

Summary
Studies on bacterial plant diseases have thus far been focused on the single bacterial species causing the disease, with very little attention given to the many other microorganisms present in the microbiome. This study intends to use pathobiome analysis of the rice foot rot disease, caused by Dickeya zeae, as a case study to investigate the effects of this bacterial pathogen to the total resident microbiome and to highlight possible interactions between the pathogen and the members of the community involved in the disease process. The microbiome of asymptomatic and the pathobiome of foot‐rot symptomatic field‐grown rice plants over two growing periods and belonging to two rice cultivars were determined via 16S rRNA gene amplicon sequencing. Results showed that the presence of D. zeae is associated with an alteration of the resident bacterial community in terms of species composition, abundance and richness, leading to the formation of microbial consortia linked to the disease state. Several bacterial species were significantly co‐presented with the pathogen in the two growing periods suggesting that they could be involved in the disease process. Besides, culture‐dependent isolation and in planta inoculation studies of a bacterial member of the pathobiome, identified as positive correlated with the pathogen in our in silico analysis, indicated that it benefits from the presence of D. zeae. A similar microbiome/pathobiome experiment was also performed in a symptomatically different rice disease evidencing that not all plant diseases have the same consequence/relationship with the plant microbiome. This study moves away from a pathogen‐focused stance and goes towards a more ecological perception considering the effect of the entire microbial community which could be involved in the pathogenesis, persistence, transmission and evolution of plant pathogens.
Introduction
It has been long accepted that disease severity is a multifaceted mechanism, being the outcome of interactions between the pathogen, host and the environment (Brader et al., 2017). Plants are colonized and live in association with a large number of different microorganisms which play important roles in plant health and resistance to biotic and abiotic stresses (Schlaeppi and Bulgarelli, 2015; Vannier et al., 2019; Liu et al., 2020). Members of the plant microbiome undergo direct interactions such as predation, parasitism, mutualism and competition (Faust and Raes, 2012). What happens then to the plant microbiome when an incoming pathogen establishes itself and causes disease? Does the microbial environment change, tolerate and/or assist the colonization of the pathogen? There are several reports, especially in humans and animals, demonstrating that a successful pathogen invasion disrupts the resident microbiota, resulting in a shift from a healthy to a dysbiotic unstable state (Anna Karenina principle; Clemente et al., 2012; Gilbert, 2016; Zaneveld et al., 2017; Proctor, 2019). We now need to take into consideration that interactions between the pathogen and other microorganisms (harmless, neutral or even beneficial) of the microbiome can occur and affect positively or negatively the virulence thus adding a fourth dimension to the disease triangle (Brader et al., 2017). Besides, certain plant diseases are complex, involving the interaction/cooperation of different pathogens (Lamichhane and Venturi, 2015). The recent perception that the microbiome contributes to disease formation and severity, along with the discovery of complex diseases has led to the introduction of the term ‘pathobiome’ (Vayssier‐Taussat et al., 2014), which is the pathogen(s) integrated with the host‐associated microorganisms (encompassing prokaryotes, eukaryotes and viruses). Elucidation of the members of the pathobiome could identify the key biomarker species that can team‐up with the pathogen (Agler et al., 2016). This could become important for understanding pathogenesis, persistence, transmission and evolution of several plant pathogens and for developing microbiome‐based plant protection strategies (Schlaeppi and Bulgarelli, 2015).
Dickeya spp. is one of the top 10 important bacterial phytopathogens in the world (Mansfield et al., 2012). There are currently eight species in this genus, including Dickeya chrysanthemi, Dickeya dadantii, Dickeya dianthicola, Dickeya dieffenbachiae, Dickeya paradisiaca, Dickeya zeae, Dickeya aquatica, Dickeya solani (Samson et al., 2005; Brady et al., 2012) and among them D. dadantii, D. solani and D. zeae cause devasting disease, resulting in a considerable loss in crop yield. Dickeya spp. causes soft rot disease in a wide variety of economically important crops and ornamental plants such as Zea mays, Oryza sativa, Solanum tuberosum and Musa spp. in different parts of the world (Sabet, 1954; Jafra et al., 2009; Sławiak et al., 2009; Laurila et al., 2010). Infections by Dickeya spp. usually results in maceration and rotting of parenchymatous tissue of the affected organ. In particular, infected rice plants by D. zeae present a dark brown decay of the tillers at the site of infection, which later lead to the collapse of the entire plant (Goto, 1979; Barras et al., 1994; Collmer and Bauer, 1994; Nassar et al., 1994; Hussain et al., 2008; Pu et al., 2012). D. zeae are often present in latent infections on many host crops and they can persist overwinter in contaminated plant residues. Under certain conditions, such as in decreasing O2 concentration, high temperature, high humidity and water film on the surface of the plant organs, latency is broken and the bacteria start to grow and cause decay. In these situations, the inoculum produced in one growing season persists to the next, and increases in load over a period of years. A change in spatial distribution of infected plants occurs; it appears as ‘spots’ with the highest density in the centre of infected plants. In most cases, penetration by bacteria occurs via breaks in the periderm caused by bruising in harvesting, insects, nematodes, or fungal infections (Perombelon and Kelman, 1980). Unlike the other members of the genus, D. zeae can infect both monocotyledons and dicotyledons; it produces large quantities of pectic enzymes, phytotoxins and bacteriocins that enable it to macerate the plant parenchymatous tissue and result in disease (Samson et al., 2005; Zhou et al., 2011; Cheng et al., 2013). A quorum sensing regulated zms gene cluster in D. zeae rice isolates, which encodes the biosynthesis of zeamine phytotoxins, is responsible of inhibiting rice seeds germination and growth; no other virulence associated mechanisms is currently known (Zhou et al., 2011; Zhou et al., 2016).
This study performs pathobiome analysis of the rice foot rot disease, caused by D. zeae, as a model in order to investigate the effects of this bacterial pathogen to the total resident stem microbiome and to highlight possible interactions between the pathogen and the members of the microbiota. The bacterial communities of field‐grown rice plants with or without symptoms of foot rot over two rice‐growing periods and belonging to two different rice cultivars have been characterized via 16S rRNA gene amplicon sequencing. Three different factors (host‐related, abiotic and biotic) known to affect the plant microbiota composition were investigated. The main focus was to identify possible interactions and co‐operations between the pathogen and bacterial members of the pathobiome. Using network of interactions and differential abundance analysis we identified likely positive interactions between the pathogen and members of the pathobiome that are consistent in the two growing periods and is independent of the two rice cultivars sampled. Culture‐dependent methods and in planta studies have been performed to support some of the results obtained from the microbiome/pathobiome analysis.
In summary, this study highlights that the plant microbiome shows marked changes in its composition and structure during the foot rot disease and that this disease can be viewed in part as the result of the interactions between a primary plant pathogen with members of the pathobiome.
Results
Rice sampling for pathobiome and microbiome 16S rRNA gene community sequencing
In order to unveil how the plant microbiome can be affected by pathogen establishment, pathobiome analysis in symptomatic rice plants and microbiome analysis in asymptomatic plants was performed and compared. Rice plants at the same developmental stage (second phase of the vegetative rice growth cycle according to IRRI scale) were sampled in two growing periods (2017 and 2018) from two separate rice growing fields cultivated with two rice cultivars (cv. Barone and cv. Sole). An overview on the sampling scheme is presented in Table 1. The symptomatic plants were selected according to the visible presence of the typical foot‐rot symptoms, characterized by yellow and dry leaves, black rot and foul‐smelling base and roots. The asymptomatic plants were collected according to the absence of any visible infection signs and as close as possible to the symptomatic plants, in order to decrease the microbial variability due to the soil and other environmental factors. The disease severity between the majority of rice plants exhibiting foot‐rot symptoms was comparable (2–3 disease severity index) in each sampling year (Table S1 and Fig. S1). The disease incidence was lower in 2017 compared with 2018; thus, during the 2018 sampling, the number of symptomatic plants collected increased.
Table 1.
Sample design description.
| July 2017 | July 2018 | |||
|---|---|---|---|---|
| cv. Barone | cv. Sole | cv. Barone | cv. Sole | |
| Asymptomatic | 5 | 5 | 9 | 13 |
| Symptomatic | 4 | 6 | 16 | 16 |
| Total number samples | 20 | 54 | ||
Description of the total number of rice plants sampled in this study arranged according to the symptomatology (asymptomatic/symptomatic), year (2017/2018) and rice cultivar (Barone/Sole).
DNA was purified from infected stems 3–5 cm around the symptoms lesion site, close to the crown roots and from the same plant zone of healthy plants, in order to reduced bacterial community variability due to the plant compartment. Bacterial communities were studied via 16S rRNA gene amplicon sequencing.
Bacterial community compositional shifts between asymptomatic and symptomatic rice plants
The number of reads passing the quality filter was on average 4082.06 (ranging 70–18 557). The average number of OTUs was 66.69 (ranging 15–168). Immediately after the quality check, the reads annotated as Chloroplast (on average 47.1% ± 29.5 – Table S2) were removed from the dataset. The number of OTUs was higher in samples of 2018 (P < 0.01) compared with the samples of 2017 and in symptomatic samples compared with the asymptomatic ones (P < 0.001; Table 2). The Shannon diversity values were higher in symptomatic samples compared with the asymptomatic ones in 2018 (P < 0.001), while no significant differences were detected in 2017. (Fig. 1A and B; Table S3). The PCoA based on the weighted UniFrac distance measure showed that symptomatic and asymptomatic samples formed two distinct clusters (Fig. 1C). Asymptomatic and symptomatic samples from the year 2018 were significantly (P = 0.001) different and clustering separately, while the differences in 2017 were not evident (P = 0.1). Asymptomatic and symptomatic samples from the year 2017 were partially overlapping, suggesting that their microbiomes were more similar than the ones from the year 2018.
