

Summary
Despite a much higher proportion of intragenic heterochromatin‐containing genes in crop genomes, the importance of intragenic heterochromatin in crop development remains unclear. Intragenic heterochromatin can be recognised by a protein complex, ASI1–AIPP1–EDM2 (AAE) complex, to regulate alternative polyadenylation.
Here, we investigated the impact of rice ASI1 on global poly(A) site usage through poly(A) sequencing and ASI1‐dependent regulation on rice development.
We found that OsASI1 is essential for rice pollen development and flowering. OsASI1 dysfunction has an important impact on global poly(A) site usage, which is closely related to heterochromatin marks. Intriguingly, OsASI1 interacts with the intronic heterochromatin of OsXRNL, a nuclear XRN family exonuclease gene involved in the processing of an miRNA precursor, to promote the processing of full‐length OsXRNL and regulate miRNA abundance. We found that OsASI1‐mediated regulation of pollen development partially depends on OsXRNL. Finally, we characterised the rice AAE complex and its involvement in alternative polyadenylation and pollen development.
Our findings help to elucidate an epigenetic mechanism governing miRNA abundance and rice development, and provide a valuable resource for studying the epigenetic mechanisms of many important processes in crops.
Keywords: alternative polyadenylation, epigenetics, heterochromatin, microRNA, rice development
Introduction
As conserved regulatory mechanisms of gene expression in eukaryotes, epigenetic mechanisms play essential roles in multiple biological processes in higher plants, ranging from basic development, allelic variation and heterosis to environmental stress responses and immunity (Varshney et al., 2005; Schnable & Springer, 2013; Quint et al., 2016; Chang et al., 2020). Despite extensive investigations in the model plant Arabidopsis, epigenetic mechanism‐based regulation of agronomic traits in crop plants remains largely elusive. Heterochromatin is a tightly packed form of chromatin. Although heterochromatin only represents a lower proportion of chromatin compared with lightly packed euchromatin in eukaryotic genomes, its importance has been increasingly articulated. Repressive epigenetic marks, including methylated DNA cytosine, histone H3 lysine 9 di‐methylation (H3K9me2) and H3 lysine 27 monomethylation (H3K27me1) are required for the formation of heterochromatin (Liu et al., 2010; Duan et al., 2018; Zhang et al., 2018). In addition to the distribution in constitutively silenced centromeres and telomeres, repressive heterochromatin elements within gene bodies are also observed in eukaryotic genomes, particularly in crops (To et al., 2015; J. Zhang et al., 2020), and the insertions of transposable elements (TEs) are one of the major causes of these genic heterochromatin. The impacts of heterochromatin on gene expression depend on its genetic context. For example, a recent study has reported that RNA‐directed DNA methylation (RdDM)‐mediated CHH (where H represents A, T or C) methylation in the miniature inverted‐repeat TEs (MITEs) of OsMIR156d/OsMIR156j promoter regions, represses the expression of these two miRNA genes, thereby negatively regulating rice tillering (Xu et al., 2020). By contrast, the D14 gene, which is a suppressor of rice tillering, is activated by CHH methylation at a MITE site in its downstream region (Xu et al., 2020). Compared with the extensively documented transcriptional repression effect of promoter heterochromatic elements to downstream genes due to the inaccessibility of RNA polymerase to DNA, the role of genic heterochromatin in gene expression and basic biological processes in plants, particularly in crop plants considering the high proportion of genic heterochromatin (To et al., 2015), has long been elusive. This mystery has been partially solved by some recent advances. A study of cash crop oil palm revealed a key role of intronic DNA methylation in the generation of somaclonal variation epialleles (Ong‐Abdullah et al., 2015; Paszkowski, 2015). In this case, loss of DNA methylation and RdDM‐related small RNAs for the Karma transposon, which is inserted in the intron of the homeotic gene DEFICIENS (DEF1), led to ectopic splicing and premature termination of the DEF1 gene, thereby contributing to the origin of mantled oil palm (Ong‐Abdullah et al., 2015).
In the last few years, several groups have reported that some chromatin regulators participate in the polyadenylation regulation of intronic heterochromatin‐containing gene, and are required for processing of full‐length transcripts. These factors include ANTI‐SILENCING 1 (ASI1) (Wang et al., 2013), also named INCREASE IN BONSAI METHYLATION 2 (IBM2) (Saze et al., 2013) and SHORT GROWTH 1 (SG1) (Coustham et al., 2014), and EDM2 (Tsuchiya & Eulgem, 2013; Lei et al., 2014). A recent study further revealed that ASI1 and EDM2 associate together through ASI1‐IMMUNOPRECIPITATED PROTEIN 1 (AIPP1), also named EMD3 (Lai et al., 2019), to form a protein complex (the AAE complex) and function in the same genetic pathway (Duan et al., 2017b). In this mechanism, the AAE complex interacts with intronic heterochromatin to promote distal polyadenylation. The proportion of intronic heterochromatin‐containing genes is very low in Arabidopsis. Considering the high proportion of genic heterochromatin‐containing genes in crop plants (To et al., 2015), it can be expected that the AAE‐mediated alternative polyadenylation pathway would play a more important role in crop plants. This notion is supported by a more recent study in which OsIBM2, the rice orthologue of Arabidopsis IBM2 protein, was required for the proper expression of intronic heterochromatin‐containing genes (Espinas et al., 2020). However, both the regulation of the OsASI1/IBM2 pathway on global poly(A) site usage and its contribution to specific biological processes in crops still remain unclear.
In this study, we investigated the effect of OsASI1 dysfunction on global poly(A) site usage and its biological functions in rice. We found that OsASI1 was indispensable for pollen development and flowering. OsASI1 dysfunction led to severe developmental defects including dramatically reduced fertility and late flowering. Strand‐specific sequencing of poly(A) site usage revealed that knockout of OsASI1 had an important impact on poly(A) site usage of a large subset of genes, especially heterochromatic element‐marked genes. Alternative polyadenylation (APA) events occurred not only in the coding regions but also in 3′ untranslated regions (3′UTR). We further revealed that an OsASI1‐mediated APA mechanism participated in the regulation of miRNA abundance through modulation of the polyadenylation of an XRN exonuclease‐encoding gene, OsXRNL, which was partially responsible for OsASI1‐mediated developmental regulation. We finally demonstrated that OsASI1 acted in a protein complex to regulate rice development. Our results uncovered an epigenetic mechanism in terms of the regulation of miRNA abundance and rice development, and provide a valuable resource for the study of epigenetic mechanisms of many important processes in crops.
Materials and Methods
Plant materials and growth conditions
All rice materials used in this study were on the Japonica rice (Oryza sativa) variety Nipponbare background. For phenotype observation and statistical analysis of agronomic traits, the plants were cultivated under local growing conditions in Shanghai, China (30°N, 121°E) during the normal rice‐growing season from mid‐May to mid‐October. For the plant materials used for molecular experiments and high‐throughput sequencing, plants were grown in chambers under conditions of a photoperiod of 14 h : 10 h, light : dark, 30°C : 25°C.
Pollen fertility analysis
Mature pollen grains were stained with a 2% (w/v) iodine–potassium iodide (I2–KI) solution and photographed using a Leica DM6000B microscope.
mRNA‐seq and gene expression analysis
For mRNA‐seq, total RNAs were purified from 2‐wk‐old seedlings and 60‐d plants (including inflorescence tissues) after germination using the RNeasy Plant Mini Kit (Qiagen, 74904). Library construction and Illumina sequencing were performed by the Beijing Genome Institute (Wuhan, China). Oligo(dT)‐attached magnetic beads were used to purify mRNA. Sequencing reads were mapped to the rice genome (RGAP7.0) using the Tophat program. Clean reads were mapped to the assembled unique genes by Bowtie2 (v.2.2.5) and the expression levels of genes were calculated using rsem (v.1.2.8) and normalised to fragments per kilobase of transcript per million mapped reads (FPKM).
For gene expression analysis, total RNAs were isolated with TRIzol reagent (Sangon Biotech, Shanghai, China), and first‐strand cDNA was reverse transcribed from 5 μg of total RNA using the One‐Step gDNA Removal and cDNA Synthesis SuperMix Kit (Transgen, Beijing, China). RT‐qPCR analysis was performed using the PerfectStart Green qPCR SuperMix (Transgen). Primers for qRT–PCR are listed in Supporting Information Table S1.
PAT‐seq assay and poly(A) site usage analysis
Total RNAs were extracted from 2‐wk‐old seedlings using the RNeasy Plant Mini Kit (74904; Qiagen). PAT‐seq was conducted according to a previous report (Yu et al., 2019), and sequenced on an Illumina HiSeq 2500 single‐end mode, at the core facility of the College of Environment and Ecology, Xiamen University. For PAT‐seq data processing, raw reads were pretreated as previously described (Yu et al., 2019). Poly(A) site usage (PSU) was calculated by reads of one poly(A) site divided by reads of the corresponding gene. ΔPSU represents the difference for PSU between the mutant and wild‐type. The deAPA events were identified with thresholds of absolute ΔPSU ≥ 0.05 and DEseq2 Padj < 0.05. Folded Cumulative Distribution Function in the mountainplot R package was used for folded cumulative curve plotting, which folded the top half of the cumulative curve over (Yu et al., 2019). The scale of the upslope was present on the left (from 0 to 0.5), the scale of downslope was present on the right (from 0.5 to 1.0). The top of the folded cumulative curve represents the median of the corresponding dataset. For proximal/distal poly(A) site switching analysis, the top two expressed poly(A) sites were retained for analysis. The 3′UTR length was calculated according to a previous publication (Lin et al., 2020), and padj < 0.05 was considered as significant 3′UTR lengthening or shortening events. Published histone modification data (PRJNA597065) was downloaded and mapped to the MSU7 genome using STAR alignment, and further deduplicated using the Picard tool. For ChIP‐seq density plotting, regions were divided into 2‐bp bins. The average read density in each bin was calculated for plotting. Genes containing transposable elements or retrotransposons were extracted according to MSU7 locus annotation for TE‐overlapping and TE‐excluding analyses.
Small RNA expression and sequencing analysis
Small RNA blotting was conducted according to a previous report with minor modifications (Duan et al., 2012). In brief, 50 μg of total RNAs were separated by 15% PAGE with 7 M urea, and blotted onto a Hybond N+ membrane (GE, Amersham) using a transblot semidry transfer cell (Bio‐Rad). Membranes were subjected to chemical crosslinking with EDC crosslinking buffer (0.16 M EDC, 0.13 M 1‐methylimidazole at pH 8.0) at 65°C for 90 min. Biotin‐labelled probes were added to the commercial PerfectHyb (Sigma) and incubated with the membranes at 42°C overnight. A Chemiluminescent Nucleic Acid Detection Module Kit (89880; Thermo Fisher, Waltham, MA, USA) was used to detect the hybridisation signals according to the instruction manual. For the quantitative PCR analysis of small RNAs, total RNAs from rice panicles before heading were reverse transcribed using TransScript® miRNA First‐Strand cDNA Synthesis SuperMix (Transgen). RT‐qPCR analysis was performed using the PerfectStart Green qPCR SuperMix (Transgen). Primers are listed in Table S1.
