

Abstract
The shift from fossil carbon sources to renewable ones is vital for developing sustainable chemical processes to produce valuable chemicals. In this work, value‐added formamides were synthesized in good yields by the reaction of amines with C2 and C3 biomass‐based platform molecules such as glycolic acid, 1,3‐dihydroxyacetone and glyceraldehyde. These feedstocks were selectively converted by catalysts based on Cu‐containing zeolite 5A through the in situ formation of carbonyl‐containing intermediates. To the best of our knowledge, this is the first example in which all the carbon atoms in biomass‐based feedstocks could be amidated to produce formamide. Combined catalyst characterization results revealed preferably single CuII sites on the surface of Cu/5A, some of which form small clusters, but without direct linking via oxygen bridges. By combining the results of electron paramagnetic resonance (EPR) spin‐trapping, operando attenuated total reflection (ATR) IR spectroscopy and control experiments, it was found that the formation of formamides might involve a HCOOH‐like intermediate and .NHPh radicals, in which the selective formation of .OOH radicals might play a key role.
Keywords: amines, biomass-based feedstocks, C−C bond cleavage, electron paramagnetic resonance spectroscopy, formylation, selective oxidation
Waste not, want not: Oxidative N‐formylation of amines to formamides by sustainable C2 and C3 biomass‐based carbonyl sources such as glycolic acid, 1,3‐dihydroxyacetone and glyceraldehyde is catalyzed by very few CuII single sites in zeolite 5A with unprecedented 100 % utilization efficiency of carbon atoms.

Introduction
Dwindling fossil resources and increasing environmental concerns have stimulated growing research activities towards efficient use of renewable biomass as the most promising candidate for the production of fuels and chemicals. [1] Different from fossil sources consisting of carbon and hydrogen, biomass has a rich content of oxygen atoms, which requires new technologies and processes based on carbohydrates. Production of syngas (CO+H2) from biomass by high‐temperature reforming has attracted increasing attention in the past decades due to the beneficial atomic ratio of carbon and oxygen in biomass molecules and the high importance of syngas in the production of hydrocarbon fuels or chemicals. [2] However, this process is limited by high energy consumption and low selectivity. In comparison with this, the direct transformation of biomass and its platform molecules into building blocks under mild conditions is a more attractive approach. As a successful example, formic acid has been recently generated from biomass and its derivatives by oxidative cleavage of C−C bonds; [3] this means that biomass could be a potential carbonyl source in oxidative carbonylation. Thus, it would be highly desired if biobased feedstocks were directly used as carbonyl sources in the synthesis of valuable formamides. However, until now, reports about such type of synthesis processes are few. [4]
Formamides are important nitrogen‐containing chemicals with widespread applications in organic synthesis as solvents, building blocks and reaction intermediates. [5] Various methods have been developed for the production of formamides, [6] among which N‐formylation of amines with CO has been successfully applied in industrial production due to high atomic economy. [7] However, the high toxicity of CO brings about an increasing concern on production safety. Therefore, the development of new carbonyl sources to replace the use of toxic CO in N‐formylation of amines is highly desired. Molecules often used for this purpose include methanol, formaldehyde, formic acid and CO2.[ 6 , 8 ] Although such protocols have been successful in some cases, they suffer from problems such as expensive cost, challenging equipment requirements, harsh reaction conditions, low product selectivity and poor economy. From a sustainability point of view, it would be ideal if biomass‐based feedstocks can be directly used as carbonyl sources for the synthesis formamides by N‐formylation of amines. Apart from the construction of C−N bonds, this reaction involves the selective cleavage of C−C bonds, which is a key step in the formation of carbonyl‐constructing intermediates. Remarkably, this C−C splitting can be achieved already at room temperature, when copper‐based catalysts and hydrogen peroxide as oxidant are used.[ 4a , 9 ]
Extending our previous efforts in the production of nitrogen‐containing fine chemicals from biomass‐based feedstocks, [10] we have developed copper‐containing catalytic systems in which C2 and C3 biomass‐based feedstocks are directly used in situ as carbonyl sources for the efficient production of formamide. Different from our previous works with still insufficient carbon atom efficiency, [4] the total utilization of carbon atoms was achieved for the first time with the best catalyst of this series.
