

ABSTRACT
Chronic liver inflammation (CLI) is a risk factor for development of hepatocellular carcinoma (HCC). Galectin‐1 (Gal1) is involved in the regulation of inflammation, angiogenesis, and tumorigenesis, exhibiting multiple anti‐inflammatory and protumorigenic activities. We aimed to explore its regulatory role in CLI and HCC progression using an established model of CLI‐mediated HCC development, Abcb4 [multidrug‐resistance 2 (Mdr2)]‐knockout (KO) mice, which express high levels of Gal1 in the liver. We generated double‐KO (dKO) Gal1‐KO/Mdr2‐KO mice on C57BL/6 and FVB/N genetic backgrounds and compared HCC development in the generated strains with their parental Mdr2‐KO strains. Loss of Gal1 increased liver injury, inflammation, fibrosis, and ductular reaction in dKO mice of both strains starting from an early age. Aged dKO mutants displayed earlier hepatocarcinogenesis and increased tumor size compared with control Mdr2‐KO mice. We found that osteopontin, a well‐known modulator of HCC development, and oncogenic proteins Ntrk2 (TrkB) and S100A4 were overexpressed in dKO compared with Mdr2‐KO livers. Our results demonstrate that in Mdr2‐KO mice, a model of CLI‐mediated HCC, Gal1‐mediated protection from hepatitis, liver fibrosis, and HCC initiation dominates over its known procarcinogenic activities at later stages of HCC development. These findings suggest that anti‐Gal1 treatments may not be applicable at all stages of CLI‐mediated HCC.—Potikha, T., Pappo, O., Mizrahi, L., Olam, D., Mailer, S. M., Rabinovich, G. A., Galun, E., Goldenberg, D. S. Lack of galectin‐1 exacerbates chronic hepatitis, liver fibrosis, and carcinogenesis in murine hepatocellular carcinoma model. FASEB J. 33, 7995–8007 (2019). www.fasebj.org
Keywords: HCC, Mdr2, Spp1, Ntrk2, S100a4
ABBREVIATIONS
- Abcb4
multidrug-resistance 2 (Mdr2)
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- B6
C57BL/6
- CLI
chronic liver inflammation
- dKO
double‐knockout
- FVB
FVB/NJ
- Gal1
galectin‐1
- HCC
hepatocellular carcinoma
- IHC
immunohistochemistry
- KO
knockout
- Mdr2
multidrug‐resistance 2 (Abcb4)
- Opn
osteopontin
- Spp1
Opn gene
About 25% of human cancers are associated with chronic inflammation (1). To investigate the role of chronic inflammation in the development of hepatocellular carcinoma (HCC), we use the Abcb4 [multidrug‐resistance 2 (Mdr2)]‐knockout (KO) mice as a model. These mice lack the Mdr2 P‐glycoprotein responsible for the phosphatidylcholine transport across the canalicular membrane and develop chronic hepatitis and cholangitis at an early age, inevitably followed by HCC at a later age (2). The gene expression profiling that we performed on the Mdr2‐KO livers at the precancerous stages of liver disease revealed aberrant expression of several immune and inflammatory regulatory genes that have an important role in hepatocarcinogenesis, including up‐regulation of Lgals1, which encodes galectin‐1 (Gal1) (3). Gal1 is widely expressed in epithelial and immune cells and acts both extracellularly and intracellularly, modulating innate and adaptive immune responses and acting as a homeostatic agent (4). It promotes apoptosis of activated CD8 T cells, T helper (Th)1, and Th17 cells; suppresses the secretion of proinflammatory IL‐2 and IFN‐γ; and favors the secretion of anti‐inflammatory IL‐10 and TGF‐β1 (5–7). Gal1 is a key effector of regulatory T cells (8) and is required for the regulatory function of B cells (9). Gal1 overexpression in many types of tumors and/or surrounding tissues promotes tumor progression by different mechanisms, including inhibition of antitumor immune responses (5, 10, 11), by augmenting Ras activation (12) and by stimulating tumor angiogenesis (13–15). Gal1 promotes HCC cell adhesion (16); it is overexpressed in human HCC (17, 18) and in Mdr2‐KO livers (at all tested ages and in most tested tumors) (3, 19). Gal1 also modulates cell survival and proliferation, acting in different model systems as either a mitogen or an inhibitor of cell proliferation (20). Thus, Gal1 may have a dual effect on tumor development mediated by chronic inflammation, acting either as an inhibitory mediator at the early inflammatory stage of a disease or protumorigenic at the later stages. Importantly, we have previously demonstrated that loss of Gal1 differentially affects response of the FVB/NJ (FVB) and C57BL/6 (B6) inbred mouse strains to acute liver inflammation (21). In order to reveal the role of Gal1 in HCC development, we generated double mutants Mdr2‐KO/Lgals1‐KO on the FVB and B6 genetic backgrounds and compared HCC development in these strains with their parental Mdr2‐KO strains for both sexes.
MATERIALS AND METHODS
Mice
Mice were housed at the specific pathogen‐free unit of the Animal Facility at the Hebrew University Medical School, under a 12‐h light/dark cycle and were provided with food (Teklad 2918, 18% protein diet; Envigo, Somerset, NJ, USA) and water ad libitum. All animal studies were approved by the Hebrew University–Hadassah Medical School Ethics Review Board [the Animal Care Unit holds National Institutes of Health (NIH; Bethesda, MD, USA) approval OPRR‐A01‐5011 and the American Association for the Accreditation of Laboratory Animal Care International accreditation 1285]. Wild‐type B6 mice were obtained from Envigo RMS Ltd. (Jerusalem, Israel); the Lgals1‐KO mutants of the B6 strain were kindly provided by Prof. Francoise Poirier (Institut Jacques Monod, Universités P6 and P7, Paris, France) and propagated in our facility. The Mdr2‐KO/B6 and Gal1‐KO/FVB mice were generated in our laboratory (21, 22). The dKO/FVB and dKO/B6 strains we generated by mating Gal1‐KO with Mdr2‐KO mice of either B6 or FVB backgrounds to generate F1 double heterozygous mice. Then, F1 mice were intermated to get the F2 progeny from which the appropriate dKO genotypes were established. To select the required Gal1‐KO and Mdr2‐KO genotypes, DNA from mouse tails was purified by D‐Tail Extraction Kit (Synthezza Bioscience, Jerusalem, Israel). Genotyping was carried out by PCR analysis using primers specific to Gal1, Mdr2, and Neo genes as previously described (21). Amplicons were electrophoresed on 2% agarose gels.
Harvesting mouse liver tissue
At the indicated ages, mice were anesthetized with isoflurane, blood was collected from the periorbital sinus, and anesthetized mice were then euthanized by cervical dislocation. All mice were operated at the same time interval between 12:00 am and 3:00 pm immediately after they were euthanized. The intact livers were excised for tissue processing and weighed, and one part was fixed in 4% neutral buffered formaldehyde for histology, while the remaining liver tissue was snap‐frozen for RNA and protein analyses. The formalin‐fixed livers were embedded in paraffin.
Blood analysis
Serum was obtained by centrifugation of total blood at 3200 g for 10 min. Liver damage was assessed through the alanine aminotransferase (ALT) and alkaline phosphatase (ALP) activities (U/L) in the serum by an enzyme assay using Reflotron (Roche, Basel, Switzerland).
Gene expression analysis
Total RNA was isolated from frozen (−80°C) liver tissues with the Trizol reagent (Thermo Fisher Scientific, Waltham, MA, USA) as described by the manufacturer and used for either Nanostring mRNA assay (NanoString Technologies, Seattle, WA, USA) or quantitative RT‐PCR. Reverse transcription of total RNA was performed using the qScript cDNA Synthesis Kit (95047), and quantitative RT‐PCR was performed with Perfecta Sybr Green Fast Mix Rox (95073; both Quanta Biosciences, Gaithersburg, MD, USA). Reactions were run on a CFX384 Real‐Time System with C1000 Touch Thermal Cycle (Bio‐Rad, Hercules, CA, USA) and consisted of an initial denaturation for 20 s at 95°C followed by 40 cycles of 1 s at 95°C and 20 s at 60°C Following each run, melting curves were generated to verify specific product formation. PCR primers specific for genes of interest were designed by Primer‐Blast (https://www.ncbi.nlm.nih.gov/tools/primer‐blast) and synthesized by Integrated DNA Technologies (Skokie, IL, USA). Primers used in this study are shown in Supplemental Table S1. The primer's specificity was ensured using 2 types of controls, the first without reverse transcriptase (1 for each primer pair) and the second with no DNA template (1 for each primer pair). All quantitative PCRs were run in triplicates. Data were analyzed using CFX Manager software (Bio‐Rad), and the expression of each target gene was quantified relative to the expression of the Hprt reference gene.
Cell culture and proliferation analysis
HepG2 hepatoblastoma cells (American Type Culture Collection, Manassas, VA, USA) were maintained as a monolayer culture in DMEM (Thermo Fisher Scientific) and supplemented with 10% fetal bovine serum in a 5% CO2 incubator at 37°C Subculturing of cells was carried out using 0.25% trypsin‐EDTA (Thermo Fisher Scientific). Cell proliferation was analyzed by using the xCEL‐Ligence System (Roche) that measures electrical impedance across microelectrodes integrated into the bottom of tissue culture E‐Plates.