For small RNA sequencing, a library was prepared with 1 μg of total RNAs for each sample. Total RNAs were purified by electrophoretic separation on a 15% urea denaturing polyacrylamide gel electrophoresis (PAGE) gel and the 18–30 nt small RNAs were ligated to adenylated 3′ adapters annealed to unique molecular identifiers (UMI), followed by the ligation of 5′ adapters. The adapter‐ligated small RNAs were subsequently transcribed into cDNA using Superscript II Reverse Transcriptase (Invitrogen) and then several rounds of PCR amplification were performed to enrich the cDNA fragments. The library was qualified and quantified by two methods: check the distribution of the fragments size using the Agilent 2100 bioanalyser, and quantify the library using real‐time quantitative PCR (qPCR) (TaqMan Probe). The final ligation PCR products were sequenced using the DNBSEQ platform (BGI‐Shenzhen, Shenzhen, China).
Nuclear run‐on assay
Nuclear run‐on assay was performed as previously reported (Y‐Z. Zhang et al., 2020). First, nuclei were isolated using lysis buffer. After two washes with resuspension buffer, nuclei were resuspended in storage buffer and then mixed with transcription buffer. After 15 min incubation at 30°C, TRIzol reagent was added to stop the reaction. The DNA‐free RNAs were isolated, and then inoculated with 2 μg anti‐BrdU antibody (Abcam) at room temperature for 10 min and precipitated with Dynabeads (Thermo Fisher). Then, blocking buffer was added and the mixture was incubated for 30 min at room temperature. Finally, RNA was extracted using TRIzol reagent (Thermo Fisher). cDNAs were synthesised using SuperScript IV Reverse Transcriptase (Thermo Fisher) and subjected to qPCR analysis.
IP‐MS and protein interaction analysis
An IP‐MS assay was performed according to a previous report (Duan et al., 2017a). In brief, total proteins were extracted from 3 g of rice leaves using lysis buffer (50 mM Tris HCl, pH 7.6, 150 mM NaCl, 5 mM MgCl2, 10% glycerol, 0.1% NP‐40, 0.5 mM dithiothreitol and 1% protease inhibitor cocktail) and precipitated for 3 h at 4°C with Dynabeads protein G (Invitrogen) conjugated to anti‐Myc (05‐724; Millipore) antibody. After sequential washing with lysis buffer and PBS buffer, the precipitated proteins were subjected to LC‐MS/MS analysis. For yeast‐two‐hybrid (Y2H) analysis, the coding sequences of tested proteins were inserted into pGBKT7 and pGADT7 vectors (Clontech). Recombinant plasmids were cotransformed into yeast strain AH109. Transformants were grown on SD−Leu−Trp dropout medium and selected on SD−Leu−Trp−His medium plates containing 3‐amino‐1,2,4‐triazole. All Y2H experiments were repeated three times independently.
Bimolecular fluorescence complementation (BiFC) and subcellular localisation assays in rice protoplasts
For BiFC assays, the coding sequences of tested proteins were inserted into PSAT6‐nYFP and PSAT6‐cYFP vectors. The NLS sequence was fused in‐frame with RFP into PA7‐35S‐RFP as a nuclear marker. For subcellular location, the coding sequences of OsASI1, OsAIPP1a, OsAIPP1b and OsEDM2 were inserted into PA7‐YFP. Rice protoplast preparation and PEG‐mediated transformation were performed as described previously (Chen et al., 2010). After incubation in the dark for 16–20 h, fluorescence was observed using a confocal laser scanning microscope (Leica, TCS SP8).
ChIP‐qPCR analysis
ChIP assay was performed as previously described (Zhao et al., 2020). Here, c. 3 g of 2‐wk‐old seedlings were crosslinked in PBS with 1% formaldehyde under vacuum. Chromatin was extracted and fragmented into 200–600 bp by sonication using a Bioruptor (Diagenode, Liege, Belgium), and immunoprecipitation was performed using the following antibodies: anti‐H3K9me2 (ab1220, Abcam) and anti‐Myc (05‐724; Millipore). After incubation with Dynabeads protein G, the antibody–bead complexes were incubated with precleared chromatin overnight at 4°C on a rotator to immunoprecipitate the target chromatin. After washing and reverse crosslinking, the precipitated and input DNAs were purified for qPCR analysis.
Results
OsASI1 is essential for rice fertility and flowering time control
In rice, OsASI1 was encoded by a single copy gene LOC_Os01g42460 (Fig. S1). To investigate its role in rice development, CRISPR/Cas9‐mediated gene editing of the OsASI1 gene was conducted and two mutant alleles were generated, osasi1‐1 and osasi1‐2, in which nucleic acid deletion of ‘A’ and ‘AA’ were observed, respectively; both caused a premature termination upstream of the BAH domain (Fig. S2). By searching alternative open reading frames (ORFs), we found that both osasi1‐1 and osasi1‐2 mutations may encode a truncated protein with an intact BAH domain but different sizes of N‐terminal regions (Fig. S2). OsASI1 dysfunction did not lead to obvious abnormalities in the development of rice pistil and stamen (Fig. S3). By contrast, we found that pollen development was greatly affected by osasi1 mutations (Fig. 1a). When stained with iodine–potassium iodide (I2–KI), the pollen grains of both osasi1 mutants appeared much lighter in colour compared with wild‐type (WT) pollen grains. Further analysis of the pollen defects indicated that a significantly greater proportion of aborted pollen grains was observed in osasi1 mutants compared with that in WT plants (Fig. 1b). The defective pollen rate of the osasi1 mutant was c. 90%, suggesting that osasi1‐1 is a more severe mutant allele than osasi1‐2. This result indicated that the development of mostly pollen grains was aborted due to mutations of the OsASI1 gene. In line with the higher defective pollen rate, seed‐setting rates of osasi1 mutants were dramatically reduced compared with that of WT plants (Fig. 1b). The higher defective pollen rate and lower seed‐setting rate observed in osasi1 mutants indicated that OsASI1 was indispensable for the proper regulation of rice fertility. In addition to regulating rice fertility, we found that OsASI1 also participated in the regulation of flowering time. osasi1 mutants showed a late flowering phenotype (Fig. 1a). The heading dates of osasi1 mutants were c. 15–22 d later than that of WT plants (Fig. 1b). To further clarify the role of OsASI1 in regulation of rice fertility and flowering time, OsASI1 overexpression plants, in which a four‐Myc tag was fused to the C‐terminus of OsASI1, were generated on the WT background under the control of the Cauliflower Mosaic Virus 35S promoter (from this point forwards named OsASI1ox). OsASI1ox plants exhibited similar normal pollen development and flowering time as those of WT plants (Figs 1, S4). The seed‐setting rate was as high as that of WT plants, indicating that OsASI1 overexpression did not have a severe effect on rice development.
Fig. 1.
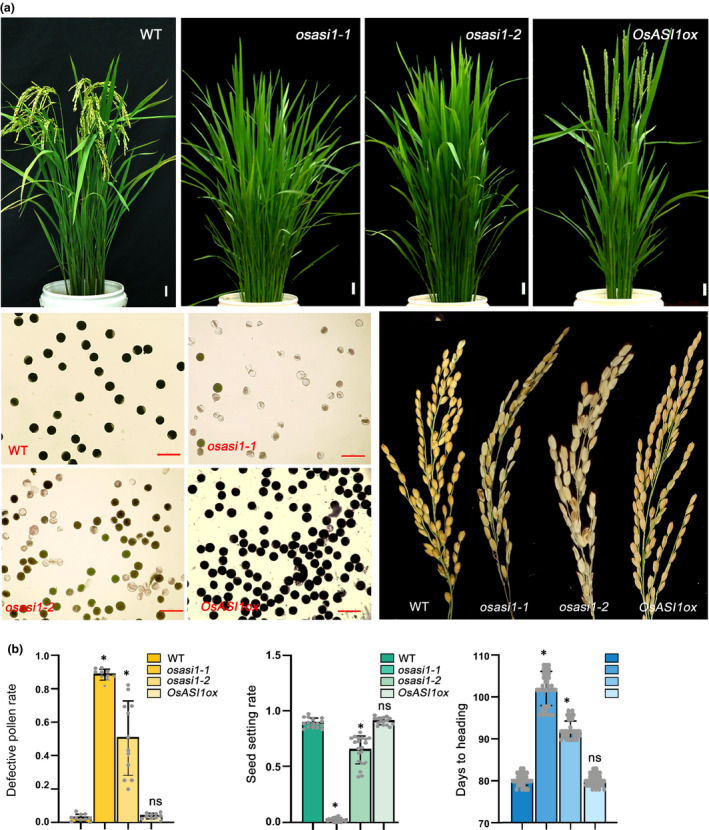
OsASI1 regulates rice fertility and flowering time. (a) Upper panel, the flowering time phenotype of osasi1 mutants and OsASI1‐overexpression plants. Photographs were taken at the heading time for wild‐type (WT) plants. Bars, 3 cm. Lower left panel, iodine–potassium iodide (I2–KI) staining results showing the effect of osasi1 mutations and OsASI1 overexpression on the development of rice pollen grains. Bars, 70 μm. Lower right panel, the morphological phenotype of rice spikes. (b) Defective pollen rates, seed‐setting rate and enumeration of days to heading of different genotypes. For the defective pollen rate, anthers from different plant parts were collected for microscope observation. Photographs of 15–30 samples were subjected to counting of normal and defective pollen. Each grey dot represents a counting event from one photograph (sample). Black horizontal lines represent the mean, and the error bars indicate ± SD from the number of counting events (n = 15–30). For seed‐setting rate, spikelets from different plant parts were collected to count the seed‐setting rate. Each grey dot represents a counting event from one spikelet. Black horizontal lines represent the mean, and the error bars indicate the ± SD from the number of counting events (n = 15–40). Unpaired two‐tailed Student’s t‐test was performed. *, P < 0.01. ns, no significance.
We found that OsASI1 dysfunction had a significant effect on global gene expression. mRNA‐seq analysis identified 1554 differentially expressed genes (DEGs; with a two‐fold cutoff) in the osasi1‐1 mutant compared with WT (Fig. S5a). These genes covered multiple biological processes (Figs S5b,c, S6). For example, the flowering time‐related gene HEADING DATE 3A (OsHd3a), a rice orthologue of the Arabidopsis florigen gene FLOWERING LOCUS T (FT), was downregulated in the osasi1‐1 mutant. Intriguingly, the RICE FLOWRING LOCUS T1 (OsRFT1) gene, which encodes the closest homologue of OsHd3a, also exhibited reduced expression in the osasi1‐1 mutant (Fig. S5c). Both OsHd3a and OsRFT1 have been shown to act as floral activators to promote flowering under short photoperiod conditions (Kojima et al., 2002; Komiya et al., 2008). Downregulation of OsHd3a and OsRFT1 was confirmed by quantitative reverse transcription PCR (RT‐qPCR) analysis (Fig. S5d). This result was consistent with the late flowering phenotype observed in the osasi1 mutants.