Results and Discussion
Catalytic tests
The N‐formylation of aniline with glycolic acid (GA) as carbonyl source and aqueous H2O2 (35 wt %) as oxidant over different 0.5 wt % Cu‐containing catalysts (Table 1) was chosen as a model reaction in dioxane as solvent. Remarkably, all catalysts showed excellent activity in the screening tests for the formation of the desired formamide product with byproducts being azobenzene and azoxybenzene formed by oxidation of excess aniline (Table 1, entries 1–3). In the case of Cu/MCM and Cu/HY, the obtained product yields (calculated on the basis of the number of carbon atoms in GA) were even higher than 100 %. This is due to oxidation of the dioxane solvent [11] which has been proven by blank experiments without GA (Table 1, entries 4–6). Interestingly, Cu/5A is not able to form the desired products in the absence of GA, however, it is the most active catalyst in converting the carbon atoms of GA to the formamide product. Therefore, it has been used for optimization of the reaction conditions (Table S1 in the Supporting Information). The solvent screening results showed that dioxane is the best reaction medium because much lower product yields were obtained when using H2O, CH3CN, toluene, xylene and octane as solvents (Table S1, entries 1–6). The optimized combination of 3 mmol aniline and 6 mmol H2O2 are required for excellent product yields (Table S1, entries 7–14). Besides, 25 mg catalyst and a reaction time of 9 h were sufficient to obtain 98 % product yield (Table S1, entries 15 and 16). No higher yield has been obtained after 9 h and again it is confirmed that the Cu/5A catalyst cannot catalyze the reaction of excess aniline with dioxane to formamides. Much lower product yields were obtained when reducing the reaction temperature to 50 °C (Table 1, entry 7), and this is true also for the Cu‐free parent zeolite, by which only 19–37 % formamide yield were obtained (Table 1, entries 8 and 9), indicating that it is the Cu centers accounting for the good catalytic activity. Excellent product yields could be maintained even when the Cu content was reduced to 0.2 wt %, but the product yield decreased slightly when the Cu content was raised to 1.0 wt % in Cu/5A, which might be due to a loss of Cu dispersion (entries 10 and 11).
Table 1.
Catalysts screening and reaction conditions optimization.[a]
|
| ||||||
|---|---|---|---|---|---|---|
|
|
Catalysts |
GA [mmol] |
PhNH2 [mmol] |
H2O2 [mmol] |
Yield [%] |
UECA [%] |
|
1 |
Cu/5A |
0.5 |
5 |
6 |
99 |
99 |
|
2 |
Cu/MCM‐41 |
0.5 |
5 |
6 |
113 |
25 |
|
3 |
Cu/HY |
0.5 |
5 |
6 |
108 |
17 |
|
4 |
Cu/5A |
– |
5 |
6 |
0 |
– |
|
5 |
Cu/MCM‐41 |
– |
5 |
6 |
88 |
– |
|
6 |
Cu/HY |
– |
5 |
6 |
91 |
– |
|
7[b,c] |
Cu/5A |
0.5 |
3 |
6 |
64 |
– |
|
8[b,d] |
5A |
0.5 |
3 |
6 |
37 |
– |
|
9[c] |
– |
0.5 |
3 |
6 |
19 |
– |
|
10[b,d,e] |
Cu/5A |
0.5 |
3 |
6 |
98 |
|
|
11[b,d,f] |
Cu/5A |
0.5 |
3 |
6 |
88 |
|

[a] Reaction conditions: 100 mg catalyst, 0.5 mmol GA, 5 mmol aniline, 6 mmol H2O2 (35 wt %), dioxane 2 mL, 70 °C, 12 h. Yields were determined by GC‐FID with biphenyl as the external standard. UECA means the utilization efficiency of carbon atoms in GA. UECA=100 %×(n formamide product yields with GA−n formamide product yields without GA)/n carbon atoms in GA (mol/mol). [b] 25 mg catalyst. [c] 50 °C. [d] 9 h. [e] 0.2 wt % Cu/5A catalyst. [f] 1.0 wt % Cu/5A catalyst.
The reusability, being a vital parameter for the performance of heterogeneous catalysts, was tested for the most active catalyst Cu/5A (Figure S1). In order to accurately evaluate the stability, the reaction time was shortened to 1.5 h to analyze the initial reaction activity. After each run, the catalyst was recovered by simple centrifugation, washing by ethanol (3×8 mL) and drying at 60 °C for 2 h, and reused without further treatment. Product yields of 27, 15, 13 and 15 % were obtained in the 1st, 2nd, 3rd and 4th. This means that the catalyst lost stability only during the 1st run but remained stable afterwards. The decline in activity is most probably due to the loss in the Cu content, which decreased from 0.50 wt % in the fresh to 0.13 wt % after the 1st run, but then maintained stable. To test if the leached Cu is responsible for the reaction, the catalyst was removed by filtration after reacting for 3 h when the product yield was 73 % (Scheme S1a). When the reaction was run for another 6 h only with the separated Cu containing solution, no further increase of the product yield was obtained. This shows clearly that Cu supported on zeolite 5A is the real active species.
After the reaction conditions had been optimized, the substrate scope of amines and its tolerance towards functional groups were evaluated (Table 2). Good to excellent product yields were obtained when aniline derivatives with different groups such as −CH3, −F and −Cl were used as substrates. Besides, a variety of other amines such as benzylic amines, aliphatic primary amines, dibenzylamine, aromatic as well as aliphatic cyclic secondary amines were well tolerated in this system and moderate to good product yields were obtained although in some cases higher amounts of amines were required.
Table 2.
N‐formylation of different amines with GA.[a]
|
|
|---|
|
|


[a] Yields were determined by GC‐FID with biphenyl as the external standard. [b] 5 mmol amine.
