

Summary
We describe 36 patients with splenic marginal zone lymphoma (SMZL) with transformation (SMZL‐T), including 15 from a series of 84 patients with SMZL diagnosed at the Hospital Clinic of Barcelona (HCB) and 21 diagnosed with SMZL‐T in other centres. In the HCB cohort, the cumulative incidence of transformation at 5 years was 15%. Predictors for transformation were cytopenias, hypoalbuminaemia, complex karyotype (CK) and both the Intergruppo Italiano Linfomi (ILL) and simplified Haemoglobin, Platelet count, lactate dehydrogenase (LDH) and extrahilar Lymphadenopathy (HPLL)/ABC scores (P < 0·05). The only independent predictor for transformation in multivariate analysis was CK [hazard ratio (HR) 4·025, P = 0·05]. Patients with SMZL‐T had a significantly higher risk of death than the remainder (HR 3·89, P < 0·001). Of the 36 patients with SMZL‐T, one developed Hodgkin lymphoma and 35 a diffuse large B‐cell lymphoma, 71% with a non‐germinal centre phenotype. The main features were B symptoms, lymphadenopathy, and high serum LDH. CK was observed in 12/22 (55%) SMZL‐T and fluorescence in situ hybridisation detected abnormalities of MYC proto‐oncogene, basic helix‐loop‐helix transcription factor (MYC), B‐cell leukaemia/lymphoma 2 (BCL2) and/or BCL6 in six of 14 (43%). In all, 21 patients received immunochemotherapy, six chemotherapy, one radiotherapy and three splenectomy. The complete response (CR) rate was 61% and the median survival from transformation was 4·92 years. Predictors for a worse survival in multivariate analysis were high‐risk International Prognostic Index (HR 5·294, P = 0·016) and lack of CR (HR 2·67, P < 0·001).
Keywords: splenic marginal zone lymphoma, histological transformation, complex karyotype, prognostic factors, survival
Introduction
Splenic marginal zone lymphoma (SMZL) is an uncommon low‐grade lymphoma that accounts for ~2% of all lymphoproliferative disorders. 1 , 2 SMZL usually has an indolent course and a favourable outcome with a median overall survival (OS) of >10 years. 2 , 3 , 4 Risk stratification is based on the scores proposed by the Intergruppo Italiano Linfomi (IIL) 5 and the International Splenic Lymphoma Study Group (ISLSG) simplified Haemoglobin, Platelet count, lactate dehydrogenase (LDH) and extrahilar Lymphadenopathy (HPLL/ABC), 6 which consider low‐, intermediate‐ and high‐risk groups.
Despite the indolent behaviour of SMZL, about 10–15% of patients manifest aggressively and undergo transformation, usually to a diffuse large B‐cell lymphoma (DLBCL). 7 , 8 , 9 , 10 Although transformation of low‐grade lymphomas to high‐grade B‐cell neoplasms is well‐known, the information of such a scenario in SMZL regarding the incidence and factors associated with transformation and the clinico‐biological features and outcome of these patients is limited. 10 , 11 There have been few reports of patients with SMZL with confirmed transformation, 10 , 11 , 12 , 13 but either these included a small number of patients, or all types of marginal zone‐derived lymphomas with only a few of splenic origin. In addition, some reports were focussed on the pathological features or only described the incidence with few details on the clinico‐biological characteristics at transformation. 12 , 13
In the present study, we retrospectively analysed 36 patients with SMZL who underwent transformation (SMZL‐T), during the disease course or rarely at diagnosis. The aims of the present study were: (i) to describe the clinical and cytogenetic features, response to therapy and outcome from the time of transformation of these patients, and (ii) to assess the incidence and clinical and biological factors that could predict transformation in a cohort of 84 patients consecutively diagnosed with SMZL in a single institution.
Patients and methods
Patients
A total of 36 patients from different centres with histologically confirmed transformation of SMZL (SMZL‐T) were included in the study. In all, 15 were retrieved from a series of 84 patients with SMZL consecutively diagnosed at the Hospital Clinic of Barcelona (HCB) between March 1994 and August 2017, and 21 were referred from other institutions of the Spanish Lymphoma Group (GELTAMO) and UK. The cohort from the HCB was used to estimate the incidence and predictors of transformation. The following clinical and biological data at diagnosis and at the time of transformation were recorded: age; gender; performance status according to the Eastern Cooperative Oncology Group Performance Status (ECOG PS) scale and presence of B symptoms; haematological and biochemical parameters, serum LDH and β2‐microglobulin (B2M) levels, monoclonal component and autoimmune screen; number and size of nodal and extra‐nodal involvement; presence of splenomegaly; degree of bone marrow (BM) infiltration; Ann Arbor Stage and presence of bulky disease (defined as a tumour diameter >7 cm); prognostic scores according to the HPLL/ABC and IIL for SMZL, and the International Prognostic Index (IPI) in patients with SMZL‐T; cytology, immunophenotype, histology; conventional cytogenetics and fluorescence in situ hybridisation (FISH); treatment modality; relapse, survival and causes of death.
Response at the end of initial treatment of SMZL, and after each line of therapy was assessed according to conventional criteria based on the 2008 SMZL guidelines by computed tomography (CT) scans, BM biopsy and aspirate, peripheral blood (PB) cytology and flow cytometry. 14 Complete response (CR) was defined as the complete resolution of organomegaly and disease‐related symptoms, normalisation of blood counts without evidence of BM or PB infiltration by clonal B cells. Partial response (PR) was considered when there was a ≥50% improvement in all features. All the other situations were considered as no response (NR). Response duration was calculated from the time of response assessment for patients in CR and PR to relapse/progression. The responses to therapy in patients with SMZL‐T were evaluated with a CT scan and BM biopsy or aspirate according to conventional criteria for lymphomas. 15 In 16 (44%) of the patients with SMZL‐T, a positron emission tomography scan was also available to assess the response. CR was considered as the total disappearance of tumour masses, disease‐related symptoms, and normalisation of the initial abnormal tests for ≥1 month. PR was defined when the tumour mass decreased by ≥50% along with the disappearance of disease‐related symptoms. Disease relapse or progression was defined as the appearance of new symptoms or signs of the disease demonstrated by tissue biopsy, or other appropriate studies. OS and progression‐free survival (PFS) were calculated according to standard definitions. 16 All patients gave informed consent, according to the ethical standards of the ethic committees of the different institutions.