OsASI1 controls global poly(A) site usage and prefers to regulate alternative polyadenylation of intragenic heterochromatin‐containing genes
Similar to its Arabidopsis orthologue, OsASI1 resides in the nucleus (Fig. S7). Here, to investigate the effects of OsASI1 dysfunction on global poly(A) site usage, we performed strand‐specific poly(A) tag sequencing (PAT‐seq), which could accurately identify the poly(A) sites in a strand‐specific manner (Yu et al., 2019) in the WT and osasi1‐1 mutant plants, with two replicates. PAT‐seq analysis revealed that 657 poly(A) sites, residing in 573 genes, were differentially used (absolute ΔPSU ≥ 0.05, and padj < 0.05) in the osasi1‐1 mutant compared with WT plants (Fig. 2a). To explore the genome‐wide impact of OsASI1 dysfunction on the poly(A) usage pattern, the top two used poly(A) sites from each gene were retained for switching poly(A) site usage analysis. The results showed that >1000 poly(A) sites exhibited ectopic usage following the osasi1‐1 mutation, among which 2331 poly(A) sites switched to distal sites and 821 poly(A) sites switched to proximal sites (Fig. 2b). Intriguingly, we found that the knockout of OsASI1 resulted in 3′UTR lengthening of genes compared with WT plants (Fig. S8a,b). Overall, these results suggested that OsASI1 dysfunction affected the poly(A) site usage within both 3′UTR and coding regions.
Fig. 2.
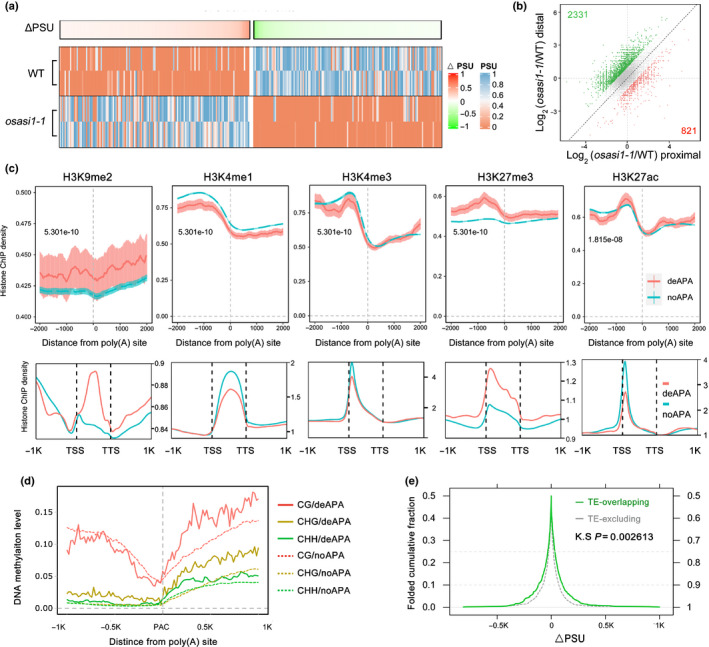
OsASI1 controls global poly(A) site usage and preferentially targets heterochromatin‐containing genes. (a) Knockout of OsASI1 results in alternative polyadenylation. Poly(A) site usage (PSU) of each deAPA site was plotted and clustered by ΔPSU. The colour key from green to red represents the ΔPSU scale between WT and osasi1‐1. The colour key from orange to blue indicates the PSU scale of samples. (b) Knockout of OsASI1 results in switching of poly(A) site usage. For each gene, the top two used poly(A) sites were retained for analysis. Green dots: PSU variance of the distal site is two‐fold the PSU variance of the proximal site. Red dots: PSU variance of the proximal site is two‐fold the PSU variance of the distal site. (c) Metaplots showing the distribution patterns of different histone marks around poly(A) sites (upper panel) and the bodies (lower panel) of deAPA genes. NoAPA genes serve as negative controls. (d) Plots showing DNA methylation levels in WT plants in the poly(A) sites and flanking regions of deAPA genes. NoAPA genes serve as negative controls. (e) Folded cumulative fraction plot showing the ΔPSU profiles of poly(A) sites located in TE‐overlapping/TE‐excluding genes.
To further uncover the molecular features of OsASI1 target genes, the chromatin states of the differentially alternative polyadenylated genes (deAPA) were analysed. Based on the public rice database (Zhao et al., 2020), we first examined the distribution patterns of different histone modifications, including H3K9me2, H3K4me1, H3K4me3, H3K27me3 and H3K27ac, around deAPA sites and on the bodies of deAPA genes. As shown in Fig. 2(c, upper panel), compared with noAPA genes, a higher density of H3K9me2 and H3K27me3 was observed in the poly(A) site and flanking region of deAPA genes, whereas H3K4me3 and H3K27ac displayed similar densities in the poly(A) sites of noAPA and deAPA genes. H3K4me1 density was slightly lower in deAPA genes compared with noAPA genes. Similar patterns were also observed in the bodies of the deAPA genes (Fig. 2c, lower panel). We next investigated the DNA methylation patterns of the deAPA genes using a published rice DNA methylome (Xu et al., 2020). Interestingly, compared with noAPA genes, the DNA cytosine methylation (5mC) level of deAPA genes in poly(A) site downstream regions was much higher in all cytosine contexts (Fig. 2d), supporting the notion that DNA methylation levels have an important influence on OsASI1‐mediated regulation of poly(A) site usage.
The high density of H3K9me2 and DNA methylation observed in OsASI1‐mediated deAPA genes suggested that OsASI1‐mediated polyadenylation regulation has a preference for those heterochromatin‐containing genes. Given that these heterochromatic marks were heavily enriched in TEs, we next compared the preference of OsASI1‐mediated polyadenylation regulation for TE‐containing and TE‐excluding genes. The results indicated that poly(A) site usage in TE‐containing genes was more variable compared with that of TE‐excluding genes in the osasi1‐1 mutant (Fig. 2e). Intriguingly, most of the 3′UTR lengthened genes did not contain or overlap with TE (Fig. S8c). Therefore, OsASI1‐mediated polyadenylation may experience different regulatory mechanisms in 3′UTR regions.
OsASI1‐mediated epigenetic regulation of polyadenylation participates in multiple biological processes by directly binding to genic heterochromatin
PAT‐seq identified hundreds of deAPA genes (Table S2). Gene Ontology (GO) analysis indicated that these deAPA genes were enriched in diverse molecular processes such as nucleotide binding, posttranslational protein modification and oxidation reduction, and participated in multiple biological processes ranging from basic development and metabolism to environmental responses (Fig. S9). For instance, LOC_Os03g37411, which encodes a multiantimicrobial extrusion family MATE efflux protein and is predicted to have antiporter activity, was a direct target of OsASI1‐mediated polyadenylation regulation. The LOC_Os03g37411 gene bears a large intron with TE insertions (Fig. 3a,b). DNA is hypermethylated in the intronic TE region, and both the public H3K9me2 database (Fig. 3b) and individual chromatin immunoprecipitation (ChIP)‐qPCR analysis (Fig. S10) indicated that repressive H3K9me2 marks were enriched in this region. PAT‐seq analysis revealed a distal poly(A) site, which corresponded to the full‐length transcript, and many proximal poly(A) sites within the largest intron (Fig. 3a). In the WT, the distal poly(A) site had the highest usage. Mutation of osasi1‐1 led to a dramatic increase in the usage of intronic proximal poly(A) site, whereas the distal poly(A) site signal was markedly reduced compared with WT (Fig. 3a). This pattern was confirmed by mRNA‐seq analysis, which clearly indicated a reduction in 3′‐end reads downstream of the largest intron and an obvious increase in 5′‐end reads in the osasi1‐1 mutant compared with WT (Fig. 3a). In line with the switch of poly(A) site usage, RT‐qPCR results showed that osasi1 mutations led to a significant reduction in full‐length transcripts (Long), but increased the accumulation of short transcripts (Short) (Fig. 3c). Similar ectopic usage of poly(A) sites was also observed in many deAPA genes such as the LOC_Os02g28074 gene encoding an XRN‐like exonuclease (Figs 3a–c, S10) and the LOC_Os01g46169 gene encoding a GDSL‐like lipase/acylhydrolase OsGELP21 (Fig. 3a) and others.
Fig. 3.
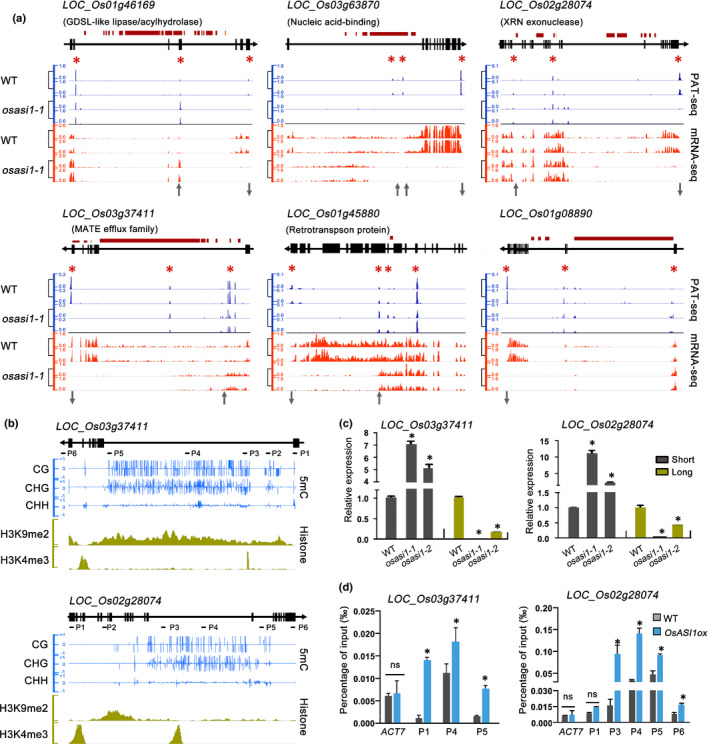
OsASI1 directly binds genic heterochromatin to regulate poly(A) site usage. (a) Integrative Genomics Viewer (IGV) snapshots of PAT‐seq and mRNA‐seq showing the distinct polyadenylation patterns and gene expression of representative deAPA genes in WT and the osasi1‐1 mutant. Two replicates were shown for each analysis. Black and red boxes represent exons and transposable and repetitive elements (TREs), respectively. The red asterisks represent major poly(A) sites identified by PAT‐seq. The grey upwards and downwards arrows represent upregulated and downregulated poly(A) site usage, respectively, in the osasi1‐1 mutant compared with WT. (b) IGV snapshots of DNA methylation and histone density showing the DNA methylation levels and H3K9me2/H3K4me3 distributions at two representative OsASI1 target genes. (c) RT‐qPCR results showing the ectopic expression of poly(A) site‐specific short and long transcripts of two selected target genes in WT and the osasi1‐1 mutant. Specific primer pairs (as labelled in (b)) were designed to determine the expression of different transcripts. Error bars represent the SD of three biological replicates. *, P < 0.01. (d) ChIP‐qPCR results showing the relative density of OsASI1 protein at different regions of two selected OsASI1 target genes in WT and OsASI1ox transgenic plants. The relative density of OsASI1 was normalised by the percentage relative to input. Error bars represent the SD of three biological replicates. Unpaired two‐tailed Student’s t‐test was performed. *, P < 0.01. ns, no significance.
