

Abstract
Introduction
Plasma proteins affect biological processes and are common drug targets but their role in the development of Alzheimer's disease and related dementias remains unclear. We examined associations between 4953 plasma proteins and cognitive decline and risk of dementia in two cohort studies with 20‐year follow‐ups.
Methods
In the Whitehall II prospective cohort study proteins were measured using SOMAscan technology. Cognitive performance was tested five times over 20 years. Linkage to electronic health records identified incident dementia. The results were replicated in the Atherosclerosis Risk in Communities (ARIC) study.
Results
Fifteen non‐amyloid/non‐tau–related proteins were associated with cognitive decline and dementia, were consistently identified in both cohorts, and were not explained by known dementia risk factors. Levels of six of the proteins are modifiable by currently approved medications for other conditions.
Discussion
This study identified several plasma proteins in dementia‐free people that are associated with long‐term risk of cognitive decline and dementia.
Keywords: cognitive decline, cohort study, dementia, longitudinal study, proteomics
1. BACKGROUND
Alzheimer's disease (AD) and related dementias pose an increasing challenge with considerable costs to the individual and to health and social care services. 1 Amyloid beta (Aβ) and tau proteins have dominated pathophysiological research on dementia etiology, 2 but to date prevention and treatment trials targeting these biomarkers have been unsuccessful. 3 , 4 In addition, longitudinal studies suggest that most cognitively normal amyloid‐positive people never develop clinical dementia. 5 Given this and the high prevalence of mixed types of dementia in the general population, there is a need to expand research on early biomarkers for dementia beyond amyloid and tau.
Causes of dementia are increasingly thought to be systemic. 6 , 7 Recent development of scalable platforms allows simultaneous assessment of thousands of circulating proteins. 8 , 9 These may have the potential to identify novel drivers of dementia. For several reasons, circulating proteins are promising targets for biomarker and drug discovery. Proteins are regulators and effectors of biological processes and are also imprints of the effects of genes, the environment, age, current comorbidities, behaviors, and medications, all of which may affect dementia development. 10 Proteins are also highly druggable as they can be targeted by monoclonal antibodies, small‐molecule drugs, or proteolysis‐targeting chimaeras. 11 , 12 , 13 , 14 Indeed, of all currently approved medications, ≈96% target proteins. 11 Animal studies support a causal role of circulating plasma proteins in neurodegeneration. Plasma from young mice, via injection or through parabiosis (joining two animals so they share blood circulation), has been shown to restore memory and stimulate synaptic plasticity in the aged mouse hippocampus. 15 , 16 , 17 In humans, multiple circulating proteins have been linked with dementia. 18 However, human studies to date have been mainly cross‐sectional, based on small samples (N <1000), or lacked replication.
In this report from two large prospective cohort studies, the British Whitehall II and US Atherosclerosis Risk in Communities (ARIC) study, we used SOMAscan technology to examine 4953 plasma proteins as risk factors for cognitive decline and dementia. To determine the role of proteins in the early stages of neurodegeneration when dementia may still be preventable, we assessed plasma proteins in late middle age and followed subsequent cognitive decline and dementia over two decades. Our study design thus explicitly takes into consideration the long preclinical phase of dementia.
2. METHODS
2.1. Study design and participants
We used the Whitehall II study as our discovery cohort and the ARIC study as the replication cohort (Figure 1). In 1985 to 1988, all civil servants aged 35 to 55 years based in 20 departments in London, UK, were invited to participate in the Whitehall II cohort study, and 73% (n = 10,308) agreed. 19 Blood samples for proteomic analyses were collected from a random subsample of 2274 dementia‐free individuals in 1997 to 1999. 20 Cognitive performance measurements were conducted at this and four subsequent clinical examinations in 2002 to 2004, 2007 to 2009, 2012 to 2013, and 2015 to 2016. Follow‐up started from 1997 to 1999 and ended at death, dementia, or in October 2019.
FIGURE 1.
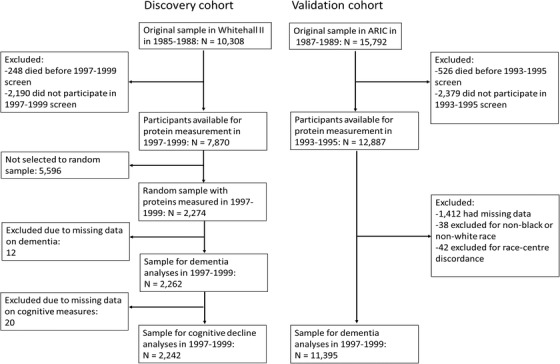
Flowchart of sample selection in discovery and validation cohorts. ARIC, the Atherosclerosis Risk in Communities study
ARIC 21 is a community‐based cohort study of 15,792 participants from four US communities: Washington County, Maryland; Forsyth County, North Carolina; northwestern suburbs of Minneapolis, Minnesota; and Jackson, Mississippi. Participants were aged 44 to 66 years at study entry in 1987 to 1989. Blood samples for protein analysis were drawn in 1993 to 1995 and analyzed for 11,395 individuals free of dementia. Prevalent dementia cases were identified from hospital discharge and by contacting participants annually via telephone or by administering a brief cognitive screener. Follow‐up for incident dementia ended at death, dementia, or in December 2017.
RESEARCH IN CONTEXT
Systematic Review: We searched PubMed for studies on plasma and serum proteome and dementia etiology, without language or date restrictions, up to March 11, 2021. Most of the identified studies had small (N <1000) sample size, lacked replication cohort, or were cross‐sectional and thus did not allow identification of proteins associated with early disease development when individuals are asymptomatic.
Interpretation: In this follow‐up of two large cohorts with a total of 13,657 participants, we identified and replicated 15 plasma protein biomarkers in asymptomatic individuals that were associated with cognitive decline and 20‐year risk of dementia. These proteins are involved in innate and adaptive immunity, blood‐brain barrier dysfunction, vascular pathology, and central insulin resistance providing evidence of the systemic pathogenesis of dementia.
Future Directions: Future research should examine whether the identified proteins are causal risk factors for dementia and whether they could provide clues for drug development or repurposing of existing medications.
2.2. Assessment of plasma proteins
In Whitehall II and ARIC, proteins were analyzed using the SOMAscan version 4 assay. The analyses used plasma ethylenediaminetetraacetic acid samples stored at –80°C. Earlier studies and Supplementary Methods in supporting information describe performance of the SOMAscan assay and the modified aptamer binding in detail. 22 , 23 , 24 In brief, the assay uses a mix of thousands of slow off‐rate modified aptamers (SOMAmers). The aptamers bind to proteins in participants’ plasma samples and the specificity is ensured with a two‐step process analogous to a conventional immunoassay. Median intra‐ and inter‐assay coefficients of variation for SOMAscan version 4 are ≈5% and assay sensitivity is comparable to that of typical immunoassays, with a median lower limit of detection in the femtomolar range. 23 The specificity of the aptamer reagents is good, has been tested in several ways, 10 , 20 , 24 , 25 , 26 , 27 and was also assessed in this study for protein hits using Olink Explore panel as a reference in a subset of 543 Whitehall II participants.
2.3. Cognitive testing
At least two complete measurements of the Whitehall II cognitive test battery were available for 2242 (99%) participants. The battery is sensitive to any decrease in cognitive performance and covers four domains: executive function, memory, and phonemic and semantic fluency (Supplementary Methods). Based on these measures, we constructed a global cognitive score by first standardizing the distribution of each test domain measured in follow‐up visits to the baseline score to create z‐scores with mean 0 and standard deviation (SD) 1. We then summed the domain specific scores at each phase and standardized the summary score to the baseline summary score; this approach minimizes measurement error inherent in individual tests. 28
2.4. Dementia follow‐up
Whitehall II study participants were linked to the National Health Services (NHS) Hospital Episode Statistics (HES) database, and the British National mortality register using individual NHS identification numbers for linkage. 29 The NHS provides nearly complete health‐care coverage for all individuals legally resident in the UK. Incident dementia was defined using the World Health Organization International Classification of Diseases, version 10 (ICD‐10) codes F00, F01, F03, G30, and G31 and ICD‐9 codes 290.0–290.4, 331.0–331.2, 331.82, and 331.9. Sensitivity and specificity of dementia assessment based on HES data are 0.78 and 0.92, respectively. 30 We also conducted informant interviews and checked participants’ medications at each clinical examination for dementia‐related medication. In sensitivity analyses on dementia subtypes, those with prevalent atherosclerotic cardiovascular disease (coronary heart disease, heart failure, peripheral artery disease, or stroke) at the time of dementia diagnosis were classified as having vascular dementia and those without as having non‐vascular dementia.