Table 2.
Average number of OTUs subset by cultivar, symptom and year.
| Parameter | Classes | Avg. N° OTUs | SD | P‐value a |
|---|---|---|---|---|
| Cultivar | Sole | 65,15 | 44,15 | N.S. |
| Barone | 68,82 | 34,79 | ||
| Symptom | Healthy | 35,87 | 23,07 | < 0.001 |
| Sick | 79,33 | 39,01 | ||
| Year | 2017 | 33,81 | 15,65 | < 0.01 |
| 2018 | 74,9 | 40,32 |
Average number of different OTUs subset by parameters (cultivar, symptom and year) and split in classes. OTU numbers differed significantly with the symptomatology (healthy vs. sick) and the year (2017 vs. 2018). Significance between classes was calculated by Wilcoxon–Mann–Whitney test.
After Wilcoxon–Mann–Whitney test.
Fig 1.
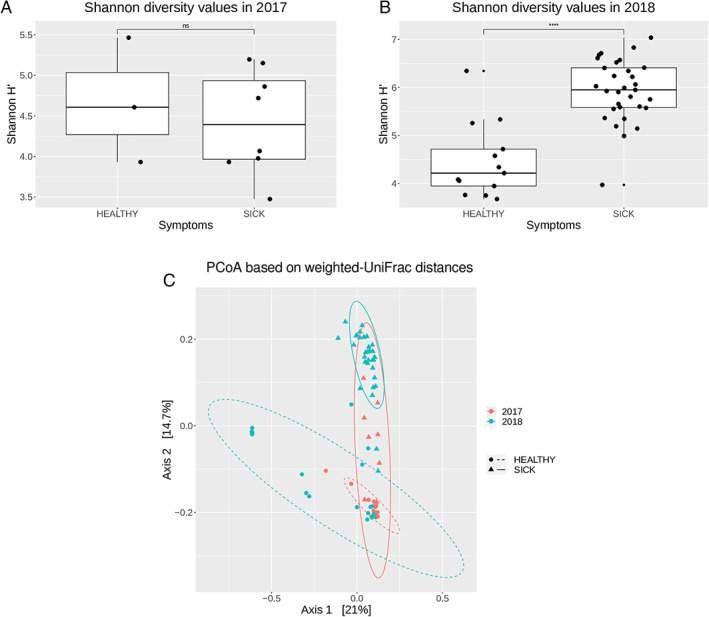
Complexity and composition of the microbial communities of asymptomatic and symptomatic samples.
A. Alpha diversity estimations of the bacterial communities associated with asymptomatic (healthy) and symptomatic (sick) samples, using Shannon's H index for year 2017; and (B) for year 2018; box plot depict medians (central horizontal lines), the inter‐quartile ranges (boxes), 95% confidence intervals (whiskers) and outliers (black dots). Asterisk indicate significant differences between two groups of samples (*P < 0.05). Statistical analyses were calculated based on Wilcoxon‐test.
C. Principal Coordinate Analysis (PCoA) based on weighted UniFrac distances. Ellipses show confidence intervals (CI) of 95% for each sample type. Statistical significance has been inferred using PERMANOVA (see also Fig. S2 and Table S4).
The sampling scheme had three factors: symptomatology (healthy/sick), year (2017/2018) and rice cultivar (Sole/Barone). To determine whether there were significant differences driven by these three factors, we performed a PERMANOVA test on the UniFrac distances within each independent set (with 999 permutations in all tests). In all three factors, the differences in microbiome composition between the two sets were significant, however, the symptomatology was the stronger driver of the diversity among the samples (pseudo‐F: 10.48), whereas the year (5.49) and the rice cultivar (3.06) displayed considerably lower effects (Fig. S2). To further determine the effect of the symptomatology on the cultivar in each year we compared the asymptomatic and symptomatic plants microbiome in each year for each cultivar (Table S4). The results confirmed that the samples from 2018 were significantly different for both cultivars, although in 2017 this difference was not significant.
To ensure that the patterns observed were not an effect of the low sampling depth, we performed the same analysis at a rarefaction of 1894 reads. This rarefaction resulted in the removal of the 50% of the samples, mostly from healthy samples, yet keeping all set of factors. The alpha‐ and beta‐ diversity patterns described in the analysis were consistent with the ones found at the sampling depth of 600 reads (Fig. S3).
The OTUs were annotated using the Silva database (Quast et al., 2012); in total, we identified 20 phyla, 36 classes, 95 orders, 158 families and 278 genera. The microbiomes were overall dominated by Proteobacteria of the alpha‐ and gamma‐class and the Bacteroidetes and Negativicutes (Fig. 2A, Tables S5 and S6). The phyla Firmicutes and Campylobacteria were found at higher average abundance in symptomatic samples (20.82% versus 0.67% in healthy and 3.33% vs. 0.17% in healthy respectively). On the other hand, the phylum Acidobacteria was detected at significant higher average abundance in asymptomatic samples (5.25% vs. 0.47% in infected plants; Table S6). At the family level, the most prevalent taxa were Pectobacteriaceae, Sphingomonadaceae, Comamonadaceae, Azospirillaceae and Arcobacteraceae among the Proteobacteria. Firmicutes were dominated by Clostridiaceae and Lachnospiraceae. Bacteroidetes were mainly represented by the family Prevotellaceae, and Acidobacteria were mainly composed of OTUs assigned to the Blastocatellaceae family. All families presented distinct differences among asymptomatic and symptomatic microbiomes (Fig. 2B, Table S5 and S6). The number of shared and unique genera between asymptomatic and symptomatic samples is shown in the Venn diagram (Fig. S4). One hundred and fifty‐eight genera were found only in the bacterial communities of symptomatic samples, whereas 40 genera were found exclusively in asymptomatic samples only, while 140 genera were shared (i.e., found in both conditions).
Fig 2.
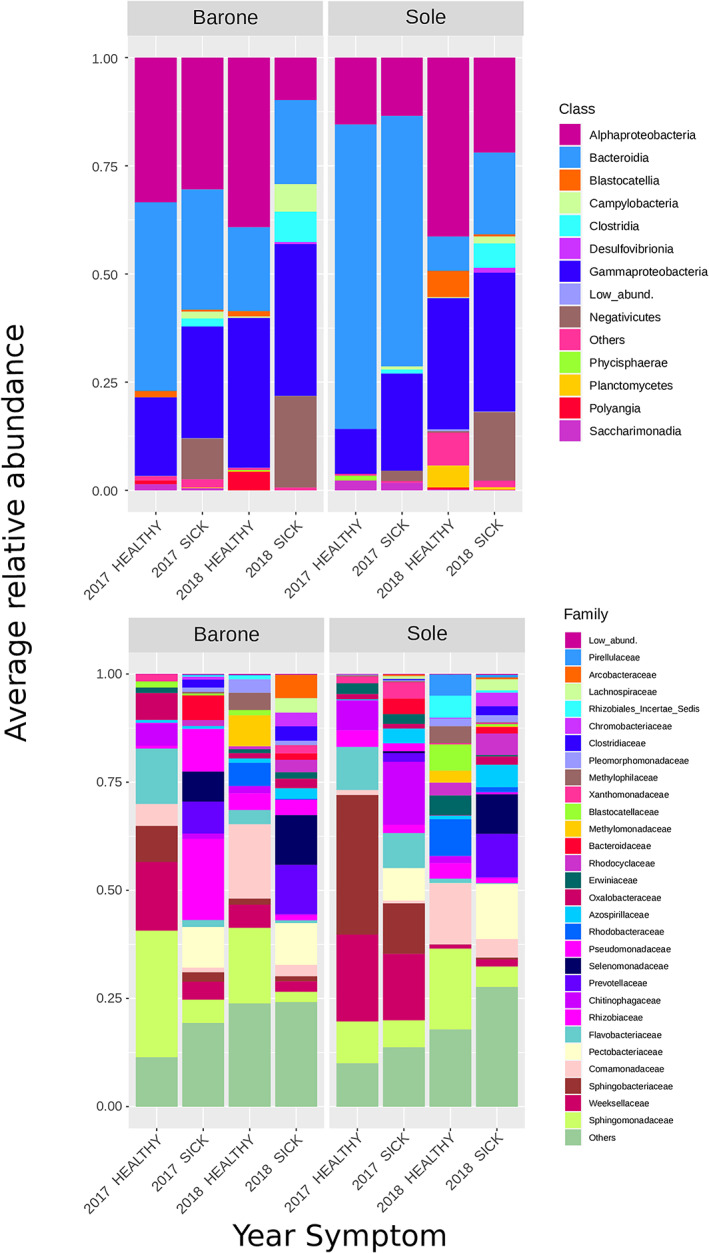
Average relative abundance (% of sequencing reads) of predominant OTUs. Stacked bar charts showing the relative abundance at the level of (A) Class and (B) Family, divided according to the symptoms and year. Taxa whose abundance was < 1% have been lumped into ‘Low abundance’ category; taxa that did not classify at that specific taxonomic level was lumped into the category ‘Others’. The exact relative abundance values for each taxon in each category are presented in Table S5.
The network analysis, based on the Sparse Correlations for Compositional (SparCC) correlation values, revealed that the networks in asymptomatic and symptomatic samples were different. The network deriving from asymptomatic samples microbiome featured a more distinct clustering pattern, a lower number of nodes and edges and a lower average degree compared with the network derived from symptomatic samples (Fig. 3). The two networks had a comparable number of average neighbours per node, but the network derived from the asymptomatic samples had higher clustering coefficient and density, whereas the network of the symptomatic samples was sparser.