To address the question of whether OsASI1 directly targeted the genic heterochromatin to regulate polyadenylation, OsASI1 binding at selected target genes was determined in WT and OsASI1ox transgenic plants via ChIP‐qPCR assay. The results indicated that OsASI1 had specific binding to the genic heterochromatin of the selected LOC_Os03g37411 and LOC_Os02g28074 genes (Fig. 3d). Combined with the above evidence, it is reasonable to conclude that OsASI1‐mediated polyadenylation is an important mechanism in the epigenetic regulation of multiple biological processes in rice.
OsASI1 regulates miRNA abundance by modulating alternative polyadenylation of an XRN‐like exonuclease gene
As mentioned above, an XRN‐like exonuclease‐encoding gene LOC_Os02g28074 (from this point forwards named OsXRNL), was subjected to OsASI1‐mediated APA regulation (Figs 3, S9). XRN family proteins, the homologues of yeast and human Rat1/Xrn2 proteins, are highly conserved 5′→3′ exonucleases in eukaryotes (Kurihara, 2017). It has been extensively documented that XRN family exonucleases function in multiple processes depending on their subcellular localisation. Cytoplasmic XRN4 in Arabidopsis is involved in multiple decay pathways including the degradation of 3′ intermediates of miRNA cleavage and the turnover of miRNA* strands (Souret et al., 2004; Nagarajan et al., 2019; Liu et al., 2020). The nuclear exonucleases XRN2 and XRN3 in Arabidopsis have been proven to function in transcription termination of RNA polymerase II and act as RNA silencing suppressors via multiple mechanisms (Gy et al., 2007; Kurihara et al., 2012; Krzyszton et al., 2018; You et al., 2019). Depletion of XRN2 can lead to bias miRNA strand loading into Ago, which in turn affects miRNA biogenesis in C. elegans (Chatterjee et al., 2011). In the rice genome, at least four XRN domain‐containing proteins were encoded (Fig. 4a). A fluorescence localisation assay in rice protoplast cells indicated that the OsXRNL protein mainly resided in the nucleus (Fig. S11a). The nuclear localisation prompts us to investigate whether OsXRNL dysfunction had certain impacts on miRNA abundance, such as what the Arabidopsis AtXRN2 and AtXRN3 proteins do. To this end, two mutant alleles were generated using CRISPR/Cas9‐mediated editing within the first exon (Fig. S11b). Small RNA sequencing was performed in WT plants, osasi1‐1 and osxrnl‐1 mutants using 60‐d‐old plants. Analysis of the length distribution of mapped small RNAs indicated that the distribution patterns of osxrnl‐1 and osasi1‐1 mutants were similar to that of WT plants, with 24‐nt and 21‐nt small RNAs accounting for the similar highest proportion (Fig. S12). Importantly, analysis of the fold changes for all detected miRNAs indicated that average miRNA levels were obviously reduced in the osxrnl‐1 mutant compared with WT plants (Fig. 4b). Intriguingly, a moderate reduction of miRNA levels was also observed in the osasi1‐1 mutant (Fig. 4b).
Fig. 4.
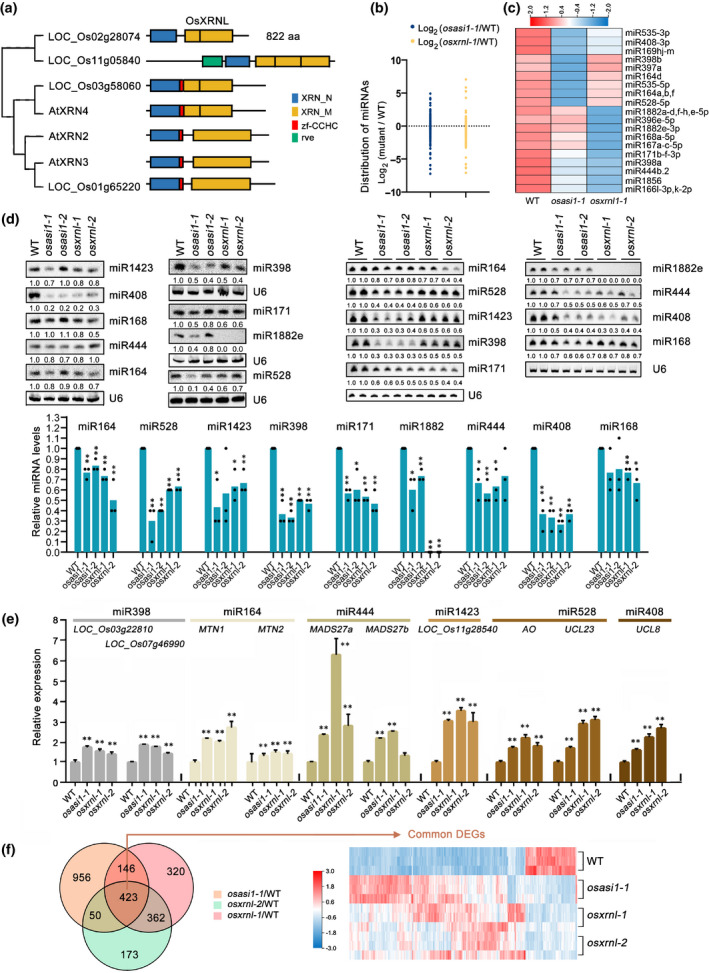
OsASI1 regulates miRNA biogenesis by modulating alternative polyadenylation of an XRN‐like exonuclease gene. (a) Phylogenetic analysis of the XRN family proteins in Arabidopsis and rice. Domain structures are shown. aa, amino acid. (b) Distribution of the fold changes for all detected miRNAs in osasi1‐1 and osxrnl‐1 mutants compared with WT plants. The fragments per kilobase of exon model per million (FPKM) mapped fragments of all miRNA abundance were calculated. Each circle represents one miRNA. (c) Heatmap of small RNA sequencing showing the expression pattern of selected miRNAs in different genotypes. (d) Upper panel: small RNA blotting analysis showing the accumulation of selected miRNAs in osasi1 and osxrnl mutants. Results of three biological replicates are shown. U6 serves as an RNA loading control. Lower panel: column diagrams showing the relative miRNA levels estimated from band signals (also indicated at the bottom of each lane), with miRNA levels in WT plants set to 1.0. Unpaired two‐tailed Student’s t‐test was performed. *, P < 0.05; **, P < 0.01. (e) RT‐qPCR results showing the increased expression of target mRNAs of selected miRNAs in osasi1 and osxrnl mutants compared with that of WT plants. Error bars represent the ± SD of three biological replicates. **, P < 0.01. (f) Left panel, Venn diagram showing the overlap of DEGs between osasi1‐1, osxrnl‐1 and osxrnl‐2 mutants. Right panel, heatmap of mRNA‐seq showing the expression pattern of common DEGs between osasi1‐1, osxrnl‐1 and osxrnl‐2 mutants and WT plants, each with three replicates.
In line with this trend, we found that some annotated miRNAs were downregulated in both osxrnl‐1 and osasi1‐1 mutants (Fig. 4c; Table S3). To confirm this pattern, accumulation of representative miRNAs was measured using a small RNA blotting assay with three biological replicates (Fig. 4d). The results indicated that most of the selected miRNAs were significantly downregulated in two osxrnl mutant alleles compared with WT. Importantly, accumulation of these miRNAs in the osasi1‐1 mutant was also reduced compared with that in WT plants (Fig. 4d). The downregulation of miRNA abundance was consistent with the significant reduction of OsXRNL‐L transcripts in the osasi1 mutant (Fig. 3).
To further verify the downregulation of these miRNAs, the relative expression of selected miRNA target genes was measured by RT‐qPCR using both 60‐d‐old plants. In line with the reduction of miRNA abundance, most of the selected mRNAs were significantly increased in the osxrnl and osasi1 mutants compared with WT plants (Fig. 4e). Therefore, these results suggested that OsXRNL plays an important role in the regulation of miRNA abundance and OsASI1 influences miRNA abundance through modulation of poly(A) site usage of the OsXRNL gene.
In Arabidopsis, nuclear XRNs have been shown to modulate miRNA abundance through distinct mechanisms. AtXRN3 and AtXRN2 are involved in catalysing the elimination of the MIRNA loop and the 3′ remnant (Gy et al., 2007; Kurihara et al., 2012). Overaccumulation of MIRNA loops were observed in xrn3 and xrn2/xrn3 mutants. To investigate whether OsXRNL utilised similar mechanism to modulate miRNA abundance, we compared the relative expression of pri‐miRNA and stem‐loop. The RT‐qPCR results indicated that, contrary to the reduction in mature miR1882e and miR398 levels, accumulation of stem‐loops of MIR1882e and MIR398 were significantly increased in osxrnl and osasi1 mutants, whereas the pri‐miRNA levels were not significantly changed (Fig. S13), demonstrating an involvement of OsXRNL in the elimination of MIRNA stem‐loops.
OsASI1 dysfunction partially phenocopies osxrnl mutants in terms of gene expression and developmental defects
It is well known that miRNAs have an important impact on gene expression and play essential roles in plant development and responses to environmental stimuli (Jones‐Rhoades et al., 2006; Chen, 2009; Cui et al., 2020). The ectopic accumulation of miRNAs in osxrnl and osasi1 mutants inspired us to compare the phenotypes of these two mutants in terms of global gene expression and developmental defects. To this end, mRNA‐seq analysis was performed in the osxrnl‐1 and osxrnl‐2 mutants using 60‐d‐old plants with three biological replicates. The data indicated that there were 1251 and 1008 genes in the osxrnl‐1 and osxrnl‐2 mutants, respectively, showing more than two‐fold expression changes compared with WT plants, and 785 genes were commonly regulated between two mutant alleles (Fig. 4f). Intriguingly, among the 785 common DEGs, 423 genes (53.88%) were also differentially expressed in the osasi1‐1 mutant (Fig. 4f), and these common target genes displayed a similar expression pattern in osasi1‐1, osxrnl‐1, and osxrnl‐2 mutants compared with WT plants (Fig. 4f), suggesting that a substantial number of genes were commonly regulated by OsASI1 and OsXRNL.