Inspired by the success of glycolic acid as in situ carbonyl source in the N‐formylation of amines, other biomass‐based C3 feedstocks were also explored as carbonyl source to synthesize formamides from aniline. As expected, excellent product yields were obtained when 1,3‐dihydroxyacetone and glyceraldehyde were used as the carbonyl source (Table 3). Based on this and the result that the formamide product can't be formed by the reaction of aniline and dioxane, a possible conclusion is that all the carbon atoms in the two C3 molecules were transferred into the formamide products, which means that our catalytic system is be very active for the total utilization of carbon atoms in C2 and C3 biomass‐based feedstocks.
Table 3.
N‐formylation of aniline with C3 biomass‐based feedstocks.[a]

[a] Yields were determined by GC‐FID with biphenyl as the external standard.
Catalyst characterization
In order to derive structure‐reactivity correlations, the catalysts were characterized by a variety of standard techniques such as elemental analysis, BET surface area, X‐ray diffraction (XRD), and electron paramagnetic resonance (EPR). Inductively coupled plasma optical emission spectroscopy (ICP‐OES) showed that the actual Cu content in all catalysts is between 0.49–0.51 wt % and, thus, very close to the targeted value of 0.5 wt % (Table 4). However, the Cu content in the used Cu/5a catalyst decreased greatly from 0.50 to 0.14 wt % after four runs, suggesting an obvious Cu leaching. N2‐adorption–desorption analysis revealed that the introduction of Cu in zeolite 5A increased the specific surface area and the total pore volume, while the reverse effect was observed in the case of MCM‐41 and HY (Table 4). The higher specific surface area of Cu/5A may be due to the fact that introduction of Cu breaks some of the 5A zeolite's cages and provide more sites to adsorb N2 molecules. [12] By comparing the pore size distribution of the 5A support before and after introducing Cu, more micropores (0.69–0.90 nm) can be seen after introducing Cu ions (Figure S2). Compared to fresh Cu/5A catalyst, the BET surface area of the used Cu/5A catalyst declined significantly but the total pore volume and average pore size increased, which can be attributed to the destruction of the 5A support and the formation of more large pores confirmed by the pore size distribution curve (Figure S2). XRD powder patterns of the supports before and after Cu ion exchange are virtually the same, confirming that the crystal structure is preserved (Figure S3). As expected for Cu contents as low as 0.50 wt %, no XRD peaks of Cu species were observed, suggesting that the supported Cu species are highly dispersed or amorphous. The used Cu/5A catalyst showed a strongly changed XRD pattern with much weaker reflections, which indicates a clear structure change of 5A support. This result is consistent with N2‐adorption‐desorption analysis. The XPS spectrum of fresh Cu/5A catalyst (Figure S4) showed a weak CuII satellite peak with a binding energy around 943 eV and a typical Cu 2p3/2 binding energy of 933.9 eV, [13] thus suggesting the presence of surface CuII species. No Cu XPS signals were obtained for the used Cu/5A, which might be due to the low Cu content (0.14 wt %) resulting from Cu leaching during the reaction. Aberration corrected scanning transmission electron micrographs of catalyst Cu/5A obtained by a high angle annular dark field detector (HAADF‐STEM) showed a hollow needle‐like shape of the zeolite crystals. In very thin regions, a few small structures with extensions below 1 nm could be observed that might fit with Cu (marked by arrows in Figure S5). This agrees with EDX data, showing small copper peaks in large agglomerations of support particles.
Table 4.
Basic physical property of catalysts.
|
|
Catalysts |
Cu content[a] |
SA[b] |
PV[c] |
APD[d] |
|---|---|---|---|---|---|
|
|
|
[wt %] |
[m2 g−1] |
[cc g−1] |
[nm] |
|
1 |
Cu/5A |
0.50 |
521.6 |
0.51 |
3.9 |
|
2 |
5A |
– |
355.7 |
0.39 |
4.4 |
|
3 |
Cu/MCM‐41 |
0.51 |
834.1 |
0.98 |
4.6 |
|
4 |
MCM‐41 |
– |
949.8 |
1.09 |
4.6 |
|
5 |
Cu/HY |
0.49 |
912.1 |
0.56 |
2.5 |
|
6 |
HY |
– |
936.6 |
0.57 |
2.5 |
|
7[e] |
Cu/5A |
0.14 |
181.6 |
0.69 |
15.3 |
[a] Determined by ICP‐OES. [b] Specific surface area. [c] Total pore volume. [d] Average pore diameter. [e] After the 4th run.