Fig 3.
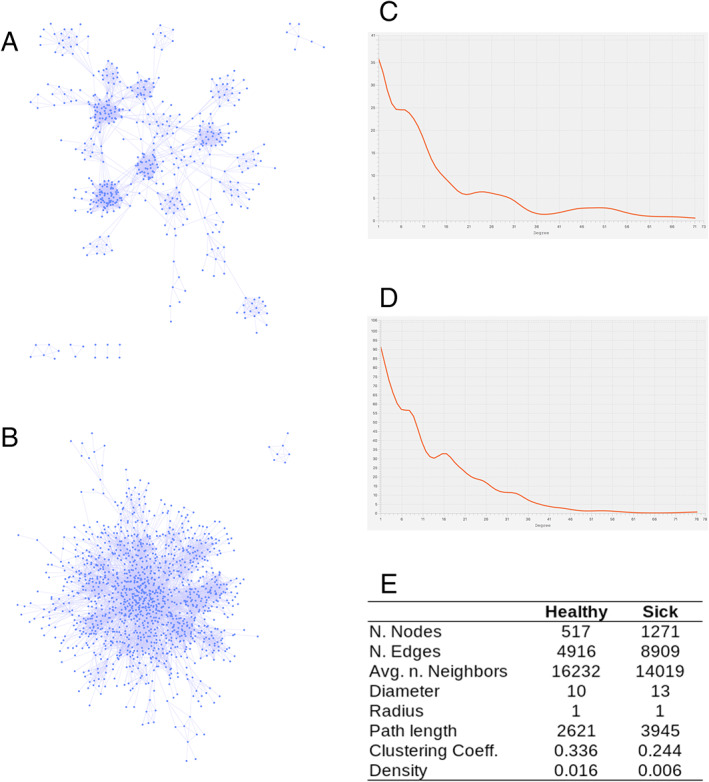
Network analysis of microbiomes and pathobiomes.
A. OTUs interactions network based on the SparCC values (nodes are the OTUs and the links were established when the SparCC absolute correlation value was above 0.3) among the OTUs in asymptomatic microbiomes and B. in symptomatic microbiomes.
C. and D. Nodes degree distributions for asymptomatic and symptomatic microbiomes respectively.
E. Main topological statistics on the two networks.
Dickeya zeae is present only in foot rot symptomatic rice plants
The average relative abundance of Dickeya genera reads for symptomatic plants was 10.53% (ranging 0.0%–31.42% Fig. 4A; Table S1). Reads annotated as Dickeya were detected only in one asymptomatic plant at the abundance of 0.39%. The relative abundance of Dickeya reads was slightly higher in the symptomatic samples from Sole cultivar; however, the difference was not statistically significant (Fig. S5). To further confirm the presence of D. zeae, the virulence zsmA gene, which is unique to D. zeae (Zhou et al., 2011), was amplified using as template purified DNA from plant material. The zsmA gene was only successfully amplified from symptomatic samples, demonstrating again the presence of D. zeae and its involvement in foot‐rot disease (Fig. S6).
Fig 4.
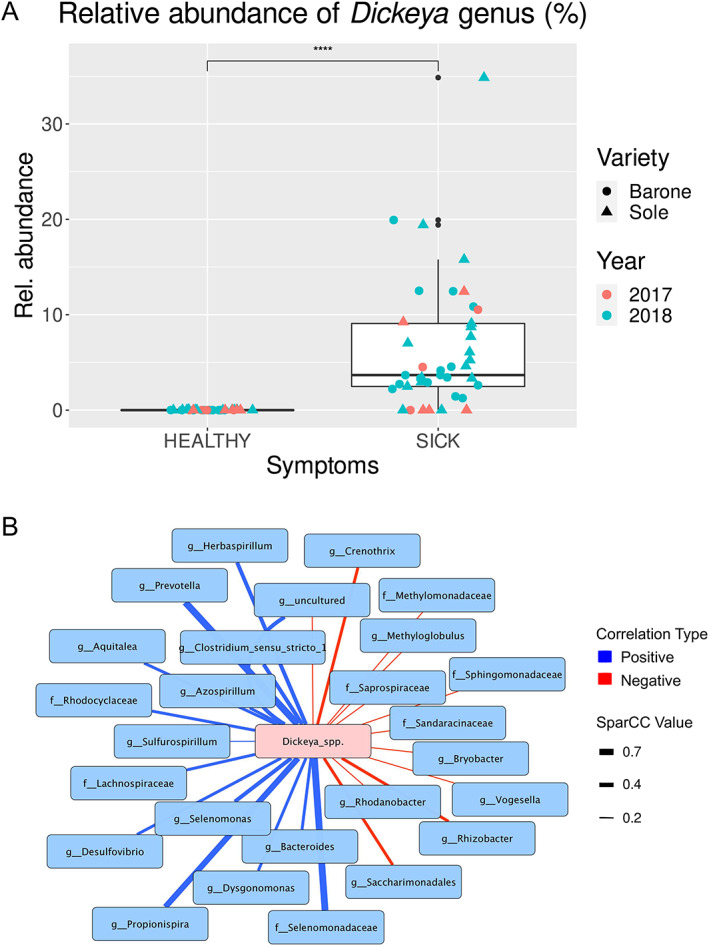
Comparison of Dickeya genus abundance between asymptomatic (HEALTHY) and symptomatic (SICK) plants, and interactions network.
A. Box plot depicting medians (central horizontal lines), the inter‐quartile ranges (boxes), 95% confidence intervals (whiskers). Asterisks indicate significant differences between the two groups of samples. Statistical analyses were calculated based on Wilcoxon‐test.
B. Interactions network based on the SparCC values among the OTUs classified as Dickeya genus and the most significant taxa showing stronger correlation values. Negative correlations are depicted as red edges, while positive correlations are marked as blue edges.
In order to explore the possible interactions between the pathogen and the other members of the pathobiome, we performed a co‐occurrence network analysis; this analysis revealed 27 significant correlations (r ≥ 0.5 for co‐occurrence and r ≤ −0.2 for co‐exclusion); between Dickeya spp. and different OTUs classified at a various taxonomic rank (Fig. 4B). The strongest positive correlations identified were with OTUs belonging to the family Selenomonadaceae, genus Prevotella, Propionispira, Clostridium_sensu_stricto_1 and genus Azospirillum. On the other hand, the strongest negative correlations observed were with OTUs belonging to the genus Crenothrix, Rhizobacter and Saccharimonadales Fig. 4B).
Several bacterial taxa significantly correlate with D. zeae presence in symptomatic plants
A linear discriminant analysis (LDA) between the bacterial community composition of symptomatic samples (pathobiome) and asymptomatic samples (microbiome) was performed, in order to identify potential biomarker taxa between the two conditions. A graphical representation of the differentially represented‐taxa (P value < 0.05) from the two growing periods is shown in Fig. 5 and in the extensive version (Fig. S7). Among the significantly differentially represented‐taxa, 76 were more abundant in symptomatic samples and 16 in the asymptomatic ones (Fig. S7). The genera Prevotella, Propionispira, Clostridium_sensu_stricto_1 and Azospirillum, identified in the network analysis as positively interacting with Dickeya spp. (Fig. 4B), were also found to be statistically correlated with the disease condition (Fig. 5). On the other hand, Crenothrix and Rhizobacter, negatively interacting with Dickeya spp. (Fig. 4B), were statistically correlated with the asymptomatic condition (Fig. 5). Notably, several taxa such as Pseudomonas, Azospirillum, Burkholderia, Sulfurospirillum, Pleomorphomonas and Magnetospirillum were present in both asymptomatic and symptomatic samples but were significantly enriched in the samples affected by the disease. Some taxa were only present in the symptomatic samples, in particular Prevotella, Propionispira, Selenomonas, Clostridium_sensu_stricto_1 and Clostridium_sensu_stricto_12, suggesting that their establishment was significantly stimulated/influenced by the presence of D. zeae.
Fig 5.
