

Abstract
Tuberous sclerosis complex (TSC) is a rare genetic disease of autosomal dominant transmission that, in most cases, results from the presence of pathogenic variants of the TSC1 or TSC2 genes, encoding hamartin and tuberin, respectively. It is a multisystemic disease, affecting most frequently the brain, skin, kidney, and heart. The wide variety of possible clinical manifestations, given this multisystem dimension, makes the follow-up of patients with TSC an exercise of multidisciplinarity. In fact, these patients may require the intervention of various medical specialties, which thus have to combine their efforts to practice a medicine that is truly holistic. The past few years have witnessed a dramatic leap not only in the diagnosis and management of TSC patients, with standard monitoring recommendations, but also in the therapeutic field, with the use of mTORC1 inhibitors. In this article, we review the clinical manifestations associated with TSC, as well as the treatment and follow-up strategies that should be implemented, from a multidisciplinary perspective.
Keywords: tuberous sclerosis complex, multidisciplinary communications, care management, patient
Introduction
Tuberous sclerosis complex (TSC) is a rare autosomal dominant genetic disease, with an estimated incidence at birth of 1/6.000 to 1/10.000.1–3 It represents a multisystem condition, defined by the development of benign, noninvasive tumors called hamartomas in several organ systems, most commonly in the brain, kidney, heart and skin. Clinical presentation and severity are highly variable, even between closely related individuals.1–7
TSC results from pathogenic variants in TSC1 (loci 9p34) or TSC2 (16p13) genes that encode hamartin and tuberin, respectively.8,9 These proteins are involved in complex interactions with the mammalian target of rapamycin (mTOR)-signaling pathway, which controls a variety of cell functions, such as cell growth, proliferation, and survival.10–18 Hamartin and tuberin form a regulatory complex responsible for limiting the activity of an important intracellular regulator of cell growth and metabolism known as mammalian target of rapamycin complex 1 (mTORC1) via inhibition of the small GTPase Ras homolog enriched in brain (Rheb) (Figure 1).5,11–18 In TSC individuals, there is a permanent activation of the mTOR pathway.
Figure 1.

Overview of mTOR-TSC regulation and upstream and downstream mediators. Mutations in either TSC1 or TSC2 genes lead to hyperactivation of the downstream mTOR pathway by suppression of Rheb-mediated mTORC1 inhibition. The TSC1-TSC2 protein complex integrates cues such as nutrients, growth factors, hormones, and mitogens to regulate the activity of mTOR. mTOR complexes 1 and 2 are mediators of important cellular functions. Some of the downstream targets of mTORC1 were already clearly identified, including S6K1 and 4E-BP1 proteins, which play a major role in the regulation of cell growth, proliferation, and metabolism. TSC1 and TSC2 proteins have additional roles besides the modulation of mTOR, since inhibition of B-Raf kinase via Rheb is an mTOR-independent function of tuberin. Sirolimus and everolimus are effective inhibitors of mTORC1 via FKBP12.
Despite being a hereditary disease, most TSC patients (70%) result from somatic mutations, representing sporadic cases. Those patients more frequently show a mutation in TSC2.6,19
Accurate diagnosis is fundamental to the implementation of long-term surveillance programs and personalized treatment.2 Diagnostic criteria for TSC were firstly established in 1998, but in 2012, the International Tuberous Sclerosis Complex Consensus Group reviewed and established updated criteria, based on major and minor features (Table 1A). For a definite diagnosis, two major features are required or, alternatively, one major and two or more minor features should be present. The identification of either a TSC1 or TSC2 pathogenic variant20,21 is also sufficient to make a definite diagnosis of TSC.2
Table 1.
