

Abstract
The correlation of mechanical properties of polymer materials with those of their molecular constituents is the foundation for their holistic comprehension and eventually for improved material designs and syntheses. Over the last decade, optical force probes (OFPs) were developed, shedding light on various unique mechanical behaviors of materials. The properties of polymers are diverse, ranging from soft hydrogels to ultra‐tough composites, from purely elastic rubbers to viscous colloidal solutions, and from transparent glasses to super black dyed coatings. Only very recently, researchers started to develop tailored OFP solutions that account for such material requirements in energy (both light and force), in time, and in their spatially detectable resolution. We here highlight notable recent examples and identify future challenges in this emergent field.
Keywords: chemiluminescence, fluorescence, mechanochemistry, polymers, sensors
In recent years, notable works on adjusting optical force probes in their responses to energy (both light and force), in time, and in their spatially detectable resolution have been published and help unraveling the macroscopic with relation to the molecular mechanical properties of polymer materials. Here, notable examples that put a dent in the universe of optical force sensing are highlighted.
1. Introduction
The mechanical deformation of a polymer material usually starts with the macroscopic application of force and leads to conformational, configurational, and constitutional covalent and non‐covalent bond rearrangements on the molecular level. This process hence covers around seven to ten orders of magnitude in length scale, rendering its analysis and understanding a conceptual and instrumental challenge. A comprehensive insight into the mechanical properties of a polymer material requires suitable analytical tools and techniques that allow collecting and mapping mechanical information across these scales.
In polymer mechanochemistry,[ 1 , 2 ] functional molecular motifs (mechanophores)[ 3 , 4 ] are designed to undergo faster bond dissociation or isomerization reactions under force compared to the polymers they are incorporated within. The force‐induced activation of the latent mechanophore function can be tailored to cause an alteration in the optical properties, rendering the mechanophore an OFP.[ 5 , 6 , 7 , 8 ] Such OFPs allow real‐time in situ or post mortem monitoring of force‐induced events from the molecular to the macroscopic scale, making them ideal tools to investigate material mechanics.
OFPs undergo diverse optical transformations in response to force. Changes in absorption, fluorescence, and chemiluminescence are most widely used in this context ‐ each with their own associated advantages and disadvantages. [6] Although absorption alterations can be easily detected by eye, their sensitivity and spatial resolution are limited by Lambert‐Beer's law and the diffraction limit. [9] Fluorescence modulation in combination with highly resolved microscopy techniques offers advantages in both sensitivity and spatial resolution[ 9 , 10 ] but is less advantageous for spatiotemporally resolved measurements, as the cumulative increase of the signal can interfere with the detection of single events. Conversely, chemiluminescence is a transient phenomenon that gives spatiotemporally resolved insights, yet it requires in situ observation, as post mortem analysis is not possible. [11]
However, OFPs are not a one‐size‐fits‐all solution for every polymer material and research problem. The identity, capabilities, and localization of an OFP within a material determine the mechanical information that is accessible regarding the spectral properties, force range, and spatiotemporal resolution in solution, the homogenous bulk, and at interfaces as well as interphases. Polymer materials are vastly different, for example, in their time‐dependent mechanical properties (creep, stress‐relaxation) or in their optical properties (opacity, intrinsic fluorescence). Hence, tailoring OFPs to a specific demand of the parent material under investigation is a necessary step to obtain meaningful and representative data.
In recent years, brilliant works on adjusting OFPs in their responses to energy (both light and force), in time, and in their spatially detectable resolution have been published, and we here highlight notable examples that put a dent in the universe of optical force sensing.
2. Tailoring the Optical Response
The targeted design of the optical responses of OFPs is the basis to achieve optical orthogonality and complementarity in their applications. Fine‐tuning of absorption, excitation, and emission wavelengths including the associated Stokes shifts enables, for example, the separate detection of non‐activated and activated OFP states. Moreover, complex and compartmentalized materials call for the parallel use of multiple OFPs that must be addressed and resolved independently. OFPs must have specific properties which depend on the requirements for the quality of information. For example, an OFP tasked to simply visualize a deformation process must meet different criteria than those for local resolution of mechanical processes on different scales, for the quantitative characterization of mechanical events, or for real‐time in situ bond scission detection.
2.1. Absorbing force probes
OFPs that alter their absorption (Figure 1a, left) generally exhibit color changes that are visible by eye and hence are popular for the straightforward visualization of force‐induced events in materials. In more quantitative approaches, the absorption changes are recorded by solution and solid‐state spectroscopy or by high‐speed cameras for real‐time, qualitative, macroscopic monitoring (cf. Section 5.1).[ 12 , 13 ]
Figure 1.
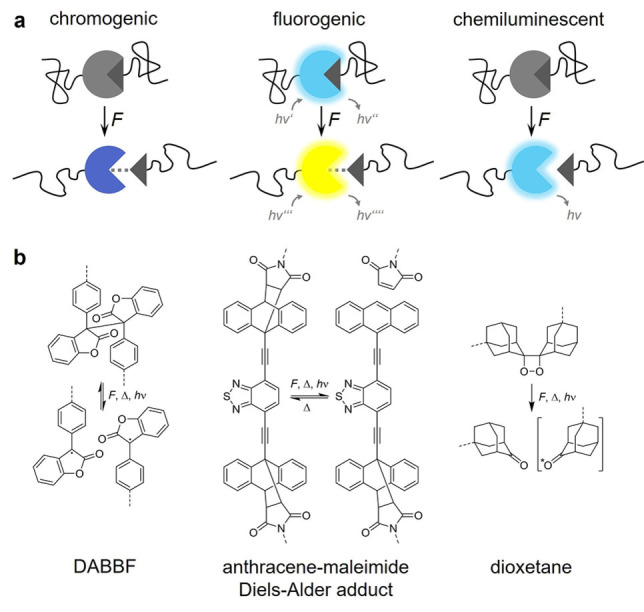
Different optical responses of OFPs that alter either absorption, fluorescence, or chemiluminescence. (a) Schematic depiction and (b) structural formula of exemplary OFPs. DABBF is a scissile, reversible OFP that becomes blue colored upon activation. [17] Anthracene‐maleimide Diels‐Alder adducts are scissile, quasi‐static OFPs that can either turn “on” their fluorescence or are dual‐fluorescent. [18] Dioxetanes are scissile, quasi‐static, mechanoluminescent OFPs. [11] Note that force‐induced transitions can be either scissile or non‐scissile as indicated by the dashed lines in (a). Dashed lines in (b) indicate the attachment points to the macromolecular framework.