In ARIC, incident dementia cases between 1993 and 2011 were identified by contacting participants annually via telephone or by administering a brief cognitive screener, and if applicable their caregivers completed a questionnaire. This information, supplemented by surveillance of dementia‐related hospital discharge and death certificate codes, was used to estimate the dementia onset date. Clinical and neuropsychological examinations to ascertain dementia cases were conducted in visits five (2011 to 2013) and six (2016 to 2017). 31 , 32 Using all available data, suspected cases were adjudicated by a committee of clinicians.
2.5. Measurement of baseline covariates
In the Whitehall II study, standard self‐administered questionnaires provided data on age, sex, ethnicity, socioeconomic status, education, medication, alcohol consumption, and smoking. Depressive symptoms were ascertained using the General Health Questionnaire (GHQ). 33 ICD codes used to measure comorbidities (atrial fibrillation, coronary heart disease, depression, diabetes, heart failure, peripheral artery disease, and stroke) are provided in Supplementary Methods. Experienced clinical nurses measured body mass index (BMI) and systolic blood pressure and took blood samples for lipid and glucose measurements. 19 Using DNA extracted from whole blood, a standard polymerase chain reaction (PCR) assay determined apolipoprotein E (APOE) genotype using the salting out method. 34 , 35 Two blinded independent observers read the genotype and any discrepancies were resolved by repeating the PCR analysis. In ARIC, baseline covariates included age, sex, and a combined race–study center variable, measured using standard operating protocols.
2.6. Statistical analysis
Proteins were transformed to a normal distribution by inverse rank‐based normal transformation. We used Spearman correlation to test protein‐protein correlations. To maximize power for selection of candidate protein predictors, we followed a two‐step protocol Figure S1a and S1b in supporting information and Supplementary Methods). The first step included a linear regression to examine associations between each protein and the cognitive decline slope derived from mixed‐effects linear regression of repeated cognitive assessments. The assumptions of linear regression were assessed by plotting the residuals in residuals versus fitted, normal Q‐Q, scale‐location, and residual versus leverage plots. 36 We used false discovery rate (FDR) correction of 5% to select proteins from linear regression for the second step. This translated to a P‐value cut‐off of .002. In the second step, the proteins that survived FDR correction were analyzed in age‐, sex‐, and ethnicity‐adjusted Cox regression models 37 with incident dementia as the outcome. P‐value = .05 was the cut‐off for statistical significance. The proportionality assumption in all Cox models was assessed with Schoenfeld residuals, log‐log plots, and time‐interaction coefficients 37 and was not violated.
To examine reproducibility, proteins that were robustly associated with cognitive decline and dementia in the Whitehall II study were also analyzed in the ARIC study using dementia as an outcome. These Cox models were adjusted for age, sex and race‐study center. In sensitivity analyses conducted in the Whitehall II study, we ran Cox regression adjusted for age, sex, ethnicity, systolic blood pressure, total cholesterol, antihypertensive medication, smoking, diabetes, APOE genotype, BMI, alcohol consumption, education, and GHQ score to study the effect of confounders. No continuous covariate was categorized. We used this model to study whether the associations between proteins and dementia were attributable to reverse causation by excluding the first 10 years of follow‐up. To address potential survival bias, we conducted adjusted Fine and Gray competing risk analysis 38 with dementia and death as outcomes. The effect of comorbidities on the protein–dementia associations was studied with time‐varying covariates and the effect of missing data was examined with multiple imputation (Supplementary Methods). To minimize bias due to ethnic admixture in ARIC, we performed a sensitivity analyses after excluding non‐White participants from the analyses.
For identified and replicated proteins, we searched the Genotype‐Tissue Expression (GTEx); 39 Human Protein Atlas; 40 , 41 Database for Annotation, Visualization and Integrated Discovery (DAVID); 42 , 43 UniProt; 44 and ChEMBL 45 databases to characterize protein expression profiles, their cellular localization, and drugs that can target them. We used statistical software R (3.6.0) and Stata (version 16.1 MP; Stata Corp) for all analyses.
3. RESULTS
Mean age of the 2274 Whitehall II participants was 56.1 (SD 5.9), and 1653 (73.0%) were men (Table 1). During a mean follow‐up of 20.4 years, 106 individuals developed dementia. After FDR correction of 5%, 246 of the 4953 proteins were associated with an increased rate of cognitive decline. None of the amyloid‐, tau‐, or neurofilament‐related proteins were associated with accelerated cognitive decline after FDR correction (Table S1 in supporting information).
TABLE 1.
Participant characteristics at the time of blood collection for protein measurement in the Whitehall II study and the Atherosclerosis Risk in Communities study
| Characteristic, n (%) or mean (SD) | Whitehall II N = 2274 | ARIC N = 11,395 |
|---|---|---|
| Demographic variables | ||
| Age, mean (SD) | 56.1 (5.9) | 60.2 (5.7) |
| Men, No. (%) | 1653 (73.0) | 5190 (45.6) |
| White, No. (%) | 2089 (92.3) | 8991 (78.9) |
| Education, No. (%) | ||
| Less than high school | 522 (31.1) | 2311 (20.3) |
| High school/vocational | 427 (25.4) | 4813 (42.3) |
| College/graduate/ professional | 731 (43.5) | 4255 (37.4) |
| Apolipoprotein E ε4 alleles, No. (%) | ||
| 0 | 1432 (73.4) | 7678 (67.4) |
| 1 | 469 (24.1) | 3358 (29.5) |
| 2 | 49 (2.5) | 359 (3.2) |
| Physiological and lab variables, mean (SD) | ||
| Body mass index, kg/m2 | 26,6 (4,1) | 28.5 (5.6) |
| Total cholesterol, mg/dL | 232.1 (40.4) | 208.8 (38.7) |
| Cardiovascular risk factors, No. (%) | ||
| Hypertension | 286 (12.6) | 4686 (41.3) |
| Diabetes mellitus | 13 (0.6) | 1806 (15.9) |
| Cigarette smoking, current | 204 (9.1) | 2042 (18.0) |
| Mean follow‐up time (SD) | 20.4 (3.2) | 17.7 (6.1) |
| Median follow‐up time (IQR) | 21.5 (21.0, 22.1) | 20.0 (14.1, 22.4) |
Abbreviations: ARIC, Atherosclerosis Risk in Communities study; IQR, interquartile range; SD, standard deviation.
Notes: Values are displayed as means (SD) for continuous variables, frequencies (column percentages) for categorical variables, and median (IQR) for follow‐up time.
Of the 246 proteins, 21 were also associated with incident dementia in the Whitehall II study (Figure 2). Thus, replication analyses in ARIC were based on these 21 proteins. In ARIC, 1942 of the 11,395 participants developed dementia during a mean follow‐up of 17.4 years. Of the 21 proteins associated with cognitive decline and dementia in the Whitehall II study, 15 (71%) were also associated with dementia in the ARIC cohort. Levels of all 15 proteins were higher in individuals who developed dementia (Figures S2‐S4 in supporting information). Hazard ratios for a 1 SD higher level in each of the 15 proteins ranged from 1.08 to 1.64 in age‐, sex‐, and ethnicity/race‐adjusted analyses. A cognitive domain–specific analysis showed that phonemic fluency and executive functioning were the main drivers of the associations between the 15 proteins and a higher rate of cognitive decline (Table S2‐S3 in supporting information).
FIGURE 2.
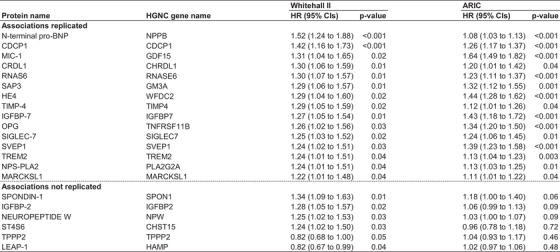
Proteins associated with dementia in Whitehall II and ARIC cohorts. HR, hazard ratio; 95% CI, 95% confidence interval; ARIC, the Atherosclerosis Risk in Communities study; SD, standard deviation; FDR, false discovery rate of 5%; Protein names: N‐terminal pro‐BNP, N‐terminal pro‐BNP; CDCP1, CUB domain‐containing protein 1. MIC‐1, growth/differentiation factor 15. CRDL1, Chordin‐like protein 1; RNAS6, ribonuclease K6; SAP3, ganglioside GM2 activator; HE4, WAP four‐disulfide core domain protein 2; TIMP‐4, metalloproteinase inhibitor 4; IGFBP‐7, insulin‐like growth factor‐binding protein 7; OPG, tumor necrosis factor receptor superfamily member 11B; Siglec‐7, sialic acid‐binding Ig‐like lectin 7; SVEP1, Sushi, von Willebrand factor type A, EGF and pentraxin domain‐containing protein 1; TREM2, triggering receptor expressed on myeloid cells 2; NPS‐PLA2, phospholipase A2, membrane associated; MARCKSL1, MARCKS‐related protein; Spondin‐1, spondin‐1; IGFBP‐2, insulin‐like growth factor‐binding protein 2; Neuropeptide W, neuropeptide W; ST4S6, Carbohydrate sulfotransferase 15; TPPP2, tubulin polymerization‐promoting protein family member 2; LEAP‐1, hepcidin
Of the 15 identified and replicated proteins, 14 (N‐terminal pro‐BNP, CDCP1, MIC‐1, CRDL1, RNAS6, SAP3, HE4, TIMP‐4, IGFBP‐7, OPG, SVEP1, TREM2, NPS‐PLA2, MARCKSL1) were secreted and one (SIGLEC‐7) was a cell membrane protein. They were mostly expressed in tissues other than the brain (Figure 3 and Table S4 in supporting information). The proteins were related to one or more biological systems that are relevant to dementia etiology: activation of the immune system, blood‐brain barrier (BBB) breakdown, vascular pathology, and central insulin resistance (Table 2). For six proteins, a medication that can influence the protein's function (NPS‐PLA2, CDCP1, MIC‐1, and IGFBP‐7) or reduce its levels (N‐terminal pro‐BNP, and OPG) is available. The medications are used to treat diabetes, cancer, and inflammatory or cardiovascular diseases and all except the one on MIC‐1 have passed phase III trials (Table 3).