Diagnostic Criteria for Tuberous Sclerosis and Testing Recommendations of Asymptomatic Patients (Adapted from Northrup H et al2)
| A. Diagnostic Criteria for Tuberous Sclerosis | ||
|
1. Genetic diagnostic criteria The presence of pathogenic variants in TSC1 or TSC2 genes (eg, out-of-frame indel or nonsense mutation; large genomic deletion; missense mutation). |
2. Clinical diagnostic criteria 2.1) Major features ♦ Hypomelanotic macules (≥3, at least 5-mm diameter) ♦ Angiofibromas (≥3) or fibrous cephalic plaque ♦ Ungual fibromas (≥2) ♦ Shagreen patch ♦ Retinal hamartomas (multiple) ♦ Cortical dysplasias (≥3, including tubers and brain white matter radial migration lines) ♦ Subependymal nodules ♦ Subependymal giant cell astrocytoma ♦ Cardiac rhabdomyoma ♦ Lymphangioleiomyomatosis* ♦ Angiomyolipomas (≥2) * 2.2) Minor features ♦ “Confetti” skin lesions ♦ Dental enamel pits (>3) ♦ Intraoral fibromas (≥2) ♦ Retinal achromic patch ♦ Renal cysts (multiple) ♦ Nonrenal hamartomas *A combination of lymphangioleiomyomatosis and angiomyolipomas with no other clinical features does not meet criteria for a definite diagnosis (it is considered as only 1 major feature). Definite diagnosis: Two major features or one major feature with ≥2 minor features. Possible diagnosis: Either one major feature or ≥2 minor features. |
|
| B. Testing Recommendations for Asymptomatic Patients | ||
| Assessment | Initial Testing | Follow-Up Testing |
| Dermatologic examination | At diagnosis | As indicated |
| Ophthalmic examination | At diagnosis | As indicated |
| Electrocardiography | At diagnosis | At indicated |
| Echocardiography | If cardiac symptoms occur | If cardiac dysfunction occurs |
| Brain MRI | At diagnosis | Children and adolescents: every 1–3 years |
| Electroencephalography | At diagnosis | As indicated for seizure management |
| Neurodevelopmental assessment | At diagnosis and at school entry | As indicated |
| Chest computed tomography | In adulthood (women only) | If pulmonary dysfunction occurs |
| Renal Ultrasonography | At diagnosis | Every 1–3 years |
Notes: Adapted from Northrup H, Krueger DA, International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49(4):243–254. Copyright © 2013 Elsevier Inc. Creative Commons CC-BY-NC-ND.2
In the past few years, research and new technologies allowed the advancement in diagnosis and management of TSC patients, promoting not only a multidisciplinary approach, but also the clinical use of mTORC1 inhibitors, such as sirolimus and everolimus, for the treatment of several clinical manifestations associated to the condition. These drugs proved sustained regression of brain tumors, renal angiomyolipomas, liver angiomyolipomas, and pulmonary lymphangioleiomyomatosis (LAM).22–24 We aimed to provide a clinical and multidisciplinary approach to TSC patients, describing monitoring and managing perspectives for the respective clinical manifestations.
Methods
A review of the literature regarding the current aspects of TSC monitoring and managing was conducted. A comprehensive search strategy was applied on PubMed, using a combination of free text words relevant to the topic and subject headings specific to the database. High-quality peer-reviewed original manuscripts and review articles with full-text in English, were included. Results were screened based on titles and abstracts to ascertain suitability for inclusion, and when addressed the topic of the review a full text reading was performed. Book chapters that provided important data for this review were also added. In total, 85 references, published between 1993 and 2022, were identified and data extraction was performed through a qualitative content analysis approach, including genomic and biological background, clinical features, disease’s diagnosis and managing, evidence-based treatment options, and monitoring indications. The images that illustrate the article and that refer to diagnostic tests were obtained from patients who are part of our own series, after signing an informed consent, in use in our institution after approval by the local Ethics Committee.
Clinical Features and Their Management
TSC is a very heterogeneous condition with a phenotypic spectrum ranging from asymptomatic skin lesions, such as hypomelanotic macules, to drug-resistant epilepsy and cognitive deficits.2 Both genders are equally affected, but women can have more manifestations. Also, TSC1 variants are associated with milder disease, and some features (renal and liver angiomyolipomas, retinal hamartomas) are very rare or not seen at all in TSC1 patients.19,25,26 Neurological and renal manifestations, namely subependymal giant cell astrocytomas (SEGAs) (Figure 2) and renal angiomyolipomas (Figure 3), are the most feared complications, representing the main cause of significant morbidity and mortality associated with the disease.1,2,7 For SEGAs, morbidity complicating surgical treatment is estimated between 5% and 50% of the cases.27
Figure 2.