Notable examples include a bis‐naphthopyran reported by McFadden and Robb. [14] This double‐mechanophore OFP shows gradient multicolor mechanochromism and resolves the amount of force delivered to the OFP by activation of either one or two of the conjugated naphthopyran moieties, leading to a gradual bathochromic shift in the latter scenario. Robb and coworkers recently enhanced this OFP system by color tuning and tailoring of the fading kinetics for higher versatility. [15]
Multicolor mechanochromism can also be achieved by simple mixing of different mechanochromic polymers, as Otsuka and coworkers have demonstrated.[ 16 , 17 ] Polymers with tetraarylsuccinonitrile and diarylbibenzothiophenonyl OFPs are activated simultaneously and the polymer blends differentiate between stretching and grinding. [16] In another work, they have shown how a diarylbibenzofuranone (DABBF) OFP (Figure 1b, left) in a polymer/silica composite is placed in the silica‐rich domains and a naphthopyran OFP in the polymer‐rich domain. [17] Depending on the magnitude of force, solvent addition, and time, different compartments of the material are activated, showing a gradual color evolution of absorption.
A future challenge is to further shift the absorption changes to the red and infrared (IR) region. While Stauch and Dreuw suggest NIR, IR, and Raman OFPs by computation,[ 21 , 22 ] a respective synthetic implementation remains to be realized.
2.2. Fluorescent force probes
Fluorogenic OFPs rely on the conversion of an optically silent state into a fluorescent one (or vice versa), or their fluorescence is modulated between a non‐activated and activated dual‐fluorescent OFP (Figure 1a, middle). Compared with purely chromogenic OFPs, fluorogenic OFPs offer higher sensitivity and spatial resolution (cf. Section 5). [9] Dual‐fluorescent OFPs are advantageous for tracking of the non‐activated material by fluorescence microscopy, which is helpful for non‐uniform, compartmentalized, or mobile materials, such as nanoparticles and colloids. [23]
Many examples for fluorogenic OFPs have been reported. Amongst them, we highlight a dual‐fluorescent benzoxazole OFP recently prepared by Weder and coworkers. [24] Upon force‐induced ester scission, an excited state intramolecular proton transfer is activated, leading to a fluorescence shift from blue to green. Göstl and coworkers have improved the optical orthogonality between the non‐activated and activated states of dual‐fluorescent OFPs. [18] By the preparation of benzothiadiazole‐ (BTD) and naphthothiadiazole‐based anthracene‐maleimide Diels‐Alder adducts, they have realized a distinctive fluorescence band transition at different wavelengths without spectral interference between the two OFP states (Figure 1b, middle and Figure 2a).
Figure 2.

A selection of bond scission mapping visualizations in OFP‐crosslinked polymer networks: (a) Overlay of CLSM micrographs showing non‐activated (cyan) and activated (yellow) BTD OFP in crosslinked rubber networks, and pixel‐by‐pixel contour map representation of left panel showing the percentage of activated relative to remaining non‐activated BTD upon fracture. Reproduced from Ref. [18]. Copyright 2021, the authors. (b) Progression and extent of a crack front in time and space for dioxetane‐crosslinked PMMA networks under chloroform ingress and swelling‐induced mechanoluminescence. Reproduced from Ref. [19]. Copyright 2017, American Chemical Society. (c) Intensity‐based mapping of bond scission in notched samples of single, double, and triple networks, containing dioxetane crosslinker in the first network, around the tip of a propagating crack and schematic of the bond scission mechanism. Reproduced from Ref. [20]. Copyright 2014, American Association for the Advancement of Science. (d) Stress map around the tip of a propagating crack in acrylate networks crosslinked with spiropyran converted to different merocyanine isomers. Reproduced from Ref. [13]. Copyright 2021, Royal Society of Chemistry.
Further shifting excitation and emission wavelengths into the red and IR region is also a future challenge for fluorogenic OFPs. In addition to spectral tuning, this might be realized through photophysical effects, such as the force‐activated triplet‐triplet annihilation photon upconversion where low‐energy excitation light is anti‐Stokes converted to high‐energy emission light. [25]
2.3. Mechanoluminescent force probes
In contrast to the permanent optical signals of absorbing and fluorescent OFPs, mechanoluminescent OFPs show ephemeral chemiluminescence when subjected to force (Figure 1a, right and Figure 2b and c). This is of great use for the temporal resolution of force‐induced transitions in materials (cf. Section 4.2). The only mechanoluminescent OFP developed up today is bis(adamantyl)‐1,2‐dioxetane (Figure 1b, right). As bond scission generates an adamantone in an energetically defined T1 state, spectral tuning of the emission wavelengths can only be achieved by the introduction of triplet acceptors. [26]
Chen, Sijbesma, and coworkers have introduced rare‐earth EuIII complexes alongside dioxetane OFPs in a polymer to harvest intense red chemiluminescence upon OFP activation. [27] Notably, Chen and coworkers also use a rhodamine OFP as triplet acceptor alongside the dioxetane OFP. [28] This allows the combination of mechanoluminescence and mechanochromism, where Förster resonance energy transfer (FRET) from the activated dioxetanes to the activated rhodamines also red‐shifts the dioxetane emission.
