

Abstract
Background
The leucine‐rich repeat kinase 2 (LRRK2) gene harbors both rare highly damaging missense variants (eg, p.G2019S) and common noncoding variants (eg, rs76904798) with lower effect sizes that are associated with Parkinson's disease (PD) risk.
Objectives
This study aimed to investigate in a large meta‐analysis whether the LRRK2 Genome‐Wide Association Study (GWAS) signal represented by rs76904798 is independently associated with PD risk from LRRK2 coding variation and whether complex linkage disequilibrium structures with p.G2019S and the 5′ noncoding haplotype account for the association of LRRK2 coding variants.
Methods
We performed a meta‐analysis using imputed genotypes from 17,838 patients, 13,404 proxy patients, and 173,639 healthy controls of European ancestry. We excluded carriers of p.G2019S and/or rs76904798 to clarify the role of LRRK2 coding variation in mediating disease risk and excluded carriers of relatively rare LRRK2 coding variants to assess the independence of rs76904798. We also investigated the co‐inheritance of LRRK2 coding variants with p.G2019S, rs76904798, and p.N2081D.
Results
LRRK2 rs76904798 remained significantly associated with PD after excluding the carriers of relatively rare LRRK2 coding variants. LRRK2 p.R1514Q and p.N2081D were frequently co‐inherited with rs76904798, and the allele distribution of p.S1647T significantly changed among patients after removing rs76904798 carriers.
Conclusions
These data suggest that the LRRK2 coding variants previously related to PD (p.N551K, p.R1398H, p.M1646T, and p.N2081D) do not drive the 5′ noncoding GWAS signal. These data, however, do not preclude the independent association of the haplotype p.N551K‐p.R1398H and p.M1646T with altered disease risk. © 2021 The Authors. Movement Disorders published by Wiley Periodicals LLC on behalf of International Parkinson Movement Disorder Society. This article has been contributed to by US Government employees and their work is in the public domain in the USA.
Keywords: Parkinson's disease, genetics, association, LRRK2
The leucine‐rich repeat kinase 2 (LRRK2) gene has been a focus of Parkinson's disease (PD) research since the discovery that pathogenic mutations in the gene are related to autosomal‐dominant PD. 1 , 2 The most common pathogenic mutation, p.G2019S, has a relatively high frequency in Ashkenazi Jews and North African Arabs and is also found in about 1% of Europeans with PD. 3 Although the underlying mechanism of toxicity is not fully elucidated, the p.G2019S mutation has been shown to increase LRRK2 kinase activity, which is considered to cause a toxic gain of function. 4 Hyperactive mutations in LRRK2 have been shown to impact lysosomal and endocytic regulation by increasing LRRK2 recruitment to lysosomes. 5 Some neuropathological changes and clinical features caused by LRRK2‐PD are similar to those of idiopathic PD, suggesting that there could be common mechanisms of pathogenesis and potentially common therapies. 6
Several studies have nominated LRRK2 missense variants as either causal for or associated with an increased risk of PD in individuals of Asian (eg, p.A419V, p.R1628P, and p.G2385R), Arab‐Berber (eg, p.Y2189C), and European ancestries (eg, p.M1646T, p.N2081D, and p.R1441C/G/H). 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 A common protective haplotype (p.N551K‐p.R1398H‐p.K1423K) has also been observed in individuals of both Asian and European ancestries. 7 In addition, common noncoding variation (rs76904798, odds ratio [OR]: 1.15, 95% confidence interval [CI]: 1.13–1.18, P = 1.52e‐28) upstream of LRRK2 is associated with increased PD risk, an effect that is independent of the p.G2019S mutation. 15
Although the exact causal variant at the 5′ GWAS signal of LRRK2 is unknown, 16 , 17 it has been shown in a relatively small cohort of 1381 PD patients and 1328 controls that the 5′ signal had a low degree of correlation with known coding susceptibility variants, including p.M1646T and protective haplotype p.N551K‐R1398H‐K1423K, indicating the independence of this GWAS signal. 18 The independence of this signal from LRRK2 coding variation suggests that changes in the expression or splicing of LRRK2 could mediate PD risk. There is evidence that the GWAS‐nominated noncoding variation tagged by rs76904798 could affect LRRK2 expression. The allele rs76904798‐T has been associated with increased LRRK2 expression in monocytes, 19 in monocyte‐derived microglia‐like cells,20 and in both human brain microglia and induced pluripotent stem cell (iPSC)‐derived models. 21 The same allele has also been associated with faster development of motor symptoms. 22 To further investigate the pattern of association at the LRRK2 locus and determine in a significantly larger data set whether rs76904798 is independently associated with PD from LRRK2 coding variation, we performed a conditional association analysis using individual‐level genotype data from 17,838 PD patients, 13,404 proxy patients (ie, individuals with a first‐degree relative who has PD but does not have PD themselves), and 173,639 healthy controls of European ancestry.
Patients and Methods
Genotyping Data
International Parkinson's Disease Genomics Consortium Genotyping Data
Genotype data were obtained as previously described. 15 Only data sets with participants who had high‐quality imputation scores (R2 > 0.8) for p.G2019S and the 5′ noncoding variant rs76904798 were included. Quality control parameters can be found in the Supplementary Information. In total, 13 data sets were included from the International Parkinson's Disease Genomics Consortium (IPDGC), with individual‐level genotypes from 16,309 PD patients and 17,705 healthy controls of European ancestry (Table S1). Following quality filtering, data sets were imputed using the Haplotype Reference Consortium imputation panel r1.1 2016 through the Michigan imputation server with default settings with phasing using the EAGLE v2.3 option. 23 Genotypes were filtered for imputation quality R2 > 0.8, with the exception of rs10847864 (HIP1R GWAS signal) that was included as an independent signal on chromosome 12 (located ~82 mb upstream of LRRK2) despite slightly lower imputation quality in the Myers‐Foroud (MF) 24 and Spanish Parkinson's (IPDGC) cohorts (SPAIN3 and SPAIN4) (Table S2). Principal components (PCs) were calculated as described in the Supplementary Information.
UK Biobank Data
The UK Biobank is a large study of ~500,000 individuals from the United Kingdom with a variety of phenotypic information, including information on PD status such as ICD‐10 designation and self‐report. 25 PD patients were identified using field code 42033, and proxy patients were included based on their paternal PD status (data field 20107) and maternal PD status (data field 20110). We have previously shown that these proxy patients share genetic risk with PD patients. 15 Genotype data were obtained as previously described 15 and split into case–control and proxy–control data sets for separate GWAS. Genotypes were filtered for imputation quality R2 > 0.8. Quality control parameters are available in the Supplementary Information. After quality control, the case–control data set consisted of individual‐level genotypes from 1529 patients and 15,279 healthy controls, with the controls selected randomly from the pool of nonaffected, nonproxy individuals in the biobank. The proxy–control data set consisted of 13,404 proxy patients and 140,655 healthy controls (Table S1). PCs were calculated as described in the Supplementary Information.
Association Analyses
Summary statistics were generated for each study using a logistic regression model on imputed genotypes from chromosome 12, followed by Firth regression when logistic regression failed to converge. IPDGC data were adjusted for biological sex, age, and the first five PCs representing population substructure. Age at onset was used for PD cases and age at study for healthy controls, except in the Vance (dbGap phs000394) and MF 24 cohorts due to missing data. The UK Biobank data were adjusted for biological sex, age at recruitment, Townsend, and the first five PCs. Genome‐wide association study by proxy was carried out as previously described on UK Biobank proxy‐case data 15 because these individuals are at‐risk but not true cases. A fixed‐effects model was fitted using METAL v2018‐08‐28 under default settings 26 to combine summary statistics across the 13 cohorts from IPDGC and the UK Biobank case–control and proxy‐control data sets. After GWAS filtering of multi‐allelic variants, regional plots of the meta‐analysis results were made around the LRRK2 gene using LocusZoom v1.3. 27 Forest plots were generated using the metafor package in R. A summary of the association results for the LRRK2 variants examined in this study is presented in Table 2 and Table S3. We included an independent association signal on chromosome 12 at the HIP1R locus (rs10847864), identified in a previous PD meta‐analysis, 15 in all LRRK2 conditional analyses. This independent locus is included for comparison to the LRRK2 variants because it is not expected to be affected by the complex linkage disequilibrium (LD) patterns within the LRRK2 region. Statistical power was calculated as described in the Supplementary Information.