FIGURE 3.
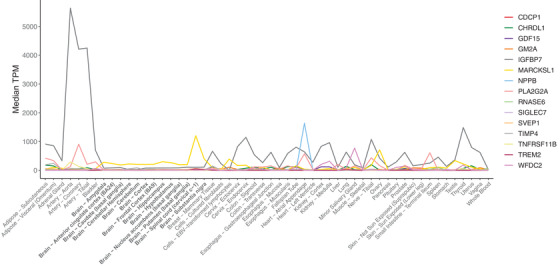
Expression profile of the genes coding the 15 proteins associated with rate of cognitive decline and dementia derived from Genotype‐Tissue Expression (GTEx) database. Y‐axis shows the extent to which these genes are expressed by the organs and tissues listed in X‐axis. For clarity, tissues in the brain are marked in bold. EBV, Ebstein‐Barr virus; TPM, transcripts per million
TABLE 2.
Association between 15 proteins and dementia‐related pathologies
| Association with | |||||
|---|---|---|---|---|---|
| Protein | Immune system | BBB dysfunction | Vascular pathology | Central insulin resistance | Previous studies with dementia outcome |
| N‐terminal pro‐BNP | No | No | Yes | No | Prospective cohort 56 , 57 , 103 , 104 |
| CDCP1 | Yes | Yes | No | No | None |
| MIC‐1 | Yes | Yes | Yes | No | Case control 49 , 50 |
| CRDL1 | No | No | Yes | No | None |
| RNAS6 | Yes | No | No | No | None |
| SAP3 | No | Yes | Yes | No | Case‐control 27 |
| HE4 | Yes | Yes | Yes | No | None |
| TIMP‐4 | Yes | Yes | Yes | No | Case‐control 51 |
| IGFBP‐7 | No | No | Yes | Yes | Case‐control 48 |
| OPG | Yes | Yes | Yes | No | Case‐control 52 |
| SIGLEC‐7 | Yes | No | No | No | None |
| SVEP1 | Yes | Yes | Yes | No | None |
| TREM2 | Yes | No | No | No | Prospective cohort 55 |
| NPS‐PLA2 | Yes | No | Yes | No | Case‐control 54 |
| MARCKSL1 | Yes | Yes | No | No | None |
| Total N of proteins | 11 | 8 | 10 | 1 | 8 |
Notes: In the first four columns, "Yes" indicates that the protein has been linked to the pathology and "No" indicates no evidence of such a link is available. Row "Total" provides the total number of protein‐pathology associations for each dementia‐related pathology. The fifth column provides reference and type of evidence linking these proteins to dementia risk.
Abbreviation: BBB, blood‐brain barrier.
TABLE 3.
Drugs that can influence currently identified proteins associated with dementia, their potential mechanisms of action, and current indications
| Protein | Medication | Action | Phase passed | Indications |
|---|---|---|---|---|
| CDCP1 | Itolizumab 118 | Prevents CDCP1 binding to CD6 and down regulates T cell activation and infiltration. It also reduces synthesis of pro‐inflammatory cytokines reducing T cell infiltration at sites of inflammation. | III | Psoriasis |
| NPS‐PLA2 | Varespladib and Varespladib Methyl 117 | Inhibits arachidonic acid pathway in inflammation by inhibiting NPS‐PLA2 activity and subsequent leukocyte activation. | III | Atherosclerotic cardiovascular diseases, inflammatory diseases, snake venom antidote |
| IGFBP‐7 | Intranasal insulin, metformin, and GLP‐1 receptor agonists 113 | Insulin nasal spray restores central insulin levels that may be downregulated by elevated IGFBP‐7 levels. Metformin acts as insulin sensitizer and GLP‐1 agonists stimulate insulin secretion. | III | Cognitive decline and Alzheimer´s disease |
| N‐terminal pro‐BNP | Antihypertensive medications 103 | Reduce N‐terminal pro‐BNP levels in circulation by reducing atrial and ventricular overload. | III | Hypertension |
| OPG | Atorvastatin, metformin, pioglitazone, rosiglitazone 88 , 119 , 120 | Reduce OPG levels possibly by reducing inflammation and by stabilizing atherosclerotic plaques. | III | Atherosclerotic cardiovascular diseases, diabetes |
| MIC‐1 | Monoclonal antibody CTL‐002 121 | Neutralizes MIC‐1 | ‐ | Advanced cancer |
A series of sensitivity analyses based on the Whitehall data suggest that our findings are robust. The decrease in hazard ratios was between 0.00 and 0.07 for the 15 protein–dementia associations after multivariable adjustment for age, sex, ethnicity, education, systolic blood pressure, antihypertensive medication, total cholesterol, BMI, alcohol consumption, smoking, diabetes, GHQ score, and APOE status; after adjusting comorbidities (atrial fibrillation, coronary heart disease, depression, diabetes, heart failure, peripheral artery disease, and stroke, all treated as time‐varying covariates); after reducing the possibility of reverse causation bias by excluding dementia cases that occurred during the first 10 years of follow‐up; after taking into account the competing risk of death; and after imputation of missing covariates (Table S5 in supporting information). The results did not change markedly after excluding non‐White participants in the ARIC study (Table S6 in supporting information). There were no sex differences in observed protein–dementia associations (range of P‐values for interaction from 0.11 to 0.99). Associations between standard risk factors and dementia were similar to those reported in recent meta‐analyses, 46 , 47 suggesting that Whitehall is not an exceptional cohort study (Table S7 in supporting information).
There were no strong correlations between the 15 proteins (all correlations ≤|0.53|, Figure S5 in supporting information). Nine proteins had higher HR for non‐vascular and six for vascular dementia but the differences were not statistically significant and could not be studied reliably due to lack of power (Table S8 in supporting information).
Correlations between the 11 proteins measured with SOMAscan version 4 and Olink Explore platform ranged between 0.46 and 0.87 (Table S9 in supporting information). Previous studies also suggest the measurement of proteins based on SOMAScan aptamers is reliable and specific (Table S10 in supporting information). Of the 15 proteins identified by aptamers, eight (N‐terminal pro‐BNP, MIC‐1, HE4, TIMP‐4, IGFBP‐7, SVEP1, TREM2, and NPS‐PLA2) have been previously cross‐validated with more than one method including mass spectrometry, immunoassay, Olink protein panel, and by identification of genetic variants influencing the concentration of the protein measured using the Somalogic platform in the vicinity of the encoding gene (i.e., cis protein quantitative trait loci data). Six other proteins have been cross‐validated using one method, four (CDCP1, RNAS6, OPG, and Siglec‐7) using cis protein quantitative trait loci data, one (CRDL1) using mass spectrometry, one (SAP3) indirectly from an association between SAP3 and AD observed using mass spectrometry, while one of the 15 proteins (MARCKSL1) remains unvalidated.
4. DISCUSSION
To our knowledge, this is the largest proteome‐wide study with replicated results on long‐term associations between plasma proteins, cognitive decline, and risk of dementia to date. We identified 15 plasma proteins that were associated with increased rate of cognitive decline and an increased 20‐year risk of dementia in the British Whitehall II study. Associations with dementia were replicated in an independent US cohort study (ARIC); were robust to adjustments for known dementia risk factors; and were not explained by competing risk of death, cardiometabolic comorbidities, or reverse causation bias. The 15 proteins, of which 14 were secreted and 1 was a cell membrane protein, were mostly expressed outside the central nervous system suggesting that they relate to systemic processes that increase dementia risk.