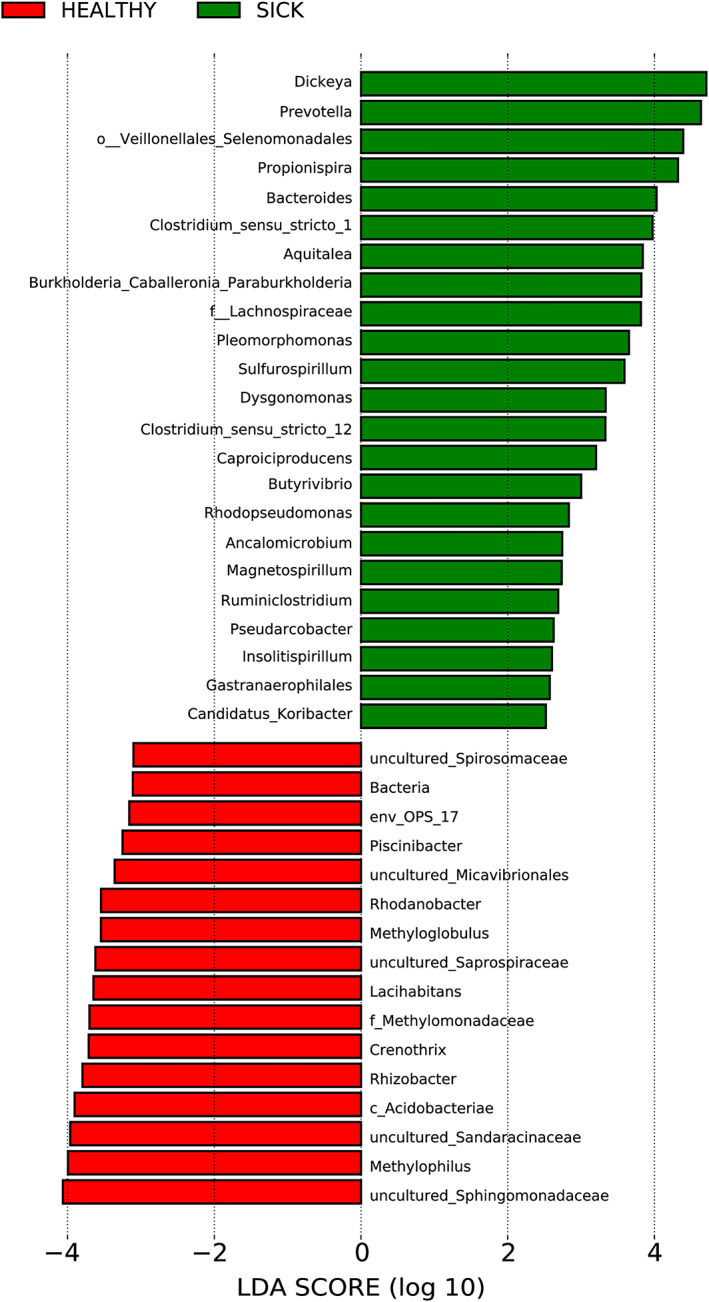
Linear discriminant analysis (LDA) scores of the main bacterial genera which significantly differ in their relative abundance between asymptomatic (HEALTHY) and symptomatic (SICK) samples. Differential abundance of bacterial genera between two groups of samples tested was assessed by performing a linear discriminant analysis (LDA). The two‐symptomatology conditions were used as class to test. Only genera with a P value < 0.05 for the Kruskal–Wallis test and an LDA score > 2 are displayed. An extended version of this plot is available as Fig. S7.
The culturable fraction of bacteria from rice foot‐rot symptomatic plants
To begin to investigate whether some members of the pathobiome are cooperating with the pathogen in the disease process, it was important to isolate some bacterial strains that belong to taxa which were significantly enriched by the presence of D. zeae (see above). Five representative samples of symptomatic plants from the 2017 and 10 representative samples from the 2018, were used for performing a culturable fraction study via 16S rRNA gene amplicon sequencing. This allowed us to determine which bacterial genera/species were cultivable under laboratory conditions. Four different growth solid media (including both rich and minimal media; Table S7) and two different time points were used for growing bacteria from the macerated plant samples; all the bacteria colonies grown were then collected en masse, resuspended and diluted and the total DNA purified as described in the Materials and Methods section. 16S rRNA gene community sequencing was then performed and results are summarized in Table 3. Among the 298 different taxa detected in the pathobiome (see above, Venn diagram in Fig. S4), the fraction of bacteria culturable under the conditions tested here was 13%.
Table 3.
List of genera detected via 16S rRNA gene amplicon sequencing deriving from the cultivable fraction of the pathobiome. Four different media and two‐time points have been used for the isolation.
| Culturable taxa | 869‐medium | M9‐medium | PDA‐medium | TSA‐medium |
|---|---|---|---|---|
| Achromobacter | ✓ | ✓ | ✕ | ✓ |
| Acinetobacter | ✓ | ✓ | ✓ | ✓ |
| Acidovorax | ✓ | ✓ | ✕ | ✓ |
| Aeromonas | ✓ | ✓ | ✓ | ✓ |
| Azospirillum | ✓ | ✓ | ✕ | ✓ |
| Aquitalea | ✓ | ✓ | ✕ | ✓ |
| Burkholderia | ✓ | ✓ | ✓ | ✓ |
| Brevundimonas | ✓ | ✓ | ✕ | ✓ |
| Chryseobacterium | ✓ | ✓ | ✓ | ✓ |
| Citrobacter | ✓ | ✓ | ✓ | ✓ |
| Comamonas | ✓ | ✓ | ✕ | ✓ |
| Caulobacter | ✓ | ✓ | ✓ | ✓ |
| Delftia | ✓ | ✓ | ✕ | ✓ |
| Dickeya | ✓ | ✓ | ✓ | ✓ |
| Elizabethkingia | ✓ | ✓ | ✓ | ✓ |
| Enterobacter | ✓ | ✓ | ✓ | ✓ |
| Enterococcus | ✓ | ✓ | ✓ | ✓ |
| Exiguobacterium | ✓ | ✓ | ✕ | ✓ |
| Flavobacterium | ✓ | ✓ | ✕ | ✓ |
| Herbaspirillum | ✓ | ✓ | ✕ | ✓ |
| Klebsiella | ✓ | ✓ | ✓ | ✓ |
| Kosakonia | ✓ | ✓ | ✓ | ✓ |
| Microbacterium | ✓ | ✓ | ✕ | ✓ |
| Microvirgula | ✓ | ✓ | ✕ | ✓ |
| Morganella | ✓ | ✓ | ✕ | ✓ |
| Novosphingobium | ✓ | ✓ | ✕ | ✓ |
| Paenibacillus | ✓ | ✓ | ✕ | ✓ |
| Pandoraea | ✓ | ✓ | ✕ | ✓ |
| Pantoea | ✓ | ✓ | ✓ | ✓ |
| Providencia | ✓ | ✓ | ✕ | ✓ |
| Pseudomonas | ✓ | ✓ | ✓ | ✓ |
| Rhizobium | ✓ | ✓ | ✕ | ✓ |
| Salmonella | ✓ | ✓ | ✓ | ✓ |
| Serratia | ✓ | ✓ | ✓ | ✓ |
| Sphingobacterium | ✓ | ✓ | ✕ | ✓ |
| Sphingomonas | ✓ | ✓ | ✕ | ✓ |
| Stenotrophomonas | ✓ | ✓ | ✓ | ✓ |
| Vagococcus | ✓ | ✓ | ✕ | ✓ |
| Xanthomonas | ✓ | ✓ | ✕ | ✓ |
The first column of the table shown the list of genera detected in the culturable pathobiome study via 16S rRNA gene amplicon sequencing. The ability of each culturable bacteria genus to grown in the media used in this experiment is defined by presence (✓) or absence (✕).
From the same symptomatic samples selected for the culturable pathobiome fraction analysis, a bacterial collection of 100 pure strains was generated (Table S8) and molecular characterized via 16S rRNA complete gene amplification. In planta virulence tests were then performed with one isolate, namely Burkholderia spp. A56; the reason was that the Burkholderia genus significantly correlated with the D. zeae presence (see above LDA, Fig. 5) and it was considerably enriched in the pathobiome culturable fraction. The virulence test was performed using single inoculations as well as co‐inoculations along with D. zeae. No significant differences in the size of the lesion caused by D. zeae were observed between plants treated only by the pathogen and those co‐inoculated by the pathogen and with the Burkholderia spp. isolate (Fig. S8). Bacterial CFU of D. zeae recovered from the infection site was on average of 1.2 × 107 and 3.2 × 107 CFU g−1 in both plants inoculated alone or co‐inoculated, indicating that this strain does not affect the growth of D. zeae in planta experiment (Fig. 6). A significant difference (P value < 0.001) however was observed in bacterial CFU of Burkholderia spp. A56 comparing the single inoculation (on average 6.3 × 105 CFU g−1) versus the co‐inoculation with D. zeae (1.1 × 107 CFU g−1; Fig. 6). This indicated that Burkholderia spp. A56 colonized the rice plant significantly more resulting in a higher bacterial load when D. zeae was present.
Fig 6.
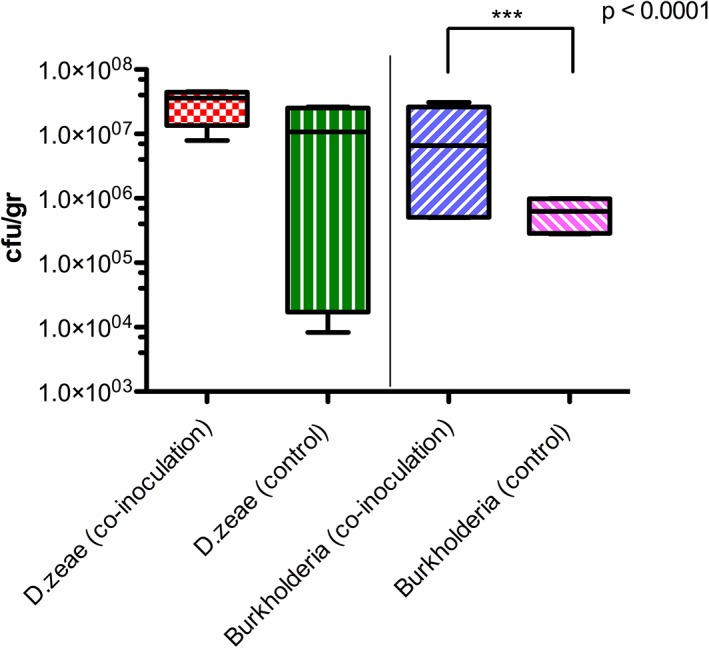
Co‐infection test on rice plants by Dickeya zeae and Burkholderia sp. A56 as possible co‐operator. The putative co‐operator was inoculated independently and in co‐presence of the pathogen. CFU/g of D. zeae and Burkholderia sp. A56 recovered after the co‐inoculation was evaluated in rice plant stem's lesion site/section at 15 dpi by plating serial dilutions from plant tissue and by antibiotic selection (D. zeae Rif and Burkholderia Amp100, Gm40, Nf100). For each single and co‐inoculation treatment, eight plants were used. The control treatment was realized by single inoculation of pathogen or co‐operators independently and PBS. Significance between groups was calculated using t‐test; no significant differences were found between the CFU/g of Dickeya recovered.