TABLE 2.
Association results of LRRK2 variants
| RS‐ID | Protein consequence | Unconditioned, OR (95 CI) | Unconditioned, P‐value | Δ p.G2019S Δ rs76904798, OR (95 CI) | Δ p.G2019S Δ rs76904798, P‐value | Δ p.G2019S Δ p.N2081D, OR (95 CI) | Δ p.G2019S Δ p.N2081D, P‐value | HetISq | HetPVal |
|---|---|---|---|---|---|---|---|---|---|
| rs76904798 | NA | 1.12 (1.08–1.16) | 4.01E‐09 | NA | NA | 1.11 (1.07–1.16) | 1.40E‐07 | 23.793 | 0.04853 |
| rs33995463 | L119P | 0.94 (0.69–1.29) | 0.7223 | 0.91 (0.64–1.29) | 0.5976 | 0.91 (0.66–1.25) | 0.5521 | 5.687 | 0.7708 |
| rs7308720 | N551K | 0.9 (0.85–0.95) | 1.13E‐04 | 0.93 (0.88–0.99) | 0.01506 | 0.91 (0.86–0.96) | 4.55E‐04 | 8.957 | 0.8338 |
| rs10878307 | I723V | 1 (0.95–1.06) | 0.8648 | 1.03 (0.97–1.09) | 0.3545 | 1.03 (0.98–1.08) | 0.3042 | 23.402 | 0.05403 |
| rs7133914 | R1398H | 0.9 (0.86–0.95) | 1.31E‐04 | 0.93 (0.87–0.98) | 0.01029 | 0.91 (0.86–0.96) | 6.92E‐04 | 7.938 | 0.8925 |
| rs35507033 | R1514Q | 1.13 (0.98–1.31) | 0.1002 | 0.98 (0.66–1.47) | 0.9402 | 1.14 (0.98–1.32) | 0.08066 | 9.926 | 0.7676 |
| rs33958906 | P1542S | 0.93 (0.86–1.01) | 0.0807 | 0.97 (0.88–1.05) | 0.4304 | 0.96 (0.88–1.04) | 0.2771 | 17.997 | 0.2069 |
| rs35303786 | M1646T | 1.18 (1.07–1.3) | 9.15E‐04 | 1.16 (1.04–1.3) | 6.59E‐03 | 1.19 (1.08–1.32) | 6.32E‐04 | 21.122 | 0.09855 |
| rs11564148 | S1647T | 0.99 (0.96–1.02) | 0.4799 | 0.99 (0.96–1.03) | 0.7366 | 0.98 (0.95–1.01) | 0.1157 | 10.287 | 0.7409 |
| rs34637584 | G2019S | 9.02 (6.14–13.25) | 3.66E‐29 | NA | NA | NA | NA | 4.34 | 0.993 |
| rs33995883 | N2081D | 1.18 (1.07–1.29) | 6.59E‐04 | 1.2 (0.9–1.61) | 0.2054 | NA | NA | 17.291 | 0.241 |
| rs3761863 | M2397T | 1.01 (0.98–1.04) | 0.5904 | 1.04 (1–1.07) | 0.03988 | 1.01 (0.98–1.04) | 0.7049 | 20.418 | 0.1175 |
| rs10847864 | NA | 1.1 (1.07–1.13) | 8.76E‐11 | 1.09 (1.06–1.13) | 1.65E‐07 | 1.1 (1.06–1.13) | 3.42E‐10 | 32.267 | 3.67E‐03 |
Odds ratios (OR), 95% confidence intervals (CI), and P‐values from the IPDGC and UK Biobank meta‐analyses are reported for all LRRK2 variants examined in this study and the independent chromosome 12 signal in HIP1R (rs10847864). Heterozygosity estimates are based on a meta‐analysis of the unconditioned data sets. “HetISq” and “HetPVal” refer to the I2 and P‐value in the heterogeneity test, respectively. LRRK2, leucine‐rich repeat kinase 2; IPDGC, International Parkinson's Disease Genomics Consortium.
Co‐inheritance Analysis
We used two methods to compare the allelic distributions of the single nucleotide polymorphisms (SNPs) provided in Table 1 before and after removing the carriers of p.G2019S, rs76904798, and p.N2081D or a combination of these variants. First, we aggregated all samples used in the meta‐analysis and calculated the percentage of carriers of a particular SNP that were excluded in the conditioned data sets (Table S4). This was used as a disease‐independent measure of the co‐inheritance of the two variants. Second, we performed Fisher's exact test in R with Bonferroni correction to assess whether there were significant differences in the allelic distributions of each SNP between the unconditioned and conditioned data sets. For this comparison, only patients and proxy patients were included due to the large imbalance between the number of patients and controls in the UK Biobank. This analysis was performed separately for the IPDGC case, UK Biobank case, and proxy‐case data sets (Table S5).
TABLE 1.
LRRK2 variants examined in this study
| MarkerName (hg19) | REF | ALT | RS‐ID | Region | Gene | Protein consequence | MAF unconditioned | MAF conditioned |
|---|---|---|---|---|---|---|---|---|
| 12:40614434 | C | T | rs76904798 | Intergenic | LINC02471;LRRK2 | NA | 0.1451 | 0 |
| 12:40629436 | T | C | rs33995463 | Exonic | LRRK2 | L119P | 0.0026 | 0.0031 |
| 12:40657700 | C | G | rs7308720 | Exonic | LRRK2 | N551K | 0.0667 | 0.0767 |
| 12:40671989 | A | G | rs10878307 | Exonic | LRRK2 | I723V | 0.0684 | 0.0787 |
| 12:40702911 | G | A | rs7133914 | Exonic | LRRK2 | R1398H | 0.0692 | 0.0794 |
| 12:40707778 | G | A | rs35507033 | Exonic | LRRK2 | R1514Q | 0.0089 | 0.0026 |
| 12:40707861 | C | T | rs33958906 | Exonic | LRRK2 | P1542S | 0.029 | 0.0338 |
| 12:40713899 | T | C | rs35303786 | Exonic | LRRK2 | M1646T | 0.0169 | 0.0196 |
| 12:40713901 | T | A | rs11564148 | Exonic | LRRK2 | S1647T | 0.2979 | 0.3442 |
| 12:40734202 | G | A | rs34637584 | Exonic | LRRK2 | G2019S | 0.0009 | 0 |
| 12:40740686 | A | G | rs33995883 | Exonic | LRRK2 | N2081D | 0.0168 | 0.003 |
| 12:40758652 a | C | T | rs3761863 | Exonic | LRRK2 | M2397T | 0.3318 | 0.3536 |
| 12:123326598 | G | T | rs10847864 | Intronic | HIP1R | NA | 0.3563 | 0.3558 |
All LRRK2 missense variants with frequency >0.001 in the IPDGC, UK Biobank case–control and proxy‐control data sets, in addition to rs76904798, p.G2019S, and the independent chromosome 12 signal in HIP1R (rs10847864). MarkerName denotes the chromosome and base pair position with respect to reference assembly GRCh37/hg19. “REF” and “ALT” denote the reference and alternate alleles used in the association analysis. "RS‐ID" denotes the reference SNP cluster ID used by databases and researchers to refer to specific SNPs. “MAF unconditioned” and “MAF conditioned” denote the minor allele frequency from the combined IPDGC and UK Biobank data sets used in the unconditioned and conditioned (Δ p.G2019S Δ rs76904798) analyses, respectively. LRRK2, leucine‐rich repeat kinase 2; IPDGC, International Parkinson's Disease Genomics Consortium.