3. Tailoring the Force Range
The force under which an OFP responds with a signal must be smaller than the force required for non‐selective mechanochemical transformations in the material under investigation. Hence, a high‐performance structural polymer with a stiffness in the GPa region requires a different OFP than a biomimetic hydrogel where the stiffness may be only hundreds of Pa. In addition, OFPs can be divided into those that undergo threshold transformations at a critical force (mostly relying on bond scission, cf. Figure 1) and those that undergo gradual optical changes and can be used as “force rulers” (Figure 3).
Figure 3.
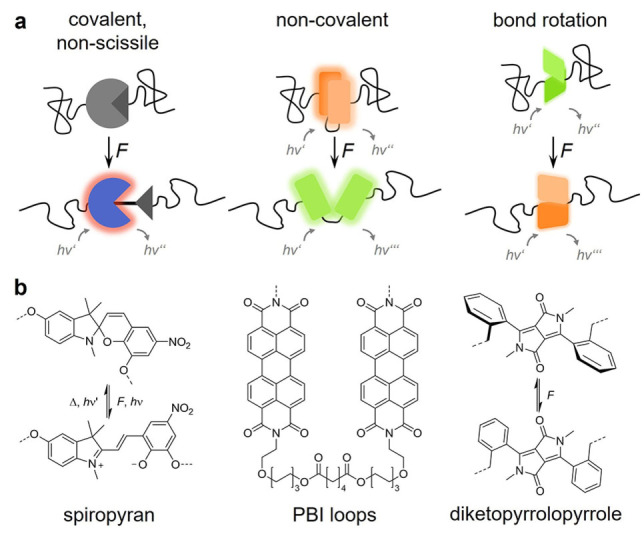
Examples for different force responses of OFPs undergoing non‐scissile covalent bond isomerization, non‐covalent bond rearrangement, or bond rotation. (a) Schematic depiction and (b) structural formula of exemplary OFPs. Spiropyran is a non‐scissile, reversible OFP that undergoes covalent bond isomerization to merocyanine, which has purple color and is red fluorescent. [31] PBI loops vary between excimer (associated) and monomer (dissociated) emission. [38] Diketopyrrolopyrroles change the emission wavelength based on charge transfer from the planarization of the twisted phenyl moiety. [45]
Adaption of the force response is generally achieved by tuning the bond dissociation energy (BDE) of the OFP through transition from strong, covalent to weak, non‐covalent bonds. Very low forces may even be displayed by force‐induced bond rotations. However, since the mechanochemical BDE usually correlates to the thermal BDE, the display of very high or very low forces is a challenging endeavor. For example, industrial manufacturing conditions of many high‐performance polymers entail processing temperatures of more than 200 °C, which is beyond the thermal stability of most OFPs. On the other hand, the force response of an OFP designed for very low forces may become indistinguishable from its response to slight temperature changes during the mechanical deformation.
3.1. Probes for covalent bond scission
OFPs undergoing covalent bond scission withstand forces in the nN range and are ideal for applications in stiff and tough materials (Figure 3a, left). Most OFPs described above are of this variety, because they tend to be thermally bistable with ΔG ≠ far above operating temperature, which facilitates post mortem analysis. As covalent bond scission reactions are discrete events, OFPs of this variety are threshold probes and provide an optical signal that is proportional to the number of broken bonds within a material. Examples for popular OFPs cleaving in the nN range are anthracene‐maleimide Diels‐Alder adducts or coumarin dimers.[ 29 , 30 ] Spiro compounds, [31] such as spiropyran, naphthopyran, or rhodamine, are notable exceptions as these isomerize reversibly at hundreds of pN (Figure 3b, left and Figure 2d).[ 32 , 33 ] Craig and coworkers have demonstrated that the force required for the isomerization of spiropyran can be tuned by varying the attachment points of the OFP to the polymer chain.[ 34 , 35 ] However, this has only a minimal effect on the mechanical activation in bulk polymeric materials due to the elusive nature of the interplay between molecular OFP and macroscopic polymer system.[ 36 , 37 ]
3.2. Non‐covalent force probes
Non‐covalent force probes rely on the alteration of, for example, van der Waals, π‐π, H‐bond, or hydrophobic interactions (Figure 3a, middle). Since such interactions (individually) possess much smaller BDEs compared to covalent bonds, they are useful for the detection of pN forces.
Perylene, perylene bisimide, or oligo(phenylenevinylene) are examples for popular aggregachromic OFPs that are blendable into a polymer matrix and alter their aggregation state, i. e., the interaction of their π‐systems upon force‐induced material deformation.[ 5 , 8 ] Generally, an alteration of the fluorescence signals corresponding to the aggregated (excimer emission) and de‐aggregated (single dye emission) OFPs is correlated to the deformation of the matrix polymer. While the macroscopic mechanical state is visualized well, the depth of obtainable molecular mechanical information is limited, as aggregachromic OFPs are not anchored within their matrix polymer. Notably, Weder, Schrettl, Sagara, and coworkers as well as De Bo and coworkers have converged the principles of aggregachromic and discrete mechanophore OFPs by synthesizing folded perylene bisimide loop structures, [38] pyrene‐containing cyclophanes, [39] and rotaxane‐based emitters.[ 40 , 41 , 42 , 43 ] In these, the formerly intermolecular transitions are now rendered reversible intramolecular deaggregation/aggregation events and the structures are covalently anchored within the polymer material (Figure 3b, middle). This allows the analysis of such OFPs in analogy to a traditional mechanophore and opens a new alley towards spectral tuning of OFPs.