Cytology, histology review, immunohistochemistry (IHC) and cytogenetics
The diagnosis of SMZL was made according to the International Extranodal Lymphoma Study Group (IELSG) guidelines and World Health Organization (WHO), 14 , 17 based on morphology and flow cytometry of PB lymphocytes and BM aspirate and biopsy and/or splenic histology, with the aid of IHC. The diagnostic material in the HCB cohort was splenic histology in 52; lymph node biopsy in three; and BM and PB cytology, flow cytometry and cytogenetics in 57.
The diagnosis of SMZL‐T was made on tissue histology and IHC of the involved site in all patients, according to the updated WHO classification criteria. 17 All the histological material was reviewed by D.M., E.C. and E.M. To assess the clonality of the transformed samples, the FR1 region of the immunoglobulin heavy‐chain variable‐region (IGHV) was amplified using primers corresponding to the BIOMED‐2 multiplex polymerase chain reaction protocol.
Cytogenetic studies were carried out on G‐banded chromosomes in 77 patients. Karyotypes were described according to the International System for Human Cytogenetic Nomenclature (ISCN). 18 Complex karyotype (CK) was defined by the presence of at least three clonal chromosomal abnormalities, the abnormalities detected only by FISH were not considered for the definition of CK. FISH studies were performed in 56 patients using MetaSystems FISH probes and hybridisation and detection were performed according to the manufacturer’s protocols.
Statistical analysis
Differences among the patients’ characteristics were assessed using the chi‐square test (two‐tailed), the Student’s t‐test, or non‐parametric tests, when necessary. The actuarial survival analysis was performed using the Kaplan–Meier method and the differences assessed by the log‐rank test. Multivariate analyses were performed with the step‐wise proportional hazards Cox model. Cumulative incidence was used to assess the risk of transformation in the 84 patients with SMZL from the HCB and compared by use of Gray’s test. Multivariate analysis was performed by Fine–Gray test. CR achievement and transformation were considered time‐dependent variables and as such analysed by means of landmark, Mantel–Byar test or Cox model with time‐dependent variable. A P < 0·05 was considered statistically significant.
Results
Incidence and risk of transformation
The incidence of and risk factors for transformation were evaluated in the 84 patients in the HCB cohort, where 15 patients eventually developed SMZL‐T at a median (range) of 6·0 (0–22·8) years from diagnosis. The main disease features of these 84 patients at diagnosis are detailed in (Table SI). The median (range) age was 63 (32‐91) years. Prognostic scores according to the IIL and HPLL/ABC stratification showed that two‐thirds of the patients had intermediate‐ or high‐risk SMZL scores.
Cytogenetic studies at diagnosis in the patients that developed transformation are summarised in Table I. The cytogenetic results for the 54 patients who did not develop transformation are shown in Table SII. Aberrant karyotypes were found in 32/54 (59%) patients. The recurrent 7q deletion was detected in 18 (37%) patients by karyotype and as a single abnormality in 10/18 (56%). In case 43, with a t(1;18;14)(q41;q21;q32), in which IGH‐B‐cell leukaemia/lymphoma 2 (BCL2) rearrangement was demonstrated by FISH in 93% of cells, the spleen histology at diagnosis was studied exhaustively and other diagnosis such as follicular lymphoma (FL), mucosa‐associated lymphoid tissue (MALT) and nodal marginal zone lymphomas were ruled out. A CK was found in only seven of 54 (13%) patients.
Table 1.
Cytogenetics at diagnostic and at the time of transformation in the 36 patients with splenic marginal zone lymphoma with transformation (SMZL‐T).
| Case | Sample | Sample time | Cytogenetics | CK | FISH |
|---|---|---|---|---|---|
| 1 | PB | DX | 46,XY,t(1;14)(p36;q32),t(1;14;16)(q11;q32;p12),del(7)(q31q36)[4]/46,idem, der(5)t(5;6)(q21;q25), der(6)ins(6;5)(q25; q21q35)[6]/46,idem,del(5)(q21q23)[4]/46,XY[6] | Yes | |
| 1 | PB/LN | T | (*PB) 47,XY,add(1)(q11),del(1)(p?32p?36),‐5,‐6,del(7)(q31q36),‐9, der(12)t(5;12)(q13;p11),add(14)(q32)x2,+4mar[9] | Yes | (LN) MYC, BCL2: Normal |
| 2 | PB | DX | 47,XY,del(6)(q23q24),del(7)(q31q32),del(13)(q12q14),+mar[cp19]/46,XY[1] | Yes | IGH‐CCND1: Normal |
| 3 | PB | T | 47,X,‐X,del(1)(p22),+add(3)(p13),+5,add(6)(q24),del(7)(q32q36),t(14;18)(q32;q21)[16] | Yes | |
| 4 | PB | DX | 46,XX,trp(1)(q23q32)[19]/46,XX[1] | ||
| 4 | Spleen | DX | 46,XX,trp(1)(q23q32),add(3)(p25),add(16)(q?23)[cp5] | Yes | |
| 4 | *PB | T |
*46,XX,trp(1)(q23q32),der(2),add(3)(p25),de(11)(q22q23),t(14;18)(q32;q21)[cp20] *46,XX,trp(1)(q23q32),der(2),add(3)(p25),de(11)(q22q23),t(14;18)(q32;q21)[cp20] |
Yes | |
| 5 | BM | DX | 46,XX(20) | ||
| 5 | LN/*PB | T | (PB) 46‐47,XX,?dup(1)(q21q32),del(7)(q21q31),add(8)(p23),add(8)(q24),+10,del(11)(q22q23),del(12)(q23q24),del(18)(p11),+mar1,+mar2[cp8]/46,XX,inv(17)(p11q21)[2] | Yes | (*PB) 7q31: IGH‐CCND1, MYC: Normal |