Immunohistochemistry
Immunostaining was done on 4‐μm–thick formalin‐fixed paraffin‐embedded liver tissue sections by standard procedures. Haematoxylin and eosin–stained sections were used to examine liver morphology. Masson's trichrome staining was performed for evaluating liver fibrosis. Pathology review was performed in a blind fashion by an experienced pathologist (O.P.) with the histologic scoring system for tumors.
The following antibodies to specific markers were used for immunohistochemical staining: CD3 (1:200, MCA1477, CD34; Sm1603; Acris, Herford, Germany), F4/80 (1:300; Serotec, Oxford, United Kingdom), Ly6B.2 (1:300, MCA771GA; Serotec), Ki67 (275R‐14; Thermo Fisher Scientific), Ntrk2 (1:150, sc‐377218; Santa Cruz Biotechnology, Dallas, TX, USA), panCK (1:400Z0622; DakoCytomation, Glostrup, Denmark), S100A4 (1:200, ab40722; Abcam, Cambridge, MA, USA), Opn gene (Spp1) (1:200, sc21742; Santa Cruz Biotechnology), and vimentin (1:200, EPR3776; Abcam). The following Horseradish Peroxidase‐conjugated secondary antibodies were used: anti‐rabbit (K4003), anti‐mouse (K4001; both EnVision, Glostrup, Denmark), and anti‐rat (Histo‐fine, Nishirei, Japan). Color was developed using either 3‐amino‐9‐ethylcarbazole (AEC, 00‐1111; Thermo Fisher Scientific) for 10 min (30 min in the case of CD3) or 3,3′‐diaminobenzidene using a Zymed Super Picture Kit (87‐9663; Thermo Fisher Scientific) for 5 min (ß‐catenin, F4/80). Counterstain was performed with filtered Cat‐Hematoxylin (Pharmatrade, Dubai, United Arab Emirates). Negative controls were used by omitting the primary antibody. Stainings were visualized with the Nikon Eclipse E600 microscope equipped with the CellSens software imaging applications (Olympus, Tokyo, Japan). The number of F4/80+‐ and Ki67+‐expressing cells was counted on 10 random fields at ×200 magnification per slide, and a total of 4 or 5 mice per group were used. For the morphometric assessment of Spp1, Cd34, panCK, and Masson's trichrome stained area, the integrated optical density was calculated using ImageJ Software (NIH) in 10 random fields per section containing as similar a size as possible of portal tract or hepatic vein at ×200 magnification, and the results were averaged.
Statistical analysis
Statistical significance between groups was estimated using either the Fisher's exact test (for tumor incidence) or the 2‐tailed, unpaired Student's t test (in all other cases). Results are expressed as means ± sem; differences were considered significant at P < 0.05.
RESULTS
In order to identify possible molecular mechanisms underlying enhanced tumor development in Gal1‐deficient Mdr2‐KO mice, we compared the intensity of liver injury, fibrosis, inflammation, and cell proliferation in Mdr2‐KO/Gal1‐KO (dKO) with Mdr2‐KO mice at early (1–3 mo), middle (6 and 9 mo), and terminal (12–18 mo) ages (Supplemental Fig. S1). Mice were terminated at the age when most animals develop tumors larger than 3 mm in diameter. In both B6 and FVB strains, females develop tumors earlier than males; thus, females were terminated earlier. Notably, on the FVB genetic background, tumors appeared earlier than on the B6 genetic background (21); thus, FVB mice were terminated earlier than B6 mice of both sexes. There was no difference in animal survival between experimental groups (unpublished results).
Enhanced tumor development in the Mdr2‐KO/Gal1‐KO compared with Mdr2‐KO mice
Loss of Gal1 exacerbated HCC development in the Mdr2‐KO mice on both genetic backgrounds (Fig. 1 ). The most prominent effect caused by Gal1 loss was observed on the B6 genetic background; dKO mice had increased tumor incidence in both females (for tumors with diameter of at least 3 mm) and males (for tumors with diameter of at least 8 mm; Fig. 1A ). The percent of tumor‐bearing mice at the age of 16 mo was increased in B6 dKO vs. Mdr2‐KO mice of both sexes (Supplemental Fig. S3A). Tumor load was also increased in the B6 dKO mice for small tumors in females and for large tumors in males (Supplemental Fig. S2A–C). Tumors of B6 dKO mice had higher differentiation grades and higher grade of cell pleomorphism (Supplemental Fig. S3C, D, respectively) and had a tendency toward a higher mitosis rate (Supplemental Fig. S3B). On the FVB genetic background, loss of Gal1 had no effect on tumor incidence (Fig. 1B ) but caused increased tumor load both in females and males at the age of 1 yr (Supplemental Fig. S2D, E, respectively).
Figure 1.
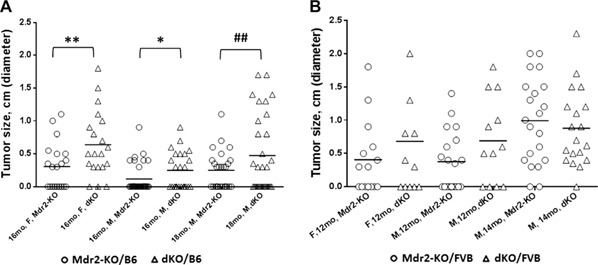
Loss of Gal1 enhances HCC development in Mdr2‐KO/B6 mice. Incidence of tumors with diameters ≥0.3 cm. A) Tumor incidence of the following experimental groups of B6 mice: 16‐mo‐old females (Mdr2‐KO, n = 21 and dKO, n = 20), 16‐mo‐old males (Mdr2‐KO, n = 32 and dKO, n = 24), and 18‐mo‐old males (Mdr2‐KO, n = 26 and dKO, n = 31). B) Tumor incidence of the following experimental groups of FVB mice: 12‐mo‐old females (Mdr2‐KO, n = 15 and dKO, n = 12), 12‐mo‐old males (Mdr2‐KO, n= 19 and dKO, n= 12), and 14‐mo‐old males (Mdr2‐KO, n= 21 and dKO, n= 19). Only nodules with diameters ≥0.3 cm were counted. F, females; M, males. Statistical significance was calculated using Fisher's exact test. *P = 0.05 for 1‐tailed test and for tumors ≥0.3 cm, **P = 0.043 for 2‐tailed test and for tumors ≥0.3 cm, ## P = 0.0076 for 2‐tailed test for large tumors ≥0.8 cm.
Enhanced severity of the chronic inflammatory disease in the Mdr2‐KO/Gal1‐KO compared with Mdr2‐KO mice at early age
At an early age, loss of Gal1 in Mdr2‐KO mice of both tested strains increased bile duct proliferation as shown by enlarged ductular reactions (Fig. 2A, B ) and hepatocyte injury evidenced by higher levels of ALT activity in the serum (Fig. 2C, D ). Immunohistochemical analysis of immune cell infiltration revealed increased recruitment of monocytes and macrophages in the B6 dKO compared with Mdr2‐KO mice at 2 and 3 mo of age (Fig. 3A, B ). Infiltration of T cells and neutrophils was low and comparable between both tested groups (unpublished results). Increased chronic liver inflammation (CLI) was also reflected by augmented proliferation of hepatocytes and nonparenchimal cells in the livers of double mutants compared with B6 Mdr2‐KO mice (Fig. 3C, D ). To understand the molecular mechanisms underlying increased liver injury and cell proliferation of Gal1‐deficient B6 Mdr2‐KO mice, we compared transcript levels of multiple known regulators of immune response and inflammation in the B6 dKO and Mdr2‐KO mice by quantitative RT‐PCR and Nanostring technology. Genes that were differently expressed between experimental groups are represented in Fig. 3E, F , whereas genes that were expressed at similar levels are listed in Supplemental Tables S2 and S3.
Figure 2.
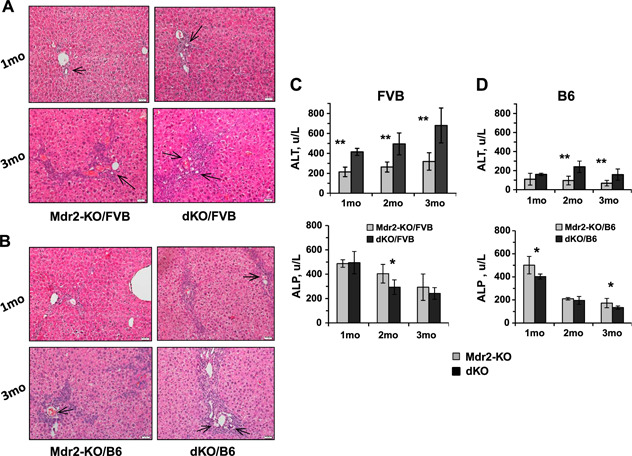
Increased bile duct proliferation, ductular reactions, and hepatocyte injury in the livers of dKO compared with Mdr2‐KO mice at young age. A, B) Hematoxylin and eosin staining of representative liver sections from Mdr2‐KO and dKO mice from FVB (A) and B6 (B) genetic background, 1 (upper panels) or 3 (bottom panels) mo of age. Ductal reactions are shown by arrows. Representative images for animals (n = 6–8) of each particular group are shown. Original magnification, ×200; scale bars, 20 μm. C, D) Liver enzyme activities (units per liter) in the serum of young FVB (C) and B6 (D) males. For each time point, 6–9 mice were used. The results are shown as means ± se. *P < 0.05, **P < 0.005 for dKO vs. Mdr2‐KO.