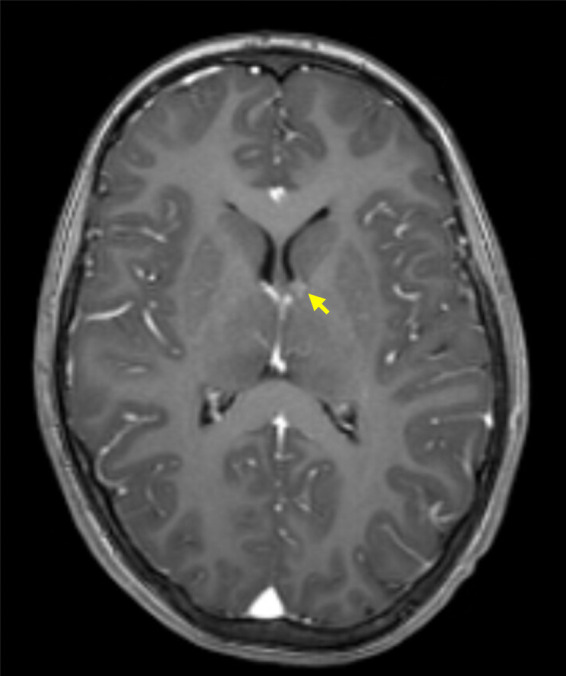
Small SEGA of the left hemisphere, in a child diagnosed with TSC. The image represents a T1-weighted MRI axial section after intravenous gadolinium administration (arrow points to SEGA).
Figure 3.
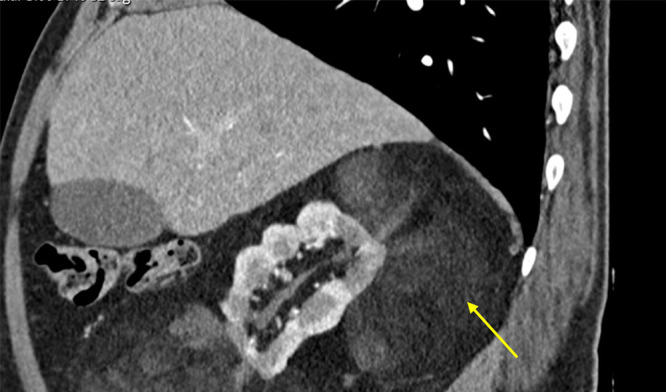
Large angiomyolipoma adjacent to the right kidney. The image is a sagittal section of a contrast-enhanced Computed Tomography scan obtained from an adult patient diagnosed with TSC, which reveals a lobulated right kidney and a voluminous mass (arrow), consisting essentially of fat, adjacent to it.
In this way, it is easily understood that TSC is a multisystem and progressive disease, requiring long-term care from a multidisciplinary team. Even for asymptomatic patients, there are specific recommendations regarding disease surveillance (Table 1B).2,7,28 The discovery of the regulation of the mTOR pathway has resulted in important clinical advances and the current therapeutic challenge is the use of mTORC1 inhibitors (Table 2) for the treatment of several TSC manifestations, with benefits already highlighted in numerous studies.2,29,30 Of the different mTOR inhibitors, only rapamycin (also known as sirolimus) and their active derivative everolimus have been clinically evaluated for the management of TSC patients.32,33
Table 2.
Pharmacological Characterization, Clinical Indications and Adverse Effects of mTOR Inhibitors for Tuberous Sclerosis Complex Treatment (Adapted from Palavra F et al5)
| Sirolimus (Rapamycin) | Everolimus | |
|---|---|---|
| Molecular weight | 914.2 g/mol | 958.2 g/mol |
| Biochemically functional form | Active form | Active derivative (hydroxyethyl ester) of sirolimus |
| Route of administration | Orally, once daily Topic (concentrations 0.1% to 1%) |
Orally, once daily |
| Protein binding | ∼92% | ∼75% |
| Bioavailability and distribution | Low oral bioavailability (solution:14%; tablet: 18%) Large distribution (around 12 L/kg), ∼95% into RBCs |
Tablet: 20% Wide distribution into RBCs; good blood-brain partition coefficient |
| Metabolization | Hepatic CYP3A4, glycoprotein 1 | CYP3A4, CYP3A5, CYP2C8 |
| Terminal half-life | 46–78 h | 26–30 h |
| Elimination | Feces (91%), urine (2%) | Feces (>90%), urine (2%) |
| Clinical indications for TSC | Approved for severe-to-moderate LAM. Proved regression of kidney and liver angiomyolipomas, SEGAs, and seizures frequency. Topical formulation for facial angiofibromas. |
Approved for SEGA and adult angiomyolipoma associated with TSC. |
| Common adverse reactions | GI effects (constipation, abdominal pain, diarrhea), peripheral oedema, hypertension, hypertriglyeridaemia, hypercholesterolaemia, creatinine increase, anaemia, thrombocytopaenia, urinary tract infection, arthralgia, headache, fever Irritation and burning sensation after topical administration. |
Stomatitis, infections, rash, fatigue, diarrhea, hypercholesterolaemia, hyperglycaemia, hypertriglyceridaemia. elevated transaminases, anaemia, leukopenia, thrombocytopenia and lymphopenia. |
Notes: Adapted from Palavra F, Robalo C, Reis F. Recent advances and challenges of mTOR inhibitors use in the treatment of patients with tuberous sclerosis complex. Oxid Med Cell Longev. 2017;2017:9820181. Copyright © 2017 Filipe Palavra et al. This is an open access article distributed under the Creative Commons Attribution License.5
Abbreviations: RBCs, red blood cells; CYP3A, intestinal cytochrome p450 3 A enzymes; SEGA, subependymal giant cell astrocytomas; GI, gastrointestinal.