| 7 | PB | DX | 46,XX[20] | ||
| 7 | Spleen | DX | 46,XX,del(7)(q22q32)[1],add14(q32)[3][cp3]/46,XX[27] | 7q31:deletion | |
| 7 | *PB | T | *46,XX,?dup(3)(q26q26),del(7)(q22q32),del(11)(q22q23)[cp6]/46,XX[6] | Yes | |
| 7 | LN | T | 46,XX,?dup(3)(q26q26),del(7)(q22q32),del(11)(q22q23)[cp6]/46,XX[6] | Yes | 7q31: deletion; BCL2, BCL6, MYC: Normal |
| 8 | LN | T | 46,XY,del(7)(q22q32)[4]/48,XY,+3,del(3)(p12p14),+5,del(7)(q22q32)[2]/46,XY[14] | Yes | MYC: amplification; BCL2, BCL6: Normal |
| 9 | PB | DX | 48,XY,+3,del(3)(q23q27),i(6)(p10),+18[17]/46,XY[3] | Yes | |
| 9 | *PB | T | *48,XY,+3,del(3)(q23q27), i(6)(p10),add(12)(p13),del(14)(q24q32),+18,del(20)(q?11q?13)[15] | Yes | BCL2, BCL6, MYC: Normal |
| 10 | PB | DX | 46,XX[20] | ATM, 13q14·3.3, TP53: Normal; CEP12: trisomy | |
| 10 | *PB | T | *46,XX[20] | CEP12: trisomy 12; del 6q; BCL2, BCL6, MYC, ATM, TP53, 13q14·3, IGH‐CCND1, IGH‐BCL2: Normal | |
| 11 | PB | DX | 46,XY,del(20)(q12q13)[4]/46,XY[16] | 7q32·1: Normal | |
| 11 | LN/*BM | T | (*BM) 46,XY,del(20)(q12q13)[4]/46,XY[16] | (LN) MYC rearranged: BCL2, BCL6: gain/amplification | |
| 12 | *PB | T | *46,XY[20] | TP53: deletion; BCL2, BCL6, MYC, 7q31: Normal | |
| 13 | PB | DX | 46,XX,dup(1)(q21q24)[3]/47,idem,+add(3)(q26),add(14)(q32)[3]/46,XX[8] | Yes | IGH: 30% rearranged; IGH‐BCL2: normal |
| 13 | PB | T | *49‐50,XX,dup(1)(q21q24)x2,+3,+5,+7,+add(14)(q32)[cp7]/46,XX[7] | Yes | |
| 14 | *PB | T | *46,XY[20] | IGH‐CCND1: Normal | |
| 15 | PB | DX | 46,XX[20] | ||
| 15 | PB | T | *46,XX,?del(11)(q22q23)[3]/45,idem,‐X[1]/45,X,‐X[5]/46,XX[12] | Yes | ATM, TP53, CEP12, 13q14·3: Normal |
| 16 | PB | DX | 46,XY[18] | IGH‐CCND1, ATM, TP53, CEP12, 13q14·3: Normal | |
| 16 | *PB | T | *46,XY[18] | BCL2, BCL6, MYC: Normal | |
| 17 | PB | DX | 46,XX,i(3)(q10)[4]/46,XX[16] | ||
| 17 | *PB | T | *46,XX,i(3)(q10)[4]/46,XX[4] | ||
| 18 | PB | DX | 46,XX,i(3)(q10)[4]/46,XX[12] | ||
| 18 | *PB | T | *46,XX,i(3)(q10)[6]/46,XX[17] | BCL6: gain; 9p21·3: biallelic deletion; IGH‐CCND1 and IGH‐BCL2: Normal | |
| 19 | Spleen/ LN | T | ND | BCL2, BCL6, MYC: Normal | |
| 20 | PB | DX | 47,XX,del(7)(q21q32),+19[15]/46,XX[4] | 7q31: deletion | |
| 21 | Spleen | DX | 46,XX,del(6)(q21q27)[3]/46,XX[17] | 7q31, IGH‐CCND1, IGH‐BCL2: Normal | |
| 21 | LN | T | ND | MYC: gain, BCL6: gain/rearranged; IGH: rearranged, TP53: Normal | |
| 23 | LN | T | MYC: amplification; BCL6: Normal | ||
| 24 | PB | DX | 50,XY,+5,+12,+19,+22[5]/46,XY[15] | Yes | |
| 25 | LN | DX | 46,XY,del(7)(q21q36)[1]/46,XY[22] | 7q32: 47% deletion; IGH: Normal | |
| 25 | Spleen | T | ND | 7q32: 22% deletion | |
| 26 | Spleen | DX | ND | IGH‐CCND1, API‐MALT1 BCL2: Normal | |
| 26 | Spleen | T | ND | 7q31: deletion; BCL2, BCL6, MYC: Normal | |
| 27 | LN | T | BCL6, BCL2, MYC: Normal | ||
| 28 | BM | DX | IGH‐CCND1, ATM, TP53, CEP12, 13q14·3: Normal | ||
| 33 | PB | DX | 51,XX,+3,+8,+11,+18,+18[4]/51,XX,+3,+8,+der(11)add(11)(q23),+18,+18[4]/46,XX[29] | Yes | CCND1: gain; 7q31: Normal |
| 33 | PB | T | *50,X,+der(3),+8,+11,+18[2]/51,idem,+18[1]/46,XX[17] | Yes | BCL2, BCL6, MYC: amplified; 7q31, CCND1‐IGH, ATM, TP53, CEP12, 13q14·3: Normal |
| 35 | Spleen | T | BCL2, MYC: Normal |
BM, bone marrow; CK, complex karyotype; DX, diagnosis; LN, lymph node; ND, not done; PB, peripheral blood; T, transformation; WCP, whole‐chromosome painting.
7q deletions/alterations are highlighted in bold font.
Indicates that the karyotype or FISH have been performed at the time of transformation but in the non‐transformed sample.
At diagnosis of SMZL, 33 of the 84 (39%) patients required treatment, while a watchful‐waiting policy was adopted in the remaining 51 (61%). Front‐line therapy is detailed in Table SI. After that, 27 of 33 (82%) patients achieved a response: nine (27%) CR, 18 (54%) PR; and six (18%) NR.
After a median (range) follow up of 6·67 (0·2–22·8) years, 37 of 84 (44%) patients with SMZL have died. The median OS was 12·2 years with a 10‐year OS of 63% [95% confidence interval (CI) 51–75] (Fig S1). Factors predicting for a shorter OS in univariate analysis are shown in Table SIII. In multivariate analysis using the Cox regression test, the initial variables that significantly predicted a shorter OS were age >60 years [hazard ratio (HR) 7·48, P = 0·001] and CK (HR 3·74, P = 0·011).
The cumulative incidence of transformation at 5 years from diagnosis was 15% (95% CI 7–23) (Figure 1A). Variables predicting a higher risk of transformation in the univariate analysis were haemoglobin level of <100 g/l (P = 0·03), lymphocyte count of <0.8 × 109/l (P = 0·032), platelet count of <100 × 109/l (P = 0·03), albumin of <35 g/l (P < 0·05) and CK (P = 0·026). In addition, intermediate‐ or high‐risk IIL (P = 0·008) and high‐risk HPLL (P = 0·038) predicted higher risk of transformation (Figure 1B, C). A watchful‐waiting policy at diagnosis did not significantly influence the risk of transformation. In multivariate analysis the only initial variable that significantly predicted risk of transformation was having a CK at diagnosis (HR 4·02, P = 0·05).