Figure 3.
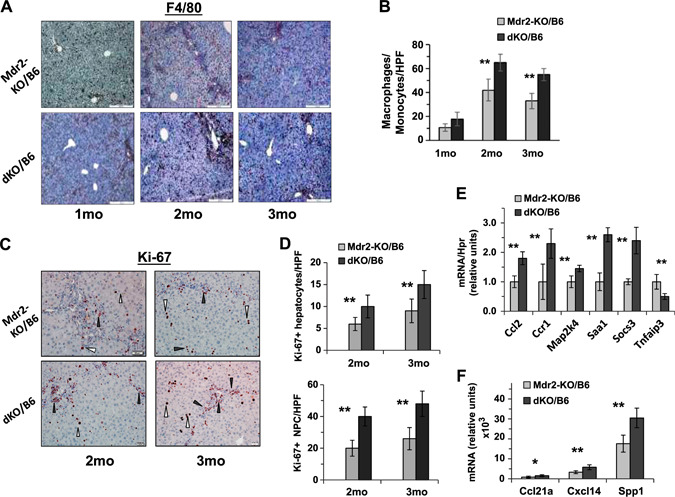
Increased inflammation and proliferation of liver cells in dKO/B6 vs. Mdr2‐KO/B6 livers. A) F4/80 immunostaining in the Mdr2‐KO/B6 (top panel) and dKO/B6 (bottom panel) livers. Red‐brown signals distributed in the tissue represent F4/80+ monocytes and macrophages. Original magnification, ×100; scale bars, 50 μm. B) Quantification of F4/80+ cells as presented in A in Mdr2‐KO and dKO livers (mean of 5–6 males per each experimental group; 10 images were collected and analyzed per mouse). C) Ki67 immunostaining in the livers of 2‐ and 3‐mo‐old Mdr2‐KO/B6 and dKO/B6 mice. White triangles point to hepatocytes and black triangles to nonparenchymal (NPC) cells. Original magnification, ×200; scale bars, 20 μm. D) Quantification of Ki67‐positive hepatocytes (upper graphs) and Ki67‐positive nonparenchymal cells (bottom graphs) as presented in C. The counting was performed in the liver zone 2 for hepatocytes and zone 1 (periportal region) for NPC in the original magnification, ×400 field; n = 3–5 mice/genotype. Means ± sd. *P < 0.05, **P < 0.01 in dKO vs. Mdr2‐KO mice (Student's t test). E, F) Expression of genes associated with inflammation in the liver of 2‐mo‐old Mdr2‐KO and dKO mice assigned by either quantitative RT‐PCR (E) or by Nanostring assay (F). There were 4–6 mice for each time point. Results are presented as a means ± sd. *P < 0.05, **P < 0.005 (Student's t test).
Liver fibrosis was significantly more prominent in B6 dKO vs. Mdr2‐KO at 2 and 3 mo of age (Fig. 4A ). This was accompanied by an increased expression of fibrosis‐associated genes Col1a, Mmp9, Sma1, and Spp1 in the liver of B6 dKO vs. Mdr2‐KO mice at the age of 2 mo (Fig. 4B ). Remarkably, the number of CD34‐positive cells in fibrotic septs and in the periphery of bile ducts was significantly increased in the livers of dKO vs. Mdr2‐KO young mice of both FVB and B6 strains (Fig. 4C, D , respectively). A key role of bone marrow‐derived CD34‐positive fibrocytes in the development of hepatic fibrosis in Mdr2‐KO mice has been demonstrated (23). A significant increase of ductular reaction in dKO vs. Mdr2‐KO livers of both mouse strains was detected at 1 and 3 mo of age (Fig. 5A, B ). The increased Spp1 expression in dKO vs. Mdr2‐KO livers of both mouse strains was confirmed at the protein level by immunostaining (Fig. 5C, D ). Remarkably, osteopontin (Opn) in the liver of all tested Mdr2‐KO mice was expressed mainly in cholangiocytes.
Figure 4.
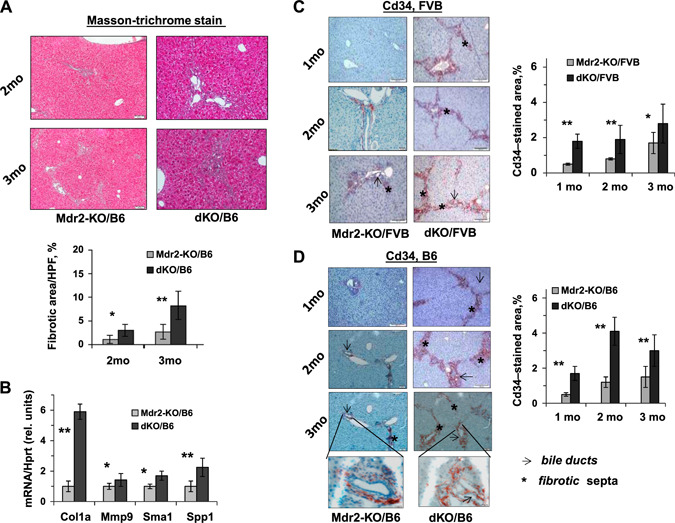
Increased severity of fibrogenesis in dKO/B6 vs. Mdr2‐KO/B6 livers at young age. A) Masson's trichrome stain and morphometric assessment of Masson's trichrome stained areas in Mdr2‐KO and dKO livers using ImageJ Software. Original magnification, ×100; scale bars, 50 μm; n= 4–5 mice/genotype. The values are means ± sem. *P < 0.05, **P < 0.01 for dKO vs. age‐matched Mdr2‐KO mice. B) Increased hepatic expression of the fibrogenesis‐ and billiary‐related genes in dKO compared with Mdr2‐KO/B6 murine livers at the age of 2 mo (n = 3–5 mice/genotype). Means ± sd. *P < 0.05, **P < 0.01 for dKO vs. Mdr2‐KO mice (Student's t test). C, D) IHC of CD34 and its quantification by morphometric analysis in the Mdr2‐KO and dKO mouse livers from FVB (C) and B6 (D) genetic backgrounds. Multiple CD34+ cells were observed in fibrotic septa (stars) and around bile ducts (arrows) in dKO vs. Mdr2‐KO livers (red‐brown color); n = 4–6 mice/genotype. Counting per ×20 power field. Original magnification: ×100; scale bars, 50 μM; inserts (D), ×600; scale bars, 10 μM.
Figure 5.
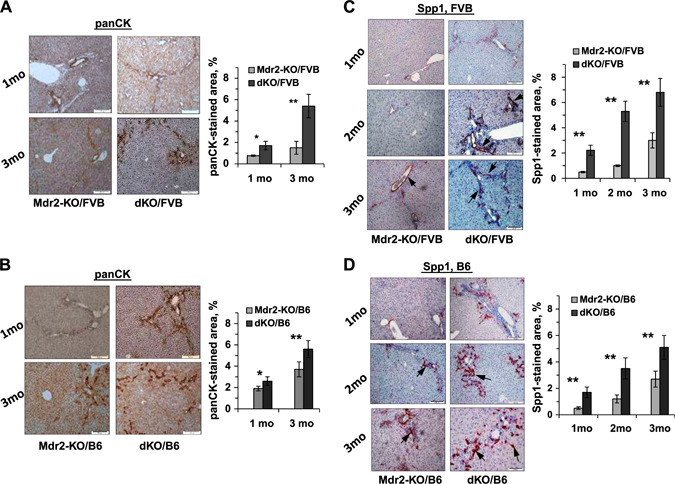
Increased severity of cholangiopathy in dKO/B6 vs. Mdr2‐KO/B6 livers at young age. A, B) PanCK immunostaining and its quantification by morphometric analysis in the livers of dKO and Mdr2‐KO mice from FVB (A) and B6 (B) genetic backgrounds. PanCK expression is mainly confined to the biliary epithelial cells. C, D) IHC of Spp1 expression and its quantification by morphometric analysis in the livers of dKO and Mdr2‐KO mice from FVB (C) and B6 (D) genetic backgrounds. The Spp1 gene was expressed mainly in the biliary epithelial cells (arrows with closed head). Original magnification, ×100; scale bars, 50 μm.
We then tested the effect of Gal1 loss on the expression of the Ntrk2 (TrkB) protein, one of the known Gal1 regulators (24); this protein is frequently elevated in HCC and associated with shorter patient survival (25). Ntrk2 was significantly overexpressed in the liver of young dKO vs. Mdr2‐KO mice, both at the mRNA and protein levels (Fig. 6A, B , respectively), and was localized mainly at the cytoplasm of hepatocytes, especially in the first layer of pericentral hepatocytes (Fig. 6B ).