Discussion
Studies on bacterial plant diseases have thus far mainly focused on single bacterial species causing the disease, with little attention given to the many other microorganisms present in the microbiome. In this study, the foot rot disease caused by D. zeae was used as model to investigate how the plant microbiome changes in response to a pathogen attack and to identify the most likely genera that could undergo interactions with the pathogen and possibly play a role in the pathosystem. This study shifts from a pathogen‐focused view towards a more ecological perception of the foot rot disease foreseeing the entire microbial community dynamics.
Comparison of the stem rice microbiome of asymptomatic and symptomatic plants in two different growing periods and belonging to two rice cultivars has evidenced the dominant presence of D. zeae only in the plants showing the symptoms, suggesting that this approach is suitable for detecting the causal/primary agent of a disease. The number of Dickeya genus reads however showed variations in relative abundance among the single symptomatic samples. Regulation of the expression of virulence associated factors in bacteria is most often cell density‐dependent (Elsayed et al., 2020); thus it is possible that the plants sampled were at different times/stages of the infection or in a latent stage for some of the asymptomatic plants. In addition, we observed an increase in the abundance of Dickeya genera reads during the year 2018 compared with the 2017. This difference was not evident in the symptom's severity, since the infected plants either from the 2017 or 2018 year showed a similar degree of symptoms. The disease incidence (the number of plants affected) was lower during the 2017 compared with the 2018 year, this was probably due to the field growing conditions (temperature, soil and water status) and to the earlier stage of the epidemic.
Significant changes were detected in the bacterial composition and abundance between symptomatic and asymptomatic plants. Our results showed that the presence of the pathogen not only had a significant impact on the plant health, but also influenced the overall structure of the plant‐associated microbiome. There was a drastic increase of the biodiversity between the asymptomatic and symptomatic plants from the 2018 year, while the difference between the two conditions in 2017 was less marked; this could be due to the lower incidence of the disease and possibly that the sampling was performed at an early phase of the epidemic. Bacterial communities from both asymptomatic and symptomatic samples clustered together independently by the year and the cultivar, suggesting that the seasonal changes and the cultivar affect only partially the microbiome composition of the asymptomatic or symptomatic plants. It is widely accepted that microbial community structures are affected by abiotic and host‐dependent factors (Dastogeer et al., 2020); however, our results suggested that these two factors affect the microbiome composition in our samples to a minor extent. Several published studies have often demonstrated that some diseases are associated with the changes occurring in the microbiome (Ma et al., 2019); however, it is still uncertain whether diversity change is a cause of microbiome‐associated disease or a consequence.
The analyses and comparison of the co‐occurrence networks between healthy and disease states revealed that the microbiome of asymptomatic plants had a higher clustering coefficient compared with symptomatic plants. This supports the hypothesis that the bacterial community in a healthy situation is featured by biologically significant associations among OTUs which could be either cooperative or functionally related. In healthy plants, the total bacterial community is formed by a high number of bacterial genera which are present in similar amounts. Moreover, the community composition in asymptomatic samples appears similar and stable. The fitness of the plant is likely to depend on the organized biodiversity of its microbiome that is effective in rapid adaptation/plasticity to environmental changes (Vandenkoornhuyse et al., 2015). The same analysis derived from symptomatic plants revealed a sparser network, with a lower number of edges and clustering coefficient (Fig. 3). Reduced connectivity of the microbial networks and an increase in heterogeneity in pathobiomes is consistent with previous studies and to the Anna Karenina principle (Zaneveld et al., 2017; Li and Convertino, 2019; Sweet et al., 2019).
Upon establishment of D. zeae, we observed (i) conversion of a Proteobacteria‐rich community to a Firmicutes‐rich community; and (ii) an increase of the overall relative bacterial diversity. The latter is an exception to the common finding that pathobiomes are associated with decreased biodiversity. It is generally accepted that high diversity defines healthy microbiomes, whereas reduction in diversity may be associated with dysbiosis, as evident in human microbiome studies (Li et al., 2012; Lozupone et al., 2012). However, the inverse relationship between biodiversity and disease status has been recently questioned (Ma et al., 2019) as other cases in which the dysbiotic state is associated with higher alpha diversity have been reported (Ceccarani et al., 2019; Lamelas et al., 2020). For example, bacterial communities associated with tropical coral‐host revealed that diseased samples were associated with a higher bacterial biodiversity index; this condition likely reflects imbalance in the community structure (Closek et al., 2014). The increase in taxa associated with the disease can also be due to opportunistic bacteria which may compete for available resources and/or benefit of the impaired host defence system.
Interestingly, we noticed that Firmicutes, such as Lachnospiraceae and Clostridiaceae, were more abundant in symptomatic samples. Moreover, the majority of the taxa that positively correlate with the disease condition such as Prevotella, Propionispira, Pleomorphomonas, Clostridium_sensu_stricto, Sulfurospirillum, Rhodopseudomonas and many others are obligate or facultative anaerobes, suggesting that under disease conditions oxygen levels changes may favour growth of anaerobic bacteria. Accordingly, the drastic decrease of Chloroplast sequences in the symptomatic samples is also correlated to the rapid depletion of O2 and to the anaerobic conditions developed. Oxygen deficiency with increased availability of nutrients, as well as the reduction of the plant immune resistance, is likely to promote rapid growth of rotting bacteria favouring the establishment of decay (Ma et al., 2007). Under these conditions, only a small amount of the pathogen (< 102) is likely to be required to initiate a lesion (Perombelon and Kelman, 1980) and D. zeae possibly undergoes several interactions with anaerobic bacteria that are present in the pathobiome as a result of plant rotting ecology.
Co‐occurrence network analysis identified several positive and negative interactions with D. zeae; these may be involved in the establishment of a stable consortium that could influence the development of the disease. Very strong positive correlations were detected between D. zeae and Prevotella and Propionispira, independent of the year or the cultivar and consistent across samples, supporting the hypothesis that D. zeae undergoes specific positive interactions with some members of the pathobiome. The highest correlation occurred with Prevotella, which is able to decompose the hemicellulose and pectin, the major constituents of plant cell walls (Ueki et al., 2007). It is therefore likely that strains belonging to this genus play an important role in some steps of the disease development and aggravation.
To begin to address possible interactions between the pathogen and other microbes in the pathobiome‐network, we isolated several bacterial strains from the pathobiome. We noticed that many isolates displayed low abundance in the pathobiome community studies, the reason for this is intrinsic to the properties of the microbial communities, which are usually dominated by very few species; therefore, high‐throughput cultivation would likely recover many species that have low abundance (Lynch and Neufeld, 2015). Among the taxa positively correlated with the disease condition, we were able to isolate as a single and pure colonies only a few strains. Unfortunately, under the conditions used here (aerobic grown condition, limited number of different media, limited time‐points) we did not succeed in isolating strains belonging to Prevotella or Propionispira genera; the reason for this is unknown, it could be that they require specific growth conditions or these strains cannot grow as pure isolates. In planta experiments were performed with a Burkholderia spp. strain, which significantly correlated with D. zeae, according to the LDA analysis and its 16S rRNA sequence match the sequence in the amplicon community analysis. We noticed that its plant colonization ability significantly increased when D. zeae is present, indicating that it benefits from the presence of the pathogen. The improved growth of Burkholderia spp. might be explained in multiple ways: it might be due to (i) the cell–cell signalling and cooperation between this member of the microbiome and the pathogen, (ii) the nutrients that become available as a result of the plant rotting and (iii) the plant derived defence molecules and metabolites, such as phytohormones, aromatic amino acids and phenolic compounds. The plant environment changes in response to a pathogen infection due to host cell wall lignification, synthesis of phytoalexins or increase in phenolics components and hormone‐like substances (Glazebrook, 2005). These molecules may serve as nutrient source, signalling molecules, or have a antimicrobial activities, and may thus influence all the members of the community (Eichmann et al., 2021). These co‐inoculations did not result in a more severe disease in our plant‐disease model. It cannot be excluded that in the wild, D. zeae could benefit from the presence of Burkholderia sp., resulting in more severe symptoms or more rapid development of the foot‐rot disease.
Additionally, the shift of the biodiversity in the pathobiome could be in part due to (i) the production by the pathogen of antimicrobial compounds, (ii) higher carbon and nitrogen source availability, (iii) the local and systemic plant immune response and (iv) plant–microbe signalling and microbe‐microbe interactions. The plant‐host can therefore be mainly involved through changes of the root exudate composition and thus causing a differential growth and/or recruitment of microbial members. A notable example of cooperative behaviour is the plant pathogen Pseudomonas savastanoi pv. savastanoi causing the olive‐knot disease which undergoes interspecies cell–cell signalling via quorum sensing signalling molecules with endophytic harmless Erwinia toletana resulting in a more aggressive disease where both bacterial species benefit (Hosni et al., 2011; Buonaurio et al., 2015). A possible mechanism of mutual benefit is via complementarity in metabolic pathways resulting in an ameliorated metabolic capacity of the consortium compared with the single species (Buonaurio et al., 2015). Future research is needed to understand how members of the pathobiome modify the activity of the primary pathogen; this will require functional studies of the pathosystem as well community profiling.