The partial phenocopy between the osasi1 and osxrnl mutants in terms of miRNA abundance and gene expression inspired us to propose a hypothesis that OsASI1‐mediated regulation of rice development may partially depend on OsXRNL‐mediated modulation of miRNA abundance. In support of this hypothesis, two miRNAs, miR528 and miR408, have been shown to participate in the regulation of multiple processes, including pollen development. OsmiR528 plays vital roles in male fertility (Y‐C. Zhang et al., 2020), antiviral defence (Yao et al., 2019; Yang et al., 2020), abiotic stress responses (Tang & Thompson, 2019; Zhu et al., 2020) and flowering time control (Yang et al., 2019). It has been shown that miR528 affects pollen development by posttranscriptional silencing of the uclacyanin gene OsUCL23 (Y‐C. Zhang et al., 2020). The mir528 knockout mutant showed aborted pollen development at the late binucleate pollen stage, leading to a significant reduction in the seed‐setting rate (Y‐C. Zhang et al., 2020). Similar to miR528, OsmiR408 is involved in the regulation of pollen development and grain yield by posttranscriptional silencing of another uclacyanin gene OsUCL8 (Zhang et al., 2017; Yang et al., 2018). Consistent with our hypothesis, we found that both miR528 and miR408 were downregulated in osasi1 and osxrnl mutants (Fig. 4c,d). Considering the severe developmental defects of pollen in osasi1 mutants, we also examined by RT‐qPCR analysis whether miRNAs were differently expressed in osasi1 and osxrnl mutants in inflorescence tissues. The data indicated that all the detected miRNAs, including miR528 and miR408, were significantly reduced in the inflorescence tissues of the osasi1‐1, osxrnl‐1 and osxrnl‐2 mutants compared with WT plants (Fig. S14), and the expression of OsUCL23 and OsUCL8, the target genes of miR528 and miR408, respectively, were significantly increased in inflorescence tissues (Fig. 4e).
In line with the ectopic expression of miRNAs and target genes, two xrnl mutants displayed similar developmental defects with osasi1 mutants, including delayed heading time, significantly increased defective pollen rate as well as reduced seed‐setting rate compared with WT plants (Fig. 5a,b). Therefore, the observations that osxrnl mutations partially phenocopy the osasi1 mutants in terms of developmental phenotypes and global gene expression strongly supported our hypothesis that OsASI1‐mediated regulation of rice development partially depends on OsXRNL‐mediated modulation of miRNA abundance.
Fig. 5.
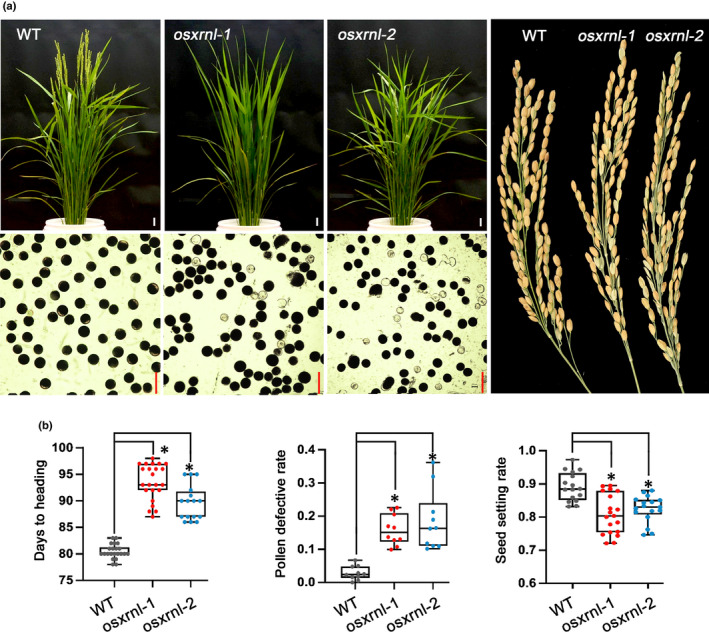
OsXRNL dysfunction partially phenocopies osasi1 mutants in developmental defects. (a) Developmental phenotype analysis of osxrnl mutants showing late heading time and reduced fertility. Left upper panel, late flowering phenotype of osxrnl mutants. Seedling photographs were taken at the heading time of WT plants, Bars, 3 cm. Left lower panel, iodine–potassium iodide (I2–KI) staining showing the developmental defects of pollen grains. Bars, 50 μm. Right panel, the morphological phenotype of rice spikes. (b) Statistical analysis of the days to heading (left panel), defective pollen rate (middle panel) and seed‐setting rate (right panel) of osxrnl mutants. The data points are shown as dots. Error bars represent the ± SD (n = 15–30). Unpaired two‐tailed Student’s t‐test was performed. *, P < 0.01.
OsASI1 regulates rice development partially through modulation of OsXRNL RNA processing
To further test our hypothesis, we then investigated whether the proper expression of OsXRNL was essential for OsASI1‐mediated developmental regulation. For this purpose, two different OsXRNL constructs were introduced into the osasi1‐1 mutant under the control of the native promoter to generate transgenic plants. One construct contained the full‐length coding sequence (from this point forwards referred to as XRNLcds), and the other one contained the full‐length coding sequence plus the 14th and 15th introns, representing a heterochromatic element‐enriched region that was bound by OsASI1 (from this point forwards referred to as XRNLint) (Fig. 6a). Both OsXRNL constructs contained a Flag‐fused epitope at the C‐terminus. According to our hypothesis, the intact OsXRNL protein would accumulate much more in the XRNLcds transgene than that in the XRNLint transgene because the absence of intronic heterochromatin will protect XRNLcds from proximal polyadenylation regulation caused by the osasi1‐1 mutation, thereby leading to successful processing of the full‐length XRNL‐L transcript. To test our hypothesis, transcriptional rate was first determined in two transgenic plants using nuclear run‐on assay. The results indicated that these two transgenes had a comparable transcriptional rate (Fig. 6b, upper panel). Next, the protein levels were examined via western blotting assay. As expected, the transgenic OsXRNL‐Flag protein accumulated normally in most of the XRNLcds transgene lines, but exhibited much lower levels in all of the XRNLint transgene lines (Fig. 6b, lower panel). Consistent with the distinct accumulation of transgenic OsXRNL‐Flag protein, the XRNLcds transgene partially rescued the developmental defect of osasi1‐1 mutant in fertility (Fig. 6c–e). The defective pollen rates of two randomly selected XRNLcds transgene lines were c. 35% and 45%, which were significantly lower than that of the osasi1‐1 mutant, but still higher than WT plants (Fig. 6d). In addition, the heading time of the XRNLcds transgene was slightly earlier than the osasi1‐1 mutant (Fig. S15). Instead, no obvious developmental defects, including fertility and heading time, were rescued in all of the XRNLint transgene plants (Figs 6c–e, S15). These findings, combined with the partial phenocopy in terms of developmental defects and gene expression, prompted us to conclude that OsASI1 regulates rice development partially through modulation of OsXRNL RNA processing.
Fig. 6.
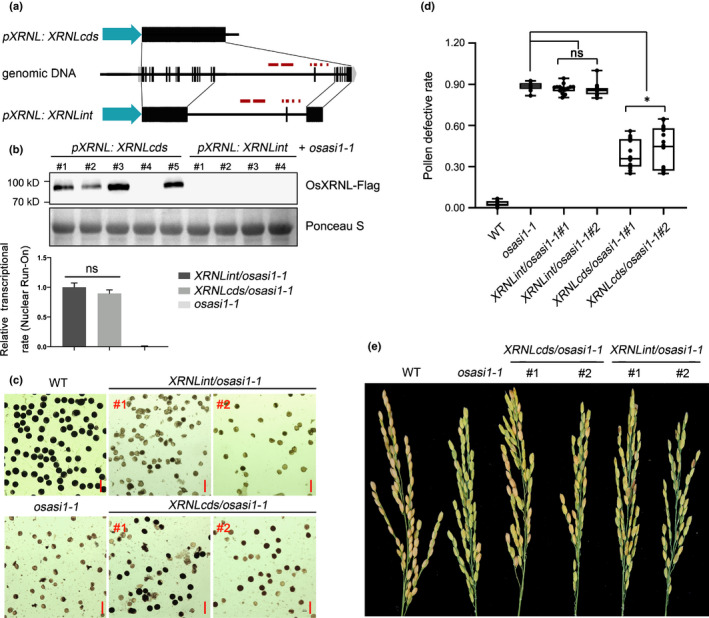
Expression of the OsXRNL coding sequence partially rescues the developmental defects of the osasi1‐1 mutant. (a) Diagram showing the structures of two transgenic constructs. Black and grey boxes represent coding sequence and untranslated regions, respectively. The red boxes represent transposable elements (TREs). (b) Upper panel, western blotting result showing the different accumulation levels of OsXRNL‐Flag protein in randomly selected lines of two transgenic plants. Ponceau S staining serves as a protein loading control. Lower panel, nuclear run‐on data showing the relative transcriptional rate of OsXRNLint and OsXRNLcds transgenes. Error bars represent the ± SD of three biological replicates. osasi1‐1 mutant serves as the nontransgene control. Unpaired two‐tailed Student’s t‐test was performed. ns, no significance. (c) Iodine–potassium iodide (I2–KI) staining showing the developmental defects of pollen grains of different genotypes. Bar, 50 µm. (d) Analysis of the defective pollen rate of different genotypes. Data points are shown as black dots. Error bars represent the ± SD (n = 10–20). Unpaired two‐tailed Student’s t‐test was performed. *, P < 0.01. ns, no significance. (e) Photograph showing the seed setting of different genotypes.
OsASI1 acts in a protein complex to regulate polyadenylation and rice development
Recently, AtASI1, a homologue protein of rice OsASI1, has been shown to associate with two other proteins, a PHD protein, EDM2, and an RRM‐containing protein, AIPP1, to form a protein complex that functions in polyadenylation regulation (Duan et al., 2017b; Zhang et al., 2021). We next asked whether there was a protein complex in OsASI1‐mediated regulation of polyadenylation and development in rice. To answer this question, OsASI1 immunoprecipitation assays coupled with mass spectrometry analysis (IP‐MS) were conducted using OsASI1ox transgenic rice. The results indicated that one EDM2‐like protein (from this point forwards named OsEDM2), which shared the same domain constituents with AtEDM2, was copurified with OsASI1 (Fig. 7a,b). In addition, two RRM proteins (from this point forwards named OsAIPP1a and OsAIPP1b), which shared high sequence similarity with AtAIPP1 (Fig. S16), were also pulled down by OsASI1 (Fig. 7a). Both OsEDM2 and the OsAIPP1a/b proteins localised to the nucleus (Fig. S7). To investigate the interaction relationship between these proteins, Y2H and BiFC assays were performed. We found that both OsAIPP1a and OsAIPP1b could directly interact with OsASI1 and OsEDM2, and no direct interaction of OsASI1–OsEDM2 was observed (Fig. 7c,d), suggesting that OsASI1 associated with OsEDM2 through bridge proteins OsAIPP1a or OsAIPP1b to form a protein complex.
Fig. 7.