Figure 6.
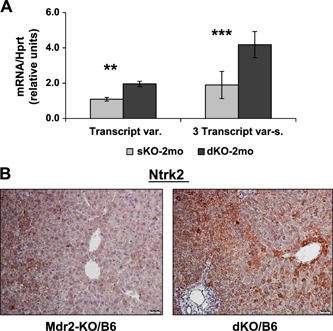
Increased Ntrk2 expression in the liver of young dKO/B6 vs. Mdr2‐KO/B6 mice. A) Ntrk2 expression in the livers of 2‐mo‐old B6 Mdr2‐KO and dKO mice assessed by quantitative RT‐qPCR; n = 3–5 mice/genotype. Three transcript variants of murine Ntrk2 were detected. Only transcript variant 2 (NM_008745.3) could be distinguished from the other 2 transcripts by quantitative RT‐PCR. Left panel shows expression of the transcript variant 2, whereas right panel shows expression of all 3 transcripts together (NM_001025074.2, NM_008745.3, NM_001282961.1); 4–5 mice/assay were used. Means ± sd. **P < 0.005, ***P < 0.0005 in dKO vs. Mdr2‐KO mice (Student's t test). B) IHC of the Ntrk2 protein in the livers of 2‐mo‐old B6 Mdr2‐KO and dKO mice. Original magnification, ×200; scale bars 20 μm.
Enhanced severity of the chronic inflammatory disease in the Mdr2‐KO/Gal1‐KO compared with Mdr2‐KO mice at middle age
In order to follow the dynamics of chronic hepatitis and cholangitis in dKO compared with Mdr2‐KO mice, we explored the extent of inflammation and fibrosis markers in the livers of 6‐ and 9‐mo‐old mice of the B6 strain only. Histologically, livers of dKO mice were characterized by a higher degree of inflammation and biliary hyperplasia (Supplemental Fig. S4A). The dKO mice had increased body and liver weights (Supplemental Fig. S4B); however, the body–to–liver weight index was similar to that of the Mdr2‐KO mice (unpublished results). At the age of 9 mo, the dKO mice showed significantly higher levels of ALT and ALP activity in blood (Fig. 7A ), indicating more severe injury to both hepatocytes and cholangiocytes, compared with the Mdr2‐KO mice. At both middle ages tested, livers of the dKO mice demonstrated higher infiltration of neutrophils and macrophages (Fig. 7B, C , and Supplemental Fig. S4C, D, respectively) and higher levels of the S100A4 and Spp1 proteins (epithelial‐mesenchymal transition markers; Supplemental Fig. S5A, B, respectively). At the age of 6 mo, dKO livers were characterized by a higher degree of fibrosis (Fig. 7D ) and higher proliferation of both hepatocytes and nonparenchymal cells (Fig. 7E ). It should be mentioned that in both tested middle ages (6 and 9 mo), expression of vimentin and Cd34 genes was similar in the liver of B6 dKO and Mdr2‐KO mice (unpublished results).
Figure 7.
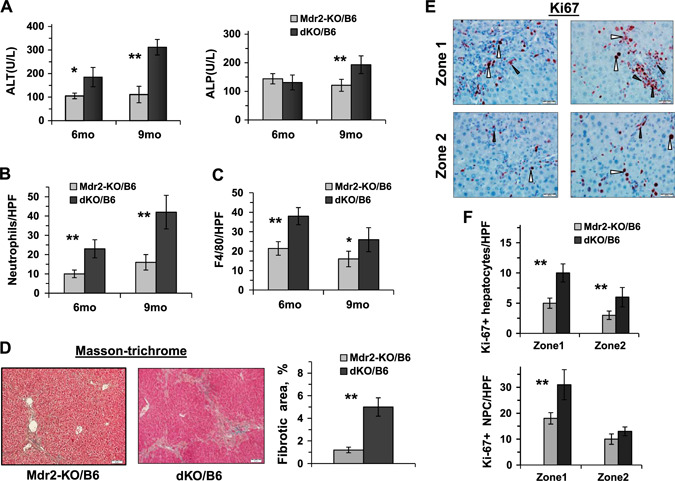
Increased liver inflammation and fibrosis in the middle‐aged dKO/B6 vs. Mdr2‐KO/B6 mice. A) ALT and ALP activity in the serum of Mdr2‐KO/B6 and dKO/B6 males 6 and 9 mo of age (5–8 mice for each time point and genotype). Means ± sd. *P < 0.05, **P < 0.01 in dKO vs. Mdr2‐KO mice (Student's t test). B, C) Quantification of Ly6G‐positive (B) and F4/80‐positive (C) leukocytes in the liver of dKO vs. Mdr2‐KO mice. The representative images are shown in Supplemental Fig. S4C and D, respectively. D) Masson's trichrome staining of the liver of 6‐mo‐old B6 dKO and Mdr2‐KO males and its quantification by morphometric analysis. Original magnification, ×100; scale bars, 50 μm. E) Representative immunohistochemical staining of the livers of 6‐mo‐old Mdr2‐KO and dKO B6 males using Ki67‐specific antibody. Hepatocytes are marked by white arrowheads and nonparenchymal cells (NPCs) by black arrowheads. Original magnification, ×400; scale bars, 20 μM. F) Quantification of Ki67‐positive hepatocytes and Ki67‐positive NPC. The counting was performed in liver zones 1 (periportal region) and 2 (middle zone) for all tested liver sections; n = 3–5 mice/genotype. Means ± sd. *P < 0.05, **P < 0.01 in dKO vs. Mdr2‐KO mice (Student's t test).
Molecular mechanisms of the enhanced hepatocarcinogenesis in the Mdr2‐KO/Gal1‐KO compared with Mdr2‐KO mice
In order to reveal the molecular and cellular mechanisms associated with the enhanced tumor development in the dKO vs. Mdr2‐KO mice, we analyzed morphologic, biochemical, and histologic parameters of the appropriate male mice at the ages when they generate tumors (Fig. 8 and Supplemental Fig. S6). In the FVB strain, dKO and Mdr2‐KO males had similar levels of the ALT activity at 12 and 14 mo of age and a similar level of the ALP activity at 12 mo of age, whereas at 14 mo, dKO males had significantly lower ALP activity in blood (Supplemental Fig. S6A, B). The FVB dKO males at the age of 12 mo also had higher infiltration of macrophages and increased expression of Opn in the liver (Fig. 8B, E , respectively). In the B6 strain, dKO males had higher ALT activity in the blood at the age of 16 and 18 mo, whereas ALP blood activity in dKO and Mdr2‐KO males was similar (Supplemental Fig. S6C, D, respectively). Liver and body mass were slightly increased in dKO males at both ages tested, whereas liver and body weight index was slightly increased in dKO males at the age of 18 mo only (Supplemental Fig. S6E). The B6 dKO mice had increased expression of proinflammatory mediators, including Ccl5, Ccr1, Cxcl2, Ltbr, and Saa1, and increased liver macrophage infiltration (Fig. 8A, B , respectively). In addition, livers of the B6 dKO mice were characterized by significantly increased fibrosis (Fig. 8C ) and enhanced biliary hyperplasia (Fig. 8D ), accompanied by up‐regulation of the Krt19 gene (Fig. 8F ) and increased expression of the fibrosis‐associated genes Spp1 (Fig. 8E , bottom panels), Cola1a, and Mmp9 (Fig. 8F ). Remarkably, Opn was expressed almost exclusively in the nontumor liver; among 20 tumors analyzed by immunohistochemistry (IHC) (10 from B6 Mdr2‐KO and 10 from B6 dKO mice of both sexes), Opn expression was detected in only 1 tumor (from B6 dKO male). Immunohistochemical staining of Gal1 in the liver of aged B6 Mdr2‐KO mice revealed no correlation (or anticorrelation) between expressions of this lectin and Opn protein (Supplemental Fig. S7). In the liver of 9‐mo‐old B6 Mdr2‐KO mice, Gal1 protein was localized mainly in the cytoplasm of nonhepatocyte cells (cholangiocyte, endothelial, and immune cells) and rarely in the cytoplasm and nuclei of hepatocytes (Supplemental Fig. S7A). In the nontumor liver of 18‐mo‐old B6 Mdr2‐KO mice, Gal1 protein was localized in the cytoplasm of hepatocytes and nonhepatocyte cells, including cholangiocyte, endothelial, and immune cells, whereas in tumors, it was localized within the cytoplasmic and membrane compartments of hepatocyte cells and rarely in the nuclei of hepatocytes (Supplemental Fig. S7B).
Figure 8.