To detect even lower forces in the fN range, transitions that exhibit a similarly low ΔG ≠, such as bond rotations, must be exploited (Figure 3a, right). Sprakel and coworkers have used a conjugated donor polymer poly(dioctylfluorene‐alt‐benzothiadiazole) that is randomly doped with dithienyl‐benzothiadiazole acceptor moieties. [44] Since the OFP essentially acts as an entropic spring, uncoiling of the chain leads to an optically detectable alteration of the energy transfer efficiency, which is readily connected to the applied force. Moreover, OFPs relying on bond rotation are often continuous, non‐threshold systems, which allows their use as “force rulers”. Sommer and coworkers have demonstrated this principle by using a diketopyrrolopyrrole donor‐acceptor OFP that acts as a torsional spring and reflects macroscopic force through a variation of its dihedral angle and consequently the emission spectrum (Figure 3b, right). [45]
Compared with de novo synthesized OFPs, DNA‐based OFPs are readily obtained, programmable, and generally rooted in the spectral modulation of known distance‐sensitive FRET dye pairs. [46] While these OFPs enable the detection of forces in the pN range, which is suitable for soft biomimetic hydrogels, their incorporation into artificial materials is synthetically challenging, which Walther and coworkers recently have tackled with acrylamide‐terminated DNA OFP crosslinkers. [47] Since the analysis of mechanical processes in biohybrid materials and the associated interaction between living systems and polymers is an emerging challenge, [48] nucleic acid and genetically engineered protein [49] OFPs could become increasingly important.
4. Tailoring the Temporal Response
Due to the viscoelastic behavior of most polymers, their mechanics depend on the timescale on which a deformation is experienced by a material. For time‐independent polymer materials, a singular, invariable, and permanent signal from a corresponding OFP can be translated into useful information. However, when time‐dependent properties, such as deformational creep, stress‐relaxation, or fatigue should be investigated, the response of the OFP must meet the corresponding requirements. [50] Hence, it is currently a challenge to develop OFPs that synchronize their response with the mechanical properties of the matrix polymer. [13]
4.1. Quasi‐static force probes
Quasi‐static OFPs enable the post mortem examination of deformed or fractured materials and are an invaluable tool for processes where force application cannot be performed and monitored in situ or where multiple analytical stages are required (Figure 4, left). Note that while the principle of microscopic reversibility applies to any given system, many OFPs become quasi‐irreversible by kinetic (low concentrations) or thermodynamic (high ΔG ≠) constraints after their activation.
Figure 4.
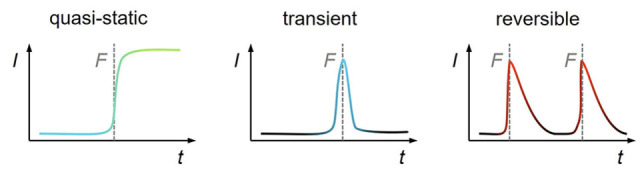
Different temporal responses of OFPs that are either quasi‐static, transient, or reversible. The measured optical signal intensity I is shown as a function of observation time t and timepoints of force application to the material are indicated with F.
A representative example for this OFP class are Diels‐Alder adducts from anthracene and maleimide by Göstl and coworkers (Figure 1b, middle). [51] When incorporated as crosslinkers into hydrogel networks, mapping of bond scission events and crack propagation by confocal laser scanning microscopy (CLSM) is possible. [52] With dual‐fluorescent Diels‐Alder adduct OFPs (cf. Section 2.2), the non‐activated and activated states can be visualized independently by CLSM and a direct pixel‐by‐pixel quantification of the mechanochemical activation process can be achieved (Figure 2a). [18] The irreversibility of the OFP is important, as the instrumental requirements of highly resolved optical microscopy are incompatible with most mechanical deformation techniques, rendering the in situ monitoring an open challenge in such scenarios.
4.2. Transient force probes
When real‐time tracking of individual force‐induced events is desired, the cumulative signal increase of quasi‐static OFPs can decrease the spatial detectability of such events (Figure 4, middle). Dioxetane OFPs (cf. Section 2.3, Figure 1, right) are especially useful in such scenarios.
For example, Clough, van der Gucht, and Sijbesma have recorded a cascade of discrete bond scission events in glassy dioxetane‐crosslinked poly(methyl methacrylate) (PMMA) networks upon chloroform ingress with temporal resolution and spatial correlation (Figure 2b). They discern flashes of light over several minutes caused by the tensile stresses exerted from the swollen part of the polymer onto the non‐swollen part. [19] Also in multinetworks, Creton, Sijbesma, and coworkers have shown the real‐time mapping of bond scission ahead of a propagating crack by using the dioxetane OFP (Figure 2c). [20] Notably, these authors investigate the complex mechanical behavior of silica‐filled polydimethylsiloxane (PDMS) regarding its irreversible, history‐dependent stress‐softening, i. e., the Mullins effect. [53] They revealed a complex, possibly delayed stress transfer from the bulk material to the OFPs upon unloading of the networks previously subjected to cyclic uniaxial tensile testing.