Fig 1.
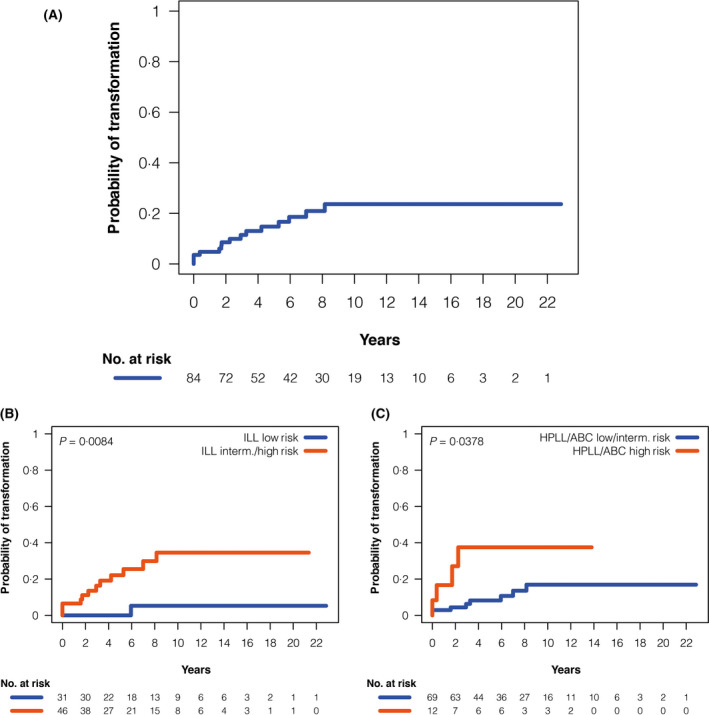
Risk of transformation from diagnosis in the 84 patients with SMZL from the HCB . (A) Cumulative incidence of histological transformation, (B) Cumulative incidence according to ILL risk score, and (C) Cumulative incidence according to HPLL/ABC risk score. HBC, Hospital Clinic of Barcelona; HPLL, Haemoglobin, Platelet count, lactate dehydrogenase and extrahilar Lymphadenopathy; ILL, Intergruppo Italiano Linfomi; SMZL, splenic marginal zone lymphoma.
Finally, when comparing the survival of SMZL‐T with that of the patients that did not transform, patients with SMZL‐T had a significantly shorter OS (HR 3·894, 95% CI 1·816–8·350; P < 0·001) as estimated by the Mantel–Byar test.
Clinico‐biological features of SMZL at transformation
As already mentioned, 21 patients with SMZL‐T were referred from other institutions (whose initial features at diagnosis are detailed in Table SIV) and 15 from the HCB cohort constituted the 36 patients with SMZL‐T whose main clinico‐biological features at the time of transformation are detailed in Table II. In five patients, the transformation occurred at diagnosis and in the remaining 31 at follow‐up at one or several sites (detailed in Table II).
Table 2.
Clinico‐biological features of the 36 patients with SMZL‐T at the time of transformation.
| Characteristics | N | n (%) |
|---|---|---|
| Age >60 years | 36 | 25 (69) |
| Females | 36 | 23 (64) |
| B symptoms | 35 | 24 (69) |
| ECOG Performance Status ≥2 | 35 | 11 (69) |
| Ann Arbor Stage III–IV | 36 | 33 (92) |
| Bulky disease (>7 cm) in lymph nodes | 30 | 8 (27) |
| Haemoglobin level <100 g/l | 36 | 31 (86) |
| Platelet count <100 × 109/l | 36 | 5 (14) |
| Raised LDH | 33 | 23 (70) |
| Raised B2M | 28 | 22 (79) |
| IPI high risk | 35 | 29 (83) |
| Transformation time | 36 | |
| At diagnosis | 5 (14) | |
| During follow‐up* | 31 (86) |
B2M, β2‐microglobulin; ECOG, Eastern Cooperative Oncology Group; LDH, lactate dehydrogenase.
Sites of transformation: 17 in one site: 11 in lymph node, four in bone marrow, one in spleen and one in nasal septum; 14 in more than one site: lymph node and bone marrow in six (four of them with also extra‐nodal involvement), lymph node and pleural effusion in three, spleen, liver, pleural effusion and ascites in two, lymph node and lip in one, spleen and bone marrow in one, and spleen, bone marrow and lymph node in one.
Most patients were aged >60 years, had anaemia and advanced stage with lymph node enlargement, B symptoms and high serum LDH as well as B2M levels (Table II). Histology of the tissue where transformation occurred showed a DLBCL in 35 patients and Hodgkin lymphoma (HL) in one. By IHC, the neoplastic cells in the patients with DLBCL expressed B‐cell antigens [cluster of differentiation (CD)20, CD79, paired box 5 (PAX5)] and were negative for CD3 and CD5. CD10 was positive in four patients, CD43 in five, and CD23 in one. Immunoglobulin (Ig)D expression was tested in 13 patients and seven were negative. Four of these seven IgD‐negative patients were previously positive in the spleen of the low‐grade SMZL. The Hans algorithm was used to determine the cell of origin in 17 (47%) of the patients with DLBCL with available data, and 12 (71%) showed a non‐germinal centre (non‐GCB) B‐cell phenotype and the remaining five (29%) a GCB phenotype. Tumour cell proliferation was high, with a median (range) Ki‐67 expression of 80 (40–100)%. p53 expression was positive in four of nine (44%) patients and Epstein–Barr virus (EBV) was demonstrated by EBV‐encoded small nuclear RNAs (EBERs), Epstein–Barr nuclear antigen (EBNA) and/or latent membrane protein 1 (LMP1) in one of the nine (11%). A clonal relationship could be demonstrated in the 29 patients analysed. In the three patients in whom transformation was diagnosed in the spleen, there was a concomitant remaining low‐grade SMZL component. One of these three patients was diagnosed as HL with CD30+, CD15+, CD79+, CD20– and EBV– Reed–Sternberg cells. The other two patients had large areas of DLBCL in the spleen with a residual minor small cell component. Therefore, these patients were not merely SMZL with the presence of scattered large cells.