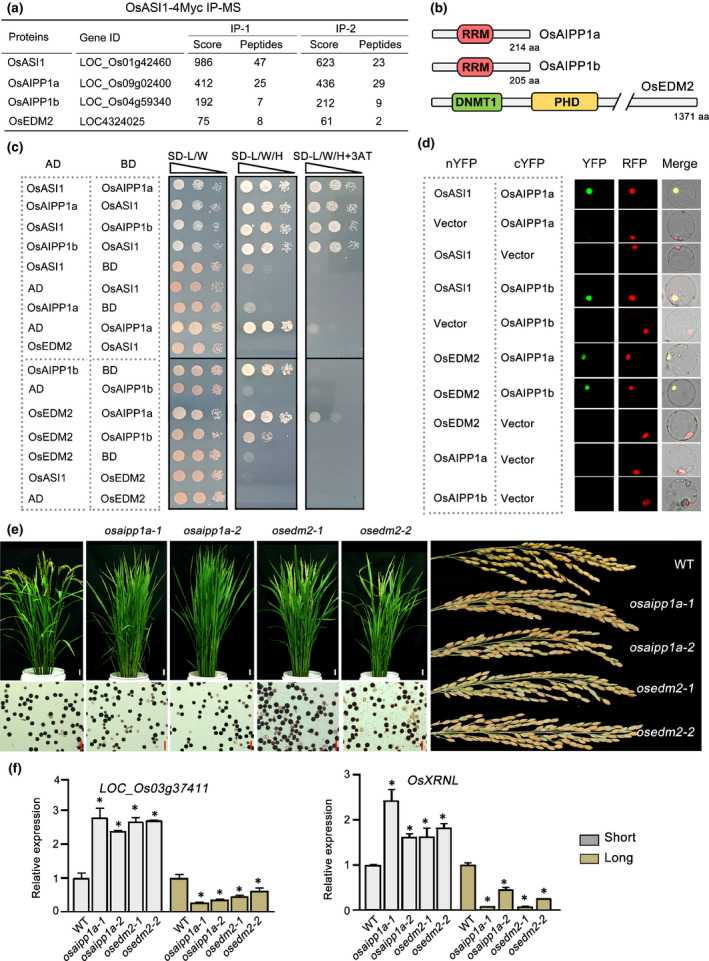
OsASI1 acts in a protein complex to regulate polyadenylation and rice development. (a) Mass spectrometry analysis showing the copurification of OsASIPP1a, OsAIPP1b and OsEDM2 with transgenic OsASI1‐4 Myc protein in vivo. (b) Domain structures of OsAIPP1a, OsAIPP1b and OsEDM2. (c) Y2H assay showing the protein interaction between OsASI1 and the copurified proteins. (d) BiFC assay was performed in rice protoplast cells. The tested proteins were fused with YFP under the control of the CaMV 35S promoter. RFP, a known nucleus localisation sequence was fused with RFP to indicate the nucleus. (e) Developmental defects of osedm2 and osaipp1a mutants. Upper left panel, the flowering time phenotype. Photograph was taken at the heading time of WT plants. Bars, 3 cm. Lower left panel, iodine–potassium iodide (I2–KI) staining showing the development of pollen grains of the selected genotypes. Right panel, seed setting phenotype of different genotypes. Bars, 50 µm. (f) RT‐qPCR results showing the ectopic expression of poly(A) site‐specific short and long transcripts of two selected target genes in different genotypes. Error bars represent the SD of three biological replicates. Unpaired two‐tailed Student’s t‐test was performed. *, P < 0.01.
We next asked whether knocking out OsEDM2 and OsAIPP1s could cause similar developmental defects as those observed in the osasi1 mutants. To this end, two mutant alleles were generated for each gene based on CRISPR/Cas9‐mediated editing (Fig. S2). Unfortunately, we failed to obtain a knockout mutant of the OsAIPP1b gene. Both the osedm2 and osaipp1a mutants exhibited obvious developmental defects in fertility (Fig. 7e). The defective pollen rates were obviously higher, and seed‐setting rates were much lower, in mutants compared with WT (Fig. S17). Moreover, enumeration of the days to heading indicated that osedm2 and osaipp1a mutants also displayed late flowering phenotypes (Figs 7e, S17). The late flowering phenotype of osaipp1a mutants was slightly weaker than that of osedm2 mutants (Fig. S18). We speculate that this may be due to potential functional redundancy between OsAIPP1a and OsAIPP1b. To investigate whether OsEDM2 and OsAIPP1a possess similar functions with OsASI1 in polyadenylation regulation, the expression patterns of representative OsASI1 target genes, LOC_Os03g37411 and OsXRNL, were examined. RT‐qPCR results indicated that the levels of full‐length transcripts of these two genes were significantly reduced in osedm2 and osaipp1a mutants. Instead, short transcript levels were obviously increased compared with WT (Fig. 7f). This result suggested that OsEDM2 and OsAIPP1a dysfunctions led to a similar APA phenotype with the osasi1 mutants at the selected genes. Therefore, combined with the above evidence, we concluded that OsASI1 associates with OsEDM2 and OsAIPP1a/OsAIPP1b to form a protein complex and regulates alternative polyadenylation and rice development.
Based on the above data, we propose a working model of OsASI1 complex‐mediated epigenetic regulation of miRNA abundance and rice development (Fig. 8). In this model, the OsASI1 complex recognises and binds to a genic heterochromatin region, which contains a high density of DNA methylation and H3K9me2 repressive marks, to promote distal polyadenylation, thereby facilitating the processing of full‐length transcripts. These genic heterochromatin‐containing genes participate in diverse biological processes. OsXRNL, one target gene of OsASI1 complex‐mediated polyadenylation regulation, is required for the proper accumulation of miRNAs via diverse mechanisms. Therefore, the OsASI1 complex regulates gene expression and associated biological processes not only through direct regulation of full‐length transcript processing but also through OsXRNL‐mediated modulation of miRNA abundance, thereby participating in miRNA‐related biological processes such as development and stress responses.
Fig. 8.
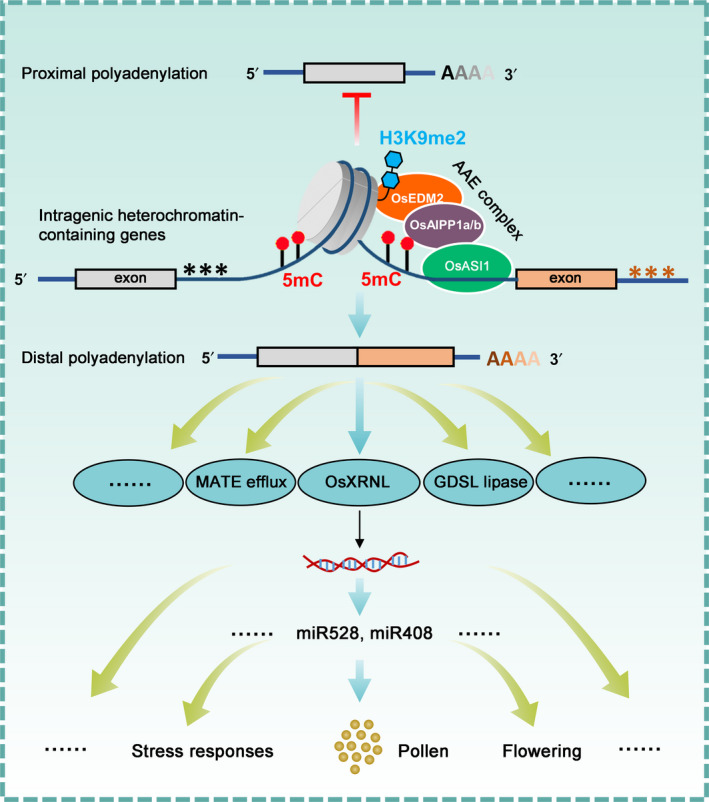
A working model of OsASI1 complex‐mediated regulation of rice development and miRNA abundance. Black and orange asterisks represent proximal and distal poly(A) sites, respectively. Grey and orange boxes represent exons.
Discussion
In this study, we reported a mechanism of miRNA abundance and rice development regulation. This mechanism was achieved through alternative polyadenylation regulation depending on the interplay between OsASI1 complex and genic heterochromatin. We found that OsASI1 complex‐mediated polyadenylation regulation was widely involved in important biological processes in rice, ranging from basic development to environmental responses.
In eukaryotic genomes, substantial amounts of genic heterochromatin come from TE insertion in genic regions. It has been estimated that 60% of TEs reside within introns, which comprise only 24% of the genome (To et al., 2015). Compared with the Arabidopsis genome, which is estimated to bear only 3% of intragenic TEs, crop plants have a higher proportion of intragenic TEs (West et al., 2014; Le et al., 2015). Therefore, it is reasonable to speculate that genic heterochromatin may have more important functions in crop plants.
In this study, we showed that OsASI1 targets genic heterochromatin to regulate poly(A) site usage. Consistent with the above speculation and a recent report that OsIBM2 is required for the expression of intronic heterochromatin‐containing genes (Espinas et al., 2020), a large subset of genes were differentially polyadenylated in the osasi1 mutant, and these deAPA genes were predicted to be involved in diverse vital processes ranging from development and metabolism to environmental responses, suggesting that the interplay between genic heterochromatin and OsASI1 is a vital mechanism in the regulation of fundamental processes in rice. In view of the diversity of deAPA genes caused by OsASI1 dysfunction, it can be expected that more biological processes are subjected to OsASI1‐mediated epigenetic regulation of polyadenylation than that those shown in this study.
As an important regulator of diverse biological processes, miRNA has been extensively explored. Recently, several studies have revealed the important roles of miRNAs in rice pollen development and fertility regulation (Zhang et al., 2017; Yang et al., 2018; Y‐C. Zhang et al., 2020). In this study, we showed that OsASI1 regulates miRNA abundance through polyadenylation regulation of an XRN family exonuclease OsXRNL. In this case, OsASI1 binds to the intronic heterochromatin of OsXRNL to inhibit the usage of proximal poly(A) site, thereby facilitating the production of full‐length functional transcripts. We found that the heterochromatic status was maintained in the inflorescence (Fig. S10b), and the ectopic expression of miRNAs, including pollen development‐related miR528 and miR408, was also present in the inflorescence tissues (Fig. S14), suggesting that the ASI1‐XRNL regulatory module is conserved in these tissues. In line with the reduced accumulation of miR528 and miR408 (Fig. 4d), the expression of their target genes OsUCL23 and OsUCL8, which have been shown to be required for pollen development, was significantly increased in osasi1 and osxrnl mutants (Fig. 4e). Therefore, we concluded that OsASI1 regulates pollen development partially through an OsASI1–OsXRNL–miRNA pathway. Our finding of the importance of OsASI1 in rice fertility is supported by a very recent report that loss‐of‐function mutant of OsASI1 (OsIBM2) is embryonic lethal and could not produce homozygous mutant seeds (Espinas et al., 2020). More importantly, we found that the OsAAE complex components had the highest expression at anther tissues (Fig. S17), further demonstrating that the OsAAE complex may play essential roles in rice fertility.
In addition to affecting rice development, it is reasonable to predict that OsASI1 also participates in more important processes than we showed in this study. On the one hand, the differentially accumulated miRNAs are involved in diverse regulation pathways. For example, in addition to fertility regulation, OsmiR528 also plays important roles in antiviral defence (Yang et al., 2020) and abiotic stress responses (Tang & Thompson, 2019; Zhu et al., 2020). Indeed, we found that the L‐ASCORBATE OXIDASE (AO) gene, which is a target gene of OsmiR528‐mediated posttranscriptional silencing and has been shown to play a key role in plant defence against Rice stripe virus (RSV) (Yao et al., 2019), was increased in osasi1 and osxrnl mutants. This result suggests that OsASI1 and OsXRNL may have a role in rice antiviral defence. On the other hand, OsASI1 controls RNA processing of a large subsets of genes that participate in distinct processes (Fig. S9). These genes cover from development to lipase metabolism and antimicrobe defence. Therefore, our data provide a valuable basis for studying the epigenetic mechanisms of these processes.