Our findings are consistent with previous research. Proteins SAP3, NPS‐PLA2, IGFBP‐7, MIC‐1, TIMP‐4, and OPG have been reported to be associated with dementia in case‐control studies, 27 , 48 , 49 , 50 , 51 , 52 , 53 , 54 and TREM2 and N‐terminal pro‐BNP in prospective cohort studies, 55 , 56 , 57 with all associations in the same direction as in this study. The increased dementia risk in individuals with high levels of plasma SVEP1, HE4, CDCP1, SIGLEC‐7, MARCKSL1, CRDL1, or RNAS6 is a novel finding. Our results also provide new evidence that midlife circulating TREM2 predicts dementia at older ages.
This study did not sytematically examine processes underlying the associations between proteins and dementia. However, the observed associations are biologically plausible as there are several mechanisms that could link the 15 proteins to dementia pathologies, including immune dysfunction, BBB dysfunction, vascular damage, and central insulin resistance. More specifically, hyper‐activation of the innate and adaptive immune system can cause endothelium damage, neuroinflammation, and amyloid and tau accumulation in the brain (Figure S6a and Sb in supporting information). 58 , 59 , 60 , 61 SVEP1 contributes to this process by activating the complement system, which, if prolonged, is detrimental to endothelial cells 62 and may contribute to hippocampal synapse loss observed in dementia. 63 , 64 SIGLEC‐7 is an inhibitory receptor in natural killer cells that recognizes “self” antigens; 65 its cleavage to plasma is thought to mark uncontrolled inflammation. 66 Increased plasma concentrations of other proteins could represent responses to pathogens (e.g., RNAS6 67 , 68 and HE4), 69 or be part of the increased immune response (e.g., NPS‐PLA2, TREM2, MIC‐1, OPG, TIMP‐4, SVEP1, MARCKSL1, and CDCP1). 49 , 52 , 53 , 55 , 66 , 70 , 71 , 72 , 73 , 74 , 75 , 76 , 77 , 78 , 79
Degeneration of the BBB provides a further potential mechanism underlying the observed protein–dementia associations. The subsequent passage of toxins and proteins into the central nervous system contributes to reduced neuronal plasticity, activation of microglia, disruptions in lipid metabolism, increased neuroinflammation, amyloid and tau accumulation, and neurodegeneration (Figure S6c). 6 , 7 , 71 , 72 , 80 , 81 , 82 , 83 Elevated SAP3 levels, also found in the cerebrospinal fluid of AD and Parkinson's disease patients, 27 , 84 have been linked to the degeneration of pericytes that are crucial for the function of BBB; 85 , 86 increased MIC‐1 and OPG are associated with endothelial dysfunction; 87 , 88 , 89 increased CDCP1 with tight junction degeneration; 90 , 91 , 92 and MARCKSL1 and SVEP1 with endothelia integrity. 93 , 94 , 95
Proteins TIMP‐4, SAP3, NPS‐PLA2, OPG, and MIC‐1 have been linked to vascular damage caused by atherosclerosis and thrombosis (Figure S6d). 87 , 89 , 96 , 97 , 98 , 99 , 100 , 101 , 102 N‐terminal pro‐BNP is elevated in response to atrial and ventricular overload, 103 and in plasma and the central nervous system, it can inhibit the activity of neprilysin, an enzyme that reduces Aβ levels. 104 Elevated N‐terminal pro‐BNP levels are related to increased risk of vascular and AD dementia, 56 , 57 , 105 , 106 SVEP1 promotes thrombosis, 107 , 108 and HE4 and CRDL1 contribute to ischemia‐related abnormal angiogenesis 109 , 110 , 111 observed in vascular and AD dementia. 6 , 112 At least 1 of the 15 proteins may contribute to central insulin resistance (Figure S6e) 113 as IGFBP‐7 reduces free plasma insulin levels and also prevents free insulin from binding to its receptor, 114 , 115 both linked to reduced insulin responsiveness. 116
For some of the identified proteins, approved protein‐modifying medication already exists, but they are for the treatment of conditions other than dementia. Varespladib, currently used to treat cardiovascular and inflammatory diseases, inhibits the arachidonic acid inflammatory pathway by inhibiting NPS‐PLA2 activity. 117 Itolizumab, used for psoriasis, prevents CDCP1 from binding to CD6, leading to downregulation in T‐cell–mediated inflammation. 118 Glucose‐lowering and statin medications reduce OPG levels. 88 , 119 , 120 Intranasal insulins 116 could counteract high levels of insulin‐binding IGFBP‐7. Antihypertensive medications reduce plasma N‐terminal pro‐BNP levels. 103 Furthermore, a monoclonal antibody that neutralizes MIC‐1 is currently being studied in a phase‐1 cancer trial. 121 Whether these drugs could play a role in future prevention or treatment of dementias (i.e., drug repurposing) needs to be investigated in genetic, systems biology, and intervention studies.
Our study has some important strengths. SOMAscan is the largest protein panel available for large‐scale studies covering proteins that enter the bloodstream by purposeful secretion to orchestrate biological processes, by cleavage from cell membranes, or by leakage from intracellular space that inform cell injury rather than biological causality. 122 In a subsample of Whitehall participants, the specificity of aptamers measuring 11 of the 15 dementia‐related proteins was supported by moderate to high inter‐assessment correlations between SOMAscan and Olink protein panels. In addition, the specificity of aptamers measuring 14 of the 15 proteins has been confirmed using mass spectrometry or orthogonal strategies. 10 , 24 , 25 , 26 , 27 This suggests our results are unlikely to be biased by measurement error due to aptamer binding to unintended target proteins. Replication of the results in an independent study with different dementia ascertainment methods, dementia conversion rate, settings, and participant characteristics supports the validity and generalizability of the findings. We were able to confirm the associations in analyses excluding dementia cases diagnosed during the first 10 years of follow‐up. This finding is consistent with the possibility that we identified proteins that play a role in the early stages of neurodegeneration when dementia may still be preventable rather than those which only mark plasma changes caused by advanced preclinical disease.
The study has several limitations. First, SOMAscan covers only ≈5000 proteins of all the nearly 20,000 proteins identified by the Human Proteome Project from genomic open reading frames. 122 In addition, our assessment was not optimized for the measurement of amyloid‐, tau‐, and neurofilament‐related proteins in plasma, which may explain that none of these proteins were significantly associated with cognitive decline and dementia. As the sample size was limited in our discovery cohort, we may have missed other important associations, such as the one between plasma apoE and AD. 123 , 124 , 125 Lack of repeat data on proteins inhibited us to assess stability of proteins over time. However, the absence of effect attenuation in protein–dementia associations during the 20‐year follow‐up suggests that SOMAscan protein measurements were stable.
Second, our discovery study, based on electronic health records, missed cases of dementia not diagnosed or treated in UK hospitals. However, previous studies on dementia ascertainment based on electronic health records in the UK suggest that our ascertainment method is valid for a study of risk factor–dementia associations. 30 Further limitations include the use of semi‐quantitative protein data that cannot determine clinically useful concentrations, limited numbers of vascular and non‐vascular dementia to study these subtypes more extensively, and the lack of information on detailed dementia subtypes based on brain imaging or cerebrospinal fluid biomarkers.
In conclusion, using large‐scale testing of plasma proteins as long‐term risk factors for cognitive decline and dementia, our results support the hypothesis that early systemic processes may drive dementia development. The protein–dementia associations identified and replicated were robust and plausible, potentially involving innate and adaptive immunity, BBB dysfunction, vascular pathology, and central insulin resistance, all of which can contribute to amyloid and tau deposition, or pathologies that characterize vascular dementia. As observational studies cannot determine causal associations, the present findings should be considered hypothesis generating. Further research is needed to determine whether the observed protein‐dementia associations are causal rather than driven by other biomarkers that have shared downstream effects with the proteins or attributable to compensatory mechanisms that modify protein levels in those who will later develop dementia.