Conventional Cytogenetics and FISH of SMZL at transformation
Information on conventional cytogenetics at the time of transformation or at diagnosis was available in 22 patients (Table I). Abnormal karyotypes were detected in 18/22 (82%) patients and these included seven with a deletion of 7q arm already present at diagnosis. Overall 7q deletion was detected in 35% of the patients (one only by FISH). Other recurrent abnormalities were add(14)(q32), t(14;18)(q32;q21), gains of 1q, 3q, trisomy 5, trisomy 18, and deletions of 6q and 11q. A CK was found in 12/22 (55%) patients and these comprised seven with CK already present at diagnosis and five that acquired the CK in the transformed sample (Table I).
FISH analysis for MYC proto‐oncogene, basic helix‐loop‐helix transcription factor (MYC), BCL2 or BCL6 genes in the transformed tissue showed abnormalities in six of 14 (43%) patients. BCL6 rearrangement plus gain (one patient), BCL6 gain/amplification (three), MYC rearrangement (one), MYC gain/amplification (four), and BCL2 gain/amplification (two). Additionally, a 17p/tumour protein p53 (TP53) deletion was detected only by FISH in one patient (case 12), and in a second patient FISH analysis showed a 9p21/CDKN2A homozygous deletion (case 18).
Therapy of SMZL‐T
Treatment data at the time of transformation was as follows: 26 patients received immunochemotherapy with rituximab (R): 21 R‐CHOP (R plus cyclophosphamide, doxorubicin, vincristine, prednisone), two R‐CVP (R plus cyclophosphamide, vincristine, and prednisone), three R‐ESHAP (R plus etoposide, methylprednisolone, cytarabine and cisplatin); six chemotherapy: four CHOP, one MINE (mesna, ifosfamide, mitoxantrone and etoposide) plus ESHAP, one FCM (fludarabine, cyclophosphamide, mitoxantrone); and one patient was treated with local radiotherapy. Four of these 33 patients who received systemic treatment had been previously splenectomised, where the diagnosis of transformation had been made. Three other patients did not receive systemic treatment and had only splenectomy. Of these three patients, one was not further treated, one died after diagnostic splenectomy (HL) and one was lost to follow‐up. The response was consolidated with autologous stem cell transplantation (ASCT) in three patients. Of the 36 patients who had systemic treatment or splenectomy, 35 were assessable for response. Of these, 22 (61%) patients achieved a CR, while 13 did not respond. Out of 22 responders, 12 continue in CR at a median follow‐up of 59 months, eight eventually relapsed and one died from therapy‐related acute myeloid leukaemia while still in remission of the lymphoma.
Outcome from transformation of the 36 patients with SMZL‐T
A total of 20 patients with SMZL‐T (56%) eventually died and the cause of death was lymphoma related in 19 and acute myeloid leukaemia in the remaining. The median survival from the time of transformation (SFT) of the 36 patients with SMZL‐T was 4·92 years, with a 5‐year SFT of 46% [95% confidence interval (CI) 27–65] (Figure 2A). The variables predicting poor SFT were age >60 years (P = 0·05), non‐ambulatory performance status (ECOG PS ≥2; P = 0·036), platelet count of <100 × 109/l (P < 0·001) and high‐risk IPI (P = 0·0053) (Figure 2B), as listed in Table III. In addition, patients not achieving a CR after treatment for transformation had a poorer survival (landmark 4 months; P < 0·001). In a multivariate analysis, including platelet counts, IPI and response (age and performance status were considered redundant with IPI), the variables that independently predicted a worse SFT in the final model with 34 patients were IPI (low/intermediate‐risk vs. high‐risk; HR 5·294; P = 0·016) and CR achievement (CR vs. no CR; HR 0·375; P < 0·001).
Fig 2.
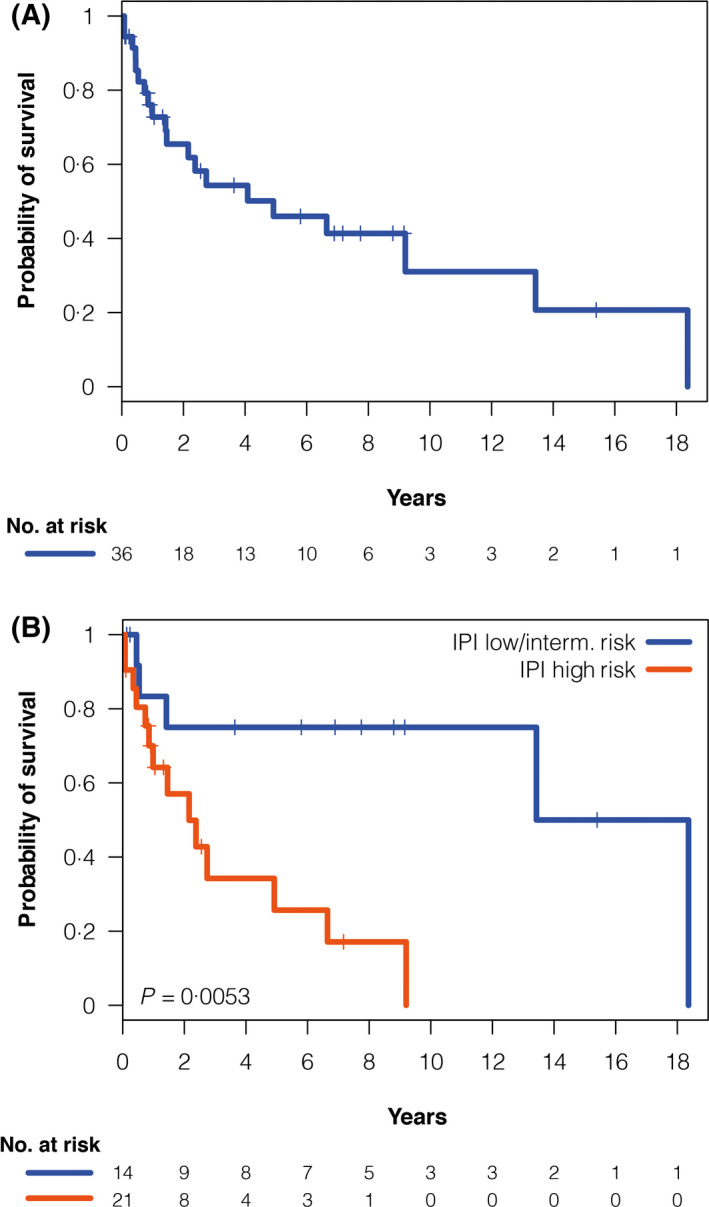
Survival from transformation in the 36 patients with SMZL‐T. (A) Overall survival from transformation, (B) Survival from transformation according to IPI. IPI, International Prognostic Index; SMZL‐T, splenic marginal zone lymphoma with transformation.