In order to determine possible commonalities among different pathosystems, a similar set of experiments of another rice pathogen was performed. The plant pathobiome and microbiome of an important bacterial disease of rice called leaf blight (BLB), caused by Xanthomonas oryzae pv. oryzae (Xoo; Mew et al., 1993; Mansfield et al., 2012), has been determined. Importantly, microbial pathobiome/microbiome experiments performed here from rice plant samples from Vietnam on the BLB vascular rice disease caused by Xoo, did not result in any perturbation/shift in the microbiome showing that the pathobiome has very low biodiversity and that this disease is very different from rice foot‐rot (Fig. S9). Xoo is a vascular pathogen colonizing in solitary the vascular system thus the microbiome does not seem to play a major role in influencing this disease. Furthermore, unlike what we have observed for the rice D. zeae pathosystem, we found no evidence to support the Anna Karenina principle, which predicts higher heterogeneity in destabilized microbial communities. Generalizations on the role of the microbiome and of possible positive biotic interactions in plant disease therefore cannot be made.
Conclusions
In summary, D. zeae alters the resident bacterial community in species composition, abundance and richness leading to the formation of a microbial consortia/pathobiome linked to the disease state. Our results have shown that the driving force of microbial community variation is the presence or absence of the foot‐rot symptoms, while the effect of the growing season and the cultivar is negligible. We detected specific anaerobic bacterial taxa, which are significantly co‐present with the pathogen over the 2 years, suggesting a possible involvement in the disease development through the facilitation of its colonization, expression of virulence factors and reduction of host resistance. It is likely that the bacterial foot rot disease is the result of a multi‐species interaction and not solely due to one single pathogen. It is important to begin shifting from the paradigm of pathogens to pathobiome in order to understand the importance of microbial communities. Deciphering the molecular basis of interbacterial relationships in the plant pathobiome will be a major future challenge in order to better understand the pathogenicity and epidemiology of microbial plant diseases and target polymicrobial infections.
Experimental procedures
Rice samples collection and processing for the microbiome/pathobiome analysis
Rice plants were collected in two different rice growing periods (2017 and 2018) from two rice fields owned by the Italian rice growers organization called SAPISE located in Vercelli. The plants were sampled at the same developmental stage (second phase of the vegetative rice growth cycle according to IRRI scale), during the 2 years, in order to avoid growth stage dependent variation in microbial community structure. Two rice cultivars were sampled for this study: cv. Barone from the rice field (45°26′33.2″N 8°21′50.0″E) and cv. Sole from the rice field (45°30′56.7″N 8°22′22.0″E). From each field, rice plants with or without symptoms were collected. The sampling scheme was based on the random presence in the field of plants clearly displaying the disease symptoms. While the asymptomatic plants, without any visible symptoms, were selected as much as possible close to the symptomatic ones.
The plant samples were washed and for each infected sample, close to the crown roots, 3–5 cm region of the brown rot stem has been selected and weighed (3 g of plant material per sample). The same region of the healthy plant stem has been cut and treated in the same way. For each sample, 2 g of plant material were macerated in liquid nitrogen using sterilized pestle and mortar and used later for bacterial community analysis. The remaining part (1 g) was resuspended in 4 ml phosphate buffered saline (PBS) solution and stored with 18% glycerol at −80°C for the culturable analyses.
Analyses of the bacterial plant community
DNA was extracted from each plant sample (0.25 g) using the PowerMax Soil DNA isolation kit (MO BIO Laboratories, Carlsbad, CA, USA) according to the manufacturer's instructions. Total DNA concentration was analysed by using a Nanodrop™ spectrophotometer (Thermofisher Scientific, Waltham, MA, USA) and normalized to a concentration of 7 ng μl−1 for preparing the 16S rRNA gene amplicon library. The DNA extracted was used to amplify the V3–V4 hypervariable region of the 16S rRNA gene for bacterial community analyses following Illumina's protocol (Illumina, San Diego, CA, USA). Briefly, individual barcoded libraries were directly prepared by PCR using long primers (Klindworth et al., 2013) incorporating the Illumina adapter sequences (16S_Amplicon_PCR_Fw: TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG; 16S_Amplicon_PCR_Rv:GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC). After the first amplification, a cleaning step was carried out with the AMPure XP bead clean up (A63880l; Beckman Coulter, Brea, CA, USA). A second PCR reaction was then performed to attach dual index and Illumina sequencing adapters using the Nextera XT Index Kit; followed by a final AMPure XP bead clean up. Amplicons size, integrity and purity were checked by the Bioanalyzer equipment (Agilent, Santa Clara, CA, USA) and the library concentration was measured by fluorimetric quantification with Qubit 2 (Invitrogen, Carlsbad, CA, USA). Finally, the libraries were pooled, adjusted to equimolar concentrations and subjected to 2 × 250 bp paired‐end high‐throughput sequencing on an Illumina Miseq platform (Illumina, San Diego, CA, USA).
Sequence data processing and statistical analysis
FASTQ files were imported into Qiime2 v2020.11 (Bolyen et al., 2019), quality filtering and OTU picking was done using DADA2 v1.18.0 (Callahan et al., 2016). OTUs consisted of groups of identical sequences. Each OTU was represented by a single sequence, named representative sequence. Representative sequences were aligned with mafft, and a phylogenetic tree was built using fasttree (Price et al., 2009; Katoh and Standley, 2014). Taxonomic assignment was based on the Silva database (release 138; Quast et al., 2012) using an ad hoc classifier trained on the region amplified by the primer pairs used in the present study. After the removal of the OTUs annotated as chloroplasts and mitochondria, we performed a rarefaction analysis using 600 reads per sample, to have a homogeneous sampling depth; on this dataset, we calculated alpha and beta diversity. This value was selected after observing the alpha rarefaction plots and witnessing that, at this sampling depth, both the Shannon diversity values and the number of OTUs approached the plateau for all samples (Fig. S10A). In addition, to ensure that the patterns found were not due to the low sampling depth, a rarefaction analysis using 1894 reads per sample was performed (Fig. S10B). After rarefaction, 59 out of 74 samples were retained (the 80.82%). Despite the loss of the 20% of the samples, the experimental design was still balanced in terms of variety (23 samples retained from Barone and 32 from Sole), symptomatology (16 healthy samples and 39 sick ones) and year (11 samples from year 2017 and 44 from 2018). Significant differences in alpha and beta diversity among categories of samples were assessed using the Kruskal‐Wallis (performed on each category) and PERMANOVA tests respectively. Subsequent statistical analyses and data visualizations were performed using the phyloseq package (McMurdie and Holmes, 2013) in R version 4.0.2 (R core team, 2013).
A Venn diagram was drawn to visualize unique and shared genera in sick and healthy plants; a Venn diagram was generated using the web tool http://bioinformatics.psb.ugent.be/webtools/Venn/.
To calculate the species co‐occurrence network, we exported two separate operational taxonomic unit (OTU) tables, one for each symptomatology category. Correlation values among the OTUs were calculated using fastspar (Watts et al., 2019), an implementation of the SparCC data algorithm (Friedman and Alm, 2012). We filtered out correlation absolute values below 0.3 and the table was imported in cytoscape for visualizations and analyses (Shannon et al., 2003).
To identify differential abundances of bacterial taxa between the two conditions tested, we performed a LefSe analysis, as implemented in the Galaxy server (Segata et al., 2011; http://huttenhower.org/galaxy). In this analysis, differences in the relative abundance of taxa between healthy and diseased conditions were calculated with the Kruskal–Wallis test and the trend identified by the Kruskal–Wallis test was then checked by the Wilcoxon test. For the comparison, symptomatology condition was used as a class of interest, with the Wilcoxon test alpha value set at 0.05, the alpha value of the Kruskal–Wallis test set at 0.05 and the threshold for the LDA analysis score was set at 2.0.
Culturable microbiome analysis
The macerated plant tissue of each independent plant sample (5 samples from the first sampling and 10 from the second sampling) displaying foot‐rot symptoms and stored at −80°C was mixed and diluted in 20 ml of PBS as described above. Serial dilutions were performed and 100 μl of each dilution was spread and grown on four different solid media [tryptic soy agar (TSA), PDA, M9 and 869‐medium; Table S7] and the plates were incubated at room temperature (RT) for 2 and 5 days. After 2 days the totality of the colonies grown were harvested with 2 ml of PBS and the same has been done after 5 days of incubation. From the bacterial suspension obtained, the genomic DNA was extracted using the Bacterial genomic DNA isolation kit (Norgen biotech corp., Thorold, Canada) following the manufacturer's instructions. The DNA was used then to amplify the V3 and V4 hypervariable region of the 16S rRNA gene using barcoded primers and PCR conditions following Illumina's protocol (Illumina, San Diego, CA, USA) as described above. Amplicon sequencing of the 16S rRNA gene was performed by using the Illumina MiSeq platform with v3‐v4 chemistry and 2 × 250 paired‐end reads on the MiSeq platform (Illumina) according to the manufacturer's instructions. Sequencing reads were processed and analysed as described above.
Isolation of culturable bacteria from infected rice samples
A collection of bacterial isolates has been created from the same infected rice samples used for the culturable microbiome analysis (see above). The plant samples stored with glycerol at −80°C were resuspended in PBS and serially diluted. The isolation of pure/single bacterial colonies was performed by plating different dilutions on 1/10 TSA (Difco, BD Laboratories, MD 21152, USA) solid medium. Plates were incubated at 20–22°C for 2 and 5 days and pure/single colonies, from the 10−2 dilution plates, showing distinct colony morphology were picked and streaked again on 1/10 TSA plates to ensure the purity of the culture and then stored at −80°C in 1/10 TSA and 18% glycerol.