In Arabidopsis, the nucleus‐localised XRN2/3 have been shown to function redundantly in modulating miRNA abundance through diverse mechanisms (Gy et al., 2007; Kurihara et al., 2012; Krzyszton et al., 2018; You et al., 2019). For OsXRNL, we found that the processing of some MIRNA precursors was affected in osxrnl mutants. However, the following two possibilities cannot be ruled out: first is that other mechanisms may also contribute to the ectopic expression of miRNA in osxrnl mutants, and the second is that other XRN family proteins may share redundant functions with the OsXRNL protein in rice. Moreover, considering that some miRNAs are mis‐expressed in osasi1 and osxrnl mutants in both vegetative and inflorescence tissues (Figs 4d, S14), the developmental defects observed in osasi1 and osxrnl mutants may reflect a comprehensive effect of the ectopic accumulation of multiple miRNAs. Although OsHd3a and OsRFT1 were also mis‐expressed in osasi1 and osxrnl mutants in both 60‐d‐old plants and inflorescence tissues, the poly(A) site usage was not obviously affected by OsASI1 dysfunction (Fig. S19), suggesting that these two genes may not be the direct targets of OsASI1‐dependent alternative polyadenylation. Intriguingly, XRNLcds, but not the XRNLint transgene, partially rescued the late flowering phenotype of the osasi1 mutant (Fig. S15). This result implied that OsASI1 regulates OsHd3a and OsRFT1 through an APA–miRNA and OsXRNL–miRNA pathway‐independent mechanism. Of course, we cannot rule out another possibility that other unidentified flowering‐related genes are targeted by OsASI1‐mediated APA regulation. This need to be elucidated in the future studies.
In Arabidopsis, the counterpart of OsASI1 associates with the RRM protein AIPP1 and PHD domain‐containing protein EDM2 to form a protein complex and regulates polyadenylation at very limited loci. We found that the OsASI1 complex was evolutionarily conserved in rice, which encodes one OsASI1 (ASI1), two AIPP1 and one EDM2 (Duan et al., 2017a). These findings suggested that the polyadenylation regulation mediated by the interplay between OsASI1 complex and genic heterochromatin is a conserved mechanism in plant species and may play more important roles than previously anticipated. In addition, there are several questions that remain unsolved: are these genic heterochromatic elements elastically regulated with different tissues and developmental stages, or in different ecotypes? As in oil palm, the polymorphism of intragenic heterochromatin in the homologue genes of different ecotypes may contribute to the formation of epialleles. Another question is whether the proximally polyadenylated transcripts have a substantial function in plants or just only truncated nonfunctional transcripts? Moreover, we found 3′UTR APA events exhibited different patterns than the APA events within the intron, implying a distinct regulatory mechanism that needs to be elucidated in a future study.
In summary, we uncovered a unique regulatory mechanism of miRNA abundance and rice development through the interplay between chromatin regulators and intragenic heterochromatin. Our findings provide a valuable resource for the study of epigenetic mechanisms of important biological processes in crop plants and facilitate an understanding of the functions of intragenic heterochromatin, which is widely distributed in crop genomes.
Author contributions
C‐GD and QQL designed the experiments. L‐YY, JL, H‐WX, C‐XC, J‐YC, Jinshan Zhang, Jian Zhang, Y‐XL, CY and HZ conducted the experiments. JJ, J‐KZ, QQL and C‐GD analysed the data. C‐GD wrote the paper. L‐YY, JL and H‐WX contributed equally to this work.
Supporting information
Fig. S1 Phylogenetic analysis of OsASI1 protein.
Fig. S2 Mutation information of OsASI1, OsEDM2 and OsAIPP1a.
Fig. S3 Morphological phenotypes of pistil and stamen of osasi1 and osxrnl mutants.
Fig. S4 Expression level of OsASI1 protein in OsASI1ox plants.
Fig. S5 Effect of OsASI1 dysfunction on global gene expression.
Fig. S6 Enrichment analysis of osasil1‐1 DEGs.
Fig. S7 Subcellular localisation of OsASI1, OsEDM2, OsAIPP1a and OsAIPP1b proteins.
Fig. S8 Effect of OsASI1 dysfunction on the alternative polyadenylation of 3′UTR.
Fig. S9 Gene Ontology analysis of deAPA genes in osasi1‐1.
Fig. S10 ChIP‐qPCR analysis of H3K9me2 density at selected target genes of OsASI1.
Fig. S11 Subcellar localisation of OsXRNL protein and mutation information of OsXRNL gene.
Fig. S12 Length distribution of mapped small RNAs from WT, osasi1‐1 and osxrnl‐1.
Fig. S13 Relative expression of representative MIRNAs and stem‐loops.
Fig. S14 Relative accumulation of miRNAs in inflorescence tissues.
Fig. S15 Flowering time phenotype of different XRNL transgenic plants.
Fig. S16 Sequence alignment between Arabidopsis and rice AIPP1 proteins.
Fig. S17 Relative expression of OsASI1, OsAIPP1a, OsAIPP1b and OsEDM2 in different tissues.
Fig. S18 Statistics analysis of flowering time, defective pollen rate and seed‐setting rate of osedm2 and osaipp1a mutants.
Fig. S19 The poly(A) site usage and expression analysis of flowering time‐ and pollen development‐related genes in different tissues.
Table S1 List of primers used in this study.
Table S2 List of differentially expressed APA genes in osasi1‐1 mutant.
Table S3 Detailed list of downregulated miRNAs in osasi1 and osxrnl mutants.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Acknowledgements
The work of C‐GD was supported by the Chinese Academy of Sciences, China and by the Strategic Priority Research Programme of the Chinese Academy of Sciences (XDB27040203). This work was supported in part by a grant from the National Key R&D Project of China (2016YFE0108800 to QQL). We thank Haidong Qu and Xiuxiu Wang for technical assistance, Shuining Yin (CAS Center for Excellence in Molecular Plant Sciences) for confocal technology assistance, and Zuhua He’s group for providing the NLS‐RFP nuclear marker.
Contributor Information
Qingshun Q. Li, Email: liqq@xmu.edu.cn.
Cheng‐Guo Duan, Email: cgduan@psc.ac.cn.
Data availability
The sequencing datasets generated in this study, including PAT‐seq, mRNA‐seq and small RNA sequencing, have been submitted to the NCBI BioProject database (http://www.ncbi.nlm.nih.gov/bioproject/) with accession no. PRJNA673072.
References
- Chang YN, Zhu C, Jiang J, Zhang H, Zhu JK, Duan CG. 2020. Epigenetic regulation in plant abiotic stress responses. Journal of Integrative Plant Biology 62: 563–580. [DOI] [PubMed] [Google Scholar]
- Chatterjee S, Fasler M, Bussing I, Grosshans H. 2011. Target‐mediated protection of endogenous microRNAs in C. elegans . Developmental Cell 20: 388–396. [DOI] [PubMed] [Google Scholar]
- Chen L, Shiotani K, Togashi T, Miki D, Aoyama M, Wong HL, Kawasaki T, Shimamoto K. 2010. Analysis of the Rac/Rop small GTPase family in rice: expression, subcellular localization and role in disease resistance. Plant and Cell Physiology 51: 585–595. [DOI] [PubMed] [Google Scholar]
- Chen X. 2009. Small RNAs and their roles in plant development. Annual Review of Cell and Developmental Biology 25: 21–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coustham V, Vlad D, Deremetz A, Gy I, Cubillos FA, Kerdaffrec E, Loudet O, Bouche N. 2014. SHOOT GROWTH1 maintains Arabidopsis epigenomes by regulating IBM1. PLoS ONE 9: e84687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui C, Wang JJ, Zhao JH, Fang YY, He XF, Guo HS, Duan CG. 2020. A Brassica miRNA regulates plant growth and immunity through distinct modes of action. Molecular Plant 13: 231–245. [DOI] [PubMed] [Google Scholar]
- Duan CG, Fang YY, Zhou BJ, Zhao JH, Hou WN, Zhu H, Ding SW, Guo HS. 2012. Suppression of Arabidopsis ARGONAUTE1‐mediated slicing, transgene‐induced RNA silencing, and DNA methylation by distinct domains of the Cucumber mosaic virus 2b protein. Plant Cell 24: 259–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan C‐G, Wang X, Xie S, Pan Li, Miki D, Tang K, Hsu C‐C, Lei M, Zhong Y, Hou Y‐J et al. 2017a. A pair of transposon‐derived proteins function in a histone acetyltransferase complex for active DNA demethylation. Cell Research 27: 226–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan CG, Wang X, Zhang L, Xiong X, Zhang Z, Tang K, Pan L, Hsu CC, Xu H, Tao WA et al. 2017b. A protein complex regulates RNA processing of intronic heterochromatin‐containing genes in Arabidopsis. Proceedings of the National Academy of Sciences, USA 114: E7377–E7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan CG, Zhu JK, Cao X. 2018. Retrospective and perspective of plant epigenetics in China. Journal of Genetics and Genomics 45: 621–638. [DOI] [PubMed] [Google Scholar]
- Espinas NA, Tu LN, Furci L, Shimajiri Y, Harukawa Y, Miura S, Takuno S, Saze H. 2020. Transcriptional regulation of genes bearing intronic heterochromatin in the rice genome. PLoS Genetics 16: e1008637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gy I, Gasciolli V, Lauressergues D, Morel JB, Gombert J, Proux F, Proux C, Vaucheret H, Mallory AC. 2007. Arabidopsis FIERY1, XRN2, and XRN3 are endogenous RNA silencing suppressors. Plant Cell 19: 3451–3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones‐Rhoades MW, Bartel DP, Bartel B. 2006. MicroRNAS and their regulatory roles in plants. Annual Review of Plant Biology 57: 19–53. [DOI] [PubMed] [Google Scholar]
- Kojima S, Takahashi Y, Kobayashi Y, Monna L, Sasaki T, Araki T, Yano M. 2002. Hd3a, a rice ortholog of the Arabidopsis FT gene, promotes transition to flowering downstream of Hd1 under short‐day conditions. Plant and Cell Physiology 43: 1096–1105. [DOI] [PubMed] [Google Scholar]
- Komiya R, Ikegami A, Tamaki S, Yokoi S, Shimamoto K. 2008. Hd3a and RFT1 are essential for flowering in rice. Development 135: 767–774. [DOI] [PubMed] [Google Scholar]
- Krzyszton M, Zakrzewska‐Placzek M, Kwasnik A, Dojer N, Karlowski W, Kufel J. 2018. Defective XRN3‐mediated transcription termination in Arabidopsis affects the expression of protein‐coding genes. The Plant Journal 93: 1017–1031. [DOI] [PubMed] [Google Scholar]
- Kurihara Y. 2017. Activity and roles of Arabidopsis thaliana XRN family exoribonucleases in noncoding RNA pathways. Journal of Plant Research 130: 25–31. [DOI] [PubMed] [Google Scholar]
