidney Int. 84, 509–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Little, M. H. , and Kairath, P. (2017) Does renal repair recapitulate kidney development? J. Am. Soc. Nephrol. 28, 34–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Humphreys, B. D. , Cantaluppi, V. , Portilla, D. , Singbartl, K. , Yang, L. , Rosner, M. H. , Kellum, J. A. , and Ronco, C. ; Acute Dialysis Quality Initiative (ADQI) XIII Work Group . (2016) Targeting endogenous repair pathways after AKI. J. Am. Soc. Nephrol. 27, 990–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhou, D. , and Liu, Y. (2016) Renal fibrosis in 2015: understanding the mechanisms of kidney fibrosis. Nat. Rev. Nephrol. 12, 68–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen, J. John, R. , Richardson, J. A. , Shelton, J. M. , Zhou, X. J. , Wang, Y. , Wu, Q. Q. , Hartono, J.R. , Winterberg, P.D. , and Lu, C. Y. (2011) Toll‐like receptor 4 regulates early endothelial activation during ischemic acute kidney injury. Kidney Int. 79, 288–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lech, M. , Gröbmayr, R. , Ryu, M. , Lorenz, G. , Hartter, I. , Mulay, S. R. , Susanti, H. E. , Kobayashi, K. S. , Flavell, R. A. , and Anders, H. J. (2014) Macrophage phenotype controls long‐term AKI outcomes–kidney regeneration versus atrophy. J. Am. Soc. Nephrol. 25, 292–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chiba, T. , Hukriede, N. , and de Caestecker, M. P. (2015) Kidney regeneration: lessons from development. Curr. Pathobiol. Rep. 3, 67–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dai, C. , Yang, J. , and Liu, Y. (2002) Single injection of naked plasmid encoding hepatocyte growth factor prevents cell death and ameliorates acute renal failure in mice. J. Am. Soc. Nephrol. 13, 411–422 [DOI] [PubMed] [Google Scholar]
- 45. Tan, R. J. , Zhou, D. , Zhou, L. , and Liu, Y. (2014) Wnt/β‐catenin signaling and kidney fibrosis. Kidney. Int. Suppl. (2011) 4, 84–90 [DOI] [PMC free article] [PubMed] [Google Scholar]