4.3. Reversible force probes
Reversible OFPs enable repeated activation and the correlation of the emerging optical signal to the deformation history of the polymer material under investigation (Figure 4, right). The most notable representatives of this class are spiro OFPs (cf. Section 3.1, Figure 3, left) whose reversibility is enabled by their non‐scissile nature and low ΔG ≠. The time‐dependent activation of the spiropyran OFP in poly(methyl acrylate) during stress relaxation [54] or in PMMA under creep loading [55] lays the foundation for mechanotransduction studies with reversible OFPs. In PDMS, which is a polymer with complex rheological behavior and time‐dependent elastic deformation, this OFP shows reversible activation over multiple cycles of tensile elongation. [56] This is in accordance with the mechanical response of the material, which regains its original shape upon force removal.
Creton and coworkers have used the differently colored merocyanine isomers generated upon spiropyran activation to measure and map stress history in acrylate networks. [13] This allows the construction of a pixel‐by‐pixel stress map around a crack tip just at its initiation and before its propagation prior failure (Figure 2d).
To further reduce OFP response time, Moore and coworkers have synthesized oxazine OFPs and integrated them into PDMS. [57] Compared to other spiro OFPs, their reversible mechanochromism fades within the ms to μs range upon force removal, owing to the absence of double‐bound isomerization compared to the spiropyran OFP. Notably, the opposite direction of temporal tuning is also possible. McFadden and Robb have prepared a permanent naphthopyran OFP that does not isomerize back to its initial form, rendering it a quasi‐static OFP. [58]
5. Spatial Resolution
The multiscale interrelation of macroscopic with molecular mechanical properties of a material is not only a challenge regarding the design and synthesis of OFPs but also an apparent instrumental analytical contradiction. The ideal spatially resolved image dataset, which an OFP could deliver, contains molecularly resolved information over a cm‐sized sample (Figure 5). From viewpoints, such as image acquisition time, reproducibility, and generated data size, this is technically not feasible. Since most optical properties are scale invariant, a workflow emerged over time where OFP alterations are initially monitored on a whole sample level and then regions of interest where a force‐induced event can be locally resolved with high resolution are identified. Naturally, the OFPs must satisfy the high demands regarding optical performance of the corresponding method.
Figure 5.
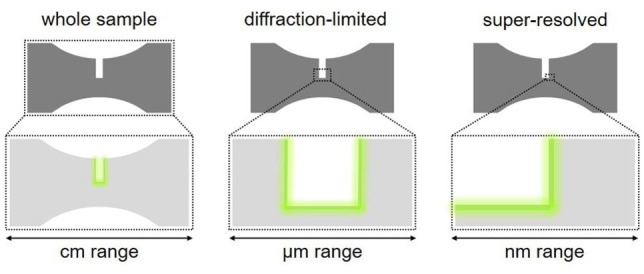
Different spatial resolutions that can be achieved during OFP analysis exemplified on a dog bone‐shaped sample widely employed for the mechanical testing of polymers. Either whole sample monitoring, diffraction‐limited microscopy, or super‐resolved microscopy techniques are used and indicate the optical signal at the region of interest.
5.1. Whole sample monitoring
Monitoring an OFP‐functionalized material over the entire dimensions of the sample is likely the method with the highest potential for in situ monitoring. Cameras, if required in combination with appropriate optics, filters, and light sources, can be set up at most mechanical testing equipment. This enables correlation of macroscopic mechanical properties and continuum simulations to the macroscopically resolved local OFP signal. This is reported for absorbing,[ 12 , 13 ] fluorescent,[ 54 , 59 , 60 , 61 ] and mechanoluminescent [20] OFPs (Figures 2c and d). Yet, such methods do not allow sub‐μm spatial resolution where fluorescence microscopy techniques become necessary.
5.2. Diffraction‐limited microscopy
Fluorescence imaging shows high sensitivity and its spatial resolution is determined by the Rayleigh criterium of ca. of the excitation wavelength when high numerical aperture optics are used. CLSM is the technique of choice when sensitivity, spatial resolution, sharp imaging, and optical sectioning are required. However, CLSM optics do not allow straightforward integration of mechanical testing equipment, which is why this is a post mortem technique and requires quasi‐static OFPs up to today.
Göstl, Sijbesma, and coworkers have shown CLSM on Diels‐Alder adduct OFPs in hydrogels to elucidate their fracture behavior depending on the water content. [52] Gong, Matsuda, and coworkers have demonstrated CLSM imaging on a crack tip of a double‐network hydrogel by using the produced mechanoradicals for latent fluorophore polymerization. [62] However, the obtained signal loses its correlation to the number of mechanochemical events in this process. Creton, Comtet, Göstl, and coworkers have tackled this issue by calibrating the CLSM with OFP reference molecules and established the first quantitative protocol. [63] There, they have quantified the number of broken bonds during crack propagation in a polymer network and find that bond scission can extend as far as 100 μm away from the fracture plane. Creton and coworkers have also applied this method in the analysis of cavitation phenomena induced in elastomers where a fast hydrogen gas decompression has been applied. [64] Göstl and coworkers have further developed the quantification approach with dual‐fluorescent OFPs (cf. Section 2.2, Figure 1, middle and Figure 2a), which enables the estimation of the number of broken bonds by quantitative comparison of the photon number of intact to cleaved OFPs per volume unit in the material. [18]
5.3. Super‐resolution microscopy
One of the most attractive goals using OFPs is to correlate optical information resolved on the molecular scale to the molecular and macroscopic mechanical properties of a polymer material. In this direction, Sprakel and coworkers have used their entropic spring OFP (cf. Section 3.2) to map forces down to the fN range on a single molecule level. [44] Yet, this method is limited to conjugated polymers and more general solutions are required.