For the bacterial taxonomic characterization 16S rRNA gene was amplified from single colonies by using fD1Funi 16S (5′‐AGAGTTTGATCCTGGCTCAG‐3′) and rP2Runi 16S (5′‐ ACGGCTACCTTGTTAGGACTT‐3′) universal primers, which flank the approximately 1505 bp region of the 16S rRNA gene (position 8–1511; Weisburg et al., 1991). Colony PCR was performed after boiling (10′ at 98°C) a colony suspension in 50 μl of sterile H2O and under the following PCR conditions: 94°C for 5 min followed by 30 cycles of 94°C for 40 s, 54°C for 40 s, 72°C for 1 min and then 5 min at 72°C before cooling down to 4°C. PCR products were purified by using Gel extraction and PCR Clean‐Up System purification kit (Euroclone S.p.A). The sequencing was performed with primers 907R (5′‐CCGTCAATTCMTTTRAGTTT‐3′) and 785F (5′‐ GGATTAGATACCCTGGTA‐3′), generating two single strand sequences that were combined and overlapped afterwards (Weisburg et al., 1991). The sequencing was realized by GATC (Eurofins Genomics Company, Germany) and identification of the isolates was obtained by BLAST analysis at NCBI (http://www.ncbi.nlm.nih.gov).
In planta test of a selected bacterial isolate to study its possible interaction with D. zeae
Among the taxa positive correlated with D. zeae and the single bacterial species present in the culture collection, we selected Burkholderia spp. A56, to test whether it might interact with D. zeae and it possibly play a role in the development of the disease. For this purpose, experiments by co‐inoculation of rice plants with the pathogen and the culturable bacteria isolate selected Burkholderia spp. A56 were performed. Ampicillin 100, Gentamicin 50 and Nitrofurantoin 100 were used for Burkholderia isolation, selection and growth. A spontaneous D. zeae rifampicin‐resistant (Rifr) was isolated by growing the strain in 1/6 TSB (Tryptic Soy Broth) medium supplemented with gradually increasing amounts of rifampicin (Rif) ranging from 15 to 100 μg ml−1. Finally, the culture was plated on TSA and single colonies were re‐inoculated in TSB containing Rif 100 μg ml−1, stored at −80°C.
The virulence degree of 20‐day rice seedlings single or co‐inoculated has been tested as follow: seeds were sterilized in 50% commercial bleaching agent (3.62% wt/vol NaOCl) for 1 h and, after abundant washes with sterile water, were incubated on sterile water‐soaked Whatman paper in the dark at 30°C for 7 days. Plantlets were transferred independently to a tube containing Hoagland's semi‐solid solution (Steindler et al., 2009) and grown for further 15 days at 27°C, 80% humidity, 16 h/8 h light–dark cycle. Inoculation was performed by injuring the stem with needles contaminated with Burkholderia spp., grown in 1/10 TSB at OD600 of 0.1. The following day, an inoculation in the same position was performed with D. zeae Rifr, grown in 1/10 TSB at OD600 of 0.1. For each treatment, eight biological replicates were performed and as control, plants were inoculated with single strains (only D. zeae or Burkholderia) at the same OD600 and PBS. Lesion appearance and development was followed for 1 week. Bacteria were re‐isolated from the dark rotten lesion in the stem of the plant by macerating it in PBS solution and serial dilutions plated in TSA containing the appropriate antibiotics followed by incubation at 30°C for 24 h and counted for CFU/g calculation.
Author contributions
Cristina Bez, Hang Dinh Thuy, Iris Bertani and Vittorio Venturi conceived the study and participated in its design. Cristina Bez, Iris Bertani and Minh Nguyen Hong conducted the experiments. Cristina Bez, Alfonso Esposito, Silvano Piazza and Danilo Licastro analysed the data. Giampiero Valè provided rice samples and rice field sites. Cristina Bez, Alfonso Esposito and Vittorio Venturi wrote the manuscript. All authors read and approved the final manuscript.
Accession numbers
The raw sequencing data discussed in this publication have been deposited in NCBI's Sequence Read Archive (SRA) and are accessible through Bioproject ID PRJNA602829 http://www.ncbi.nlm.nih.gov/bioproject/602829 .
Supporting information
Appendix S1: Supplementary Information
Acknowledgements
We gratefully acknowledge SA.PI.SE Coop.Agricola (Vercelli) and Ente Nazionale Risi (Milano) for providing rice plant samples and rice seeds. Open Access Funding provided by International Centre for Genetic Engineering and Biotechnology within the CRUI‐CARE Agreement. [Correction added on 20 May 2022, after first online publication: CRUI funding statement has been added.]
References
- Agler, M.T. , Ruhe, J. , Kroll, S. , Morhenn, C. , Kim, S.‐T. , Weigel, D. , and Kemen, E.M. (2016) Microbial hub taxa link host and abiotic factors to plant microbiome variation. PLoS Biol 14: e1002352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barras, F. , van Gijsegem, F. , and Chatterjee, A.K. (1994) Extracellular enzymes and pathogenesis of soft‐rot Erwinia . Annu Rev Phytopathol 32: 201–234. [Google Scholar]
- Bolyen, E. , Rideout, J.R. , Dillon, M.R. , Bokulich, N.A. , Abnet, C.C. , Al‐Ghalith, G.A. , et al. (2019) Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37: 852–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brader, G. , Compant, S. , Vescio, K. , Mitter, B. , Trognitz, F. , Ma, L.‐J. , and Sessitsch, A. (2017) Ecology and genomic insights into plant‐pathogenic and plant‐nonpathogenic endophytes. Annu Rev Phytopathol 55: 61–83. [DOI] [PubMed] [Google Scholar]
- Brady, C.L. , Cleenwerck, I. , Denman, S. , Venter, S.N. , Rodríguez‐Palenzuela, P. , Coutinho, T.A. , and De Vos, P. (2012) Proposal to reclassify Brenneria quercina (Hildebrand and Schroth 1967) Hauben et al. 1999 into a new genus, Lonsdalea gen. Nov., as Lonsdalea quercina comb. nov., descriptions of Lonsdalea quercina subsp. quercina comb. nov., Lonsdalea quercina subsp. iberica subsp. nov. and Lonsdalea quercina subsp. britannica subsp. nov., emendation of the description of the genus Brenneria, reclassification of Dickeya dieffenbachiae as Dickeya dadantii subsp. dieffenbachiae comb. nov., and emendation of the description of Dickeya dadantii . Int J Syst Evol Microbiol 62: 1592–1602. [DOI] [PubMed] [Google Scholar]
- Buonaurio, R. , Moretti, C. , da Silva, D.P. , Cortese, C. , Ramos, C. , and Venturi, V. (2015) The olive knot disease as a model to study the role of interspecies bacterial communities in plant disease. Front Plant Sci 6: 434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan, B.J. , McMurdie, P.J. , Rosen, M.J. , Han, A.W. , Johnson, A.J.A. , and Holmes, S.P. (2016) DADA2: high‐resolution sample inference from Illumina amplicon data. Nat Methods 13: 581–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccarani, C. , Foschi, C. , Parolin, C. , D'Antuono, A. , Gaspari, V. , Consolandi, C. , et al. (2019) Diversity of vaginal microbiome and metabolome during genital infections. Sci Rep 9: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, Y. , Liu, X. , An, S. , Chang, C. , Zou, Y. , Huang, L. , et al. (2013) A nonribosomal peptide synthase containing a stand‐alone condensation domain is essential for phytotoxin zeamine biosynthesis. Mol Plant Microbe Interact 26: 1294–1301. [DOI] [PubMed] [Google Scholar]
- Clemente, J.C. , Ursell, L.K. , Parfrey, L.W. , and Knight, R. (2012) The impact of the gut microbiota on human health: an integrative view. Cell 148: 1258–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Closek, C.J. , Sunagawa, S. , DeSalvo, M.K. , Piceno, Y.M. , DeSantis, T.Z. , Brodie, E.L. , et al. (2014) Coral transcriptome and bacterial community profiles reveal distinct yellow band disease states in Orbicella faveolata . ISME J 8: 2411–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collmer, A. , and Bauer, D. (1994) Erwinia chrysanthemi and pseudomonas syringae: plant pathogens trafficking in extracellular virulence proteins. In Bacterial Pathogenesis of Plants and Animals, Vol 192. Dangl J.L. (ed). Current topics in microbiology and immunology, Berlin/Heidelberg: Springer. pp. 43–78. [DOI] [PubMed] [Google Scholar]
- Dastogeer, K.M. , Tumpa, F.H. , Sultana, A. , Akter, M.A. , and Chakraborty, A. (2020) Plant microbiome–an account of the factors that shape community composition and diversity. Curr Plant Biol 23: 100161. [Google Scholar]
- Eichmann, R. , Richards, L. , and Schäfer, P. (2021) Hormones as go‐betweens in plant microbiome assembly. Plant J 105: 518–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsayed, T.R. , Jacquiod, S. , Nour, E.H. , Sørensen, S.J. , and Smalla, K. (2020) Biocontrol of bacterial wilt disease through complex interaction between tomato plant, antagonists, the indigenous rhizosphere microbiota, and Ralstonia solanacearum . Front Microbiol 10: 2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust, K. , and Raes, J. (2012) Microbial interactions: from networks to models. Nat Rev Microbiol 10: 538–550. [DOI] [PubMed] [Google Scholar]
- Friedman, J. , and Alm, E.J. (2012) Inferring correlation networks from genomic survey data. PLoS Comput Biol 8: e1002687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert, P. (2016) Human Nature and Suffering: London, England: Routledge. [Google Scholar]
- Glazebrook, J. (2005) Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annu Rev Phytopathol 43: 205–227. [DOI] [PubMed] [Google Scholar]