Table 3.
Univariate analysis for survival from transformation (SFT) of the 36 patients with SMZL‐T
| Variable | n | 5‐yearr SFT, % | HR (95% CI) | P |
|---|---|---|---|---|
| Age, years | ||||
| ≤60 | 11 | 67 | ||
| >60 | 25 | 34 | 3·3 (1–11·9) | 0·05 |
| ECOG performance status | ||||
| 0–1 | 25 | 58 | ||
| ≥2 | 11 | 14 | 2·7 (1·03–7·1) | 0·036 |
| Platelet count, × 109/l | ||||
| ≥100 | 5 | 30 | ||
| <100 | 31 | 50 | 12·1 (2·5–58·5) | <0·001 |
| IPI | ||||
| Low–intermediate | 14 | 75 | ||
| High | 21 | 26 | 5·2 (1·4–18·5) | 0·005 |
| Response to therapy* | ||||
| CR | 22 | 69 | ||
| No CR | 11 | 11 | 2·45 (1·5–4·0) | <0·001 |
HR, hazard ratio; CI, confidence interval; IPI, International Prognostic Index for aggressive lymphomas.
For response to therapy a landmark analysis at 4 months was established; therefore, patients with non‐evaluable response (one patient) or dying during this time (two) were excluded.
Discussion
Transformation to high‐grade lymphoma in low‐grade B‐cell neoplasms such as FL and chronic lymphocytic leukaemia/small lymphocytic lymphoma (CLL/SLL) is a well‐recognised feature in the natural history of these diseases. Data on transformation in another less frequent low‐grade lymphoma, such as SMZL, are limited and poorly documented. Information mainly derives from reports on small series of patients, and it has been mainly focussed on limited histopathological features 11 and/or clinical manifestations. 10 , 13
In the present study, we document the incidence of transformation, the risk factors responsible for this aggressive behaviour and the outcome of these patients in a series of SMZL diagnosed or referred to our Institution. To date, this is the largest series of patients with SMZL with a well‐documented histological transformation providing information not only on the incidence and clinico‐biological features of these patients, but also on the potential risk factors that predispose to this event.
As in other reports, DLBCL accounted for all our patients with SMZL‐T except for one that developed HL. IHC to determine the cell of origin in transformed tissues showed that the majority (72%) of patients studied had a non‐GCB phenotype. Rearrangements or gains of MYC, BCL6, and/or BCL2 were present in six of the 14 investigated patients. Of interest, one patient was EBV positive, a rare scenario that has been previously reported in one patient. 10 In our present patient, related clonality was proven by the detection of the same IGH rearrangement in the transformed and low‐grade tissues.
There are uncertainties regarding the incidence of transformation in SMZL. In retrospective series of patients with SMZL, transformation has been documented with a range of 11% to 19%. 3 , 7 , 10 , 11 , 12 , 19 , 20 In contrast to these data, Conconi et al. 13 documented a 5% incidence of transformation at 5 years in 85 patients with SMZL and thus lower than that previously reported. The shorter period of follow‐up in the Conconi et al. 13 series may explain the lower rate of transformation in the study. Overall, most of the data, including our present data, suggest that the incidence of transformation in SMZL is about 10–15%, intermediate of what is seen in CLL/SLL and FL. 21 , 22
The presence of extrahilar lymph node involvement has been suggested to be associated with transformation. 10 Two reports that included all types of MZL showed that raised LDH, 12 , 13 high‐risk HPLL and IPI scores, and lack of response to therapy predicted for a poor outcome in SMZL, but these variables were not predictors for transformation. 13 Early relapse has been associated with a worse outcome in FL 23 and recently in MZL 24 but to date has not been linked to transformation.
Our present data show that several clinical and biological features at diagnosis such as cytopenias, hypoalbuminaemia, high IIL and HPLL scores, lack of response to first‐line treatment, relapse, and CK significantly predicted transformation in univariate analysis. However, the only feature that remained significant in multivariate analysis was a CK. Furthermore, a CK at diagnosis was also significantly associated with a worse OS from diagnosis in the whole series. It is known that a CK (three or more chromosomal aberrations) or a high‐risk CK (five or more chromosomal aberrations) are very unfavourable prognostic factors in other B‐cell neoplasms, such as CLL/SLL. 25 The Splenic B‐cell Lymphoma Group retrospectively analysed the cytogenetic abnormalities in 330 patients with SMZL and documented that an aberrant karyotype with two or more abnormalities, 14q aberrations, and 17p deletion were associated with a worse prognosis in univariate analysis, but these abnormalities lost significance in the multivariate analysis and their impact as a risk factor for transformation was not investigated. 26 Our present finding on the association between a CK and poor outcome in SMZL parallels that documented in CLL. 25 For the first time, we demonstrate that a CK at diagnosis is a strong predictor for transformation and for a shorter OS in SMZL. This raises the issue as to whether conventional cytogenetics should be carried out in SMZL at diagnosis due to its major prognostic impact in the disease course of this lymphoma.
Information on the clinical features and outcome of SMZL‐T is scarce. The clinical manifestations in our present patients were similar to those previously documented by others. 13 We could not confirm that patients who transformed exclusively in PB and BM had a poorer prognosis as referred by Dungarwalla et al. 10 However, this scenario was very rare in our present series with only four patients having isolated BM transformation.
Immunochemotherapy is the standard therapy for de novo or transformed DLBCL. There are few reports on the management of SMZL‐T in the rituximab era. Dungarwalla et al. 10 described two of nine patients receiving immunochemotherapy and both achieved a CR. In the Else et al. 20 series, of the seven patients treated with rituximab and anthracycline‐based chemotherapy six achieved a CR and one a PR. In the Conconi et al. 13 series, responses were not recorded in the four patients with SMZL‐T that received regimens with rituximab and consolidation with an ASCT in two of them.