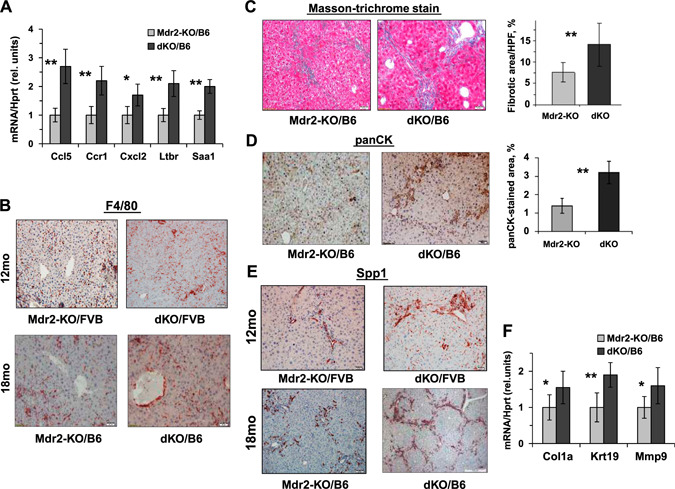
Gal1 loss enhances hepatic inflammation, fibrosis, and cholangiopathy in aged Mdr2‐KO mice. A) quantitative RT‐PCR demonstrates increased expression of inflammation‐related genes in hepatic nontumor tissues of 16‐mo‐old dKO/B6 mice (4–6 mice for each experimental group). Values for dKO mice were normalized to those of age‐matched Mdr2‐KO mice. Means ± sd. *P < 0.05, **P < 0.01 (Student's t test). B) Immunostaining of F4/80+ monocytes and macrophages infiltrating nontumor hepatic tissues of Mdr2‐KO and dKO males from FVB and B6 genetic backgrounds. Original magnification, ×100; scale bars, 50 μM. C–E) Phenotypically, livers of dKO mice were much more stiff, hard, and often not smooth compared with the Mdr2‐KO livers of the both B6 and FVB backgrounds. Masson's trichrome stain and fibrotic area quantification by morphometry in Mdr2‐KO and dKO nontumor liver tissues of 16‐mo‐old B6 mice using ImageJ software (C). IHC for panCK and its quantification confirms high hepatic fibrogenesis in 16‐mo‐old dKO/B6 mice (D). Expression of Opn revealed by immunohistochemical staining (E). Original magnification, ×100; scale bars, 50 μm. F) quantitative RT‐PCR demonstrates increased expression of hepatic genes related to fibrosis processing in nontumor liver tissues of 16‐mo‐old dKO/B6 vs. Mdr2‐KO/B6 mice. Results are presented as fold changes relative to Mdr2‐KO mice. Means ± sd. *P < 0.05, **P < 0.01 in dKO vs. Mdr2‐KO mice; 4–6 mice for each experimental group (Student's t test).
In order to reveal molecular mechanisms associated to enhanced HCC development in B6 dKO mice, we have compared expression of multiple genes in the liver of B6 dKO vs. Mdr2‐KO young and old males by quantitative RT‐PCR (Supplemental Table S2) and Nanostring assay (Supplemental Table S3). These genes include cytokines, chemokines, and immune regulators, including known regulators of the protumorigenic microenvironment and markers of liver progenitor cells (Epcam, Prom1, Cd24a, Trop2). All genes listed in Supplemental Tables S2 and S3 were not differentially expressed in dKO and Mdr2‐KO livers; their expression was similar irrespective of the genotype or undetectable.
Spp1 knockdown impairs proliferation of HepG2 cells in vitro
Human HCC cell lines tested, HuH7 and Hep3B, did not express Spp1, whereas hepatoblastoma HepG2 cells did (unpublished results). In order to examine the cell‐autonomous effect of Spp1 on liver tumor cell proliferation, we knocked down its expression with specific small interfering RNAs in HepG2 cells in vitro and followed cell proliferation using the xcCELLigence technology (Roche). In parallel, the same experiment was performed in 24‐cell plates for testing gene expression at 38 h post‐transfection. Spp1 knockdown resulted in a significant retardation of HepG2 cell proliferation during 38–63 h starting from the cell plating (Supplemental Fig. S8A). Gene expression analysis by quantitative RT‐PCR showed significant down‐regulation of SPP1, CCND1, and CCNG1, encoding cyclins D1 and G1, respectively (Supplemental Fig. S8B).
Expression of alternatively spliced Spp1 variants in the murine liver
The Spp1 gene expresses several alternatively spliced variants both in the murine and human tissues (Supplemental Fig. S9). In order to explore which Spp1 isoforms function in the Mdr2‐KO mice, we tested expression of these isoforms in the mouse liver at different stages of CLI and in liver tumors. There is only 60–70% similarity between mouse and human Spp1 genes and proteins. We detected the silent mutation CTG → CTA in the Leucine codon (is underlined in the Supplemental Fig. S9C). Using RT‐PCR followed by sequencing of the PCR product, we found that in the liver of B6 Mdr2‐KO mice, only variants Spp1 1–3 and 1–5 are expressed (Supplemental Fig. S10). Remarkably, 1–3 is the major Spp1 variant in murine liver, whose expression is at least 2 orders of magnitude higher than that of the 1–5 variant (Supplemental Fig. S10). At the age of 2 mo, expression of the Spp1‐3 variant was significantly higher in the B6 dKO vs. Mdr2‐KO mice (Supplemental Fig. S10A), in agreement with IHC data (Fig. 5D ). At the age of 16 mo, expression of the Spp1‐3 variant was significantly higher in the nontumor liver tissue from B6 dKO vs. Mdr2‐KO mice (Supplemental Fig. S10B). Expression of the Spp1‐3 in tumors was significantly lower than in nontumor liver tissue for both Mdr2‐KO and dKO B6 mice (Supplemental Fig. S10B). Nevertheless, the Spp1 transcript expression was detectable in tumors in contrast to Spp1 protein (for all but one of the analyzed tumors, as mentioned above).
DISCUSSION
Gal1, a prototype member of the galectin family widely expressed at sites of inflammation and tumor growth, can influence tumor progression through multiple mechanisms, including promotion of tumor inflammation, immunosuppression, angiogenesis, and metastasis (11, 26). The marked up‐regulation of Gal1 in the liver, its protumorigenic effects, and its broad anti‐inflammatory activity in a wide variety of pathologic settings prompted us to investigate the role of this glycan‐binding protein in CLI‐driven HCC development. Targeted inhibition of Lgals1 expression in a variety of tumors, including melanoma cells, Hodgkin lymphoma, lung adenocarcinoma, pancreatic adenocarcinoma, breast adenocarcinoma and glioblastoma, resulted in unleashed antitumor immunity, leading to inhibition of tumor growth and metastasis in syngeneic mice (26–30). From an immunologic standpoint, this lectin impairs CD8, Th1, and Th17; deactivates antigen‐presenting cells; inhibits NK cells; and promotes expansion of regulatory T cells (7, 29, 31–33). Recently, Rutkowski and colleagues (34) found that this lectin links tumor‐promoting inflammation, immunosuppression, and distant tumor growth. Although Gal1 is typically up‐regulated in cancer cells, in some tumor types, immune and/or stromal cells appear to be the main Gal1 source (26, 34, 35). Moreover, Gal1 promotes tumor angiogenesis (13–15) and augments Ras activation (12). Taking all these data into consideration, the results of our study demonstrating that Gal1 loss caused enhanced hepatocarcinogenesis in the Mdr2‐KO model are unexpected. The exacerbating effect of Gal1 loss on CLI‐mediated HCC development in this mouse model was more prominent in the B6 strain, in agreement with our previous finding that endogenous Gal1 efficiently protects murine liver from acute inflammatory insult in the B6 but not in the FVB genetic background (21). We suggest that enhanced HCC development in the Mdr2‐KO model could be explained by the highly increased CLI in the absence of the Gal1 protein. Exacerbated CLI resulted in more severe liver injury, fibrosis, and enhanced proliferation of hepatocytes and nonparenchymal cells (Figs. 2 and 3). The accelerated processes of liver tissue regeneration and remodeling could generate more tumor‐initiating cells (and at an earlier stage) in dKO compared with Mdr2‐KO mice. Thus, the protumorigenic effect associated with Gal1 loss dominated over the abovementioned antitumorigenic effects linked to Gal1 blockade at later stages of hepatocarcinogenesis. Importantly, the effect of Gal1 loss on hepatocarcinogenesis was prominent during a relatively short time period at the early cancerous stage (about 2‐mo time window at average), which was specific for each mouse strain and sex (e.g., see Supplemental Fig. S2E, F). At older ages, there was no statistically significant difference in liver tumor incidence and load between Gal1‐positive and Gal1‐negative mice of both strains (unpublished results). These results are consistent with the fact that Gal1 is just one of multiple factors dictating the outcome of CLI‐mediated hepatocarcinogenesis. Likewise, loss of Gal1 resulted in only a 1‐d retardation of liver regeneration (22).