- Goto, M. (1979) Bacterial foot rot of rice caused by a strain of Erwinia chrysanthemi . Phytopathology 69: 213–216. [Google Scholar]
- Hosni, T. , Moretti, C. , Devescovi, G. , Suarez‐Moreno, Z.R. , Fatmi, M.B. , Guarnaccia, C. , et al. (2011) Sharing of quorum‐sensing signals and role of interspecies communities in a bacterial plant disease. ISME J 5: 1857–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain, M.B. , Zhang, H.‐B. , Xu, J.‐L. , Liu, Q. , Jiang, Z. , and Zhang, L.‐H. (2008) The acyl‐homoserine lactone‐type quorum‐sensing system modulates cell motility and virulence of Erwinia chrysanthemi pv. zeae . J Bacteriol 190: 1045–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafra, S. , Przysowa, J. , Gwizdek‐Wiśniewska, A. , and Van Der Wolf, J. (2009) Potential of bulb‐associated bacteria for biocontrol of hyacinth soft rot caused by Dickeya zeae . J Appl Microbiol 106: 268–277. [DOI] [PubMed] [Google Scholar]
- Katoh, K. , and Standley, D.M. (2014) MAFFT: iterative refinement and additional methods. In Multiple Sequence Alignment Methods: Totowa, NJ: Humana Press, 1079: pp. 131–146. [DOI] [PubMed] [Google Scholar]
- Klindworth, A. , Pruesse, E. , Schweer, T. , Peplies, J. , Quast, C. , Horn, M. , and Glöckner, F. O. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next‐generation sequencing‐based diversity studies. Nucleic acids research 41: e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamelas, A. , Desgarennes, D. , López‐Lima, D. , Villain, L. , Alonso‐Sánchez, A. , Artacho, A. , et al. (2020) The bacterial microbiome of meloidogyne‐based disease complex in coffee and tomato. Front Plant Sci 11: 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamichhane, J.R. , and Venturi, V. (2015) Synergisms between microbial pathogens in plant disease complexes: a growing trend. Front Plant Sci 6: 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurila, J. , Hannukkala, A. , Nykyri, J. , Pasanen, M. , Hélias, V. , Garlant, L. , and Pirhonen, M. (2010) Symptoms and yield reduction caused by Dickeya spp. strains isolated from potato and river water in Finland. Eur J Plant Pathol 126: 249–262. [Google Scholar]
- Li, J. , and Convertino, M. (2019) Optimal microbiome networks: macroecology and criticality. Entropy 21: 506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, K. , Bihan, M. , Yooseph, S. , and Methe, B.A. (2012) Analyses of the microbial diversity across the human microbiome. PloS One 7: e32118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, H. , Brettell, L.E. , Qiu, Z. , and Singh, B.K. (2020) Microbiome‐mediated stress resistance in plants. Trends Plant Science 25(8), 733–743. [DOI] [PubMed] [Google Scholar]
- Lozupone, C.A. , Stombaugh, J.I. , Gordon, J.I. , Jansson, J.K. , and Knight, R. (2012) Diversity, stability and resilience of the human gut microbiota. Nature 489: 220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M.D. , and Neufeld, J.D. (2015) Ecology and exploration of the rare biosphere. Nat Rev Microbiol 13: 217–229. [DOI] [PubMed] [Google Scholar]
- Ma, B. , Hibbing, M.E. , Kim, H.‐S. , Reedy, R.M. , Yedidia, I. , Breuer, J. , et al. (2007) Host range and molecular phylogenies of the soft rot enterobacterial genera Pectobacterium and Dickeya. Phytopathology 97: 1150–1163. [DOI] [PubMed] [Google Scholar]
- Ma, Z.S. , Li, L. , and Gotelli, N.J. (2019) Diversity‐disease relationships and shared species analyses for human microbiome‐associated diseases. ISME J 13: 1911–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansfield, J. , Genin, S. , Magori, S. , Citovsky, V. , Sriariyanum, M. , Ronald, P. , et al. (2012) Top 10 plant pathogenic bacteria in molecular plant pathology. Mol Plant Pathol 13: 614–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurdie, P.J. , and Holmes, S. (2013) Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PloS One 8: e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mew, T.W. , Alvarez, A.M. , Leach, J. , and Swings, J. (1993) Focus on bacterial blight of rice. Plant Dis 77: 5–12. [Google Scholar]
- Nassar, A. , Bertheau, Y. , Dervin, C. , Narcy, J.‐P. , and Lemattre, M. (1994) Ribotyping of Erwinia chrysanthemi strains in relation to their pathogenic and geographic distribution. Appl Environ Microbiol 60: 3781–3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perombelon, M.C. , and Kelman, A. (1980) Ecology of the soft rot erwinias. Annu Rev Phytopathol 18: 361–387. [Google Scholar]
- Price, M.N. , Dehal, P.S. , and Arkin, A.P. (2009) FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol 26: 1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proctor, L. (2019) Priorities for the next 10 years of human microbiome research. Nature 569: 623–625. [DOI] [PubMed] [Google Scholar]
- Pu, X. , Zhou, J. , Lin, B. , and Shen, H. (2012) First report of bacterial foot rot of rice caused by a Dickeya zeae in China. Plant Dis 96: 1818–1818. [DOI] [PubMed] [Google Scholar]
- Quast, C. , Pruesse, E. , Yilmaz, P. , Gerken, J. , Schweer, T. , Yarza, P. , et al. (2012) The SILVA ribosomal RNA gene database project: improved data processing and web‐based tools. Nucleic Acids Res 41: D590–D596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team, (2013). R: A language and environment for statistical computing.
- Sabet, K. (1954) A new bacterial disease of maize in Egypt. Empire J Exp Agric 22: 65–67. [Google Scholar]
- Samson, R. , Legendre, J.B. , Christen, R. , Saux, M.F.‐L. , Achouak, W. , and Gardan, L. (2005) Transfer of Pectobacterium chrysanthemi (Burkholder et al. 1953) Brenner et al. 1973 and Brenneria paradisiaca to the genus Dickeya gen. nov. as Dickeya chrysanthemi comb. nov. and Dickeya paradisiaca comb. nov. and delineation of four novel species, Dickeya dadantii sp. nov., Dickeya dianthicola sp. nov., Dickeya dieffenbachiae sp. nov. and Dickeya zeae sp. nov. Int J Syst Evol Microbiol 55: 1415–1427. [DOI] [PubMed] [Google Scholar]
- Schlaeppi, K. , and Bulgarelli, D. (2015) The plant microbiome at work. Mol Plant Microbe Interact 28: 212–217. [DOI] [PubMed] [Google Scholar]
- Segata, N. , Izard, J. , Waldron, L. , Gevers, D. , Miropolsky, L. , Garrett, W.S. , and Huttenhower, C. (2011) Metagenomic biomarker discovery and explanation. Genome Biol 12: R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon, P. , Markiel, A. , Ozier, O. , Baliga, N.S. , Wang, J.T. , Ramage, D. , et al. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sławiak, M. , Łojkowska, E. , and Van der Wolf, J. (2009) First report of bacterial soft rot on potato caused by Dickeya sp.(syn. Erwinia chrysanthemi) in Poland. Plant Pathol 58: 794. [Google Scholar]
- Steindler, L. , Bertani, I. , De Sordi, L. , Schwager, S. , Eberl, L. , and Venturi, V. (2009) LasI/R and RhlI/R quorum sensing in a strain of Pseudomonas aeruginosa beneficial to plants. Appl Environ Microbiol 75: 5131–5140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet, M. , Burian, A. , Fifer, J. , Bulling, M. , Elliott, D. , and Raymundo, L. (2019) Compositional homogeneity in the pathobiome of a new, slow‐spreading coral disease. Microbiome 7: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueki, A. , Akasaka, H. , Satoh, A. , Suzuki, D. , and Ueki, K. (2007) Prevotella paludivivens sp. nov., a novel strictly anaerobic, gram‐negative, hemicellulose‐decomposing bacterium isolated from plant residue and rice roots in irrigated rice‐field soil. Int J Syst Evol Microbiol 57: 1803–1809. [DOI] [PubMed] [Google Scholar]
- Vandenkoornhuyse, P. , Quaiser, A. , Duhamel, M. , Le Van, A. , and Dufresne, A. (2015) The importance of the microbiome of the plant holobiont. New Phytol 206: 1196–1206. [DOI] [PubMed] [Google Scholar]
- Vannier, N. , Agler, M. , and Hacquard, S. (2019) Microbiota‐mediated disease resistance in plants. PLoS Pathog 15: e1007740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vayssier‐Taussat, M. , Albina, E. , Citti, C. , Cosson, J.F. , Jacques, M.‐A. , Lebrun, M.‐H. , et al. (2014) Shifting the paradigm from pathogens to pathobiome: new concepts in the light of meta‐omics. Front Cell Infect Microbiol 4: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts, S.C. , Ritchie, S.C. , Inouye, M. , and Holt, K.E. (2019) FastSpar: rapid and scalable correlation estimation for compositional data. Bioinformatics 35: 1064–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisburg, W.G. , Barns, S.M. , Pelletier, D.A. , and Lane, D.J. (1991) 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol 173: 697–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaneveld, J.R. , McMinds, R. , and Thurber, R.V. (2017) Stress and stability: applying the Anna Karenina principle to animal microbiomes. Nat Microbiol 2: 1–8. [DOI] [PubMed] [Google Scholar]
- Zhou, J. , Zhang, H. , Wu, J. , Liu, Q. , Xi, P. , Lee, J. , et al. (2011) A novel multidomain polyketide synthase is essential for zeamine production and the virulence of Dickeya zeae . Mol Plant Microbe Interact 24: 1156–1164. [DOI] [PubMed] [Google Scholar]
- Zhou, J.N. , Zhang, H.B. , Lv, M.F. , Chen, Y.F. , Liao, L.S. , Cheng, Y.Y. , et al. (2016) SlyA regulates phytotoxin production and virulence in Dickeya zeae EC1. Mol Plant Pathol 17: 1398–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supplementary Information