Liddle, Wang, and coworkers have tackled this issue by using single molecule fluorescence microscopy and resolved the localization as well as orientation of the transition dipole moment of single fluorophores blended within a PMMA matrix, entirely without OFPs. [65] Regular structures are imprinted by nanolithography, which enables the combination of signals from similar regions and improves labelling density and statistics. In this way, they identify and spatially resolve areas inside these structured regions with up to 20 nm resolution, thereby proving an anisotropic distribution of fluorophore orientations which were generated by mechanical deformations of the polymer matrix at the nanoscale.
Yet, the application of OFPs for fractographic approaches using super‐resolution techniques is an open challenge and remains to be implemented. The major super‐resolved fluorescence microscopy techniques are based on the sequential switching of fluorophores either in highly localized volumes or in a stochastic approach. They can be categorized in two groups: (i) point scanning of an engineered point spread function, such as RESOLFT (reversible saturable optical fluorescence transitions) and STED (stimulated emission depletion) microscopy. and (ii) single molecule localization methods, such as PALM (photoactivated localization microscopy) and STORM (stochastic optical reconstruction microscopy).[ 9 , 10 , 66 , 67 ] To realize the application of super‐resolution methods with OFPs, it is necessary to address the design of dedicated OFPs capable of performing under the demanding super‐resolution conditions. For instance, STED requires high photostability, high depletion quantum yield, and a sufficient shifted emission spectrum with respect to the excitation wavelength to allow depletion. [66] However, for single molecule localization approaches, even more specific aspects must be considered, such as the energy of the excited states, transition rates between states as well as quantum yields of fluorescence emission, photoswitching, and/or photobleaching. [67] We envisage that the design of such mechanophores could open an exciting door into the world of optical visualization of mechanical performance in polymer materials at the nanoscale.
6. Conclusions
Tailoring of OFPs in the context of polymer mechanochemistry is a comparably young field. Thus far, OFPs have been color‐tuned through the whole visible spectrum; force ranges from the fN to the nN range have been enabled; reversible reaction timescales of seconds have been achieved; the first steps towards highly resolved imaging have been made. However, polymers are such diverse materials that every copolymer, blend, or composite requires a tailor‐made OFP solution. Pushing the analytical limits of known OFP parameter spaces and benchmarking OFP performances against each other to warrant comparability of the produced data will be the major research efforts. While this is a synthetic chemical task at its core, it is imperative to tackle this challenge in a transdisciplinary approach. A hammer is useless without a nail, and we can ask questions that a given OFP can answer only if we understand the advantages and limitations of this OFP.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Siyang He obtained his B. Eng. in Polymer Science at Sichuan University in China. Subsequently, he received his M. Sc. in Material Chemistry at Saarland University in 2020 and is currently pursuing his doctoral research on photo‐ and mechanoresponsive microgels at DWI and RWTH.

Biographical Information
Maria Stratigaki obtained her degree in Materials Science and Technology at the University of Crete. She then received her M. Sc. in Applied Sciences at the Technical University of Crete and subsequently her Ph.D. in 2018 focusing on the structure‐property relationships of composite materials. She is currently conducting postdoctoral research developing mechanoresponsive materials at DWI.

Biographical Information
Silvia P. Centeno Benigno studied Chemistry at the University of Málaga in Spain where she also received her Ph.D. in 2004 on the study of charge transfer processes in the SERS of azines on silver electrodes in the group of Prof. Carlos Otero. She was postdoc in the groups of Prof. Hofkens at KU Leuven in Belgium and in the group of Prof. Wöll at RWTH, where she got acquainted with the application of super‐resolution fluorescence microscopy methods to of responsive polymer materials. She is currently Project Leader at DWI and responsible for the super‐resolution fluorescence microscopy and Raman microscopy facilities.

Biographical Information
Andreas Dreuw received his Ph.D. in Theoretical Chemistry from Heidelberg University in 2001. After a two‐year postdoc at the UC Berkeley, he joined the Goethe University of Frankfurt first as Emmy‐Noether fellow, then as Heisenberg‐Professor for Theoretical Chemistry. Since 2011, Andreas Dreuw holds the chair for Theoretical and Computational Chemistry at the Interdisciplinary Center for Scientific Computing of Heidelberg University. His research interests comprise the development of electronic structure methods and their application in Photochemistry, Mechanochemistry, Biophysics, and Material Science.

Biographical Information
Robert Göstl studied chemistry at Humboldt‐Universität zu Berlin. There, he obtained his diploma degree in 2011 and his doctoral degree in 2014 working on photoswichtable diarylethenes. In his postdoctoral research, he started working on polymer mechanochemistry at Eindhoven University of Technology until 2016. He is currently leading an Independent Research Group at DWI that develops molecular tools to understand and harness mechanical force in polymer materials.

Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
R.G. is grateful for support by a Freigeist‐Fellowship of the Volkswagen Foundation (No. 92888). S.H. and R.G. thank the Deutsche Forschungsgemeinschaft for financial support within the Collaborative Research Center SFB 985 (No. 191948804). M.S. and R.G. acknowledge financial support from an Exploration Grant of the Boehringer Ingelheim Foundation. Parts of the analytical investigations were performed at the Center for Chemical Polymer Technology CPT, which was supported by the European Commission and the federal state of North Rhine‐Westphalia (No. 300088302). Financial support is acknowledged from the European Commission (EUSMI, No. 731019). Open Access funding enabled and organized by Projekt DEAL.