Our present series with a significantly larger number of patients treated with rituximab‐based regimens showed a higher response rate to immunochemotherapy compared with only chemotherapy (CR 67% vs. 50%), but this did not translate into a survival benefit. The response rate to immunochemotherapy in SMZL‐T compares to that of transformed FL (FL‐T) and is superior to that in Richter transformation (RS) of CLL. However, and in contrast to SMZL‐T and RS of CLL, the use of anthracycline‐based immunochemotherapy in FL‐T was associated with an improvement in the outcome with an OS of 66% at 5 years. 27 , 28
In summary, we describe the disease features, outcome, incidence and risk factors for transformation in the most extensive series ever reported of patients with SMZL that underwent transformation to a high‐grade lymphoma. Notable, is the finding of a CK at diagnosis that was not only a significant predictive factor for a higher risk of transformation but also for worse survival in SMZL. Further studies aimed at identifying potential molecular players responsible for transformation of SMZL will be relevant and will shed some light in the oncogenic mechanisms of such aggressive behaviour.
Conflict of interest
Maria Joao Baptista is currently an AstraZeneca employee. The remaining authors declare no competing financial interests.
Author contributions
Study conceptualization and design (Gabriela Bastidas‐Mora, Sílvia Beà, Armando López‐Guillermo, Estella Matutes). Data collection and data review (Gabriela Bastidas‐Mora, Sílvia Beà, Alba Navarro, Dolors Costa, Neus Villamor, Dolors Colomer, Mónica Lopez‐Guerra, María Rozman, Olga Balagué, Daniel Martínez, Elías Campo, Estella Matutes). Analysis and interpretation (Gabriela Bastidas‐Mora, Sílvia Beà, Armando López‐Guillermo, Estella Matutes). Provision of study material or patients (Eva Gine, Julio Delgado, Tycho Baumann, Laura Magnano, Alfredo Rivas‐Delgado, Maria Joao Baptista, Lourdes Escoda, Miguel Alcoceba, Margarita Blanes, Fina Climent, Andrew Wotherspoon, Armando López‐Guillermo). All authors reviewed, edited, and approved the manuscript.
Supporting information
Fig S1. Overall survival from transformation in the 84 patients from the Hospital Clinic of Barcelona (HCB).
Table SI. Characteristics of 84 patients with splenic marginal zone lymphoma (SMZL) at diagnosis.
Table SII. Cytogenetics at diagnosis in the 54 of 69 patients with splenic marginal zone lymphoma (SMZL) from the Hospital Clinic of Barcelona (HCB) who did not develop transformation.
Table SIII. Risk factors for overall survival in the 84 splenic marginal zone lymphoma (SMZL) Hospital Clinic of Barcelona (HCB) cohort (univariate analysis).
Table SIV. Characteristics at the time of diagnosis of the 21 patients with splenic marginal zone lymphoma with transformation (SMZL‐T) referred from other centres.
Acknowledgments
This work was supported by grants from Fondo de Investigaciones Sanitarias, Instituto de Salud Carlos III (PI17/01061 to Sílvia Beà, PI19/00887 to Armando López‐Guillermo), Fundacion AECC/CIBERONC (PROYE18020BEA to Sílvia Beà), and CIBERONC (CB16/12/00334 to Dolors Colomer, and CB16/12/00225 to Elías Campo), Generalitat de Catalunya Suport Grups de Recerca AGAUR, 2017‐SGR‐709 (to Sílvia Beà) 2017‐SGR‐1009 (to Dolors Colomer), 2017‐SGR‐1142 (to Elías Campo), Ministerio de Ciencia e Innovación (MCI) [Grant No. RTI2018‐094274‐B‐I00 (to Elías Campo)] and the European Regional Development Fund ‘Una manera de fer Europa’. Elías Campo is an Academia Researcher of the ‘Institució Catalana de Recerca i Estudis Avançats’ (ICREA) of the Generalitat de Catalunya. This work was partially developed at the Centro Esther Koplowitz (CEK), Barcelona, Spain. We are grateful to Miriam Prieto, Cándida Gómez, Amparo Arias, and Silvia Martín for excellent technical assistance. We are also very grateful to all patients with SMZL who have participated in this study.
Contributor Information
Sílvia Beà, Email: sbea@clinic.cat.
Estella Matutes, Email: estella.matutes.juan@gmail.com.
References
- 1. Armitage JO, Weisenburger DD. New approach to classifying non‐Hodgkin’s lymphomas: clinical features of the major histologic subtypes. Non‐Hodgkin’s lymphoma classification project. J Clin Oncol. 1998;16:2780–95. [DOI] [PubMed] [Google Scholar]
- 2. Liu L, Wang H, Chen Y, Rustveld L, Liu G, Du XL. Splenic marginal zone lymphoma: a population‐based study on the 2001–2008 incidence and survival in the United States. Leuk Lymphoma. 2013;54:1380–6. [DOI] [PubMed] [Google Scholar]
- 3. Parry‐Jones N, Matutes E, Gruszka‐Westwood AM, Swansbury GJ, Wotherspoon AC, Catovsky D. Prognostic features of splenic lymphoma with villous lymphocytes: a report on 129 patients. Br J Haematol. 2003;120:759–64. [DOI] [PubMed] [Google Scholar]
- 4. Olszewski AJ. Survival outcomes with and without splenectomy in splenic marginal zone lymphoma. Am J Hematol. 2012;87:E119–22. [DOI] [PubMed] [Google Scholar]
- 5. Arcaini L, Lazzarino M, Colombo N, Burcheri S, Boveri E, Paulli M, et al. Splenic marginal zone lymphoma: a prognostic model for clinical use. Blood. 2006;107:4643–9. [DOI] [PubMed] [Google Scholar]
- 6. Montalbán C, Abraira V, Arcaini L, Domingo‐Domenech E, Guisado‐Vasco P, Iannitto E, et al. Risk stratification for Splenic Marginal Zone Lymphoma based on haemoglobin concentration, platelet count, high lactate dehydrogenase level and extrahilar lymphadenopathy: development and validation on 593 cases. Br J Haematol. 2012;159:164–71. [DOI] [PubMed] [Google Scholar]
- 7. Lenglet J, Traullé C, Mounier N, Benet C, Munoz‐Bongrand N, Amorin S, et al. Long‐term follow‐up analysis of 100 patients with splenic marginal zone lymphoma treated with splenectomy as first‐line treatment. Leuk Lymphoma. 2014;55:1854–60. [DOI] [PubMed] [Google Scholar]
- 8. Piris MA, Onaindía A, Mollejo M. Splenic marginal zone lymphoma. Best Pract Res Clin Haematol. 2017;30:56–64. [DOI] [PubMed] [Google Scholar]