Association was based on the minor allele rs3761863‐T that corresponds to the protein consequence T2397M.
Code Availability
All codes used in this study are available at https://github.com/neurogenetics/LRRK2_conditional_v3.
Results
Included Data Overview
We included 15 data sets, with 13 case–control cohorts (n_case = 16,309, n_control = 17,705), case–control data from the UK Biobank (n_case = 1529, n_control = 15,279), and proxy‐control data from the UK Biobank (n_proxy‐case = 13,404, n_control = 140,655). Together, these sample sets comprised 17,838 PD patients, 13,404 proxy patients, and 173,639 healthy controls of European ancestry. The mean age of onset for PD in the IPDGC cohorts, age at recruitment for the UK Biobank, and percentage of female participants for each data set are reported in Table S1, where this information was available.
LRRK2 rs76904798 Is Independently Associated with PD Risk From LRRK2 Coding Variation
In the examination of the IPDGC, UK Biobank case–control, and proxy–control data sets, we identified 10 nonsynonymous LRRK2 coding variants with a minor allele frequency (MAF) > 0.001 in all three data sets, as presented in Table 1. We verified the presence of these variants in the IPDGC data sets using whole‐genome sequencing data 28 and in the UK Biobank data sets using whole‐exome sequencing data. 29 The concordance rates were very high (average = 99.24%), showing that the imputation of these variants was highly accurate. The imputation quality and concordance values are presented in Tables S2 and S6, respectively.
Four of these variants (p.N551K, p.R1398H, p.M1646T, and p.N2081D) have been previously associated with either decreased or increased PD risk in Europeans, 7 , 8 , 30 and the haplotype p.S1647T‐p.M2397T has been suggested to confer a protective effect in Taiwanese individuals. 31 Although none of the four previously associated variants reached genome‐wide significance in our meta‐analysis (Table 2; Figs. S1–S4), the direction of the effects was consistent with previous studies. 7 , 15 We did not find any evidence of an association with PD for either p.S1647T or p.M2397T in this study (Figs. S5 and S6).
To confirm the effect of the previously identified PD GWAS loci in LRRK2, we first verified the associations of the known pathogenic variant p.G2019S (OR: 9.02, 95% CI: 6.14–13.25, P = 3.66E‐29) and the 5′ noncoding variant rs76904798 (OR: 1.12, 95% CI: 1.08–1.16, P = 4.01E‐09) in a meta‐analysis of all 15 data sets (Table 2; Fig. S7). We then confirmed the independent association of rs76904798 with PD risk from p.G2019S and p.N2081D (Δ p.G2019S Δ p.N2081D, OR: 1.11, 95% CI: 1.07–1.16, P = 1.40e−07) (Table 2; Fig. S8), the PD‐linked LRRK2 coding variants (Δ p.G2019S Δ p.N551K Δ p.R1398H Δ p.M1646T Δ p.N2081D, OR: 1.11, 95% CI: 1.06–1.16, P = 2.144e−06), and all of the relatively rare LRRK2 coding variants examined in this study (Δ p.G2019S Δ p.N551K Δ p.R1398H Δ p.M1646T Δ p.N2081D Δ p.L119P Δ p.I723V Δ p.R1514Q Δ p.P1542S, OR: 1.09, 95% CI: 1.04–1.15, P = 0.0002082) (Fig. 1). We used a Bonferroni‐corrected P‐value of 0.006 to determine the independence of rs76904798, which is based on the number of LRRK2 coding variants excluding p.G2019S that were examined in this study (Table 1), two of which (p.N551K‐p.R1398H) represent the same allele (0.05/9 = 0.006). We also confirmed the independent association of rs76904798 by including the allele counts of the relatively rare LRRK2 coding variants as covariates in a logistic regression model (OR: 1.11, 95% CI: 1.06–1.15, P = 9.90e‐07) (Table S7).
FIG 1.
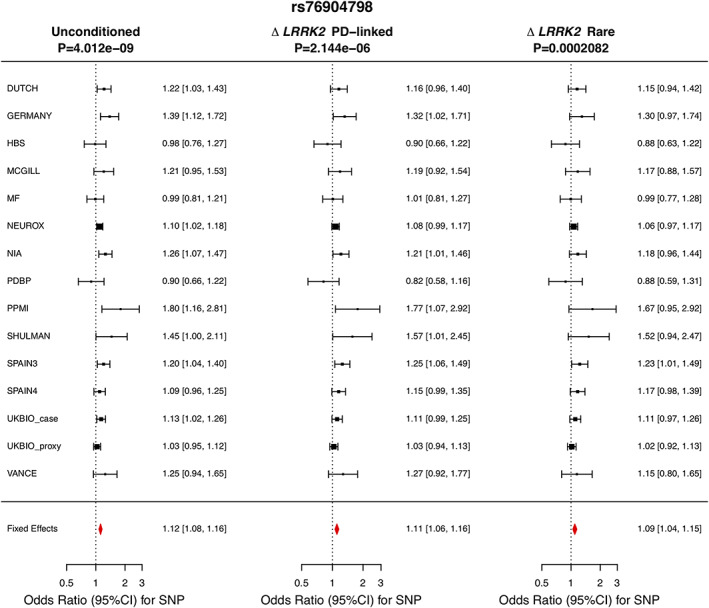
Meta‐analysis of rs76904798 in the included data sets excluding (from left to right) (1) no samples; (2) carriers of LRRK2 p.G2019S, p.N551K, p.R1398H, p.M1646T, and p.N2081D; and (3) carriers of LRRK2 p.G2019S, p.N551K, p.R1398H, p.M1646T, p.N2081D, p.L119P, p.I723V, p.R1514Q, and p.P1542S.
There were no other independent signals in the LRRK2 region that reached genome‐wide significance (P < 5e‐8). The association of the 3′ intergenic variant rs190807041 disappears after conditioning on p.G2019S, and the 5′ signal driven by noncoding LRRK2 variation explains the associations of both rs76904798 and the proximal intronic variant rs1491942 with PD risk (Fig. 2). The additional signal in the 5′ locus, rs1491942, is in complete LD with rs76904798, and this signal disappears when carriers of rs76904798 are removed.
FIG 2.
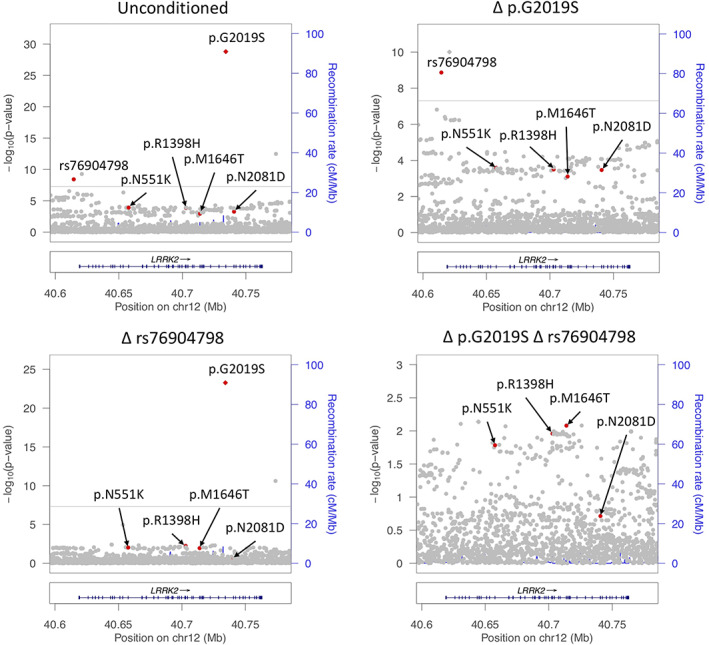
LocusZoom plot of LRRK2 association with Parkinson's disease risk.