To gain specific insight into the structure of the CuII centers, EPR spectra were measured at 298 and 93 K, respectively (Figure 1). The EPR spectrum of Cu/HY shows an axial signal characteristic of isolated CuII ions. [14] The parallel component of the g tensor (g ∥=2.346) shows hyperfine structure (hfs) splitting with a hfs constant of A ∥=133 G, due to the coupling of the single electron spin of CuII (d9, S=1/2) with the nuclear spin of 65,63CuII (I=3/2), while hfs is not resolved at the perpendicular component of the g tensor (g ⊥=2.08). The preferential formation of isolated CuII species is also suggested by a comparison of the EPR intensity (double integral of the signal) of Cu/HY measured at 298 and 93 K. For pure paramagnetic CuII species without any mutual magnetic interactions, the intensity of the EPR signal is inversely proportional to temperature. This is the case for catalyst Cu/HY, for which the intensity ratio of I 93/I 298=3.18 is nearly the same as that expected for pure paramagnetic behavior (I 93/I 298=3.20). A similar CuII signal is observed for Cu/MCM‐41, suggesting the formation of CuII single sites with similar structure, but the lower ratio of I 93/I 298=2.51 suggests minor anti‐ferromagnetic interactions that might point to closer vicinity of the CuII sites than in Cu/HY, and this is also true for sample Cu/5A. In this sample, the hfs signal of CuII single sites is least resolved due to superposition on a broad singlet resulting from magnetically interacting CuII species. The presence of the latter is also confirmed by the lowest intensity ratio of I 93/I 298=2.31. However, as discussed below in relation to the EXAFS results, we think that this broad signal does not stem from moieties in which CuII ions are directly connected by Cu−O−Cu bridges as in copper oxide. Instead, it might rather reflect dipolar interactions of closely neighboring but not directly connected single CuII ions.
Figure 1.

EPR spectra of the catalysts measured at 93 and 298 K.
To test the accessibility of the CuII ions inside the pore systems of the three catalysts, their interaction with 1,4‐dioxane was also investigated by EPR (Figure S6). Obvious changes of the EPR signals were only observed for Cu/MCM‐41 and Cu/HY after contact with 1,4‐dioxane while the spectrum of Cu/5A remained unchanged. This suggests that 1,4‐dioxane cannot enter the small pores of Cu/5A. This might be the reason for the fact that Cu/5A utilizes only the carbon atoms in glycolic acid but not in 1.4‐dioxane for the reaction of aniline.
To further analyze the valence state and the local environment of the Cu sites, catalyst Cu/5A was characterized by X‐ray absorption spectroscopy (XAS) at the Cu K‐edge. The edge energy in the X‐ray absorption near edge structure (XANES) spectrum of Cu/5A is similar to that of a CuO reference but higher than that of Cu2O and Cu foil standard samples, indicating that the Cu species in Cu/5A are essentially divalent (Figure 2a), in agreement with the EPR spectrum (Figure 1). The profile of the edge in Cu/5A is similar to that of a Cu(NO3)2 ⋅ 3 H2O reference but differs slightly from that of a CuO standard sample. This points to a difference in the local environment of CuII sites. While the CuII ions in Cu(NO3)2 are separated by the nitrate anions, [15] they are connected by Cu−O−Cu bridges in both CuO and Cu2O. In the extended X‐ray absorption fine structure (EXAFS) spectrum, a peak at 1.96 Å is observed for the first Cu−O shell in all oxidic references (Table 5, Figure 2b). However, an additional peak for the second Cu shell occurs around 2.4 Å only for Cu2O and CuO, known to contain Cu−O−Cu bridges. [16] Such peak is essentially absent in the EXAFS spectrum of catalyst Cu/5A (Figure 2b), indicating that this sample does not contain direct Cu−O−Cu connections as typical for CuO particles and, thus, is widely free of CuxOy aggregates. It means also that the small dots marked by arrows in in the HAADF‐STEM micrographs of Figure S5 might reflect ensembles of more or less single Cu ions rather than Cu x O y clusters connected by Cu−O−Cu bridges. Likewise, as mentioned above, the broad background signal in the Cu/5A EPR spectra of Figure 1 might be caused by dipolar interactions single CuII ions in sufficiently close vicinity but without direct Cu−O−Cu bridges.
Figure 2.

a) Normalized Cu K‐edge XANES spectra and b) k3‐weighted FT‐EXAFS spectra for Cu/5A.
Table 5.
EXAFS curve‐fitting results.
|
Sample |
Shell |
N |
Ri [Å] |
σ 2 |
R‐factor |
|---|---|---|---|---|---|
|
Cu foil |
Cu−Cu |
12 |
2.54 |
0.0084 |
0.00524 |
|
CuO |
Cu−O |
4 |
1.96 |
0.0038 |
0.01912 |
|
Cu(NO3)2 ⋅ 3H2O |
Cu−O |
2.5 |
1.95 |
0.0032 |
0.00471 |
|
Cu/5A |
Cu−O |
2.6 |
1.95 |
0.0049 |
0.00669 |
N: coordination number, Ri : interatomic distance, σ2: Debye‐Waller factor.