CONFLICTS OF INTEREST
In this academic–industry partnership project, academic collaborators generated the hypothesis and study design and SomaLogic, Inc. provided expertise in plasma proteins and funded the SOMAscan assays. Joni V. Lindbohm and Pyry N. Sipila have received personal lecture fees from the University of Helsinki. Nina Mars reports no conflicts of interest. Keenan A. Walker reports personal lecture fee from the Boston University Medical Center and holds the Programming Chair at the National Academy of Neuropsychology. Eric J. Brunner reports Osaka University research capacity building grant paid to employer. Archana Singh‐Manoux reports no conflicts of interest. Gill Livingston reports no conflicts of interest. Kalle Saksela reports no conflicts of interest. Jane E. Ferrie reports no conflicts of interest. Ruth C. Lovering reports personal lecture fees from the University College London, funding from a COST action grant, and is a member of the executive committee for the International Society of Biocuration (a voluntary role and no payment has been or will be made). Stephen A. Williams is employee of SomaLogic Inc., which has a commercial interest in the results and co‐inventor on multiple patents for specific proteomic models of disease. None of these patents relate to dementia (the topic of the manuscript). Aroon D. Hingorani reports no conflicts of interest. Rebecca F. Gottesman reports personal lecture fees for speaking at University of Michigan grand rounds, University of Alabama McKnight lecture, and the American College of Cardiology conference. Rebecca F. Gottesman is the secretary for the American Neurological Association. Henrik Zetterberg reports that he has served on the scientific advisory boards for Denali, Roche Diagnostics, Wave, Samumed, Siemens Healthineers, Pinteon Therapeutics, and CogRx; has given lectures in symposia sponsored by Fujirebio, Alzecure, and Biogen; and is a co‐founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program. Henrik Zetterberg is also the chair of the Alzheimer's Association Global Biomarker Standardization Consortium and the Alzheimer's Association Biolfluid‐Based Biomarker Professional Interest Area. Mika Kivimaki reports no conflicts of interest.
DATA SHARING STATEMENT
Data, protocols, and other metadata of the Whitehall II and ARIC studies are available to the scientific community. Please refer to the data sharing policies of these studies. Pre‐existing data access policies for Whitehall II and ARIC studies specify that research data requests can be submitted to each steering committee; these will be promptly reviewed for confidentiality or intellectual property restrictions and will not unreasonably be refused. Individual‐level patient or protein data may further be restricted by consent, confidentiality, or privacy laws/considerations. These policies apply to both clinical and proteomic data. Detailed information on data sharing can be found here: https://www.ucl.ac.uk/epidemiology‐health‐care/research/epidemiology‐and‐public‐health/research/whitehall‐ii/data‐sharing
Supporting information
Supporting Information
ACKNOWLEDGMENTS
We thank Jocelyn Jingsha Chen for helping in data analysis of the ARIC study. The study was supported by the UK Medical Research Council (S011676, K013351, R024227), the Wellcome Trust (221854/Z/20/Z), the National Institute on Aging (National Institutes of Health; R01AG056477 and R01AG062553), the British Heart Foundation (RG/16/11/32334), the Academy of Finland (311492), and NordForsk (75021). The Atherosclerosis Risk in Communities study has been funded in whole or in part with federal funds from the National Heart, Lung, and Blood Institute; National Institutes of Health; Department of Health and Human Services (contract numbers HHSN268201700001I, HHSN268201700002I, HHSN268201700003I, HHSN268201700004I and HHSN268201700005I), R01HL087641, R01HL086694; National Human Genome Research Institute contract U01HG004402; and National Institutes of Health contract HHSN268200625226C. Neurocognitive data collection was supported by U01 2U01HL096812, 2U01HL096814, 2U01HL096899, 2U01HL096902, 2U01HL096917 from the NIH (NHLBI, NINDS, NIA, and NIDCD). ARIC infrastructure was partially supported by grant number UL1RR025005, a component of the National Institutes of Health and NIH Roadmap for Medical Research. University College London and Johns Hopkins University have signed a collaboration agreement with SomaLogic, Inc to conduct SOMAscan of Whitehall and ARIC stored samples at no charge in exchange for the rights to analyze linked Whitehall and ARIC phenotype data. The Genotype‐Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by the following institutes: NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. Joni V. Lindbohm was supported by Academy of Finland (311492) and Helsinki Institute of Life Science (H970) grants paid to employer. Nina Mars was supported by the Academy of Finland (grant number 331671) paid to employer. Keenan A. Walker and Rebecca F. Gottesman were supported by contracts K23 AG064122 and K24 AG052573 from NIH/NIA paid to employer. Archana Singh‐Manoux was supported by the National Institute on Aging, NIH (R01AG062553;R01AG056477) grants paid to employer. Gill Livingston was supported by all to institution Wellcome collaborative grant (UNS114095), Alzheimer's Society PhD fellowship AS‐PhD‐19a‐006 NIHR HTA grant, NIHR128761 NIHR ARC PhD 1861414, NIHR senior investigator NIHR201321 NIHR HTA Project: 16/155/01 RPGF1711∖10, Dunhill Medical Trust Alzheimer's society 367 (AS‐IGF ‐16‐001), HTA 15/161/05 Wellcome Research Training Fellowship 200163/Z/15/Z AS‐SF‐15‐005, and Alzheimer's Society Senior Fellowship AS‐SF‐18b‐001. All grants were paid to employer. Eric J. Brunner was supported by the UK Medical Research Council, Economic and Social Research Council (ES/T014377/1), and UK‐China Health And Social Challenges Ageing Project (UKCHASCAP): present and future burden of dementia, and policy responses. All grants were paid to employer. Pyry N. Sipila was supported by Academy of Finland (311492 and 329202), NordForsk (75021), Helsinki Institute of Life Science (H970), and Finnish Foundation for Alcohol Studies grants paid to employer. Kalle Saksela reports no financial support. Jane E. Ferrie reports no financial support. Ruth C. Lovering was supported by Alzheimer's Research UK grant ARUK‐NAS2017A‐1 paid to employer. Stephen A. Williams is employed by SomaLogic inc. Aroon D. Hingorani was supported by NIHR Senior Investigator Competition, UKRI‐NIHR Strategic Priority Fund Multimorbidity Mechanisms, and Therapeutics Research Collaborative and the Wellcome Trust (221854/Z/20/Z) grants paid to employer. Henrik Zetterberg was a Wallenberg Scholar supported by grants from the Swedish Research Council (2018‐02532), the European Research Council (681712), Swedish State Support for Clinical Research (ALFGBG‐720931), the Alzheimer Drug Discovery Foundation, USA (201809‐2016862), and the UK Dementia Research Institute at University College London. All grants were paid to employer. Mika Kivimaki was supported by the Wellcome Trust (221854/Z/20/Z), the UK Medical Research Council (MR/S011676, MR/R024227), the National Institute on Aging (R01AG062553, R01AG056477), the Academy of Finland (311492, 329202), Helsinki Institute of Life Science (H970), NordForsk (75021), and the Finnish Work Environment Fund (190424). All grants were paid to employer.