Conditional Analysis of LRRK2 Exonic Variants Does Not Preclude an Independent Association of p.N551K‐p.R1398H and p.M1646T With Altered PD Risk
To better understand the role of coding variation at the LRRK2 locus in PD, we performed a meta‐analysis excluding the carriers of p.G2019S and rs76904798, representing the two independent GWAS signals at the locus. In total, there were 12,504 PD patients (total removed: 29.9%, p.G2019S carriers: 1.4%, rs76904798 carriers: 28.8%), 9679 proxy patients (total removed: 27.8%, p.G2019S carriers: 0.2%, rs76904798 carriers: 27.6%), and 127,254 controls (total removed: 26.7%, p.G2019S carriers: 0.1%, rs76904798 carriers: 26.6%) in the conditional analysis. After removing carriers of p.G2019S and rs76904798, there were no LRRK2 variants that reached genome‐wide significance. The results of the association analyses for the LRRK2 variants examined in this study are presented in Table 2 and Table S3, and forest plots are presented in Figures S1–S6 and S8–S12. Forest plots of LRRK2 p.K1423K, which is in high LD with p.N551K‐p.R1398H, and rs10847864 (HIP1R GWAS signal) are presented in Figures S13 and S14, respectively.
With this sample size, we had ~80% power to detect genotype relative risks ≥1.16 and ≥1.44 based on the MAFs of p.M1646T and p.N2081D, respectively, in the conditional analysis (Table 1). Similarly, we had ~80% power to detect genotype relative risks ≤0.93 based on the MAF of p.N551K and p.R1398H in the conditional analysis (Table 1). This calculation is based on the Bonferroni‐corrected significance threshold of P = 0.006.
None of the LRRK2 variants examined in this study passed Bonferroni multiple test correction after excluding the carriers of p.G2019S and rs76904798, including the previously PD‐linked variants p.N551K (OR_condi: 0.93, 95% CI: 0.88–0.99, P = 0.01506), p.R1398H (OR_condi: 0.93, 95% CI: 0.87–0.98, P = 0.01029), p.M1646T (OR_condi: 1.16, 95% CI: 1.04–1.3, P = 0.00659), and p.N2081D (OR_condi: 1.2, 95% CI: 0.91–1.61, P = 0.2054) (Table 2; Figs. S1–S4). LocusZoom plots of the stepwise conditional analysis are shown in Figure 2. However, p.N551K, p.R1398H, and p.M1646T retained Bonferroni‐corrected significance when p.G2019S and rs76904798 allele counts were included as covariates in a logistic regression model, though they still did not meet genome‐wide significance (Table S7). We also performed a meta‐analysis excluding the carriers of p.N2081D and p.G2019S because rs76904798 is often co‐inherited with p.N2081D (Fig. S15). After only the p.G2019S carriers were removed, LRRK2 p.N551K, p.R1398H, p.M1646T, and p.N2081D passed Bonferroni multiple test correction (Table S3).
Rare LRRK2 Missense Variants Are Co‐Inherited With rs76904798
To further investigate the effects of excluding rs76904798 carriers on the allele distribution of LRRK2 coding variants, we calculated the percentage of carriers that were removed after excluding the carriers of p.G2019S and rs76904798 in our data sets, as shown in Table S4. We found that 86.64% of p.N2081D carriers and 79.07% of p.R1514Q carriers were excluded after removing the carriers of rs76904798 from all 15 data sets. Only 0.19% of p.N2081D carriers and 0.08% of p.R1514Q carriers were excluded after removing p.G2019S carriers, all of which also carried rs76904798. Conversely, only 10.72% of rs76904798 carriers were excluded after removing p.N2081D carriers. Because the 5′ variant rs76904798 remained associated with PD after removing the carriers of p.G2019S and p.N2081D (Fig. S8), the GWAS signal represented by rs76904798 is independent of p.N2081D, and the previous significance reported for p.N2081D might be due to the rs76904798 signal.
We also found significant differences in the allele distributions of p.R1514Q, p.N2081D, and p.S1647T after removing rs76904798 carriers from the IPDGC case, UK Biobank case, and proxy‐case data sets (Table S5). Although p.S1647T was significantly enriched among noncarriers of rs76904798, this variant was not associated with PD, and around 17% of carriers were excluded after removing rs76904798 carriers. Only around 16% of p.N551K and p.R1398H carriers were excluded after removing rs76904798 carriers, and less than 1% after removing p.G2019S carriers. The complete results of the co‐inheritance analysis are presented in Tables S4 and S5, and the frequencies and allele distributions of the LRRK2 variants examined in this study are presented in Table S8.
Discussion
It has been established that both rare and common variations at the LRRK2 locus can influence PD susceptibility. Several large GWASs and meta‐analyses have pointed to at least two independent association signals, represented by p.G2019S and 5′ noncoding variation, that reach genome‐wide significance. 15 However, the literature has often produced conflicting or inconclusive results when it comes to PD risk associated with common variation at the LRRK2 locus. Interestingly, several LRRK2 variants have been associated with other disorders, suggesting that there are shared pathological mechanisms that result from genetic changes in LRRK2. For example, LRRK2 p.N2081D has been implicated as a shared risk variant in Crohn's disease,8 and independent LRRK2 5' variation has been shown to be a modifier of progressive supranuclear palsy phenotypes. 32 To refine our understanding of the pattern of LRRK2 association with PD, we investigated the PD risk associated with LRRK2 missense variants in a large meta‐analysis of European‐ancestry individuals.
First, we confirmed the genome‐wide associations of p.G2019S and rs76904798 and identified the potential associations of four other PD‐linked LRRK2 missense variants (p.N551K, p.R1398H, p.M1646T, and p.N2081D), all of which passed multiple test correction. After removing carriers of the common noncoding variant rs76904798, we found that these missense variants do not pass Bonferroni correction, whereas they retained Bonferroni‐corrected significance when rs76904798 was included as a covariate in a logistic regression model. These data therefore do not exclude the possibility that the putative protective haplotype p.N551K‐p.R1398H and proposed risk factor p.M1646T are independently associated with PD from rs76904798. These variants, however, did not meet genome‐wide significance in our study despite including a very large sample size.
In a recent analysis of whole‐genome sequencing data from the Accelerating Medicines Partnership‐Parkinson's Disease cohort, Bryant and colleagues found that the 5′ variant rs76904798 was associated with LRRK2 p.R1514Q and p.N2081D. 33 We confirmed the co‐inheritance of these variants with rs76904798 in a data set that is ~20 times larger and also found a significant difference in the allele distribution of p.S1647T after removing rs76904798 carriers. Our results suggest that any association between p.N2081D and PD is very likely due to linkage disequilibrium with rs76904798. Bryant and colleagues suggested that p.N2081D may be important in mediating disease risk associated with the 5′ noncoding variation. However, we establish here that rs76904798 is independently associated with PD from p.N2081D, and therefore, the role of p.N2081D in PD pathogenesis needs to be explored further and potentially reclassified.
Several LRRK2 variants have been implicated in mechanisms that are relevant to PD pathogenesis. LRRK2 rs76904798 has been previously associated with increased LRRK2 expression in monocytes 19 and in human brain and iPSC‐derived microglia 21. To further investigate the other potential effects of this variant on expression, we queried the Open Targets Platform (v. 21.06) in July 2021 for eQTL data across tissue and cell types. LRRK2 expression linked to this variant was significant across multiple tissues, including artery, skin, and thyroid, as well as monocyte cells, which is concordant with previously cited literature. The most significant expression signal was from whole‐blood eQTL data sourced from eQTLgen (beta = 0.290, P = 1.3e‐111). Increased LRRK2 expression in whole blood and monocytes is supportive of previous studies that suggest LRRK2 has a role in immune regulation and inflammation. 34 , 35 Based on public data sources and previous literature, rs76904798 has significant evidence for increasing LRRK2 expression in immune‐related cells.