S. He, M. Stratigaki, S. P. Centeno, A. Dreuw, R. Göstl, Chem. Eur. J. 2021, 27, 15889.
References
- 1. O'Neill R. T., Boulatov R., Nat. Chem. Rev. 2021, 5, 148–167. [DOI] [PubMed] [Google Scholar]
- 2. Chen Y., Mellot G., van Luijk D., Creton C., Sijbesma R. P., Chem. Soc. Rev. 2021, 50, 4100–4140. [DOI] [PubMed] [Google Scholar]
- 3. De Bo G., Macromolecules 2020, 53, 7615–7617. [Google Scholar]
- 4. Brown C. L., Craig S. L., Chem. Sci. 2015, 6, 2158–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Calvino C., Neumann L., Weder C., Schrettl S., J. Polym. Sci. Part A 2017, 55, 640–652. [Google Scholar]
- 6. Göstl R., Clough J. M., Sijbesma R. P., in Mechanochemistry in Materials, Royal Society Of Chemistry, 2017, pp. 53–75. [Google Scholar]
- 7. Stratigaki M., Göstl R., ChemPlusChem 2020, 85, 1095–1103. [DOI] [PubMed] [Google Scholar]
- 8. Traeger H., Kiebala D. J., Weder C., Schrettl S., Macromol. Rapid Commun. 2021, 42, 2000573. [DOI] [PubMed] [Google Scholar]
- 9. Chapman D. V., Du H., Lee W. Y., Wiesner U. B., Prog. Polym. Sci. 2020, 111, 101312. [Google Scholar]
- 10. Wöll D., Flors C., Small Methods 2017, 1, 1700191. [Google Scholar]
- 11. Yuan Y., Yuan W., Chen Y., Sci. China Mater. 2016, 59, 507–520. [Google Scholar]
- 12. Chen Y., Yeh C. J., Qi Y., Long R., Creton C., Sci. Adv. 2020, 6, eaaz5093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen Y., Yeh C. J., Guo Q., Qi Y., Long R., Creton C., Chem. Sci. 2021, 12, 1693–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McFadden M. E., Robb M. J., J. Am. Chem. Soc. 2019, 141, 11388–11392. [DOI] [PubMed] [Google Scholar]
- 15. Versaw B. A., McFadden M. E., Husic C. C., Robb M. J., Chem. Sci. 2020, 11, 4525–4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ishizuki K., Aoki D., Goseki R., Otsuka H., ACS Macro Lett. 2018, 7, 556–560. [DOI] [PubMed] [Google Scholar]
- 17. Kosuge T., Zhu X., Lau V. M., Aoki D., Martinez T. J., Moore J. S., Otsuka H., J. Am. Chem. Soc. 2019, 141, 1898–1902. [DOI] [PubMed] [Google Scholar]
- 18. Baumann C., Stratigaki M., Centeno S. P., Göstl R., Angew. Chem. Int. Ed. 2021, 60, 13287–13293; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 13398–13404. [Google Scholar]
- 19. Clough J. M., van der Gucht J., Sijbesma R. P., Macromolecules 2017, 50, 2043–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ducrot E., Chen Y., Bulters M., Sijbesma R. P., Creton C., Science 2014, 344, 186–189. [DOI] [PubMed] [Google Scholar]
- 21. Stauch T., Dreuw A., Angew. Chem. Int. Ed. 2014, 53, 2759–2761; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 2797–2800. [Google Scholar]
- 22. Stauch T., Hoffmann M. T., Dreuw A., ChemPhysChem 2016, 17, 1486–1492. [DOI] [PubMed] [Google Scholar]
- 23. Izak-Nau E., Demco D. E., Braun S., Baumann C., Pich A., Göstl R., ACS Appl. Polym. Mater. 2020, 2, 1682–1691. [Google Scholar]
- 24. Karman M., Verde-Sesto E., Weder C., ACS Macro Lett. 2018, 7, 1028–1033. [DOI] [PubMed] [Google Scholar]
- 25. Yildiz D., Baumann C., Mikosch A., Kuehne A. J. C., Herrmann A., Göstl R., Angew. Chem. Int. Ed. 2019, 58, 12919–12923; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 13051–13055. [Google Scholar]
- 26. Clough J. M., Sijbesma R. P., ChemPhysChem 2014, 15, 3565–3571. [DOI] [PubMed] [Google Scholar]
- 27. Yang F., Yuan Y., Sijbesma R. P., Chen Y., Macromolecules 2020, 53, 905–912. [Google Scholar]
- 28. Wu M., Guo Z., He W., Yuan W., Chen Y., Chem. Sci. 2021, 12, 1245–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Akbulatov S., Boulatov R., ChemPhysChem 2017, 18, 1422–1450. [DOI] [PubMed] [Google Scholar]
- 30. Klein I. M., Husic C. C., Kovács D. P., Choquette N. J., Robb M. J., J. Am. Chem. Soc. 2020, 142, 16364–16381. [DOI] [PubMed] [Google Scholar]
- 31. Li J., Nagamani C., Moore J. S., Acc. Chem. Res. 2015, 48, 2181–2190. [DOI] [PubMed] [Google Scholar]
- 32. Gossweiler G. R., Kouznetsova T. B., Craig S. L., J. Am. Chem. Soc. 2015, 137, 6148–6151. [DOI] [PubMed] [Google Scholar]
- 33. Pan Y., Zhang H., Xu P., Tian Y., Wang C., Xiang S., Boulatov R., Weng W., Angew. Chem. Int. Ed. 2020, 59, 21980–21985; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 22164–22169. [Google Scholar]
- 34. Barbee M. H., Kouznetsova T., Barrett S. L., Gossweiler G. R., Lin Y., Rastogi S. K., Brittain W. J., Craig S. L., J. Am. Chem. Soc. 2018, 140, 12746–12750. [DOI] [PubMed] [Google Scholar]