Lindbohm JV ,Mars N ,Walker KA, et al. Plasma proteins, cognitive decline and 20‐year risk of dementia in the Whitehall II and Atherosclerosis Risk in Communities studies. Alzheimer's Dement. 2022;18:612–624. 10.1002/alz.12419
REFERENCES
- 1. GBD 2016 Dementia Collaborators . Global, regional, and national burden of Alzheimer's disease and other dementias, 1990‐2016: a systematic analysis for the global burden of disease study 2016. Lancet Neurol. 2019;18(1):88‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jack CR, Jr , Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12(2):207‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gauthier S, Albert M, Fox N, et al. Why has therapy development for dementia failed in the last two decades? Alzheimers Dement. 2016;12(1):60‐64. [DOI] [PubMed] [Google Scholar]
- 4. Knopman DS. Lowering of amyloid‐beta by β‐secretase inhibitors—Some informative failures. N Engl J Med. 2019;380(15):1476‐1478. [DOI] [PubMed] [Google Scholar]
- 5. Brookmeyer R, Abdalla N. Estimation of lifetime risks of Alzheimer's disease dementia using biomarkers for preclinical disease. Alzheimers Dement. 2018;14(8):981‐988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sweeney MD, Zhao Z, Montagne A, Nelson AR, Zlokovic BV. Blood‐brain barrier: from physiology to disease and back. Physiol Rev. 2019;99(1):21‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shi Y, Holtzman DM. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat Rev Immunol. 2018;18(12):759‐772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Assarsson E, Lundberg M, Holmquist G, Björkesten J, Thorsen SB, Ekman D, Eriksson A, Rennel Dickens E, Ohlsson S, Edfeldt G, Andersson AC, Lindstedt P, Stenvang J, Gullberg M, Fredriksson S. Homogenous 96‐plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS One. 2014;9(4):e95192. 10.1371/journal.pone.0095192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gold L, Ayers D, Bertino J, et al. Aptamer‐based multiplexed proteomic technology for biomarker discovery. PLoS One. 2010;5(12):e15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Emilsson V, Ilkov M, Lamb JR, et al. Co‐regulatory networks of human serum proteins link genetics to disease. Science. 2018;361(6404):769‐773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Santos R, Ursu O, Gaulton A, et al. A comprehensive map of molecular drug targets. Nat Rev Drug Discov. 2017;16(1):19‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chessum NEA, Sharp SY, Caldwell JJ, et al. Demonstrating in‐cell target engagement using a pirin protein degradation probe (CCT367766). J Med Chem. 2018;61(3):918‐933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cummings J, Lee G, Ritter A, Zhong K. Alzheimer's disease drug development pipeline: 2018. Alzheimers Dement (N Y). 2018;4:195‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Scudellari M. Protein‐slaying drugs could be the next blockbuster therapies. Nature. 2019;567(7748):298‐300. [DOI] [PubMed] [Google Scholar]
- 15. Katsimpardi L, Litterman NK, Schein PA, et al. Vascular and neurogenic rejuvenation of the aging mouse brain by young systemic factors. Science. 2014;344(6184):630‐634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Middeldorp J, Lehallier B, Villeda SA, et al. Preclinical assessment of young blood plasma for Alzheimer disease. JAMA Neurol. 2016;73(11):1325‐1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sha SJ, Deutsch GK, Tian L, et al. Safety, tolerability, and feasibility of young plasma infusion in the plasma for Alzheimer symptom amelioration study: a randomized clinical trial. JAMA Neurol. 2019;76(1):35‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hampel H, O'Bryant SE, Molinuevo JL, et al. Blood‐based biomarkers for Alzheimer disease: mapping the road to the clinic. Nat Rev Neurol. 2018;14(11):639‐652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Marmot MG, Smith GD, Stansfeld S, et al. Health inequalities among British civil servants: the Whitehall II study. Lancet. 1991;337(8754):1387‐1393. [DOI] [PubMed] [Google Scholar]
- 20. Williams SA, Kivimaki M, Langenberg C, et al. Plasma protein patterns as comprehensive indicators of health. Nat Med. 2019;25(12):1851‐1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. The atherosclerosis risk in communities (ARIC) study: design and objectives. The ARIC investigators. Am J Epidemiol. 1989;129(4):687‐702. [PubMed] [Google Scholar]
- 22. Kim CH, Tworoger SS, Stampfer MJ, et al. Stability and reproducibility of proteomic profiles measured with an aptamer‐based platform. Sci Rep. 2018;8(1):8382. 10.1038/s41598-018-26640-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Candia J, Cheung F, Kotliarov Y, et al. Assessment of variability in the SOMAscan assay. Sci Rep. 2017;7(1):14248‐14755. 10.1038/s41598-017-14755-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sun BB, Maranville JC, Peters JE, et al. Genomic atlas of the human plasma proteome. Nature. 2018;558(7708):73‐79. 10.1038/s41586-018-0175-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tin A, Yu B, Ma J, et al. Reproducibility and variability of protein analytes measured using a multiplexed modified aptamer assay. J Appl Lab Med. 2019;4(1):30‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tanaka T, Biancotto A, Moaddel R, et al. Plasma proteomic signature of age in healthy humans. Aging Cell. 2018;17(5):e12799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Heywood WE, Galimberti D, Bliss E, et al. Identification of novel CSF biomarkers for neurodegeneration and their validation by a high‐throughput multiplexed targeted proteomic assay. Mol Neurodegener. 2015;10:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wilson RS, Leurgans SE, Boyle PA, Schneider JA, Bennett DA. Neurodegenerative basis of age‐related cognitive decline. Neurology. 2010;75(12):1070‐1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kivimaki M, Batty GD, Singh‐Manoux A, Britton A, Brunner EJ, Shipley MJ. Validity of cardiovascular disease event ascertainment using linkage to UK hospital records. Epidemiology. 2017;28(5):735‐739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sommerlad A, Perera G, Singh‐Manoux A, et al. Accuracy of general hospital dementia diagnoses in england: Sensitivity, specificity, and predictors of diagnostic accuracy 2008‐2016. Alzheimers Dement. 2018;14(7):933‐943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Walker KA, Sharrett AR, Wu A, et al. Association of midlife to late‐life blood pressure patterns with incident dementia. JAMA. 2019;322(6):535‐545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gottesman RF, Albert MS, Alonso A, et al. Associations between midlife vascular risk factors and 25‐year incident dementia in the atherosclerosis risk in communities (ARIC) cohort. JAMA Neurol. 2017;74(10):1246‐1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Head J, Stansfeld SA, Ebmeier KP, et al. Use of self‐administered instruments to assess psychiatric disorders in older people: Validity of the general health questionnaire, the center for epidemiologic studies depression scale and the self‐completion version of the revised clinical interview schedule. Psychol Med. 2013;43(12):2649‐2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bolla MK, Wood N, Humphries SE. Rapid determination of apolipoprotein E genotype using a heteroduplex generator. J Lipid Res. 1999;40(12):2340‐2345. [PubMed] [Google Scholar]
- 35. Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16(3):1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gareth J, Witten D, Hastie T, Tibshirani R. An Introduction to Statistical Learning: With Applications in R. Springer Publishing Company, Incorporated.; 2014. [Google Scholar]
- 37. Cox DR. Regression models and life‐tables. J R Statist Soc B. 1972;34(2):187‐220. [Google Scholar]
- 38. Zhang X ,Zhang ME, Fine J. A proportional hazards model for the subdistribution of a competing risk. J Am Stat Assoc. 1999(94):496‐509. [Google Scholar]
- 39.The data used for the analyses described in this manuscript were obtained from: [GTEx_Analysis_2017‐06‐05_v8_RNASeQCv1.1.9_gene_median_tpm.gct] the GTEx Portal https://gtexportal.org/home/datasets. Accessed September 10, 2020
- 40. The human protein atlas. http://www.proteinatlas.org. Accessed December 10, 2020.
- 41. Uhlén M, Fagerberg L, Hallström BM, et al. Proteomics. Tissue‐based map of the human proteome. Science. 2015;347(6220):1260419. [DOI] [PubMed] [Google Scholar]
- 42. Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44‐57. [DOI] [PubMed] [Google Scholar]