Previous studies of the p.G2019S mutation have shown that increased LRRK2 kinase activity, typically estimated to be approximately twofold compared to wild type, has negative effects on neuronal survival. 36 , 37 , 38 , 39 Both p.N2081D and p.M1646T are also associated with increased kinase activity compared to wild‐type LRRK2, 8 , 40 although the effect size is modest compared to p.G2019S. Despite this evidence, it is possible that prior data using transfection of plasmids in HEK293T cells are unable to disambiguate the effects of p.N2081D and rs76904798 as they are often present on the same haplotype. LRRK2 p.M1646T has also been associated with increased glucocerebrosidase activity with a larger effect than p.G2019S. 30 In addition, LRRK2 p.R1398H has been shown to affect GTPase and Wnt signaling activity contrary to pathogenic LRRK2 mutations. 8 , 41 , 42 , 43 A recent study has shown that both p.R1398H and p.N551K were able to counteract the putative pathogenic effects of p.G2019S in Drosophila models. 44 The literature therefore suggests that each of these variants has measurable effects on protein function in cells and in vivo that are consistent with the proposed direction of effects for risk versus protection. However, the relation between the functional impact and risk for PD needs to be further investigated for these coding variants.
Previous studies have also implicated the common LRRK2 variant p.M2397T in several disorders. Although this variant does not appear to affect kinase activity, 38 the allele M2397 has been reported to lower LRRK2 abundance due to protein destabilization, 45 which has been related to enhanced inflammatory responses in both Crohn's disease 45 , 46 and leprosy. 47 , 48 Despite the fact that p.M2397T has demonstrated a protective association with PD in a study consisting of 573 Taiwanese patients, 31 and with MSA—a disease that shares many clinical and pathological features with PD—in a study consisting of 177 neuropathologically confirmed patients from America and the United Kingdom, 49 we did not find any evidence that p.M2397T was associated with PD in this study. It is possible that previous studies linking LRRK2 p.M2397T with PD and MSA were underpowered due to relatively small sample sizes, and further investigation in larger and diverse cohorts is warranted.
We note several limitations of this study. First, our use of imputed genotype data limits the accuracy of some genotypes, though very high concordance of the LRRK2 coding variants with whole‐genome and whole‐exome sequence data shows that the imputation of these variants was highly accurate. Second, although we included a large amount of data in our meta‐analysis, removing over one‐quarter of the samples in the conditional analysis limited our statistical power to detect less‐common small‐effect (OR ~ 1.2) variants. Therefore, we cannot exclude that relatively common coding variants have a very small effect resulting in increased or decreased risk for PD. Further studies exploring the cumulative effects of common small‐effect variants could enhance our understanding of their genetic burden and should consider the complex linkage disequilibrium structure of the LRRK2 region. Third, our analysis was limited to individuals of European ancestry, so the results of this study may not extend beyond European‐ancestry populations. In‐depth studies of PD‐linked genes in diverse cohorts will help clarify the associations discussed here as well as broader disease mechanisms.
In conclusion, here we provide insights into the relationship between coding and noncoding variation at the LRRK2 locus and demonstrate that the 5′ LRRK2 noncoding GWAS signal represented by rs76904798 is independently associated with PD risk from LRRK2 coding variation. These coding variants are therefore unlikely to drive the LRRK2 GWAS signal in individuals of European ancestry and require additional genetic and functional studies to clarify their potential impact on disease state. Characterizing the PD susceptibility associated with LRRK2 genetic variation is critical for anticipating disease progression and response to LRRK2‐targeted therapeutics.
AuthorRoles
Concept and design: C.B., H.L.L.
Statistical analysis: J.L., M.A.N., C.B., H.L.L.
Contributed expertise, data, or DNA samples: X.R., R.L., M.R.C., Z.G.‐O.
Drafting of the manuscript: All authors
Critical revision of the manuscript: All authors
Supporting information
APPENDIX S1 Supporting Information
Figure S1. Meta‐analysis of p.N551K in the included data sets excluding (from left to right) (1) no samples, (2) carriers of rs76904798 and p.G2019S, and (3) carriers of p.N2081D and p.G2019S.
Figure S2. Meta‐analysis of p.R1398H in the included data sets excluding (from left to right) (1) no samples, (2) carriers of rs76904798 and p.G2019S, and (3) carriers of p.N2081D and p.G2019S.
Figure S3. Meta‐analysis of p.M1646T in the included data sets excluding (from left to right) (1) no samples, (2) carriers of rs76904798 and p.G2019S, and (3) carriers of p.N2081D and p.G2019S.
Figure S4. Meta‐analysis of p.N2081D in the included data sets excluding (from left to right) (1) no samples and (2) carriers of rs76904798 and p.G2019S.
Figure S5. Meta‐analysis of p.S1647T in the included data sets excluding (from left to right) (1) no samples, (2) carriers of rs76904798 and p.G2019S, and (3) carriers of p.N2081D and p.G2019S.
Figure S6. Meta‐analysis of p.M2397T in the included data sets excluding (from left to right) (1) no samples, (2) carriers of rs76904798 and p.G2019S, and (3) carriers of p.N2081D and p.G2019S.
Figure S7. Meta‐analysis of (from left to right) (1) p.G2019S and (2) rs76904798 in the included data sets.
Figure S8. Meta‐analysis of rs76904798 in the included data sets excluding (from left to right) (1) no samples and (2) carriers of p.N2081D and p.G2019S.
Figure S9. Meta‐analysis of p.L119P in the included data sets excluding (from left to right) (1) no samples, (2) carriers of rs76904798 and p.G2019S, and (3) carriers of p.N2081D and p.G2019S.
Figure S10. Meta‐analysis of p.I723V in the included data sets excluding (from left to right) (1) no samples, (2) carriers of rs76904798 and p.G2019S, and (3) carriers of p.N2081D and p.G2019S.
Figure S11. Meta‐analysis of p.R1514Q in the included data sets excluding (from left to right) (1) no samples, (2) carriers of rs76904798 and p.G2019S, and (3) carriers of p.N2081D and p.G2019S.
Figure S12. Meta‐analysis of p.P1542S in the included data sets excluding (from left to right) (1) no samples, (2) carriers of rs76904798 and p.G2019S, and (3) carriers of p.N2081D and p.G2019S.
Figure S13. Meta‐analysis of p.K1423K in the included data sets excluding (from left to right) (1) no samples, (2) carriers of rs76904798 and p.G2019S, and (3) carriers of p.N2081D and p.G2019S.
Figure S14. Meta‐analysis of rs10847864 in the included data sets excluding (from left to right) (1) no samples, (2) carriers of rs76904798 and p.G2019S, and (3) carriers of p.N2081D and p.G2019S.
Figure S15. LocusZoom plot of LRRK2 association with Parkinson's disease risk conditioned on p.N2081D. The left panel shows the association signal at the LRRK2 locus in the IPDGC and UK Biobank meta‐analysis conditioned on p.N2081D, and the right panel conditions on both p.G2019S and p.N2081D. The LRRK2 variants p.N551K, p.R1398H, p.M1646T, p.G2019S, and rs76904798 are indicated by red dots.
Table S1. Overview of included data.
Table S2. Imputation quality of LRRK2 variants examined in this study.
Table S3. Summary statistics of LRRK2 variants after removing carriers of the specified variants.