- 35. Sommer M., Macromol. Rapid Commun. 2021, 42, 2000597. [DOI] [PubMed] [Google Scholar]
- 36. Kim T. A., Robb M. J., Moore J. S., White S. R., Sottos N. R., Macromolecules 2018, 51, 9177–9183. [Google Scholar]
- 37. Lin Y., Barbee M. H., Chang C.-C., Craig S. L., J. Am. Chem. Soc. 2018, 140, 15969–15975. [DOI] [PubMed] [Google Scholar]
- 38. Traeger H., Sagara Y., Kiebala D. J., Schrettl S., Weder C., Angew. Chem. Int. Ed. 2021, 60, 16191–16199; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 16327–16335. [Google Scholar]
- 39. Sagara Y., Traeger H., Li J., Okado Y., Schrettl S., Tamaoki N., Weder C., J. Am. Chem. Soc. 2021, 143, 5519–5525. [DOI] [PubMed] [Google Scholar]
- 40. Sagara Y., Karman M., Verde-Sesto E., Matsuo K., Kim Y., Tamaoki N., Weder C., J. Am. Chem. Soc. 2018, 140, 1584–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sagara Y., Karman M., Seki A., Pannipara M., Tamaoki N., Weder C., ACS Cent. Sci. 2019, 5, 874–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Muramatsu T., Okado Y., Traeger H., Schrettl S., Tamaoki N., Weder C., Sagara Y., J. Am. Chem. Soc. 2021, 143, 9884–9892. [DOI] [PubMed] [Google Scholar]
- 43. Sandoval-Torrientes R., Carr T. R., Bo G. D., Macromol. Rapid Commun. 2021, 42, 2000447. [DOI] [PubMed] [Google Scholar]
- 44. van de Laar T., Schuurman H., van der Scheer P., Maarten van Doorn J., van der Gucht J., Sprakel J., Chem 2018, 4, 269–284. [Google Scholar]
- 45. Raisch M., Maftuhin W., Walter M., Sommer M., Nat. Commun. 2021, 12, 4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Merindol R., Delechiave G., Heinen L., Catalani L. H., Walther A., Nat. Commun. 2019, 10, 528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Creusen G., Schmidt R. S., Walther A., ACS Macro Lett. 2021, 10, 671–678. [DOI] [PubMed] [Google Scholar]
- 48. Liu Y., Galior K., Ma V. P.-Y., Salaita K., Acc. Chem. Res. 2017, 50, 2915–2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhou Y., Huo S., Loznik M., Göstl R., Boersma A. J., Herrmann A., Angew. Chem. Int. Ed. 2021, 60, 1493–1497; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 1515–1519. [Google Scholar]
- 50. Takaffoli M., Zhang T., Parks D., Zhao X., J. Appl. Mech. 2016, 83, 071007. [Google Scholar]
- 51. Göstl R., Sijbesma R. P., Chem. Sci. 2016, 7, 370–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Stratigaki M., Baumann C., van Breemen L. C. A., Heuts J. P. A., Sijbesma R. P., Göstl R., Polym. Chem. 2020, 11, 358–366. [Google Scholar]
- 53. Clough J. M., Creton C., Craig S. L., Sijbesma R. P., Adv. Funct. Mater. 2016, 26, 9063–9074. [Google Scholar]
- 54. Beiermann B. A., Kramer S. L. B., May P. A., Moore J. S., White S. R., Sottos N. R., Adv. Funct. Mater. 2014, 24, 1529–1537. [Google Scholar]
- 55. Degen C. M., May P. A., Moore J. S., White S. R., Sottos N. R., Macromolecules 2013, 46, 8917–8921. [Google Scholar]
- 56. Gossweiler G. R., Hewage G. B., Soriano G., Wang Q., Welshofer G. W., Zhao X., Craig S. L., ACS Macro Lett. 2014, 3, 216–219. [DOI] [PubMed] [Google Scholar]
- 57. Qian H., Purwanto N. S., Ivanoff D. G., Halmes A. J., Sottos N. R., Moore J. S., Chem 2021, 7, 1080–1091. [Google Scholar]
- 58. McFadden M. E., Robb M. J., J. Am. Chem. Soc. 2021, 143, 7925–7929. [DOI] [PubMed] [Google Scholar]
- 59. Celestine A.-D. N., Beiermann B. A., May P. A., Moore J. S., Sottos N. R., White S. R., Polymer 2014, 55, 4164–4171. [Google Scholar]
- 60. Wang Q., Gossweiler G. R., Craig S. L., Zhao X., J. Mech. Phys. Solids 2015, 82, 320–344. [Google Scholar]
- 61. Celestine A.-D. N., Sottos N. R., White S. R., Strain 2019, 55, e12310. [Google Scholar]
- 62. Matsuda T., Kawakami R., Nakajima T., Gong J. P., Macromolecules 2020, 53, 8787–8795. [Google Scholar]
- 63. Slootman J., Waltz V., Yeh C. J., Baumann C., Göstl R., Comtet J., Creton C., Phys. Rev. X 2020, 10, 041045. [Google Scholar]
- 64. Morelle X. P., Sanoja G. E., Castagnet S., Creton C., Soft Matter 2021, 17, 4266–4274. [DOI] [PubMed] [Google Scholar]
- 65. Wang M., Marr J. M., Davanco M., Gilman J. W., Liddle J. A., Mater. Horiz. 2019, 6, 817–825. [Google Scholar]
- 66. Blom H., Widengren J., Chem. Rev. 2017, 117, 7377–7427. [DOI] [PubMed] [Google Scholar]
- 67. Dempsey G. T., Vaughan J. C., Chen K. H., Bates M., Zhuang X., Nat. Methods 2011, 8, 1027–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information