Among possible molecular mechanisms that could potentially contribute to the enhanced HCC development in dKO mice, we found up‐regulation of Opn (encoded by the Spp1 gene) in the nontumor liver as one of the most significant factors. Interestingly, Opn was overexpressed in dKO compared with Mdr2‐KO liver (mainly in cholangiocytes) at all tested ages (Figs. 3F , 4B , 5C, D , and 8E and Supplemental Fig. S5B). Interestingly, there is still no evidence linking Gal1 and Opn expression. In a previous study exploring the role of Gal1 in liver regeneration (22), we found no differences in Opn expression between Gal1‐KO and wild‐type mice at 2 and 3 d posthepatectomy (unpublished results). Opn is a multifunctional protein, which occurs in both secreted and intracellular forms and interacts with the immune system and other cell types in the injured liver. Depending on the type of injury and the associated microenvironment, Opn may show opposing roles in liver repair and carcinogenesis (36). In the chronic liver injury accompanied by ductular reaction, Opn triggers a crosstalk between epithelial and nonparenchymal liver cells; it activates hepatic stellate cells, increases collagen deposition, and stimulates proliferation of hepatic progenitor cells while attenuating hepatocyte proliferation (37). Moreover, this protein also binds to LPSs, reducing TNF availability (38). At late stages of hepatocarcinogenesis, Opn expression is associated with accelerated tumor progression and metastasis and lower patient survival (39–41). However, the role of Opn at early stages of HCC development is still not clear, although it has been proposed to modulate the immunosuppressive activity of myeloid‐derived suppressor cells (42). Notably, 2 studies exploring the role of Opn in HCC development using the same model of Opn‐deficient mice showed opposite results (43, 44). Noncancerous hepatocytes of HCC patients did not express Opn (45). Furthermore, transgenic mice expressing Opn in hepatocytes developed autoimmune hepatitis at 24 wk of age (46). Among 240 HCCs, most of which were of high grade and stage, Opn mRNA was overexpressed in about 50% of tumors (39), whereas in the cBioPortal data set of 360 HCCs (47), it is overexpressed in 3.9% of HCC samples only (Supplemental Fig. S11A). However, in both cases, HCC patients with tumors over‐expressing Opn had significantly lower survival. In this regard, Gal1 mRNA was overexpressed in 5.8% of HCC samples, but this effect did not impact patient survival (Supplemental Fig. S11B). Expression of Opn by cancer cells control their proliferation, leading to addiction as demonstrated by our findings (Supplemental Fig. S8) and the literature (40, 41). We suggest that in the Mdr2‐KO HCC model, Opn is induced in response to CLI, and this effect is aggravated in the absence of Gal1. The effect of Opn on HCC development in Mdr2‐KO mice may be stage dependent (similar to that of Gal1), being tumor suppressive at an early stage and protumorigenic at later stages of CLI.
Similar to Gal1 and Opn, overexpression of the S100A4 protein in dKO liver could have different roles at different stages of CLI in mutant mice. On one hand, S100A4 may confer protection and show anti‐inflammatory activity in response to aberrations of lipid metabolism (48), but on the other hand, it may favor tumor progression and metastasis in different cancers (49), including HCC (50). Furthermore, overexpression of the Ntrk2 (TrkB) protein in hepatocytes of the young dKO mice (Fig. 6) may contribute to the exacerbation of hepatocarcinogenesis in the absence of Gal1. Signaling through the TrkB receptor tyrosine kinase elicits different processes essential for progression of epithelial cancers, including epithelial‐mesenchymal transition (51), angiogenesis (25), and metastasis (52).
The results of our study demonstrate that in the Mdr2‐KO mouse model of CLI‐mediated HCC, loss of Gal1 exacerbates hepatocarcinogenesis, especially in mice bearing the B6 genetic background. This effect is accompanied by increased CLI, fibrosis, and liver injury at early animal age, which in turn induces multiple protective mechanisms, including overexpression of Opn and S100A4 proteins. At the middle and late animal ages, these protective mechanisms become less effective, and some of them, such as Opn and S100A4, may trigger procarcinogenic activities, resulting in increased tumorigenesis (Fig. 9 ). Hence, unlike other cancer types, our findings suggest precaution in the indication of anti‐Gal1 treatment in CLI‐mediated HCC.
Figure 9.
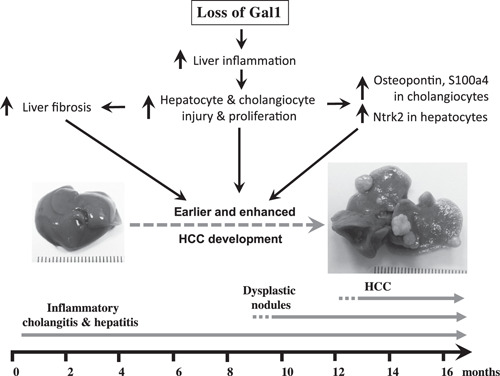
Schematic representation of the consequences of Gal1 loss in the course of CLI in Mdr2‐KO mice. Loss of Gal1 exacerbates CLI, fibrosis, and liver injury starting from the early animal age. This, in turn, induces overexpression of proteins Ntrk2 (in hepatocytes at early age), S100A4 (in cholangiocytes at the middle age), and Opn (in cholangiocytes at all ages tested). Altogether, these activities culminate in earlier and enhanced tumorigenesis.
AUTHOR CONTRIBUTIONS
T. Potikha performed the main part of the experimental work, discussions, and manuscript writing; O. Pappo conducted data analysis (pathology); L. Mizrahi performed data acquisition (part of RT‐PCR data); D. Olam provided assistance with mice and liver sample processing; S. M. Mailer performed data acquisition (Gal1 immunostaining); G. A. Rabinovich provided data analysis, discussions, manuscript writing, and editing; E. Galun performed study design and discussions and manuscript editing; and D. S. Goldenberg completed the study idea and design, data analysis, manuscript writing, and discussions.
Supporting information
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
ACKNOWLEDGMENTS
The authors thank J. P. Cerliani [Argentinean National Research Council (CONICET)] for advice, and Marion Richardson (Hadassah‐Hebrew University Medical Center) for assistance in manuscript preparation. T.P. and D.S.G. were supported by the Kamea Scientific Foundation of the Israeli Government. Work in G.A.R.'s laboratory was supported by grants from Agencia Nacional de Promoción Científica y Tecnológica (PICT 2014‐3087), Fundación Sales, Fundación Bunge & Born and the Kenneth Rainin Foundation. The work of E.G. was supported by U.S. National Institutes of Health, National Cancer Institute Grant CA197081‐02, the Ministry Of Science and Technology (MOST), the Israeli Science Foundation (ISF) collaboration with Canada (2473/2017), the personal ISF (486/2017), the Israeli Centers Of Research Excellence (ICORE)–ISF (41/2011), and by the Kron, Raskin, and Benson foundations. This work was also supported by the Deutsche Forschungsgemeinschaft, Bonn, Germany (SFB841, Project C3). The authors declare no conflicts of interest.
Potikha, T. , Pappo, O. , Mizrahi, L. , Olam, D. , Mailer, S. M. , Rabinovich, G. A. , Galun, E. , Goldenberg, D. S. Lack of galectin‐1 exacerbates chronic hepatitis, liver fibrosis, and carcinogenesis in murine hepatocellular carcinoma model. FASEB J. 33, 7995–8007 (2019). www.fasebj.org
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1. Balkwill, F. R. , and Mantovani, A. (2012) Cancer-related inflammation: common themes and therapeutic opportunities. Semin. Cancer Biol. 22, 33–40 [DOI] [PubMed] [Google Scholar]
- 2. Mauad, T. H. , van Nieuwkerk, C. M. , Dingemans, K. P. , Smit, J. J. , Schinkel, A. H. , Notenboom, R. G. , van den Bergh Weerman, M. A. , Verkruisen, R. P. , Groen, A. K. , Oude Elferink, R. P. J. , van der Valk, M. A. , Borst, P. , and Offerhaus, G. J. A. (1994) Mice with homozygous disruption of the mdr2 P-glycoprotein gene. A novel animal model for studies of nonsuppurative inflammatory cholangitis and hepatocarcinogenesis. Am. J. Pathol. 145, 1237–1245 [PMC free article] [PubMed] [Google Scholar]