- Kurihara Y, Schmitz RJ, Nery JR, Schultz MD, Okubo‐Kurihara E, Morosawa T, Tanaka M, Toyoda T, Seki M, Ecker JR. 2012. Surveillance of 3' noncoding transcripts requires FIERY1 and XRN3 in Arabidopsis. G3 (Bethesda) 2: 487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, Cuzick A, Lu XM, Wang J, Katiyar N, Tsuchiya T, Le Roch K, McDowell JM, Holub E, Eulgem T. 2019. The Arabidopsis RRM domain protein EDM3 mediates race‐specific disease resistance by controlling H3K9me2‐dependent alternative polyadenylation of RPP7 immune receptor transcripts. The Plant Journal 97: 646–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le TN, Miyazaki Y, Takuno S, Saze H. 2015. Epigenetic regulation of intragenic transposable elements impacts gene transcription in Arabidopsis thaliana . Nucleic Acids Research 43: 3911–3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei M, La H, Lu K, Wang P, Miki D, Ren Z, Duan C‐g, Wang X, Tang K, Zeng L et al. 2014. Arabidopsis EDM2 promotes IBM1 distal polyadenylation and regulates genome DNA methylation patterns. Proceedings of the National Academy of Sciences, USA 111: 527–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Hung FY, Ye C, Hong L, Shih YH, Wu K, Li QQ. 2020. HDA6‐dependent histone deacetylation regulates mRNA polyadenylation in Arabidopsis. Genome Research 30: 1407–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Lu F, Cui X, Cao X. 2010. Histone methylation in higher plants. Annual Review of Plant Biology 61: 395–420. [DOI] [PubMed] [Google Scholar]
- Liu Y, Gao W, Wu S, Lu L, Chen Y, Guo J, Men S, Zhang X. 2020. AtXRN4 affects the turnover of chosen miRNA*s in Arabidopsis. Plants 9: 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan VK, Kukulich PM, von Hagel B, Green PJ. 2019. RNA degradomes reveal substrates and importance for dark and nitrogen stress responses of Arabidopsis XRN4. Nucleic Acids Research 47: 9216–9230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong‐Abdullah M, Ordway JM, Jiang N, Ooi SE, Kok SY, Sarpan N, Azimi N, Hashim AT, Ishak Z, Rosli SK et al. 2015. Loss of Karma transposon methylation underlies the mantled somaclonal variant of oil palm. Nature 525: 533–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszkowski J. 2015. Epigenetics: The karma of oil palms. Nature 525: 466–467. [DOI] [PubMed] [Google Scholar]
- Quint M, Delker C, Franklin KA, Wigge PA, Halliday KJ, van Zanten M. 2016. Molecular and genetic control of plant thermomorphogenesis. Nature Plants 2: 15190. [DOI] [PubMed] [Google Scholar]
- Saze H, Kitayama J, Takashima K, Miura S, Harukawa Y, Ito T, Kakutani T. 2013. Mechanism for full‐length RNA processing of Arabidopsis genes containing intragenic heterochromatin. Nature Communications 4: 2301. [DOI] [PubMed] [Google Scholar]
- Schnable PS, Springer NM. 2013. Progress toward understanding heterosis in crop plants. Annual Review of Plant Biology 64: 71–88. [DOI] [PubMed] [Google Scholar]
- Souret FF, Kastenmayer JP, Green PJ. 2004. AtXRN4 degrades mRNA in Arabidopsis and its substrates include selected miRNA targets. Molecular Cell 15: 173–183. [DOI] [PubMed] [Google Scholar]
- Tang W, Thompson WA. 2019. OsmiR528 enhances cold stress tolerance by repressing expression of stress response‐related transcription factor genes in plant cells. Current Genomics 20: 100–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- To TK, Saze H, Kakutani T. 2015. DNA methylation within transcribed regions. Plant Physiology 168: 1219–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya T, Eulgem T. 2013. An alternative polyadenylation mechanism coopted to the Arabidopsis RPP7 gene through intronic retrotransposon domestication. Proceedings of the National Academy of Sciences, USA 110: E3535–3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney RK, Graner A, Sorrells ME. 2005. Genic microsatellite markers in plants: features and applications. Trends in Biotechnology 23: 48–55. [DOI] [PubMed] [Google Scholar]
- Wang X, Duan C‐g, Tang K, Wang B, Zhang H, Lei M, Lu K, Mangrauthia Sk, Wang P, Zhu G et al. 2013. RNA‐binding protein regulates plant DNA methylation by controlling mRNA processing at the intronic heterochromatin‐containing gene IBM1. Proceedings of the National Academy of Sciences, USA 110: 15467–15472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West PT, Li Q, Ji L, Eichten SR, Song J, Vaughn MW, Schmitz RJ, Springer NM. 2014. Genomic distribution of H3K9me2 and DNA methylation in a maize genome. PLoS ONE 9: e105267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Yuan K, Yuan M, Meng X, Chen M, Wu J, Li J, Qi Y. 2020. Regulation of rice tillering by RNA‐directed DNA methylation at miniature inverted‐repeat transposable elements. Molecular Plant 13: 851–863. [DOI] [PubMed] [Google Scholar]
- Yang D‐L, Zhang G, Wang L, Li J, Xu D, Di C, Tang K, Yang L, Zeng L, Miki D et al. 2018. Four putative SWI2/SNF2 chromatin remodelers have dual roles in regulating DNA methylation in Arabidopsis. Cell Discovery 4: 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R, Li P, Mei H, Wang D, Sun J, Yang C, Hao L, Cao S, Chu C, Hu S et al. 2019. Fine‐tuning of MiR528 accumulation modulates flowering time in rice. Molecular Plant 12: 1103–1113. [DOI] [PubMed] [Google Scholar]
- Yang Z, Huang Yu, Yang J, Yao S, Zhao K, Wang D, Qin Q, Bian Z, Li Y, Lan Y et al. 2020. Jasmonate signaling enhances RNA silencing and antiviral defense in rice. Cell Host & Microbe 28: 89–103 e108. [DOI] [PubMed] [Google Scholar]
- Yao S, Yang Z, Yang R, Huang Yu, Guo Ge, Kong X, Lan Y, Zhou T, Wang He, Wang W et al. 2019. Transcriptional regulation of miR528 by OsSPL9 orchestrates antiviral response in rice. Molecular Plant 12: 1114–1122. [DOI] [PubMed] [Google Scholar]
- You C, He W, Hang R, Zhang C, Cao X, Guo H, Chen X, Cui J, Mo B. 2019. FIERY1 promotes microRNA accumulation by suppressing rRNA‐derived small interfering RNAs in Arabidopsis. Nature Communications 10: 4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Lin J, Li QQ. 2019. Transcriptome analyses of FY mutants reveal its role in mRNA alternative polyadenylation. Plant Cell 31: 2332–2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Lang Z, Zhu JK. 2018. Dynamics and function of DNA methylation in plants. Nature Reviews Molecular Cell Biology 19: 489–506. [DOI] [PubMed] [Google Scholar]
- Zhang JP, Yu Y, Feng YZ, Zhou YF, Zhang F, Yang YW, Lei MQ, Zhang YC, Chen YQ. 2017. MiR408 regulates grain yield and photosynthesis via a phytocyanin protein. Plant Physiology 175: 1175–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zhang YZ, Jiang J, Duan CG. 2020. The crosstalk between epigenetic mechanisms and alternative RNA processing regulation. Frontiers in Genetics 11: 998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y‐C, He R‐R, Lian J‐P, Zhou Y‐F, Zhang F, Li Q‐F, Yu Y, Feng Y‐Z, Yang Y‐W, Lei M‐Q et al. 2020. OsmiR528 regulates rice‐pollen intine formation by targeting an uclacyanin to influence flavonoid metabolism. Proceedings of the National Academy of Sciences, USA 117: 727–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YZ, Lin J, Ren Z, Chen CX, Miki D, Xie SS, Zhang J, Chang YN, Jiang J, Yan J et al. 2021. Genome‐wide distribution and functions of the AAE complex in epigenetic regulation in Arabidopsis. Journal of Integrative Plant Biology 63: 707–722. [DOI] [PubMed] [Google Scholar]
- Zhang Y‐Z, Yuan J, Zhang L, Chen C, Wang Y, Zhang G, Peng Li, Xie S‐S, Jiang J, Zhu J‐K et al. 2020. Coupling of H3K27me3 recognition with transcriptional repression through the BAH‐PHD‐CPL2 complex in Arabidopsis. Nature Communications 11: 6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Xie L, Zhang Q, Ouyang W, Deng Li, Guan P, Ma M, Li Y, Zhang Y, Xiao Q et al. 2020. Integrative analysis of reference epigenomes in 20 rice varieties. Nature Communications 11: 2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Chen C, Zeng J, Yun Z, Liu Y, Qu H, Jiang Y, Duan X, Xia R. 2020. MicroRNA528, a hub regulator modulating ROS homeostasis via targeting of a diverse set of genes encoding copper‐containing proteins in monocots. New Phytologist 225: 385–399. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Phylogenetic analysis of OsASI1 protein.
Fig. S2 Mutation information of OsASI1, OsEDM2 and OsAIPP1a.
Fig. S3 Morphological phenotypes of pistil and stamen of osasi1 and osxrnl mutants.
Fig. S4 Expression level of OsASI1 protein in OsASI1ox plants.
Fig. S5 Effect of OsASI1 dysfunction on global gene expression.
Fig. S6 Enrichment analysis of osasil1‐1 DEGs.
Fig. S7 Subcellular localisation of OsASI1, OsEDM2, OsAIPP1a and OsAIPP1b proteins.
Fig. S8 Effect of OsASI1 dysfunction on the alternative polyadenylation of 3′UTR.
Fig. S9 Gene Ontology analysis of deAPA genes in osasi1‐1.
Fig. S10 ChIP‐qPCR analysis of H3K9me2 density at selected target genes of OsASI1.
Fig. S11 Subcellar localisation of OsXRNL protein and mutation information of OsXRNL gene.
Fig. S12 Length distribution of mapped small RNAs from WT, osasi1‐1 and osxrnl‐1.
Fig. S13 Relative expression of representative MIRNAs and stem‐loops.
Fig. S14 Relative accumulation of miRNAs in inflorescence tissues.
Fig. S15 Flowering time phenotype of different XRNL transgenic plants.
Fig. S16 Sequence alignment between Arabidopsis and rice AIPP1 proteins.
Fig. S17 Relative expression of OsASI1, OsAIPP1a, OsAIPP1b and OsEDM2 in different tissues.
Fig. S18 Statistics analysis of flowering time, defective pollen rate and seed‐setting rate of osedm2 and osaipp1a mutants.
Fig. S19 The poly(A) site usage and expression analysis of flowering time‐ and pollen development‐related genes in different tissues.
Table S1 List of primers used in this study.
Table S2 List of differentially expressed APA genes in osasi1‐1 mutant.
Table S3 Detailed list of downregulated miRNAs in osasi1 and osxrnl mutants.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Data Availability Statement
The sequencing datasets generated in this study, including PAT‐seq, mRNA‐seq and small RNA sequencing, have been submitted to the NCBI BioProject database (http://www.ncbi.nlm.nih.gov/bioproject/) with accession no. PRJNA673072.