Bringing together the results of STEM (only very few ensembles of single Cu atoms <1nm; Figure S5), XANES/EXAFS (single CuII species with no direct Cu−O−Cu bridges, Figure 2) and EPR (single CuII species, partly showing weak antiferromagnetic interactions, Figure 1), it can be concluded that the most active catalyst Cu/5A contains preferentially single CuII sites whereby a certain part of them is in closer vicinity than in catalysts Cu/HY and Cu/MCM‐41, yet formation of direct Cu−O−Cu bridges is negligible.
Mechanistic studies by EPR spin trapping experiments and operando ATR‐IR
To gain insight into the reaction mechanism of N‐formylation of aniline with GA, EPR spin trapping experiments with 5,5‐dimethyl‐1‐pyrroline N‐oxide (DMPO) as a spin trap were performed. [17] Upon adding H2O2 to a suspension of the Cu/5A catalyst in dioxane, EPR signals of DMPO‐OH, DMPO‐OOH and DMPO‐R spin adducts appeared (Figures 3a, bottom, and S7) which indicate the formation of .OH and .OOH radicals as well as a carbon‐centered organic radical .R, whereby the latter should result from dioxane. The percentage to which each spin adduct contributes to the EPR sum spectrum has been derived by spectra simulation (red traces in Figure 3a). Subsequent addition of the GA substrate changed the EPR spectrum strongly, suggesting the formation of new radicals (Figure 3a, middle). Comparison of the simulated EPR spectra of the DMPO‐R spin adducts obtained before and after adding GA (Figure S8) show slight differences, which may suggest that the .R radical after addition stems from GA rather than from dioxane. After adding aniline, a new EPR signal appeared that can be assigned to a DMPO‐NHAr spin adduct (Figure 3a, top), [18] which indicates the formation of a nitrogen‐centered organic radical intermediate (.NHPh 23.6 %). A reasonable assumption is that .OOH radicals attacked aniline since the relative amount of DMPO‐OOH decreased from 68.0 to 37.3 % immediately after adding aniline while the amount of DMPO‐OH remained almost constant (Figure 3b). Meanwhile, the increase in the relative amount of .R radicals from 12.2 to 20.2 % may be related to the reaction of .OOH radicals with GA. After 1.5 h reaction time, the concentration of both .NHPh and .R radicals decreased, probably due to the formation non‐radical formamide products (Figure 3b). Interestingly, the .NHPh radicals disappear much faster than the .R radicals suggesting that .NHPh radicals are not only involved in the synthesis of formamide products but also in the formation of byproducts such as azobenzenes. In the meantime, the relative amount of .OOH radicals increased again due to the interaction of the catalyst and excess H2O2.
Figure 3.
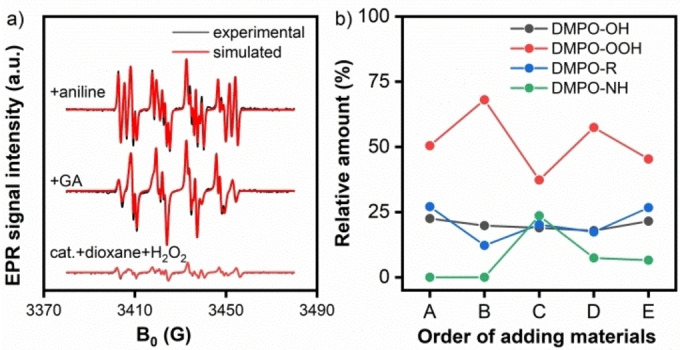
a) Normalized EPR spectra of DMPO spin adducts. Hfs parameters: A N=14.9, A H=11.0 G for .OH, A N=13.4, A ßH=11.1, A γH=1.2 G for .OOH, A N=14.9, A H=21.6 G for .R, and A N=14.9, A H=16.3 G for .NHPh. b) Relative amount of the DMPO‐X (X=OH, OOH, R, NHPh) spin adducts. A: cat.+dioxane+H2O2, B: +GA, C: +aniline, D: +1.5 h, E: +3 h.
Furthermore, the reaction of aniline and GA was investigated by operando ATR‐FTIR (Figure 4a). The IR spectra show a band at 1498 cm−1 assigned to the υ(C−C) vibration of aniline, which declined with increasing reaction time while the υ(C−N) band of formamide at 1542 cm−1 increased gradually. A new weak band appeared at 1696 cm−1 once H2O2 was added, which can be assigned to the υ(C=O) vibration of the formamide product by comparison with a reference sample of formanilide in dioxane (Figure 4b). This carbonyl band raised during 9 h of reaction due to further formation of formanilide. However, the carbonyl band of GA is hardly seen because water is formed, leading to a broad background signal around 1650 cm−1 which obscures the GA band.
Figure 4.

a) Operando ATR‐FTIR spectra. b) Second derivative of the ATR‐IR spectra of the formanilide standard sample in 1,4‐dioxane.