Rapamycin is a natural macrolide, and its mechanisms of action are common for their analogues, both working by binding to and forming a complex with FKBP12, which then inhibits mTORC1.5,34 Two major clinical trials, EXIST-1 (efficacy and safety of everolimus for subependymal giant cell astrocytomas – SEGAs – associated with TSC) and EXIST-2 (everolimus for angiomyolipoma associated with TSC or sporadic lymphangioleiomyomatosis), demonstrated beneficial effects of everolimus in both cases, allowed the approval of this drug for the treatment of those TSC-associated manifestations. In EXIST-1, treatment with everolimus (median 9.6 months) was associated with a significantly higher SEGA response rate (≥50% reduction in volume) compared with placebo (35% vs 0%; p < 0.0001).33 In EXIST-2, the angiomyolipoma response rate (≥50% reduction in volume in absence of other factors) after approximately 8 months of treatment was 42% for patients taking everolimus (vs 0% in patients receiving placebo, p < 0.0001), which increased to 54% for a median of 29 months of treatment, and 58% at the completion of the open-label extension phase (median exposure, 46.9 months).35 Sirolimus has received approval of some medicine agencies for the treatment of LAM. In MILES study (Multicenter LAM Efficacy of Sirolimus), a multicenter placebo-controlled Phase 3 study, 89 patients with LAM (8 with a co-diagnosis of TSC) receiving treatment with sirolimus (n = 46) over 12 months exhibited improvements in forced vital capacity (FVC) and quality of life, as well as stabilization of forced expiratory volume in 1 second (FEV1).36
There are various case reports and multiple prospective clinical trials showing sirolimus benefit in TSC patients,22,24,31–33 inducing regression of kidney angiomyolipomas, SEGAs and liver angiomyolipomas, but tumor volume tended to increase after therapy withdrawal.22,23,37
The oral treatment with mTOR inhibitors is associated to increased risk of infections, stomatitis, acne, amenorrhea, hypercholesterolemia, and other metabolic disturbances.5,38 It has been demonstrated that discontinuation of treatment leads to rebound growth of tumors in the majority of patients, and considerations about this subject are important, since for clinical benefit to be sustained, chronic treatment with mTORC1 inhibitors is necessary, raising concern about the long-term adverse effects described above.5,31,39
Skin and Teeth
Dermatologic manifestations are typical features of TSC, occurring in 90–100% of the patients.2,6 Hypomelanotic macules are the main dermatologic findings and are observed in 90% of people affected by the disease. They classically appear at birth or infancy, sometimes being the presenting sign of the disease, and become less evident in late adulthood.2,39,40 Poliosis is considered a variant presentation of hypomelanosis and is included in the count of hypomelanotic macules.2,40 Facial angiofibromas are hamartomatous nodules of vascular and connective tissues and occur in about 75% of cases.6,40 Typically, they onset between 2 and 5 years-old, increasing in number and size throughout adolescence and may become quite disfiguring. A fibrous cephalic plaque may occur on the forehead or other craniofacial areas in 25% of TSC patients. Other manifestations include ungual fibromas, shagreen patches, “confetti” skin lesions, dental enamel pits, and intraoral fibromas.2,39
Surveillance of dermatological findings include a detailed clinical dermatological and dental exam at the time of diagnosis, followed by annual assessment.2,40 Skin biopsy is not required for the majority of TSC patients, however it may be necessary if clinical diagnosis remains uncertain.