Table S4. Co‐inheritance analysis of LRRK2 variants examined in this study with p.G2019S, rs76904798, and p.N2081D.
Table S5. Fisher's exact test comparing the allele distributions of LRRK2 variants among cases in the unconditioned and conditioned data sets.
Table S6. Concordance of LRRK2 variants in the imputed data sets with whole‐exome sequencing data (IPDGC) and whole‐genome sequencing data (UKBIO).
Table S7. Summary statistics of LRRK2 variants, including the allele counts of the specified variants as covariates.
Table S8. Frequencies and allele distributions of LRRK2 variants examined in this study in the unconditioned and conditioned data sets.
Acknowledgements
We thank all the subjects who donated their time and biological samples to be a part of this study. We also thank all members of the International Parkinson Disease Genomics Consortium (IPDGC). See for a complete overview of members, acknowledgments, and funding http://pdgenetics.org/partners. This work was supported in part by the Intramural Research Programs of the National Institute of Neurological Disorders and Stroke (NINDS), the National Institute on Aging (NIA), and the National Institute of Environmental Health Sciences both part of the National Institutes of Health, Department of Health and Human Services: project numbers 1ZIA‐NS003154, Z01‐AG000949‐02, and Z01‐ES101986. In addition, this work was supported by the Department of Defense (award W81XWH‐09‐2‐0128) and The Michael J. Fox Foundation for Parkinson's Research. This work was supported by the National Institutes of Health grants R01NS037167, R01CA141668, P50NS071674, American Parkinson Disease Association (APDA); Barnes Jewish Hospital Foundation; and Greater St Louis Chapter of the APDA. The KORA (Cooperative Research in the Region of Augsburg) research platform was started and financed by the Forschungszentrum für Umwelt und Gesundheit, which is funded by the German Federal Ministry of Education, Science, Research, and Technology and by the State of Bavaria. This study was also funded by the German Federal Ministry of Education and Research (BMBF) under the funding code 031A430A; the EU Joint Programme—Neurodegenerative Diseases Research (JPND) project under the aegis of JPND—www.jpnd.eu—through Germany; BMBF, funding code 01ED1406; and iMed—the Helmholtz Initiative on Personalized Medicine. This study is funded by the German National Foundation grant (DFG SH599/6‐1), The Michael J. Fox Foundation, and MSA Coalition, USA. The French GWAS work was supported by the French National Agency of Research (ANR‐08‐MNP‐012). This study was also funded by France‐Parkinson Association, Fondation de France, the French program “Investissements d'avenir” funding (ANR‐10‐IAIHU‐06), and a grant from Assistance Publique‐Hôpitaux de Paris (PHRC, AOR‐08010) for the French clinical data. This study utilized the high‐performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health, Bethesda, Maryland (http://biowulf.nih.gov), and DNA panels, samples, and clinical data from the National Institute of Neurological Disorders and Stroke Human Genetics Resource Center DNA and Cell Line Repository. People who contributed samples are acknowledged in descriptions of every panel on the repository website. We thank the French Parkinson's Disease Genetics Study Group and the Drug Interaction with genes (DIGPD) study group: Y. Agid, M. Anheim, F. Artaud, A.‐M. Bonnet, C. Bonnet, F. Bourdain, J.‐P. Brandel, C. Brefel‐Courbon, M. Borg, A. Brice, E. Broussolle, F. Cormier‐Dequaire, J.‐C. Corvol, P. Damier, B. Debilly, B. Degos, P. Derkinderen, A. Destée, A. Dürr, F. Durif, A. Elbaz, D. Grabli, A. Hartmann, S. Klebe, P. Krack, J. Kraemmer, S. Leder, S. Lesage, R. Levy, E. Lohmann, L. Lacomblez, G. Mangone, L.‐L. Mariani, A.‐R. Marques, M. Martinez, V. Mesnage, J. Muellner, F. Ory‐Magne, F. Pico, V. Planté‐Bordeneuve, P. Pollak, O. Rascol, K. Tahiri, F. Tison, C. Tranchant, E. Roze, M. Tir, M. Vérin, F. Viallet, M. Vidailhet, and A. You. We also thank the members of the French 3C Consortium: A. Alpérovitch, C. Berr, C. Tzourio, and P. Amouyel for allowing us to use part of the 3C cohort and D. Zelenika for support in generating the genome‐wide molecular data. We thank P. Tienari (Molecular Neurology Programme, Biomedicum, University of Helsinki), T. Peuralinna (Department of Neurology, Helsinki University Central Hospital), L. Myllykangas (Folkhalsan Institute of Genetics and Department of Pathology, University of Helsinki), and R. Sulkava (Department of Public Health and General Practice Division of Geriatrics, University of Eastern Finland) for the Finnish controls (Vantaa85+ GWAS data). This study was also funded by the Sigrid Juselius Foundation (KM). We used genome‐wide association data generated by the Wellcome Trust Case‐Control Consortium 2 (WTCCC2) from UK patients with Parkinson's disease and UK control individuals from the 1958 Birth Cohort and National Blood Service. UK population control data were made available through WTCCC1. This study was supported by the Medical Research Council and Wellcome Trust disease center (grant WT089698/Z/09/Z). As with previous IPDGC efforts, this study makes use of data generated by the Wellcome Trust Case‐Control Consortium. A full list of the investigators who contributed to the generation of the data is available at www.wtccc.org.uk. Funding for the project was provided by the Wellcome Trust under award 076113, 085475, and 090355. This study was also supported by Parkinson's UK (grants 8047 and J‐0804) and the Medical Research Council (G0700943 and G1100643). Sequencing and genotyping done in McGill University was supported by grants from The Michael J. Fox Foundation; the Canadian Consortium on Neurodegeneration in Aging (CCNA); the Canada First Research Excellence Fund (CFREF), awarded to McGill University for the Healthy Brains for Healthy Lives (HBHL) program; and Parkinson's Society Canada. The access to part of the participants at McGill has been made possible thanks to the Quebec Parkinson's Network (http://rpq-qpn.ca/en/). DNA extraction work that was done in the United Kingdom was undertaken at University College London Hospitals, University College London, which received a proportion of funding from the Department of Health's National Institute for Health Research Biomedical Research Centre's funding. This study was supported in part by the Wellcome Trust/Medical Research Council Joint Call in Neurodegeneration award (WT089698) to the Parkinson's Disease Consortium (UKPDC), whose members are from the UCL Institute of Neurology, University of Sheffield, and the Medical Research Council Protein Phosphorylation Unit at the University of Dundee. We thank the Quebec Parkinson's Network (http://rpq-qpn.org) and its members. Harvard NeuroDiscovery Biomarker Study (HBS) is a collaboration of HBS investigators and funded through philanthropy and NIH and non‐NIH funding sources. The HBS investigators have not participated in reviewing the data analysis or content of the manuscript. PPMI—a public–private partnership—is funded by The Michael J. Fox Foundation for Parkinson's Research and funding partners, the full names of all of the PPMI funding partners are available at www.ppmi-info.org/fundingpartners. The PPMI investigators have not participated in reviewing the data analysis or content of the manuscript. For up‐to‐date information on the study, visit www.ppmi-info.org. Parkinson's Disease Biomarker Program (PDBP) consortium is supported by the National Institute of Neurological Disorders and Stroke (NINDS) at the National Institutes of Health. A full list of PDBP investigators is available at https://pdbp.ninds.nih.gov/policy. The PDBP Investigators have not participated in reviewing the data analysis or content of the manuscript.
Cornelis Blauwendraat and Hampton L. Leonard contributed equally to this study.