To explore the possible reaction intermediates, some control experiments were performed (Schemes 1 and S1). When GA and H2O2 are converted under optimized reaction conditions, yet in the absence of aniline, 7 % yield of formic acid was observed, suggesting that this is a possible intermediate (Scheme 1a). To confirm this, the control reaction of formic acid and aniline was performed under the same conditions, which led to 73 % formanilide after 3 h (Scheme 1b) and this yield is the same as that obtained in the reaction of aniline and glycolic acid (Scheme S1a). Considering the possible oxidation of formaldehyde to formic acid, a control reaction of aniline and formaldehyde was also performed. Only 17 % yield of formanilide was obtained in this case (Scheme 1c), being much lower than in the reaction of aniline and glycolic acid. Based on these results, it can be concluded that N‐formylation of aniline with glycolic acid likely passes a formic acid intermediate while a formaldehyde intermediate can be excluded.
Scheme 1.

Control experiments.
Based on the above discussions and previous literature, [4a] a possible reaction mechanism can be proposed (Scheme 2). Catalyst Cu/5A activates H2O2 to form .OOH radicals, which can attack glycolic acid and to form carbon‐centered organic .R radicals and formic acid, and also aniline to form the nitrogen‐centered radicals .NHPh. It is well known that reactions between radicals happen very easily. EPR spin trapping experiments with DMPO revealed that the relative amounts of the generated .R and .NHPh radicals decreased simultaneously with reaction time. This suggests that they react with each other to form the formamide product. As reported, formic acid easily reacts with amines to formamide products even without catalyst and oxidants.[ 8l , 19 ]
Scheme 2.
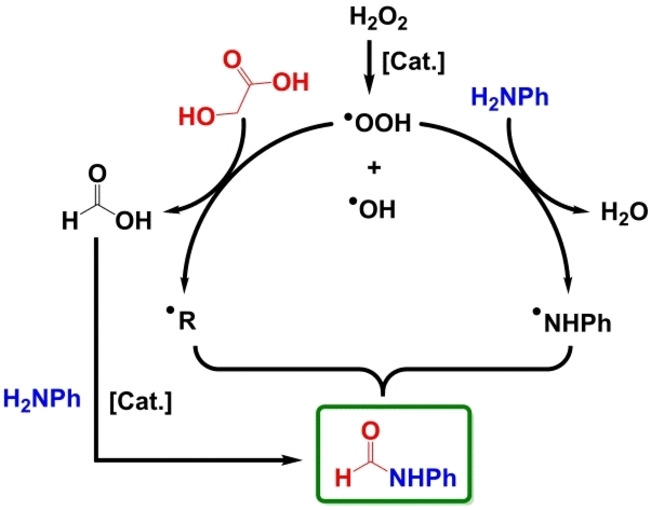
Proposed reaction mechanism.
Conclusions
In this work, we have developed a highly efficient catalyst able to make full use of the carbon atoms in C2 and C3 biomass‐based platform molecules for the N‐formylation of amines to valuable formamides in the presence of H2O2. A series of formamides with diverse structures was synthesized in good‐to‐excellent yields starting with glycolic acid, 1,3‐dihydroxyacetone and glyceraldehyde as sustainable carbonyl sources. Undesired formation of CO2 could be completely suppressed. The catalysts contain as little as 0.5 wt % CuII species, which are mainly atomically dispersed on the surface of zeolites 5A, HY, or MCM‐41. At 99 %, Cu/5A showed the highest utilization efficiency of carbon atoms in the glycolic acid substrate, whereas this value dropped to 25 and 17 % in Cu/MCM‐41 and Cu/HY, due to undesired partial conversion of the solvent. Reaction mechanism studies revealed that HCOOH is the most probable intermediate for formamide formation, whereas the selective formation of .OOH radicals might be the key for C−C bond cleavage.
Experimental Section
Typical procedure for the preparation of the Cu/5A catalyst: Cu(NO3)3 ⋅ 3 H2O (38 mg), was dissolved in 60 mL deionized water and then 2 g 5A molecular sieves (MS) were added as supports. The mixture was stirred at 60 °C for 12 h, then filtered and washed with 100 mL deionized water, and dried at 80 °C for 12 h. The obtained solid sample was calcined at 450 °C for 4 h in a muffle furnace in air with a heating ramp of 5 °C min−1 and the final catalyst sample is denoted as Cu/5A. All other catalysts such as Cu/MCM‐41 and Cu/HY were prepared in the same way. Details of characterization procedures are given in the Supporting Information.
Typical procedure for the N‐formylation of aniline with glycolic acid: The N‐formylation reaction was performed in a 38 mL pressure tube. 0.5 mmol glycolic acid (GA), 3 mmol aniline, 6 mmol H2O2 (35 wt %), 25 mg Cu/5A catalyst, and 2 mL 1,4‐dioxane were added to the reactor. Then, the reaction mixture was heated to 70 °C (oven temperature) and held at the reaction temperature for 9 h under magnetic stirring. After completion of the reaction, 77 mg biphenyl dissolved in 10 mL ethanol were added for quantitative analysis by GC‐FID. The N‐formylation of other amines with GA and the N‐formylation of aniline with 1,3‐hydroxylacetone and glyceraldehyde were performed in the same way.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
The National Natural Science Foundation of China (21961132025, 21925207), Deutsche Forschungsgemeinschaft (grant no. BR 1380/27‐1) and the “Light of West China” Program of CAS are gratefully acknowledged. We thank Christine Rautenberg, Ursula Bentrup, Stephan Bartling, Anja Simmula and Christoph Kubis for experimental support and Prof. Dr. Lirong Zheng (Institute of High Energy Physics, CAS) for helping us to perform the EXAFS (1 W1B BSRF) analysis. Open Access funding enabled and organized by Projekt DEAL.