Regarding treating dermatological manifestations, laser ablation, surgical excision and graft surgery are available as treatment options for facial angiofibromas.39 Enamel defects can be treated with restorative treatments if the patient is at high cavity risk. Oral fibromas should be excised surgically if symptomatic or if interfering with oral hygiene.1,40
Benefit of systemic mTORC1 inhibitors has been reported as secondary endpoint in clinical trials, with skin response rates significantly higher than placebo29,30,35,40 and current guidelines limit their use for individuals not eligible to surgical approaches and skin lesions with severe risk of recurrent and extensive hemorrhages. Sirolimus is available in a topical formulation and a small series of cases reported benefits using topical preparations of the drug in facial angiofibromas, with sustained improvement in erythema, size and extension of skin lesions.40,41 A recent study comparing different dosages of topical sirolimus (0.05%, 0.1%, 0.2% and 1%) in facial angiofibromas provided significant evidence of clinical improvement, in comparison to placebo, with topical sirolimus 0.2% being the most effective.42
Eye
Retinal hamartomas are observed in 30–50% of TSC patients and can be found at any age. They are considered good markers for the disease, particularly in young children.2,39,43 A retinal hamartoma can manifest morphologically as (a) translucent flat tumor, (b) whitish nodular (mulberry lesion) mass or (c) mixed variety. Other ocular features are retinal achromic patches (areas of hypopigmentation on the retina) that occur in 39% of the patients.2,39,43
A complete ophthalmologic examination is indicated at TSC diagnosis and recommended annually2 and, unless these lesions affect the macula or optic nerve, they are typically asymptomatic, and usually do not require specific treatment.1,2,39
Heart
Cardiac rhabdomyomas are the earliest sign of the disease appearing early in the fetus and newborn, around between the 20th and 30th weeks of gestation.1,2,6,39 These tumors measure approximately 3–25mm in diameter and are more commonly located within the ventricles. They are usually asymptomatic and often regress with age.39,44 However, they can be associated with obstruction of cardiac outflow, heart failure, dilated cardiomyopathy, arrhythmias1,2,39,44 and represent the most frequent cause of death in TSC infants and children aged <10 years.
In pediatric patients, especially younger than 3 years of age, an echocardiogram and ECG should be obtained.2 Until regression of cardiac rhabdomyomas is achieved, follow-up echocardiogram should be obtained every 1–3 years and ECG is recommended at minimum every 3–5 years to monitor for conduction defects.2 In asymptomatic adult patients, echocardiogram is not necessary, but a baseline ECG is still recommended.2
Currently, there is no approved pharmacological treatment for this condition, and cardiac surgery is the usual treatment for severe symptomatic patients.45,46 The upcoming randomized trial assessing the efficacy of everolimus as a specific therapy for symptomatic cardiac rhabdomyoma (ORACLE) will potentially support the first evidence-based therapy for this condition.46,47
Central Nervous System
The main neurological manifestation of TSC is epilepsy, which affects approximately 85% of the patients.2 In recent studies, TSC1 pathogenic variants were associated with less severe epilepsy phenotypes and more individuals with normal intelligence quotient (IQ).48 Epilepsy usually manifests during the first years of life and is often refractory to treatment.39,49–53 Although most patients develop multiple seizure types, infantile spasms are one of the major types of early seizures in TSC. These patients also present a higher risk of neurodevelopmental and cognitive impairment.48
The most frequent CNS lesions include cortical tubers, subependymal nodules (SENs) and SEGAs (Figure 2) that can grow into the ventricular system.2,53–55 Cortical tubers and migration lines in the white matter are identified in early neuroimaging studies and represent regions of cortical dysplasia that likely result from aberrant neuronal migration during corticogenesis.2,28,56 SENs are hamartomas that develop along the wall of the ependymal lining of the lateral and third ventricles in 80% of TSC patients and are usually asymptomatic. SEGAs, observed in 5–15% of TSC patients, in turn can lead to ventriculomegaly and hydrocephalus, resulting in marked morbidity and mortality.2,28,55
Neurocognitive and behavioral disturbances (TAND – Tuberous Sclerosis-Associated Neuropsychiatric Disorders) include aggressive behaviors, autism spectrum disorder, intellectual disability, psychiatric disorders, neuropsychological deficits, as well as school and occupational difficulties and can be identified in 40–50% of cases, with a highly variable expression and severity.52,57,58