- 3. Katzenellenbogen, M. , Pappo, O. , Barash, H. , Klopstock, N. , Mizrahi, L. , Olam, D. , Jacob-Hirsch, J. , Amariglio, N. , Rechavi, G. , Mitchell, L. A. , Kohen, R. , Domany, E. , Galun, E. , and Goldenberg, D. (2006) Multiple adaptive mechanisms to chronic liver disease revealed at early stages of liver carcinogenesis in the Mdr2-knockout mice. Cancer Res. 66, 4001–4010 [DOI] [PubMed] [Google Scholar]
- 4. Sundblad, V. , Morosi, L. G. , Geffner, J. R. , and Rabinovich, G. A. (2017) Galectin-1: a Jack-of-all-trades in the resolution of acute and chronic inflammation. J. Immunol. 199, 3721–3730 [DOI] [PubMed] [Google Scholar]
- 5. Camby, I. , Le Mercier, M. , Lefranc, F. , and Kiss, R. (2006) Galectin-1: a small protein with major functions. Glycobiobgy 16, 137R–157R [DOI] [PubMed] [Google Scholar]
- 6. Stillman, B. N. , Hsu, D. K. , Pang, M. , Brewer, C. F. , Johnson, P. , Liu, F. T. , and Baum, L. G. (2006) Galectin-3 and galectin-1 bind distinct cell surface glycoprotein receptors to induce T cell death. J. Immunol. 176, 778–789 [DOI] [PubMed] [Google Scholar]
- 7. Toscano, M. A. , Bianco, G. A. , Ilarregui, J. M. , Croci, D. O. , Correale, J. , Hernandez, J. D. , Zwirner, N. W. , Poirier, F. , Riley, E. M. , Baum, L. G. , and Rabinovich, G. A. (2007) Differential glycosylation of TH1, TH2 and TH-17 effector cells selectively regulates susceptibility to cell death. Nat. Immunol. 8, 825–834 [DOI] [PubMed] [Google Scholar]
- 8. Garín, M. I. , Chu, C. C. , Golshayan, D. , Cernuda-Morollón, E. , Wait, R. , and Lechler, R. I. (2007) Galectin-1: a key effector of regulation mediated by CD4+CD25+ T cells. Blood 109, 2058–2065 [DOI] [PubMed] [Google Scholar]
- 9. Alhabbab, R. , Blair, P. , Smyth, L. A. , Ratnasothy, K. , Peng, Q. , Moreau, A. , Lechler, R. , Elgueta, R. , and Lombardi, G. (2018) Galectin-1 is required for the regulatory function of B cells. Sci. Rep. 8, 2725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stannard, K. A. , Collins, P. M. , Ito, K. , Sullivan, E. M. , Scott, S. A. , Gabutero, E. , Darren Grice, I. , Low, P. , Nilsson, U. J. , Leffler, H. , Blanchard, H. , and Ralph, S. J. (2010) Galectin inhibitory disaccharides promote tumour immunity in a breast cancer model. Cancer Lett. 299, 95–110 [DOI] [PubMed] [Google Scholar]
- 11. Rabinovich, G. A. , and Conejo-García, J. R. (2016) Shaping the immune landscape in cancer by galectin-driven regulatory pathways. J. Mol. Biol. 428, 3266–3281 [DOI] [PubMed] [Google Scholar]
- 12. Elad-Sfadia, G. , Haklai, R. , Bailan, E. , Gabius, H. J. , and Kloog, Y. (2002) Galectin-1 augments Ras activation and diverts Ras signals to Raf-1 at the expense of phosphoinositide 3-kinase. J. Biol. Chem. 277, 37169–37175 [DOI] [PubMed] [Google Scholar]
- 13. Thyssen, V. L. , Postel, R. , Brandwijk, R. J. , Dings, R. P. , Nesmelova, I. , Satijn, S. , Verhofstad, N. , Nakabeppu, Y. , Baum, L. G. , Bakkers, J. , Mayo, K. H. , Poirier, F. , and Griffioen, A. W. (2006) Galectin-1 is essential in tumor angiogenesis and is a target for antiangiogenesis therapy. Proc. Natl. Acad. Sci. USA 103, 15975–15980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Thijssen, V. L. , Barkan, B. , Shoji, H. , Aries, I. M. , Mathieu, V. , Deltour, L. , Hackeng, T. M. , Kiss, R. , Kloog, Y. , Poirier, F. , and Griffioen, A. W. (2010) Tumor cells secrete galectin-1 to enhance endothelial cell activity. Cancer Res. 70, 6216–6224 [DOI] [PubMed] [Google Scholar]
- 15. Croci, D. O. , Cerliani, J. P. , Dalotto-Moreno, T. , Méndez-Huergo, S. P. , Mascanfroni, I. D. , Dergan-Dylon, S. , Toscano, M. A. , Caramelo, J. J. , García-Vallejo, J. J. , Ouyang, J. , Mesri, E. A. , Junttila, M. R. , Bais, C. , Shipp, M. A. , Salatino, M. , and Rabinovich, G. A. (2014) Glycosylation-dependent lectin-receptor interactions preserve angiogenesis in anti-VEGF refractory tumors. Cell 156, 744–758 [DOI] [PubMed] [Google Scholar]
- 16. Espelt, M. V. , Croci, D. O. , Bacigalupo, M. L. , Carabias, P. , Manzi, M. , Elola, M. T. , Muñoz, M. C. , Dominici, F. P. , Wolfenstein-Todel, C. , Rabinovich, G. A. , and Troncoso, M. F. (2011) Novel roles of galectin-1 in hepatocellular carcinoma cell adhesion, polarization, and in vivo tumor growth. Hepatology 53, 2097–2106 [DOI] [PubMed] [Google Scholar]
- 17. Kondoh, N. , Hada, A. , Ryo, A. , Shuda, M. , Arai, M. , Matsubara, O. , Kimura, F. , Wakatsuki, T. , and Yamamoto, M. (2003) Activation of Galectin-1 gene in human hepatocellular carcinoma involves methylation-sensitive complex formations at the transcriptional upstream and downstream elements. Int. J. Oncol. 23, 1575–1583 [PubMed] [Google Scholar]
- 18. Spano, D. , Russo, R. , Di Maso, V. , Rosso, N. , Terracciano, L. M. , Roncalli, M. , Tornillo, L. , Capasso, M. , Tiribelli, C. , and Iolascon, A. (2010) Galectin-1 and its involvement in hepatocellular carcinoma aggressiveness. Mol. Med. 16, 102–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Katzenellenbogen, M. , Mizrahi, L. , Pappo, O. , Klopstock, N. , Olam, D. , Jacob-Hirsch, J. , Amariglio, N. , Rechavi, G. , Domany, E. , Galun, E. , and Goldenberg, D. (2007) Molecular mechanisms of liver carcinogenesis in the mdr2-knockout mice. Mol. Cancer Res. 5, 1159–1170 [DOI] [PubMed] [Google Scholar]
- 20. Scott, K. , and Weinberg, C. (2002) Galectin-1 : a bifunctional regulator of cellular proliferation. Glycoconj. J. 19, 467–477 [DOI] [PubMed] [Google Scholar]
- 21. Potikha, T. , Stoyanov, E. , Pappo, O. , Frolov, A. , Mizrahi, L. , Olam, D. , Shnitzer-Perlman, T. , Weiss, I. , Barashi, N. , Peled, A. , Sass, G. , Tiegs, G. , Poirier, F. , Rabinovich, G. A. , Galun, E. , and Goldenberg, D. (2013) Inters train differences in chronic hepatitis and tumor development in a murine model of inflammation-mediated hepatocarcinogenesis. Hepatology 58, 192–204 [DOI] [PubMed] [Google Scholar]
- 22. Potikha, T. , Ella, E. , Cerliani, J. P. , Mizrahi, L. , Pappo, O. , Rabinovich, G. A. , Galun, E. , and Goldenberg, D. S. (2016) Galectin-1 is essential for efficient liver regeneration following hepatectomy. Oncotarget 7, 31738–31754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Roderfeld, M. , Rath, T. , Voswinckel, R. , Dierkes, C. , Dietrich, H. , Zahner, D. , Graf, J. , and Roeb, E. (2010) Bone marrow transplantation demonstrates medullar origin of CD34+ fibrocytes and ameliorates hepatic fibrosis in Abcb4-/- mice. Hepatology 51, 267–276 [DOI] [PubMed] [Google Scholar]
- 24. Cimmino, F. , Schulte J. H., Zoilo, M. , Koster, J. , Versteeg, R. , Iolascon, A. , Eggert, A. , and Schramm, A. (2009) Galectin-1 is a major effector of TrkB-mediated neuroblastoma aggressiveness. Oncogene 28, 2015–2023 [DOI] [PubMed] [Google Scholar]
- 25. Lam, C. T. , Yang, Z. F. , Lau, C. K. , Tam, K. H. , Fan, S. T. , and Poon, R. T. (2011) Brain-derived neurotrophic factor promotes tumorigenesis via induction of neovascularization: implication in hepatocellular carcinoma. Clin. Cancer Res. 17, 3123–3133 [DOI] [PubMed] [Google Scholar]
- 26. Orozco, C. A. , Martinez-Bosch, N. , Guerrero, P. E. , Vinaixa, J. , Dalotto-Moreno, T. , Iglesias, M. , Moreno, M. , Djurec, M. , Poirier, F. , Gabius, H. J. , Fernandez-Zapico, M. E. , Hwang, R. F. , Guerra, C. , Rabinovich, G. A. , and Navarro, P. (2018) Targeting galectin-1 inhibits pancreatic cancer progression by modulating tumor-stroma crosstalk. Proc. Natl. Acad. Sci. USA 115, E3769–E3778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rubinstein, N. , Alvarez, M. , Zwirner, N. W. , Toscano, M. A. , Ilarregui, J. M. , Bravo, A. , Mordoh, J. , Fainboim, L. , Podhajcer, O. L. , and Rabinovich, G. A. (2004) Targeted inhibition of galectin-1 gene expression in tumor cells results in heightened T cell-mediated rejection; A potential mechanism of tumor-immune privilege. Cancer Cell 5, 241–251 [DOI] [PubMed] [Google Scholar]