Relevant conflicts of interest/financial disclosures: H.L.L. and M.A.N. reported receiving support from a consulting contract between Data Tecnica International and the National Institute on Aging (NIA), National Institutes of Health (NIH), as well as ad hoc consulting for various companies. Z.G.‐O. received consultancy fees from Lysosomal Therapeutics Inc., Idorsia, Prevail Therapeutics, Inceptions Sciences (now Ventus), Ono Therapeutics, Denali, Neuron23, Handl Therapeutics, Bial Biotech, Lighthouse, Guidepoint, and Deerfield. No other disclosures were reported.
Funding agencies: This work was supported in part by the Intramural Research Programs of the National Institute of Neurological Disorders and Stroke (NINDS), the National Institute on Aging (NIA), and the National Institute of Environmental Health Sciences both part of the National Institutes of Health, Department of Health and Human Services (project numbers 1ZIA‐NS003154, Z01‐AG000949‐02, and Z01‐ES101986).
[The copyright line for this article was changed on 09 October 2021, after original online publication.]
Data Availability
UK Biobank data are available via application (https://www.ukbiobank.ac.uk/). Various GWAS summary statistics from the IPDGC data are publicly available (https://pdgenetics.org/resources).
References
- 1. Paisán‐Ruíz C, Jain S, Evans EW, et al. Cloning of the gene containing mutations that cause PARK8‐linked Parkinson's disease. Neuron 2004;44(4):595–600. [DOI] [PubMed] [Google Scholar]
- 2. Zimprich A, Biskup S, Leitner P, et al. Mutations in LRRK2 cause autosomal‐dominant parkinsonism with pleomorphic pathology. Neuron 2004;44(4):601–607. [DOI] [PubMed] [Google Scholar]
- 3. Healy DG, Falchi M, O'Sullivan SS, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2‐associated Parkinson's disease: a case‐control study. Lancet Neurol 2008;7(7):583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kluss JH, Mamais A, Cookson MR. LRRK2 links genetic and sporadic Parkinson's disease. Biochem Soc Trans 2019;47(2):651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bonet‐Ponce L, Beilina A, Williamson CD, et al. LRRK2 mediates tubulation and vesicle sorting from lysosomes. Sci Adv 2020;6(46):eabb2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tolosa E, Vila M, Klein C, Rascol O. LRRK2 in Parkinson disease: challenges of clinical trials. Nat Rev Neurol 2020;16(2):97–107. [DOI] [PubMed] [Google Scholar]
- 7. Ross OA, Soto‐Ortolaza AI, Heckman MG, et al. Association of LRRK2 exonic variants with susceptibility to Parkinson's disease: a case‐control study. Lancet Neurol 2011;10(10):898–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hui KY, Fernandez‐Hernandez H, Hu J, et al. Functional variants in the gene confer shared effects on risk for Crohn's disease and Parkinson's disease. Sci. Transl. Med 2018;10(423):eaai7795. 10.1126/scitranslmed.aai7795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wu X, Tang K‐F, Li Y, et al. Quantitative assessment of the effect of LRRK2 exonic variants on the risk of Parkinson's disease: a meta‐analysis. Parkinsonism Relat Disord 2012;18(6):722–730. 10.1016/j.parkreldis.2012.04.013 [DOI] [PubMed] [Google Scholar]
- 10. Tan EK, Peng R, Teo YY, et al. Multiple LRRK2 variants modulate risk of Parkinson disease: a Chinese multicenter study. Hum Mutat 2010;31(5):561–568. [DOI] [PubMed] [Google Scholar]
- 11. Ross OA, Wu YR, Lee MC, et al. Analysis of Lrrk2 R1628P as a risk factor for Parkinson's disease. Ann Neurol 2008;64(1):88–92. [DOI] [PubMed] [Google Scholar]
- 12. Farrer MJ, Stone JT, Lin CH, et al. Lrrk2 G2385R is an ancestral risk factor for Parkinson's disease in Asia. Parkinsonism Relat Disord 2007;13(2):89–92. [DOI] [PubMed] [Google Scholar]
- 13. Di Fonzo A, Wu‐Chou YH, Lu CS, et al. A common missense variant in the LRRK2 gene, Gly2385Arg, associated with Parkinson's disease risk in Taiwan. Neurogenetics 2006;7(3):133–138. [DOI] [PubMed] [Google Scholar]
- 14. González‐Fernández MC, Lezcano E, Ross OA, et al. Lrrk2‐associated parkinsonism is a major cause of disease in northern Spain. Parkinsonism Relat Disord 2007;13(8):509–515. [DOI] [PubMed] [Google Scholar]
- 15. Nalls MA, Blauwendraat C, Vallerga CL, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson's disease: a meta‐analysis of genome‐wide association studies. Lancet Neurol 2019;18(12):1091–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Trabzuni D, Ryten M, Emmett W, et al. Fine‐mapping, gene expression and splicing analysis of the disease associated LRRK2 locus. PLoS One 2013;8(8):e70724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Heckman MG, Labbé C, Kolicheski AL, et al. Fine‐mapping of the non‐coding variation driving the Caucasian LRRK2 GWAS signal in Parkinson's disease. Parkinsonism Relat Disord 2021;83:22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Soto‐Ortolaza AI, Heckman MG, Labbé C, et al. GWAS risk factors in Parkinson's disease: LRRK2 coding variation and genetic interaction with PARK16. Am J Neurodegener Dis 2013;2(4):287. [PMC free article] [PubMed] [Google Scholar]
- 19. Li YI, Wong G, Humphrey J, Raj T. Prioritizing Parkinson's disease genes using population‐scale transcriptomic data. Nat Commun 2019;10(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ryan KJ, White CC, Patel K, et al. Context‐specific effects of neurodegenerative disease variants in a model of human microglia. Sci Transl Med 2017;9(421):eaai7635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Langston RG, Beilina A, Reed X, et al. Association of a Common Genetic Variant with Parkinson's disease is propagated through microglia. Cold Spring Harbor Laboratory 2021; [cited 2021 Feb 23]. Available from: https://www.biorxiv.org/content/10.1101/2021.01.15.426824v1.abstract
- 22. Iwaki H, Blauwendraat C, Leonard HL, et al. Genetic risk of Parkinson disease and progression: an analysis of 13 longitudinal cohorts. Neurol Genet 2019;5(4):e354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Das S, Forer L, Schönherr S, et al. Next‐generation genotype imputation service and methods. Nat Genet 2016;48(10):1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pankratz N, Beecham GW, DeStefano AL, et al. Meta‐analysis of Parkinson's disease: identification of a novel locus, RIT2. Ann Neurol 2012;71(3):370–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bycroft C, Freeman C, Petkova D, et al. The UKbiobank resource with deep phenotyping and genomic data. Nature 2018;562(7726):203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta‐analysis of genomewide association scans. Bioinformatics 2010;26(17):2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pruim RJ, Welch RP, Sanna S, et al. LocusZoom: regional visualization of genome‐wide association scan results. Bioinformatics 2010;26(18):2336–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Iwaki H, Leonard HL, Makarious MB, et al. Accelerating medicines partnership: Parkinson's disease. Genetic resource. medRxiv 2020; [cited 2021 Feb 11]. Available from https://www.medrxiv.org/content/10.1101/2020.11.19.20235192v1 [DOI] [PMC free article] [PubMed]
- 29. Szustakowski JD, Balasubramanian S, Sasson A, et al. Advancing human genetics research and drug discovery through exome sequencing of the UKbiobank. medRxiv 2020; [cited 2021 Feb 11]. Available from https://www.medrxiv.org/content/10.1101/2020.11.02.20222232v1 [DOI] [PubMed]
- 30. Sosero YL, Yu E, Krohn L, et al. LRRK2 p.M1646T is associated with glucocerebrosidase activity and with Parkinson's disease medRxiv. Available from: 10.1101/2020.09.23.20197558 [DOI] [PMC free article] [PubMed]