All individuals with suspected TSC, regardless of age, should perform a brain magnetic resonance imaging (MRI) using conventional magnetic field strengths (1.5 or 3-Tesla), with and without gadolinium.2,59 If it is not possible or there are contraindications to perform MRI, a head computed tomography (CT) scan must be obtained. In neonates or infants, there is also the possibility to perform head ultrasound, which will disclose cortical tubers and subependymal nodules. Follow-up by MRI should be performed every 1–3 years in all individuals until the age of 25 years for SEGAs exclusion.2,31 All pediatric patients should perform a baseline electroencephalogram (EEG), even in the absence of recognized or reported clinical seizures.2 If the baseline EEG is abnormal, especially in the presence of TAND, 24-hour video-EEG to assess for electrographic or subtle clinical seizure activity is recommended.2 In fact, abnormal EEG patterns and/or subclinical seizures recorded on the EEG should urge the use of antiepileptic drug therapy without waiting for the onset of overt clinical seizures.48 TAND should be evaluated at diagnosis and follow-up appointments, at least once per year.2,48 Moreover, formal evaluations by an expert team should be performed at key time points: during the first 3 years of life, preschool, before middle school entry, during adolescence, and in early adulthood.2
Concerning treatment of CNS manifestations, surgical resection is the recommended intervention for symptomatic SEGA and cerebrospinal fluid diversion may sometimes be necessary.2,60 For growing but asymptomatic SEGA, either surgical resection or medical therapy with mTOR inhibitors can be effective.2,30 Everolimus is indicated for the treatment of patients with SEGA regardless of age, who require therapeutic intervention, but are not eligible for surgery.6,61,62
Vigabatrin, an inhibitor of gamma-aminobutyric acid (GABA) transaminase with impact on mTOR overactivation, is the first-choice for the treatment of infants with focal seizures and/or infantile spasms.30,39,63 The pre-symptomatic diagnosis strategy for epilepsy in TSC described above and emphasized in the EPISTOP study leads to recognition that preventive treatment with vigabatrin modified the natural history of seizures in TSC, reducing the risk and severity of epilepsy.48 Side effects, like retinal toxicity, should be closed monitored. Adrenocorticotrophic hormone (ACTH) or analogs can be used as second-line therapy if treatment with vigabatrin fails.2,31,39 Epilepsy surgery, ketogenic diet, or vagus nerve stimulation may be considered for refractory patients.2,31 Epilepsy surgery for TSC is a challenging procedure due to the presence of multiple tubers and the complexity of the multiplex epileptogenic network forming the epileptogenic zone. However, it is an important treatment option and data suggest that seizure freedom is achieved by 55–60% of patients. Age of seizure onset (after 1 year of life), shorter duration of epilepsy and higher IQ prior to surgery seem to be determinants of postsurgical seizure freedom.64 Both sirolimus and everolimus proved to reduce seizure frequency in TSC-related refractory epilepsy.65–68
There are also some reports of improvement in multiple aspects of social deficit behavior and neurocognition with everolimus treatment, but adequate studies to address these issues are needed.65,69–71
Lung
Lymphangioleiomyomatosis (LAM) is the replacement of alveolar tissue by cysts and proliferation of the smooth muscle, progressively affecting the pulmonary function, and affecting 40% of the female TSC patients. It is more frequently associated with TSC2 pathogenic variants and predominantly affects premenopausal women, in the third to fourth decade of life,1,2,28,39 leading to the hypothesis that estrogen regulates TSC signaling, and perhaps also the migration of TSC2-deficient cells.28,72 Early manifestations are cough, progressive dyspnea on exertion and pneumothorax.1,2,28,39 Multifocal micronodular pneumocyte hyperplasia and clear-cell tumor of the lung are rare manifestations of pulmonary involvement.2,39
Female TSC patients 18 years or older must have a baseline pulmonary function testing, 6-minute walk test, and high-resolution chest CT.2 Measuring vascular endothelial growth factor-D (VEGF-D) levels may contribute to predict future LAM progression, which can be useful to estimate the prognosis.39,73 Moreover, counseling on smoking risks and estrogen use should be achieved.2 Asymptomatic patients without normal baseline CT should perform a high-resolution chest CT every 5–10 years.2 Once cysts are detected, high-resolution chest CT every 2–3 years, annual pulmonary function testing and 6-minute walk test are recommended for evaluation of LAM progression.2
Pulmonary LAM is usually generalized and progressive, associated with a poor prognosis and there is no curative treatment, except lung transplantation.1,2,6,74 Sirolimus in now approved for use as treatment of moderate-to-severe LAM by the United States Food and Drug Administration (FDA) and by regulatory agencies in Japan, South Korea, Brazil and Russia, contributing for stabilization or improvement in pulmonary function and reduction serum VEGF-D levels and symptoms.2,23,24,36,39 Until now, no randomized studies of everolimus have been conducted for patients with LAM.