- 44. UniProt Consortium . UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47(D1):D506‐D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gaulton A, Hersey A, Nowotka M, et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017;45(D1):D945‐D954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sipilä PN, Lindbohm JV, Singh‐Manoux A, et al. Long‐term risk of dementia following hospitalization due to physical diseases: a multicohort study. Alzheimers Dement. 2020;16(12):1686‐1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stocker H, Perna L, Weigl K, et al. Prediction of clinical diagnosis of Alzheimer's disease, vascular, mixed, and all‐cause dementia by a polygenic risk score and APOE status in a community‐based cohort prospectively followed over 17 years. Mol Psychiatry. 2020. 10.1038/s41380-020-0764-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yao F, Hong X, Li S, et al. Urine‐based biomarkers for Alzheimer's disease identified through coupling computational and experimental methods. J Alzheimers Dis. 2018;65(2):421‐431. [DOI] [PubMed] [Google Scholar]
- 49. Chai YL, Hilal S, Chong JP, et al. Growth differentiation factor‐15 and white matter hyperintensities in cognitive impairment and dementia. Medicine (Baltimore). 2016;95(33):e4566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jiang J, Wen W, Sachdev PS. Macrophage inhibitory cytokine‐1/growth differentiation factor 15 as a marker of cognitive ageing and dementia. Curr Opin Psychiatry. 2016;29(2):181‐186. [DOI] [PubMed] [Google Scholar]
- 51. Qin W, Jia X, Wang F, et al. Elevated plasma angiogenesis factors in Alzheimer's disease. J Alzheimers Dis. 2015;45(1):245‐252. [DOI] [PubMed] [Google Scholar]
- 52. Emanuele E, Peros E, Scioli GA, et al. Plasma osteoprotegerin as a biochemical marker for vascular dementia and Alzheimer's disease. Int J Mol Med. 2004;13(6):849‐853. [PubMed] [Google Scholar]
- 53. Jefferson AL, Massaro JM, Wolf PA, et al. Inflammatory biomarkers are associated with total brain volume: the Framingham heart study. Neurology. 2007;68(13):1032‐1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Moses GS, Jensen MD, Lue LF, et al. Secretory PLA2‐IIA: a new inflammatory factor for Alzheimer's disease. J Neuroinflammation. 2006;3:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ohara T, Hata J, Tanaka M, et al. Serum soluble triggering receptor expressed on myeloid cells 2 as a biomarker for incident dementia: the Hisayama study. Ann Neurol. 2019;85(1):47‐58. [DOI] [PubMed] [Google Scholar]
- 56. Kerola T, Nieminen T, Hartikainen S, Sulkava R, Vuolteenaho O, Kettunen R. B‐type natriuretic peptide as a predictor of declining cognitive function and dementia–A cohort study of an elderly general population with a 5‐year follow‐up. Ann Med. 2010;42(3):207‐215. [DOI] [PubMed] [Google Scholar]
- 57. Tynkkynen J, Laatikainen T, Salomaa V, et al. NT‐proBNP and the risk of dementia: a prospective cohort study with 14 years of follow‐up. J Alzheimers Dis. 2015;44(3):1007‐1013. [DOI] [PubMed] [Google Scholar]
- 58. Moir RD, Lathe R, Tanzi RE. The antimicrobial protection hypothesis of Alzheimer's disease. Alzheimers Dement. 2018;14(12):1602‐1614. [DOI] [PubMed] [Google Scholar]
- 59. Readhead B, Haure‐Mirande JV, Funk CC, et al. Multiscale analysis of independent Alzheimer's cohorts finds disruption of molecular, genetic, and clinical networks by human herpesvirus. Neuron. 2018;99(1):64‐82.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Akiyama H, Barger S, Barnum S, et al. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21(3):383‐421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Elahi FM, Casaletto KB, La Joie R, et al. Plasma biomarkers of astrocytic and neuronal dysfunction in early‐ and late‐onset Alzheimer's disease. Alzheimers Dement. 2020;16(4):681‐695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Huber‐Lang M, Lambris JD, Ward PA. Innate immune responses to trauma. Nat Immunol. 2018;19(4):327‐341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Morgan AR, Touchard S, Leckey C, et al. Inflammatory biomarkers in Alzheimer's disease plasma. Alzheimers Dement. 2019;15(6):776‐787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ennerfelt HE, Lukens JR. The role of innate immunity in Alzheimer's disease. Immunol Rev. 2020;297(1):225‐246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Fong JJ, Tsai CM, Saha S, Nizet V, Varki A, Bui JD. Siglec‐7 engagement by GBS β‐protein suppresses pyroptotic cell death of natural killer cells. Proc Natl Acad Sci U S A. 2018;115(41):10410‐10415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Varchetta S, Mele D, Lombardi A, et al. Lack of siglec‐7 expression identifies a dysfunctional natural killer cell subset associated with liver inflammation and fibrosis in chronic HCV infection. Gut. 2016;65(12):1998‐2006. [DOI] [PubMed] [Google Scholar]
- 67. Becknell B, Eichler TE, Beceiro S, et al. Ribonucleases 6 and 7 have antimicrobial function in the human and murine urinary tract. Kidney Int. 2015;87(1):151‐161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lu L, Arranz‐Trullén J, Prats‐Ejarque G, Pulido D, Bhakta S, Boix E. Human antimicrobial RNases inhibit intracellular bacterial growth and induce autophagy in mycobacteria‐infected macrophages. Front Immunol. 2019;10:1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. LeBleu VS, Teng Y, O'Connell JT, et al. Identification of human epididymis protein‐4 as a fibroblast‐derived mediator of fibrosis. Nat Med. 2013;19(2):227‐231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Varchetta S, Lusso P, Hudspeth K, et al. Sialic acid‐binding ig‐like lectin‐7 interacts with HIV‐1 gp120 and facilitates infection of CD4pos T cells and macrophages. Retrovirology. 2013;10:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Murphy A, Sunohara JR, Sundaram M, et al. Induction of protein kinase C substrates, myristoylated alanine‐rich C kinase substrate (MARCKS) and MARCKS‐related protein (MRP), by amyloid beta‐protein in mouse BV‐2 microglial cells. Neurosci Lett. 2003;347(1):9‐12. [DOI] [PubMed] [Google Scholar]
- 72. Sunohara JR, Ridgway ND, Cook HW, Byers DM. Regulation of MARCKS and MARCKS‐related protein expression in BV‐2 microglial cells in response to lipopolysaccharide. J Neurochem. 2001;78(3):664‐672. [DOI] [PubMed] [Google Scholar]
- 73. Farooqui AA, Horrocks LA. Plasmalogens: workhorse lipids of membranes in normal and injured neurons and glia. Neuroscientist. 2001;7(3):232‐245. [DOI] [PubMed] [Google Scholar]
- 74. Fuchs T, Trollor JN, Crawford J, et al. Macrophage inhibitory cytokine‐1 is associated with cognitive impairment and predicts cognitive decline—The Sydney memory and aging study. Aging Cell. 2013;12(5):882‐889. [DOI] [PubMed] [Google Scholar]
- 75. Tsai VWW, Husaini Y, Sainsbury A, Brown DA, Breit SN. The MIC‐1/GDF15‐GFRAL pathway in energy homeostasis: implications for obesity, cachexia, and other associated diseases. Cell Metab. 2018;28(3):353‐368. [DOI] [PubMed] [Google Scholar]
- 76. Whelan CD, Mattsson N, Nagle MW, et al. Multiplex proteomics identifies novel CSF and plasma biomarkers of early Alzheimer's disease. Acta Neuropathol Commun. 2019;7(1):169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Gilgés D, Vinit MA, Callebaut I, et al. Polydom: a secreted protein with pentraxin, complement control protein, epidermal growth factor and von willebrand factor A domains. Biochem J. 2000;352(Pt 1):49‐59. [PMC free article] [PubMed] [Google Scholar]
- 78. Van Itallie CM, Tietgens AJ, Aponte A, et al. MARCKS‐related protein regulates cytoskeletal organization at cell‐cell and cell‐substrate contacts in epithelial cells. J Cell Sci. 2018;131(3):jcs210237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Enyindah‐Asonye G, Li Y, Ruth JH, et al. CD318 is a ligand for CD6. Proc Natl Acad Sci U S A 2017;114(33):E6912‐E6921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Blanco‐Suarez E, Liu TF, Kopelevich A, Allen NJ. Astrocyte‐secreted chordin‐like 1 drives synapse maturation and limits plasticity by increasing synaptic GluA2 AMPA receptors. Neuron. 2018;100(5):1116‐1132.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Sun M, Thomas MJ, Herder R, Bofenkamp ML, Selleck SB, O'Connor MB. Presynaptic contributions of chordin to hippocampal plasticity and spatial learning. J Neurosci. 2007;27(29):7740‐7750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Gruber A, Mancek M, Wagner H, Kirschning CJ, Jerala R. Structural model of MD‐2 and functional role of its basic amino acid clusters involved in cellular lipopolysaccharide recognition. J Biol Chem. 2004;279(27):28475‐28482. [DOI] [PubMed] [Google Scholar]
- 83. Villanueva EB, Little JP, Lambeau G, Klegeris A. Secreted phospholipase A(2) group IIA is a neurotoxin released by stimulated human glial cells. Mol Cell Neurosci. 2012;49(4):430‐438. [DOI] [PubMed] [Google Scholar]
- 84. Sjödin S, Brinkmalm G, Öhrfelt A, et al. Endo‐lysosomal proteins and ubiquitin CSF concentrations in Alzheimer's and Parkinson's disease. Alzheimers Res Ther. 2019;11(1):82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Caseiro A, Barros A, Ferreira R, et al. Pursuing type 1 diabetes mellitus and related complications through urinary proteomics. Transl Res. 2014;163(3):188‐199. [DOI] [PubMed] [Google Scholar]
- 86. Masson E, Wiernsperger N, Lagarde M, et al. Involvement of gangliosides in glucosamine‐induced proliferation decrease of retinal pericytes. Glycobiology. 2005;15(6):585‐591. [DOI] [PubMed] [Google Scholar]