- 31. Wu YR, Chang KH, Chang WT, et al. Genetic variants ofLRRK2 in Taiwanese Parkinson's disease. PLoS One 2013;8(12):e82001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jabbari E, Koga S, Valentino RR, et al. Genetic determinants of survival in progressive supranuclear palsy: a genome‐wide association study. Lancet Neurol 2021;20(2):107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bryant N, Malpeli N, Ziaee J, et al. Identification of LRRK2 missense variants in the accelerating medicines partnership Parkinson's disease cohort. Hum Mol Genet 2021;30(6):454–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cook DA, Kannarkat GT, Cintron AF, et al. LRRK2 levels in immune cells are increased in Parkinson's disease. NPJ Parkinsons Dis 2017;3:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wallings RL, Tansey MG. LRRK2 regulation of immune‐pathways and inflammatory disease. Biochem Soc Trans 2019;47(6):1581–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Smith WW, Pei Z, Jiang H, et al. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci 2006;9(10):1231–1233. [DOI] [PubMed] [Google Scholar]
- 37. Greggio E, Jain S, Kingsbury A, et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis 2006;23(2):329–341. [DOI] [PubMed] [Google Scholar]
- 38. West AB, Moore DJ, Biskup S, et al. Parkinson's disease‐associated mutations in leucine‐rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci USA 2005;102(46):16842–16847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Luzón‐Toro B, de la Torre ER, Delgado A, et al. Mechanistic insight into the dominant mode of the Parkinson's disease‐associated G2019S LRRK2 mutation. Hum Mol Genet 2007;16(17):2031–2039. [DOI] [PubMed] [Google Scholar]
- 40. Refai FS, Ng SH, Tan E‐K. Evaluating LRRK2 genetic variants with unclear pathogenicity. Biomed Res Int 2015;2015:678701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nixon‐Abell J, Berwick DC, Grannó S, et al. Protective LRRK2 R1398H variant enhances GTPase and Wnt signaling activity. Front Mol Neurosci 2016;9:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Berwick DC, Harvey K. LRRK2 functions as a Wnt signaling scaffold, bridging cytosolic proteins and membrane‐localized LRP6. Hum Mol Genet 2012;21(22):4966–4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Berwick DC, Javaheri B, Wetzel A, et al. Pathogenic LRRK2 variants are gain‐of‐function mutations that enhance LRRK2‐mediated repression of β‐catenin signaling. Mol Neurodegener 2017;12(1):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Toh J, Chua LL, Ho P, et al. Identification of targets from LRRK2 rescue phenotypes. Cells 2021;10(1):76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu Z, Lee J, Krummey S, et al. Leucine‐rich repeat kinase 2 (LRRK2) regulates inflammatory bowel disease through the nuclear factor of activated T cells (NFAT). Nat Immunol 2011;12(11):1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ikezu T, Koro L, Wolozin B, et al. Crohn's and Parkinson's disease‐associated LRRK2 mutations Alter type II interferon responses in human CD14 + blood monocytes ex vivo. J Neuroimmune Pharmacol 2020;15(4):794–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang D, Xu L, Lv L, et al. Association of the LRRK2 genetic polymorphisms with leprosy in Han Chinese from Southwest China. Genes Immun 2015;16(2):112–119. [DOI] [PubMed] [Google Scholar]
- 48. Fava VM, Manry J, Cobat A, et al. A missense LRRK2 variant is a risk factor for excessive inflammatory responses in leprosy. PLoS Negl Trop Dis 2016;10(2):e0004412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Heckman MG, Schottlaender L, Soto‐Ortolaza AI, et al. LRRK2 exonic variants and risk of multiple system atrophy. Neurology 2014;83(24):2256–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
APPENDIX S1 Supporting Information
Figure S1. Meta‐analysis of p.N551K in the included data sets excluding (from left to right) (1) no samples, (2) carriers of rs76904798 and p.G2019S, and (3) carriers of p.N2081D and p.G2019S.
Figure S2. Meta‐analysis of p.R1398H in the included data sets excluding (from left to right) (1) no samples, (2) carriers of rs76904798 and p.G2019S, and (3) carriers of p.N2081D and p.G2019S.
Figure S3. Meta‐analysis of p.M1646T in the included data sets excluding (from left to right) (1) no samples, (2) carriers of rs76904798 and p.G2019S, and (3) carriers of p.N2081D and p.G2019S.
Figure S4. Meta‐analysis of p.N2081D in the included data sets excluding (from left to right) (1) no samples and (2) carriers of rs76904798 and p.G2019S.
Figure S5. Meta‐analysis of p.S1647T in the included data sets excluding (from left to right) (1) no samples, (2) carriers of rs76904798 and p.G2019S, and (3) carriers of p.N2081D and p.G2019S.
Figure S6. Meta‐analysis of p.M2397T in the included data sets excluding (from left to right) (1) no samples, (2) carriers of rs76904798 and p.G2019S, and (3) carriers of p.N2081D and p.G2019S.
Figure S7. Meta‐analysis of (from left to right) (1) p.G2019S and (2) rs76904798 in the included data sets.
Figure S8. Meta‐analysis of rs76904798 in the included data sets excluding (from left to right) (1) no samples and (2) carriers of p.N2081D and p.G2019S.
Figure S9. Meta‐analysis of p.L119P in the included data sets excluding (from left to right) (1) no samples, (2) carriers of rs76904798 and p.G2019S, and (3) carriers of p.N2081D and p.G2019S.
Figure S10. Meta‐analysis of p.I723V in the included data sets excluding (from left to right) (1) no samples, (2) carriers of rs76904798 and p.G2019S, and (3) carriers of p.N2081D and p.G2019S.
Figure S11. Meta‐analysis of p.R1514Q in the included data sets excluding (from left to right) (1) no samples, (2) carriers of rs76904798 and p.G2019S, and (3) carriers of p.N2081D and p.G2019S.
Figure S12. Meta‐analysis of p.P1542S in the included data sets excluding (from left to right) (1) no samples, (2) carriers of rs76904798 and p.G2019S, and (3) carriers of p.N2081D and p.G2019S.
Figure S13. Meta‐analysis of p.K1423K in the included data sets excluding (from left to right) (1) no samples, (2) carriers of rs76904798 and p.G2019S, and (3) carriers of p.N2081D and p.G2019S.
Figure S14. Meta‐analysis of rs10847864 in the included data sets excluding (from left to right) (1) no samples, (2) carriers of rs76904798 and p.G2019S, and (3) carriers of p.N2081D and p.G2019S.
Figure S15. LocusZoom plot of LRRK2 association with Parkinson's disease risk conditioned on p.N2081D. The left panel shows the association signal at the LRRK2 locus in the IPDGC and UK Biobank meta‐analysis conditioned on p.N2081D, and the right panel conditions on both p.G2019S and p.N2081D. The LRRK2 variants p.N551K, p.R1398H, p.M1646T, p.G2019S, and rs76904798 are indicated by red dots.
Table S1. Overview of included data.
Table S2. Imputation quality of LRRK2 variants examined in this study.
Table S3. Summary statistics of LRRK2 variants after removing carriers of the specified variants.
Table S4. Co‐inheritance analysis of LRRK2 variants examined in this study with p.G2019S, rs76904798, and p.N2081D.
Table S5. Fisher's exact test comparing the allele distributions of LRRK2 variants among cases in the unconditioned and conditioned data sets.
Table S6. Concordance of LRRK2 variants in the imputed data sets with whole‐exome sequencing data (IPDGC) and whole‐genome sequencing data (UKBIO).
Table S7. Summary statistics of LRRK2 variants, including the allele counts of the specified variants as covariates.
Table S8. Frequencies and allele distributions of LRRK2 variants examined in this study in the unconditioned and conditioned data sets.
Data Availability Statement
UK Biobank data are available via application (https://www.ukbiobank.ac.uk/). Various GWAS summary statistics from the IPDGC data are publicly available (https://pdgenetics.org/resources).