Kidney
Renal angiomyolipomas are very common benign tumors in TSC patients, reported in 80% of cases.2,75 They are bilateral, multiple and tend to grow slowly from the first years of life, with an acceleration growth during adolescence or early adulthood. There is a small subgroup that can behave more aggressively and become locally invasive and tumors larger than 30 mm have highest risk of bleeding.75 Although most of them are asymptomatic, they represent one of the most frequent causes of TSC-related morbidity and mortality. In addition to angiomyolipomas, TSC patients have a higher incidence of multiple renal cysts, when compared to the general population.
There are reports of rare TSC-related renal cell carcinoma in children and young adults, with an overall incidence of 2%,52 which makes the diagnosis challenging, as they can be confused with angiomyolipomas. In these cases, if there are clinical doubts, a biopsy with histology is recommended to avoid invasive surgical procedures.
All patients, regardless of age, should perform abdominal imaging at the time of diagnosis, with MRI being the preferred modality for evaluation of angiomyolipoma and renal function must be monitored.2,76 Abdominal MRI or ultrasound should be performed to assess the progression of angiomyolipomas every 1–3 years throughout the lifetime of the patient. Posteriorly, every 1–3 years imaging evaluation and annually assessment of renal function and hypertension are required.2
Everolimus is approved for the treatment of TSC-associated angiomyolipomas and mTOR inhibitors are recommended as the first-line treatment for asymptomatic, growing renal angiomyolipomas >3 cm.2,31,34,39 Selective embolization, kidney-sparing resection or ablative therapy are acceptable second-line therapies for asymptomatic lesions.2 Emergency embolization is the first-line therapy for acute bleeding or associated aneurism.31,39 Nephrectomy should be avoided whenever possible, because of the high incidence of complications and increased risk of future renal failure.2,31,39
Other Manifestations
Various types of hamartoma lesions can occur in the endocrine system, namely adrenal angiomyolipoma, thyroid papillary adenoma, pituitary, pancreas, or gonads angiomyolipomas or fibroadenomas.2,7 Neuroendocrine tumors might be slightly more prevalent in TSC patients, but these lesions are not hamartomas.2,7,39 Hepatic multiple angiomyolipomas are reported in 10–25% of TSC patients, and they are usually asymptomatic and more common in adult women.1,2,76
Future Perspectives
TSC is a rare genetic disorder resulting from pathogenic variants in TSC1 and TSC2 genes, leading to hyperactivation of mTOR-pathway signaling which, in turn, cause the development of hamartomas.1–19 Given that this pathway controls a variety of cell functions, such as cell growth, proliferation, and survival, it affects several organs and systems in numerous ways, which are responsible for the great clinical heterogeneity of this condition.
Recent advances in research and new technologies allowed the integration of consensus in diagnosis, management and treatment of TSC, promoting a multidisciplinary approach.2,77 The approval of mTOR inhibitors for the treatment of SEGA, angiomyolipomas and LAM and their advantageous use for refractory epilepsy and other TSC manifestations represent a relevant advance in the pharmacological management of the disease.22–24,65–68 Nevertheless, despite available pre-clinical and clinical data encouraging their use in TSC treatment, more robust knowledge about how to choose the appropriate drug, dosing, formulation and long-term side effects must be considered.
Lately, second-generation mTOR inhibitors (known as mTOR kinase inhibitors or TORKinibs) have been developed. They differ from rapamycin analogues by the ability to directly inhibit the kinase by blocking the ATP catalytic site, rather than linking FKBP12, which causes inhibition of both mTORC1 and mTORC2.5,78,79 This property allows them to have distinct implications on downstream target inhibition, as well as on mechanisms of protein translation control, on regulation loops and help to differentiate TORC1- from TORC2-mediated intracellular signaling. However, if dual targeting of TORC1/TORC2 will be able to achieve higher efficacy without further relevant toxicity, comparing with everolimus, is yet to be known.5,7 These new pharmacological agents are interesting potent antiproliferative strategies and might open new research avenues for several pathological conditions, namely in oncology field.