X. Dai, X. Wang, J. Rabeah, C. Kreyenschulte, A. Brückner, F. Shi, Chem. Eur. J. 2021, 27, 16889.
Contributor Information
Prof. Dr. Angelika Brückner, Email: angelika.brueckner@catalysis.de.
Prof. Dr. Feng Shi, Email: fshi@licp.cas.cn.
References
- 1.
- 1a. Cho E. J., Trinh L. T. P., Song Y., Lee Y. G., Bae H.-J., Bioresour. Technol. 2020, 298, 122386; [DOI] [PubMed] [Google Scholar]
- 1b. Huber G. W., Iborra S., Corma A., Chem. Rev. 2006, 106, 4044–4098; [DOI] [PubMed] [Google Scholar]
- 1c. Chheda J. N., Huber G. W., Dumesic J. A., Angew. Chem. Int. Ed. 2007, 46, 7164–7183; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 7298–7318. [Google Scholar]
- 2. Göransson K., Söderlind U., He J., Zhang W., Renewable Sustainable Energy Rev. 2011, 15, 482–492. [Google Scholar]
- 3.
- 3a. Bulushev D. A., Ross J. R. H., ChemSusChem 2018, 11, 821–836; [DOI] [PubMed] [Google Scholar]
- 3b. Chen X., Liu Y., Wu J., J. Mol. Catal. 2020, 483, 110716; [Google Scholar]
- 3c. Hou Y., Niu M., Wu W., Ind. Eng. Chem. Res. 2020, 59, 16899–16910. [Google Scholar]
- 4.
- 4a. Dai X., Adomeit S., Rabeah J., Kreyenschulte C., Brückner A., Wang H., Shi F., Angew. Chem. Int. Ed. 2019, 58, 5851–5855; [DOI] [PubMed] [Google Scholar]
- 4b. Dai X., Rabeah J., Yuan H., Brückner A., Cui X., Shi F., ChemSusChem 2016, 9, 3133–3138. [DOI] [PubMed] [Google Scholar]
- 5. Weissermel K., Arpe H.-J. in Industrial Organic Chemistry, Wiley-VCH, Weinheim, 2008. [Google Scholar]
- 6.
- 6a. Allen C. L., Williams J. M. J., Chem. Soc. Rev. 2011, 40, 3405–3415; [DOI] [PubMed] [Google Scholar]
- 6b. Lundberg H., Tinnis F., Selander N., Adolfsson H., Chem. Soc. Rev. 2014, 43, 2714–2742; [DOI] [PubMed] [Google Scholar]
- 6c. Tlili A., Blondiaux E., Frogneux X., Cantat T., Green Chem. 2015, 17, 157–168; [Google Scholar]
- 6d. Valeur E., Bradley M., Chem. Soc. Rev. 2009, 38, 606–631; [DOI] [PubMed] [Google Scholar]
- 6e. Zhang Y., Dai X., Wang H., Shi F., Acta Phys. – Chim. Sin. 2018, 34, 845–857. [Google Scholar]
- 7. Bipp H., Kieczka H., in Formamides, Wiley-VCH, Weinheim, 2000. [Google Scholar]
- 8.