- 87. Lind L, Wallentin L, Kempf T, et al. Growth‐differentiation factor‐15 is an independent marker of cardiovascular dysfunction and disease in the elderly: results from the prospective investigation of the vasculature in uppsala seniors (PIVUS) study. Eur Heart J. 2009;30(19):2346‐2353. [DOI] [PubMed] [Google Scholar]
- 88. Kadoglou NP, Sailer N, Moumtzouoglou A, Kapelouzou A, Gerasimidis T, Liapis CD. Aggressive lipid‐lowering is more effective than moderate lipid‐lowering treatment in carotid plaque stabilization. J Vasc Surg. 2010;51(1):114‐121. [DOI] [PubMed] [Google Scholar]
- 89. Pérez de Ciriza C, Lawrie A, Varo N. Osteoprotegerin in cardiometabolic disorders. Int J Endocrinol. 2015;2015:564934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hynes RO. Integrins: Versatility, modulation, and signaling in cell adhesion. Cell. 1992;69(1):11‐25. [DOI] [PubMed] [Google Scholar]
- 91. Windham TC, Parikh NU, Siwak DR, et al. Src activation regulates anoikis in human colon tumor cell lines. Oncogene. 2002;21(51):7797‐7807. [DOI] [PubMed] [Google Scholar]
- 92. Thomas SM, Brugge JS. Cellular functions regulated by src family kinases. Annu Rev Cell Dev Biol. 1997;13:513‐609. [DOI] [PubMed] [Google Scholar]
- 93. Schwanzer‐Pfeiffer D, Rossmanith E, Schildberger A, Falkenhagen D. Characterization of SVEP1, KIAA, and SRPX2 in an in vitro cell culture model of endotoxemia. Cell Immunol. 2010;263(1):65‐70. [DOI] [PubMed] [Google Scholar]
- 94. Braun T, McIlhinney RA, Vergères G. Myristoylation‐dependent N‐terminal cleavage of the myristoylated alanine‐rich C kinase substrate (MARCKS) by cellular extracts. Biochimie. 2000;82(8):705‐715. [DOI] [PubMed] [Google Scholar]
- 95. Guo Y, Singleton PA, Rowshan A, et al. Quantitative proteomics analysis of human endothelial cell membrane rafts: Evidence of MARCKS and MRP regulation in the sphingosine 1‐phosphate‐induced barrier enhancement. Mol Cell Proteomics. 2007;6(4):689‐696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Ibeas E, Fuentes L, Martín R, Hernández M, Nieto ML. Secreted phospholipase A2 type IIA as a mediator connecting innate and adaptive immunity: new role in atherosclerosis. Cardiovasc Res. 2009;81(1):54‐63. [DOI] [PubMed] [Google Scholar]
- 97. Hazen SL, Ford DA, Gross RW. Activation of a membrane‐associated phospholipase A2 during rabbit myocardial ischemia which is highly selective for plasmalogen substrate. J Biol Chem. 1991;266(9):5629‐5633. [PubMed] [Google Scholar]
- 98. Yanai H, Yoshid H, Tomono Y, Tada N, Chiba H. The possible contribution of a general glycosphingolipid transporter, GM2 activator protein, to atherosclerosis. J Atheroscler Thromb. 2006;13(6):281‐285. [DOI] [PubMed] [Google Scholar]
- 99. Brew K, Dinakarpandian D, Nagase H. Tissue inhibitors of metalloproteinases: evolution, structure and function. Biochim Biophys Acta. 2000;1477(1‐2):267‐283. [DOI] [PubMed] [Google Scholar]
- 100. Melendez‐Zajgla J, Del Pozo L, Ceballos G, Maldonado V. Tissue inhibitor of metalloproteinases‐4. The road less traveled. Mol Cancer. 2008;7:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Santos‐Martínez MJ, Medina C, Jurasz P, Radomski MW. Role of metalloproteinases in platelet function. Thromb Res. 2008;121(4):535‐542. [DOI] [PubMed] [Google Scholar]
- 102. Tummalapalli CM, Heath BJ, Tyagi SC. Tissue inhibitor of metalloproteinase‐4 instigates apoptosis in transformed cardiac fibroblasts. J Cell Biochem. 2001;80(4):512‐521. [DOI] [PubMed] [Google Scholar]
- 103. Singh JSS, Burrell LM, Cherif M, Squire IB, Clark AL, Lang CC. Sacubitril/valsartan: beyond natriuretic peptides. Heart. 2017;103(20):1569‐1577. [DOI] [PubMed] [Google Scholar]
- 104. Hu WT, Holtzman DM, Fagan AM, et al. Plasma multianalyte profiling in mild cognitive impairment and Alzheimer disease. Neurology. 2012;79(9):897‐905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Hilal S, Chai YL, Ikram MK, et al. Markers of cardiac dysfunction in cognitive impairment and dementia. Medicine (Baltimore). 2015;94(1):e297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Kondziella D, Göthlin M, Fu M, Zetterberg H, Wallin A. B‐type natriuretic peptide plasma levels are elevated in subcortical vascular dementia. Neuroreport. 2009;20(9):825‐827. [DOI] [PubMed] [Google Scholar]
- 107. Dhanesha N, Nayak MK, Doddapattar P, et al. Targeting myeloid‐cell specific integrin α9β1 inhibits arterial thrombosis in mice. Blood. 2020;135(11):857‐861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Dhanesha N, Jain M, Tripathi AK, et al. Targeting myeloid‐specific integrin α9β1 improves short‐ and long‐term stroke outcomes in murine models with preexisting comorbidities by limiting thrombosis and inflammation. Circ Res. 2020;126(12):1779‐1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Sakuta H, Suzuki R, Takahashi H, et al. Ventroptin: a BMP‐4 antagonist expressed in a double‐gradient pattern in the retina. Science. 2001;293(5527):111‐115. [DOI] [PubMed] [Google Scholar]
- 110. Kane R, Godson C, O'Brien C. Chordin‐like 1, a bone morphogenetic protein‐4 antagonist, is upregulated by hypoxia in human retinal pericytes and plays a role in regulating angiogenesis. Mol Vis. 2008;14:1138‐1148. [PMC free article] [PubMed] [Google Scholar]
- 111. James NE, Emerson JB, Borgstadt AD, et al. The biomarker HE4 (WFDC2) promotes a pro‐angiogenic and immunosuppressive tumor microenvironment via regulation of STAT3 target genes. Sci Rep. 2020;10(1):8558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Iadecola C. The pathobiology of vascular dementia. Neuron. 2013;80(4):844‐866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Kullmann S, Kleinridders A, Small DM, et al. Central nervous pathways of insulin action in the control of metabolism and food intake. Lancet Diabetes Endocrinol. 2020;8(6):524‐534. [DOI] [PubMed] [Google Scholar]
- 114. Evdokimova V, Tognon CE, Benatar T, et al. IGFBP7 binds to the IGF‐1 receptor and blocks its activation by insulin‐like growth factors. Sci Signal. 2012;5(255):ra92. [DOI] [PubMed] [Google Scholar]
- 115. Yamanaka Y, Wilson EM, Rosenfeld RG, Oh Y. Inhibition of insulin receptor activation by insulin‐like growth factor binding proteins. J Biol Chem. 1997;272(49):30729‐30734. [DOI] [PubMed] [Google Scholar]
- 116. Kellar D, Craft S. Brain insulin resistance in Alzheimer's disease and related disorders: mechanisms and therapeutic approaches. Lancet Neurol. 2020;19(9):758‐766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Nicholls SJ, Kastelein JJ, Schwartz GG, et al. Varespladib and cardiovascular events in patients with an acute coronary syndrome: the VISTA‐16 randomized clinical trial. JAMA, J Am Med Assoc. 2014;311(3):252‐262. [DOI] [PubMed] [Google Scholar]
- 118. Dogra S, Uprety S, Suresh SH. Itolizumab, a novel anti‐CD6 monoclonal antibody: a safe and efficacious biologic agent for management of psoriasis. Expert Opin Biol Ther. 2017;17(3):395‐402. [DOI] [PubMed] [Google Scholar]
- 119. Nybo M, Preil SR, Juhl HF, et al. Rosiglitazone decreases plasma levels of osteoprotegerin in a randomized clinical trial with type 2 diabetes patients. Basic Clin Pharmacol Toxicol. 2011;109(6):481‐485. [DOI] [PubMed] [Google Scholar]
- 120. Esteghamati A, Afarideh M, Feyzi S, Noshad S, Nakhjavani M. Comparative effects of metformin and pioglitazone on fetuin‐A and osteoprotegerin concentrations in patients with newly diagnosed diabetes: a randomized clinical trial. Diabetes Metab Syndr. 2015;9(4):258‐265. [DOI] [PubMed] [Google Scholar]
- 121. First‐in‐human study of the GDF‐15 neutralizing antibody CTL‐002 in patients with advanced cancer (GDFATHER). https://clinicaltrials.gov/ct2/show/NCT04725474. Accessed November 5, 2021.
- 122. Suhre K, McCarthy MI, Schwenk JM. Genetics meets proteomics: perspectives for large population‐based studies. Nat Rev Genet. 2021;22(1):19‐37. [DOI] [PubMed] [Google Scholar]
- 123. Rasmussen KL, Tybjaerg‐Hansen A, Nordestgaard BG, Frikke‐Schmidt R. APOE and dementia—Resequencing and genotyping in 105,597 individuals. Alzheimers Dement. 2020;16(12):1624‐1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Rasmussen KL, Tybjærg‐Hansen A, Nordestgaard BG, Frikke‐Schmidt R. Plasma apolipoprotein E levels and risk of dementia: a mendelian randomization study of 106,562 individuals. Alzheimers Dement. 2018;14(1):71‐80. [DOI] [PubMed] [Google Scholar]
- 125. Rasmussen KL, Tybjaerg‐Hansen A, Nordestgaard BG, Frikke‐Schmidt R. Plasma levels of apolipoprotein E and risk of dementia in the general population. Ann Neurol. 2015;77(2):301‐311. [DOI] [PubMed] [Google Scholar]
- 126. Heim A. AH 4 Group Test of General Intelligence. NFER‐Nelson Publishing Company. 1970. [Google Scholar]
- 127. Borkowski J, Benton A, Spreen O. Word fluency and brain damage. Neuropsychologia. 1967;5:135‐140. [Google Scholar]
- 128. Bartlett JW, Seaman SR, White IR, Carpenter JR, Alzheimer's Disease Neuroimaging Initiative* . Multiple imputation of covariates by fully conditional specification: accommodating the substantive model. Stat Methods Med Res. 2015;24(4):462‐487. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information