- 9. Thieblemont C, Davi F, Noguera ME, Brière J, Bertoni F, Zucca E, et al. Splenic marginal zone lymphoma: current knowledge and future directions. Oncology. 2012;26 194–202. [PubMed] [Google Scholar]
- 10. Dungarwalla M, Appiah‐Cubi S, Kulkarni S, Saso R, Wotherspoon A, Osuji N, et al. High‐grade transformation in splenic marginal zone lymphoma with circulating villous lymphocytes: the site of transformation influences response to therapy and prognosis. Br J Haematol. 2008;143:71–4. [DOI] [PubMed] [Google Scholar]
- 11. Camacho FI, Mollejo M, Mateo MS, Algara P, Navas C, Hernández JM, et al. Progression to large B‐cell lymphoma in splenic marginal zone lymphoma: a description of a series of 12 cases. Am J Surg Pathol. 2001;25:1268–76. [DOI] [PubMed] [Google Scholar]
- 12. Alderuccio JP, Zhao W, Desai A, Gallastegui N, Ramdial J, Kimble E, et al. Risk factors for transformation to higher‐grade lymphoma and its impact on survival in a large cohort of patients with marginal zone lymphoma from a single institution. J Clin Oncol. 2018;36:3370–80. [DOI] [PubMed] [Google Scholar]
- 13. Conconi A, Franceschetti S, Aprile von Hohenstaufen K, Margiotta‐Casaluci G, Stathis A, Moccia AA, et al. Histologic transformation in marginal zone lymphomas. Ann Oncol. 2015;26:2329–35. [DOI] [PubMed] [Google Scholar]
- 14. Matutes E, Oscier D, Montalban C, Berger F, Callet‐Bauchu E, Dogan A, et al. Splenic marginal zone lymphoma proposals for a revision of diagnostic, staging and therapeutic criteria. Leukemia. 2008;22:487–95. [DOI] [PubMed] [Google Scholar]
- 15. Cheson BD, Horning SJ, Coiffier B, Shipp MA, Fisher RI, Connors JM, et al. Report of an international workshop to standardize response criteria for non‐Hodgkin’s lymphomas. NCI Sponsored International Working Group. J Clin Oncol. 1999;17:1244. [DOI] [PubMed] [Google Scholar]
- 16. Cheson BD, Pfistner B, Juweid ME, Gascoyne RD, Specht L, Horning SJ, et al. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25:579–86. [DOI] [PubMed] [Google Scholar]
- 17. Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. WHO classification of tumours of haematopoietic and lymphoid tissues (Revised 4th Edition). Lyon: IARC; 2017. [Google Scholar]
- 18. McGowan‐Jordan J, Hastings RJ, Moore S, Editors. ISCN 2020: an international system for human cytogenomic nomenclature (2020). Basel, Switzerland: S. Karger AG; 2020. [Google Scholar]
- 19. Xing KH, Kahlon A, Skinnider BF, Connors JM, Gascoyne RD, Sehn LH, et al. Outcomes in splenic marginal zone lymphoma: analysis of 107 patients treated in British Columbia. Br J Haematol. 2015;169:520–7. [DOI] [PubMed] [Google Scholar]
- 20. Else M, Marín‐Niebla A, de la Cruz F, Batty P, Ríos E, Dearden CE, et al. Rituximab, used alone or in combination, is superior to other treatment modalities in splenic marginal zone lymphoma. Br J Haematol. 2012;159:322–8. [DOI] [PubMed] [Google Scholar]
- 21. Alonso‐Álvarez S, Magnano L, Alcoceba M, Andrade‐Campos M, Espinosa‐Lara N, Rodríguez G, et al. Risk of, and survival following, histological transformation in follicular lymphoma in the rituximab era. A retrospective multicenter study by the Spanish GELTAMO group. Br J Haematol. 2017;178:699–708. [DOI] [PubMed] [Google Scholar]
- 22. Rossi D, Cerri M, Capello D, Deambrogi C, Rossi FM, Zucchetto A, et al. Biological and clinical risk factors of chronic lymphocytic leukaemia transformation to Richter syndrome. Br J Haematol. 2008;142:202–15. [DOI] [PubMed] [Google Scholar]
- 23. Casulo C, Byrtek M, Dawson KL, Zhou X, Farber CM, Flowers CR, et al. Early relapse of follicular lymphoma after rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone defines patients at high risk for death: an analysis from the national lymphocare study. J Clin Oncol. 2015;33:2516–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Luminari S, Merli M, Rattotti S, Tarantino V, Marcheselli L, Cavallo F, et al. Early progression as a predictor of survival in marginal zone lymphomas: an analysis from the FIL‐NF10 study. Blood. 2019;134:798–801. [DOI] [PubMed] [Google Scholar]
- 25. Baliakas P, Jeromin S, Iskas M, Puiggros A, Plevova K, Nguyen‐Khac F, et al. Cytogenetic complexity in chronic lymphocytic leukemia: definitions, associations, and clinical impact. Blood. 2019;133:1205–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Salido M, Baró C, Oscier D, Stamatopoulos K, Dierlamm J, Matutes E, et al. Cytogenetic aberrations and their prognostic value in a series of 330 splenic marginal zone B‐cell lymphomas: a multicenter study of the Splenic B‐Cell Lymphoma Group. Blood. 2010;116:1479–88. [DOI] [PubMed] [Google Scholar]
- 27. Link BK, Maurer MJ, Nowakowski GS, Ansell SM, Macon WR, Syrbu SI, et al. Rates and outcomes of follicular lymphoma transformation in the immunochemotherapy era: a report from the University of Iowa/MayoClinic specialized program of research excellence molecular epidemiology resource. J Clin Oncol. 2013;31:3272–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tsimberidou AM, O’Brien S, Khouri I, Giles FJ, Kantarjian HM, Champlin R, et al. Clinical outcomes and prognostic factors in patients with Richter’s syndrome treated with chemotherapy or chemoimmunotherapy with or without stem‐cell transplantation. J Clin Oncol. 2006;24:2343–51. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1. Overall survival from transformation in the 84 patients from the Hospital Clinic of Barcelona (HCB).
Table SI. Characteristics of 84 patients with splenic marginal zone lymphoma (SMZL) at diagnosis.
Table SII. Cytogenetics at diagnosis in the 54 of 69 patients with splenic marginal zone lymphoma (SMZL) from the Hospital Clinic of Barcelona (HCB) who did not develop transformation.
Table SIII. Risk factors for overall survival in the 84 splenic marginal zone lymphoma (SMZL) Hospital Clinic of Barcelona (HCB) cohort (univariate analysis).
Table SIV. Characteristics at the time of diagnosis of the 21 patients with splenic marginal zone lymphoma with transformation (SMZL‐T) referred from other centres.