- 8a. Wu L., Liu Q., Jackstell R., Beller M., Angew. Chem. Int. Ed. 2014, 53, 6310–6320; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 6426–6436; [Google Scholar]
- 8b. Omprakash Rathi J., Subray Shankarling G., ChemistrySelect 2020, 5, 6861–6893; [Google Scholar]
- 8c. Beydoun K., Klankermayer J. in Recent Advances on CO2 Utilization as C1 Building Block in C−N and C−O Bond Formation, Springer, Cham, 2019, pp. 39–76; [Google Scholar]
- 8d. Le Bras J., Muzart J., Molecules 2018, 23, 1939; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8e. Natte K., Neumann H., Beller M., Jagadeesh R. V., Angew. Chem. Int. Ed. 2017, 56, 6384–6394; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 6482–6492; [Google Scholar]
- 8f. Maji B., Barman M. K., Synthesis 2017, 49, 3377–3393; [Google Scholar]
- 8g. Dong K., Razzaq R., Hu Y., Ding K., Top. Curr. Chem. 2017, 375, 23; [DOI] [PubMed] [Google Scholar]
- 8h. Chen C., Verpoort F., Wu Q., RSC Adv. 2016, 6, 55599–55607; [Google Scholar]
- 8i. Vekariya R. H., Patel K. D., Patel H. D., RSC Adv. 2015, 5, 90819–90837; [Google Scholar]
- 8j. Lukasik N., Wagner-Wysiecka E., Curr. Org. Synth. 2014, 11, 592–604; [Google Scholar]
- 8k. Miyamura H., Kobayashi S., Acc. Chem. Res. 2014, 47, 1054–1066; [DOI] [PubMed] [Google Scholar]
- 8l. Gerack C. J., McElwee-White L., Molecules 2014, 19, 7689–7713; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8m. Yang X. J., Zhang Y. S., Res. Chem. Intermed. 2013, 39, 2843–2848; [Google Scholar]
- 8n. Lanigan R. M., Sheppard T. D., Eur. J. Org. Chem. 2013, 2013, 7453–7465; [Google Scholar]
- 8o. Ding S., Jiao N., Angew. Chem. Int. Ed. 2012, 51, 9226–9237; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 9360–9371; [Google Scholar]
- 8p. Muzart J., Tetrahedron 2009, 65, 8313–8323; [Google Scholar]
- 8q. Olah G. A., Ohannesian L., Arvanaghi M., Chem. Rev. 1987, 87, 671–686. [Google Scholar]
- 9.
- 9a. Chan A. P. Y., Sergeev A. G., Coord. Chem. Rev. 2020, 413, 213213; [Google Scholar]
- 9b. Lutz M. D. R., Morandi B., Chem. Rev. 2021, 121, 300–326; [DOI] [PubMed] [Google Scholar]
- 9c. Mi C., Meng X.-G., Liao X.-H., Peng X., RSC Adv. 2015, 5, 69487–69492. [Google Scholar]
- 10.
- 10a. Cui X., Yuan H., Li J.-P., De Campo F., Pera-Titus M., Deng Y., Shi F., Catal. Commun. 2015, 58, 195–199; [Google Scholar]
- 10b. Dai X., Cui X., Yuan H., Deng Y., Shi F., RSC Adv. 2015, 5, 7970–7975; [Google Scholar]
- 10c. Cui X., Deng Y., Shi F., ACS Catal. 2013, 3, 808–811; [Google Scholar]
- 10d. Cui X., Zhang C., Shi F., Deng Y., Chem. Commun. 2012, 48, 9391–9393; [DOI] [PubMed] [Google Scholar]
- 10e. Yuan H., Kusema B. T., Yan Z., Streiff S., Shi F., RSC Adv. 2019, 9, 38877–38881; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10f. Yuan H., Li J.-P., Su F., Yan Z., Kusema B. T., Streiff S., Huang Y., Pera-Titus M., Shi F., ACS Omega 2019, 4, 2510–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Mutra M. R., Dhandabani G. K., Wang J.-J., Adv. Synth. Catal. 2018, 360, 3960–3968; [Google Scholar]
- 11b. King B. H., Wang M. L., Jesse K. A., Kaur G., Tran B., Walser-Kuntz R., Iafe R. G., Wenzel A. G., J. Org. Chem. 2020, 85, 13256–13263. [DOI] [PubMed] [Google Scholar]
- 12.H. M. Magee, M. Sullivan, Nitrogen gas adsorption in zeolites 13X and 5A, Walla Walla University, College Place, WA, 2010, 1, http://www.phys.ufl.edu/reu/2008/reports/magee.pdf
- 13. Ertl G., Hierl R., Knözinger H., Thiele N., Urbach H. P., Appl. Surf. Sci. 1980, 5, 49–64. [Google Scholar]
- 14.
- 14a. Gervasini A., Manzoli M., Martra G., Ponti A., Ravasio N., Sordelli L., Zaccheria F., J. Phys. Chem. B 2006, 110, 7851–7861; [DOI] [PubMed] [Google Scholar]
- 14b. Schosseler P. M., Wehrli B., Schweiger A., Geochim. Cosmochim. Acta 1999, 63, 1955–1967. [Google Scholar]
- 15. Morosin B., Acta Crystallogr. Sect. B 1970, 26, 1203–1208. [Google Scholar]
- 16. Sharma A., Varshney M., Park J., Ha T.-K., Chae K.-H., Shin H.-J., RSC Adv. 2015, 5, 21762–21771. [Google Scholar]
- 17. Lloyd R. V., Hanna P. M., Mason R. P., Free Radical Biol. Med. 1997, 22, 885–888. [DOI] [PubMed] [Google Scholar]
- 18. Reszka K., Naghipur A., Lownz J. W., Free Radical Res. Commun. 1990, 10, 47–56. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Rahman M., Kundu D., Hajra A., Majee A., Tetrahedron Lett. 2010, 51, 2896–2899; [Google Scholar]
- 19b. Das B., Krishnaiah M., Balasubramanyam P., Veeranjaneyulu B., Nandan Kumar D., Tetrahedron Lett. 2008, 49, 2225–2227. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information