- 28. Juszczynski, P. , Ouyang, J. , Monti, S. , Rodig, S. J. , Takeyama, K. , Abramson, J. , Chen, W. , Kutok, J. L. , Rabinovich, G. A. , and Shipp, M. A. (2007) The AP1-dependent secretion of galectin-1 by Reed Sternberg cells fosters immune privilege in classical Hodgkin lymphoma. Proc. Natl. Acad. Sci. USA 104, 13134–13139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dalotto-Moreno, T. , Croci, D. O. , Cerliani, J. P. , Martinez-Allo, V. C. , Dergan-Dylon, S. , Méndez-Huergo, S. P. , Stupirski, J. C. , Mazal, D. , Osinaga, E. , Toscano, M. A. , Sundblad, V. , Rabinovich, G. A. , and Salatino, M. (2013) Targeting galectin-1 overcomes breast cancer-associated immunosuppression and prevents metastatic disease. Cancer Res. 73, 1107–1117 [DOI] [PubMed] [Google Scholar]
- 30. Van Woensel, M. , Mathivet, T. , Wauthoz, N. , Rosière, R. , Garg, A. D. , Agostinis, P. , Mathieu, V. , Kiss, R. , Lefranc, F. , Boon, L. , Belmans, J. , Van Gool, S. W. , Gerhardt, H. , Amighi, K. , and De Vleeschouwer, S. (2017) Sensitization of glioblastoma tumor micro-environment to chemo- and immunotherapy by Galectin-1 intranasal knock-down strategy. Sci. Rep. 7, 1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ilarregui, J. M. , Croci, D. O. , Bianco, G. A. , Toscano, M. A. , Salatino, M. , Vermeulen, M. E. , Geffner J. R., and Rabinovich, G. A. (2009) Tolerogenic signals delivered by dendritic cells to T cells through a galectin-1-driven immunoregulatory circuit involving interleukin 27 and interleukin 10. Nat. Immunol. 10, 981–991 [DOI] [PubMed] [Google Scholar]
- 32. Baker, G. J. , Chockley, P. , Zamler, D. , Castro, M. G. , and Lowenstein, P. R. (2016) Natural killer cells require monocytic Gr-1(+)/CD11b(+) myeloid cells to eradicate orthotopically engrafted glioma cells. OncoImmunology 5, e1163461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tesone, A. J. , Rutkowski, M. R. , Brencicova, E. , Svoronos, N. , Perales-Puchalt, A. , Stephen, T. L. , Allegrezza, M. J. , Payne, K. K. , Nguyen, J. M. , Wickramasinghe, J. , Tchou, J. , Borowsky, M. E. , Rabinovich, G. A. , Kossenkov, A. V. , and Conejo-Garcia, J. R. (2016) Satb1 overexpression drives tumor-promoting activities in cancer-associated dendritic cells. Cell Rep. 14, 1774–1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rutkowski, M. R. , Stephen, T. L. , Svoronos, N. , Allegrezza, M. J. , Tesone, A. J. , Perales-Puchalt, A. , Brencicova, E. , Escovar-Fadul, X. , Nguyen, J. M. , Cadungog, M. G. , Zhang, R. , Salatino, M. , Tchou, J. , Rabinovich, G. A. , and Conejo-Garcia, J. R. (2015) Microbially driven TLR5-dependent signaling governs distal malignant progression through tumor-promoting inflammation. Cancer Cell 27, 27–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Croci, D. O. , Morande, P. E. , Dergan-Dylon, S. , Borge, M. , Toscano, M. A. , Stupirski, J. C. , Bezares, R. F. , Avalos, J. S. , Narbaitz, M. , Gamberale, R. , Rabinovich, G. A. , and Giordano, M. (2013) Nurse-like cells control the activity of chronic lymphocytic leukemia B cells via galectin-1. Leukemia 27, 1413–1416 [DOI] [PubMed] [Google Scholar]
- 36. Wen, Y. , Jeong, S. , Xia, Q. , and Kong, X. (2016) Role of osteopontin in liver diseases. Int. J. Biol. Sci. 12, 1121–1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Strazzabosco, M. , Fabris, L. , and Albano, E. (2014) Osteopontin: a new player in regulating hepatic ductular reaction and hepatic progenitor cell responses during chronic liver injury. Gut 63, 1693–1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ge, X. , Leung, T. M. , Arriazu, E. , Lu, Y. , Urtasun, R. , Christensen, B. , Fiel, M. I. , Mochida, S. , Sorensen, E. S. , and Nieto, N. (2014) Osteopontin binding to lipopolysaccharide lowers tumor necrosis factor-α and prevents early alcohol-induced liver injury in mice. Hepatology 59, 1600–1616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pan, H. W. , Ou, Y. H. , Peng, S. Y. , Liu, S. H. , Lai, P. L. , Lee, P. H. , Sheu, J. C. , Chen, C. L. , and Hsu, H. C. (2003) Overexpression of osteopontin is associated with intrahepatic metastasis, early recurrence, and poorer prognosis of surgically resected hepatocellular carcinoma. Cancer 98, 119–127 [DOI] [PubMed] [Google Scholar]
- 40. Zhao, J. , Dong, L. , Lu, B. , Wu, G. , Xu, D. , Chen, J. , Li, K. , Tong, X. , Dai, J. , Yao, S. , Wu, M. , and Guo, Y. (2008) Down-regulation of osteopontin suppresses growth and metastasis of hepatocellular carcinoma via induction of apoptosis. Gastroenterology 135, 956–968 [DOI] [PubMed] [Google Scholar]
- 41. Sun, B. S. , Dong, Q. Z. , Ye, Q. H. , Sun, H. J. , Jia, H. L. , Zhu, X. Q. , Liu, D. Y. , Chen, J. , Xue, Q. , Zhou, H. J. , Ren, N. , and Qin, L. X. (2008) Lentiviral-mediated miRNA against osteopontin suppresses tumor growth and metastasis of human hepatocellular carcinoma. Hepatology 48, 1834–1842 [DOI] [PubMed] [Google Scholar]
- 42. Chiodoni, C. , Sangaletti, S. , Tripodo, C. , and Colombo, M. P. (2015) The ins and outs of osteopontin. OncoImmunology 4, e978711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fan, X. , He, C. , Jing, W. , Zhou, X. , Chen, R. , Cao, L. , Zhu, M. , Jia, R. , Wang, H. , Guo, Y. , and Zhao, J. (2015) Intracellular Osteopontin inhibits toll-like receptor signaling and impedes liver carcinogenesis. Cancer Res. 75, 86–97 [DOI] [PubMed] [Google Scholar]
- 44. Lee, S. H. , Park, J. W. , Woo, S. H. , Go, D. M. , Kwon, H. J. , Jang, J. J. , and Kim, D. Y. (2016) Suppression of osteopontin inhibits chemically induced hepatic carcinogenesis by induction of apoptosis in mice. Oncotarget 7, 87219–87231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gotoh, M. , Sakamoto, M. , Kanetaka, K. , Chuuma, M. , and Hirohashi, S. (2002) Overexpression of osteopontin in hepatocellular carcinoma. Pathol. Int. 52, 19–24 [DOI] [PubMed] [Google Scholar]
- 46. Mochida, S. , Yoshimoto, T. , Mimura, S. , Inao, M. , Matsui, A. , Ohno, A. , Koh, H. , Saitoh, E. , Nagoshi, S. , and Fujiwara, K. (2004) Transgenic mice expressing osteopontin in hepatocytes as a model of autoimmune hepatitis. Biochem. Biophys. Res. Commun. 317, 114–120 [DOI] [PubMed] [Google Scholar]
- 47. Gao, J. , Aksoy, B. A. , Dogrusoz, U. , Dresdner, G. , Gross, B. , Sumer, S. O. , Sun, Y. , Jacobsen, A. , Sinha, R. , Larsson, E. , Cerami, E. , Sander, C. , and Schultz, N. (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 6, pl1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hou, S. , Jiao, Y. , Yuan, Q. , Zhai, J. , Tian, T. , Sun, K. , Chen, Z. , Wu, Z. , and Zhang, J. (2018) S100A4 protects mice from high-fat diet-induced obesity and inflammation. Lab. Invest. 98, 1025–1038 [DOI] [PubMed] [Google Scholar]
- 49. Fei, F. , Qu, J. , Zhang, M. , Li, Y. , and Zhang, S. (2017) S100A4 in cancer progression and metastasis: a systematic review. Oncotarget 8, 73219–73239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yan, X. L. , Jia, Y. L. , Chen, L. , Zeng, Q. , Zhou, J. N. , Fu, C. J. , Chen, H. X. , Yuan, H. F. , Li, Z. W. , Shi, L. , Xu, Y. C. , Wang, J. X. , Zhang, X. M. , He, L. J. , Zhai, C. , Yue, W. , and Pei, X. T. (2013) Hepatocellular carcinoma-associated mesenchymal stem cells promote hepatocarcinoma progression: role of the S100A4-miR155-SOCS1-MMP9 axis. Hepatology 57, 2274–2286 [DOI] [PubMed] [Google Scholar]
- 51. Bao, W. , Qiu, H. , Yang, T. , Luo, X. , Zhang, H. , and Wan, X. (2013) Upregulation of TrkB promotes epithelial-mesenchymal transition and anoikis resistance in endometrial carcinoma. PLoS One 8, e70616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kim, M. S. , Lee, W. S. , Jeong, J. , Kim, S. J. , and Jin, W. (2015) Induction of metastatic potential by TrkB via activation of IL6/JAK2/STAT3 and PI3K/AKT signaling in breast cancer. Oncotarget 6, 40158–40171 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material