To date, other newer therapeutic strategies are under development. One exciting future direction for the treatment of mTORopathies is gene therapy, based on antisense oligonucleotides and clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9-based systems.80 Another progress is the potential use of immunotherapy for the treatment of LAM. Recent data indicate that T cells within LAM nodules and renal angiomyolipomas exhibit features of T-cell exhaustion, with coinhibitory receptor programmed cell death protein 1 (PD-1) expression on tumor-infiltrating T cells.81 Recent works on animal models highlighted on the potential utilization of immune checkpoint inhibitors for LAM, angiomyolipomas, and potentially for other TSC-associated tumors, with evidence on the improvement of T-cell exhaustion markers.81
Also, many fundamental questions regarding genotype–phenotype correlation remain unanswered. TSC is associated with developmental brain malformations, autism, and intellectual disability. Apart from those features, there are several different conditions that were already connected to mTOR dysfunction, including hypoxic-ischaemic brain injury and neurodegenerative processes.5,82 Therefore, distinct cell types and circuits may be responsible for the different neurological phenotypes.5,80 How the use of systemic mTOR inhibitors can be optimized, targeting only the affected brain area or brain signaling pathways is now a topic under debate and active research.
Conclusion
The heterogeneity of TSC manifestations represents a challenge to the implementation of appropriate treatment and follow-up protocols, which are frequently fragmented and suboptimal.83 TSC patients have a systemic and progressive disease, most of them developing significant morbidity and needing a holistic systematic monitoring by a multidisciplinary team. The definition of such a team is viewed as extremely relevant, but it may not be easy, lacking its own construct. Nevertheless, there are works that can guide its implementation in the field.84
The introduction of mTOR inhibitors in therapeutic protocols emphasizes the need for this intimate collaboration between well-prepared health professionals. Questions remain regarding to decide who, how, when, and what to treat, as well as how to manage long-term side effects of drugs. More epidemiological and clinical data like the Tuberous Sclerosis Registry to Increase Disease Awareness (TOSCA) registry and additional studies are crucial to answer these pertinent questions and to guide future clinical research.85 Yet, the key to successful therapeutic intervention in a complex systemic disease such as this is multidisciplinary collaboration.
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Curatolo P, Bombardieri R, Jozwiak S. Tuberous sclerosis. Lancet. 2008;372(9639):657–668. doi: 10.1016/S0140-6736(08)61279-9 [DOI] [PubMed] [Google Scholar]
- 2.Northrup H, Krueger DA, International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49(4):243–254. doi: 10.1016/j.pediatrneurol.2013.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Callaghan FJ, Shiell AW, Osborne JP, Martyn CN. Prevalence of tuberous sclerosis estimated by capture-recapture analysis. Lancet. 1998;351(9114):1490. doi: 10.1016/S0140-6736(05)78872-3 [DOI] [PubMed] [Google Scholar]
- 4.Lendvay TS, Marshall FF. The tuberous sclerosis complex and its highly variable manifestations. J Urol. 2003;169(5):1635–1642. doi: 10.1097/01.ju.0000058253.40352.60 [DOI] [PubMed] [Google Scholar]
- 5.Palavra F, Robalo C, Reis F. Recent advances and challenges of mTOR inhibitors use in the treatment of patients with tuberous sclerosis complex. Oxid Med Cell Longev. 2017;2017:9820181. doi: 10.1155/2017/9820181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Portocarrero LKL, Quental KN, Samorano LP, Oliveira ZNP, Rivitti-Machado MCM. Tuberous sclerosis complex: review based on new diagnostic criteria. An Bras Dermatol. 2018;93(3):323–331. doi: 10.1590/abd1806-4841.20186972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jesus-Ribeiro J, Palavra F, Reis F. Chapter 99: Tuberous Sclerosis Complex, Handbook of Tumor Syndromes. 1st ed. Boca Raton: CRC Press; 2020. DOI: 10.1201/9781351187435 [DOI] [Google Scholar]
- 8.European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell. 1993;75(7):1305–1315. doi: 10.1016/0092-8674(93)90618-Z [DOI] [PubMed] [Google Scholar]
- 9.van Slegtenhorst M, de Hoogt R, Hermans C, et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science. 1997;277(5327):805. doi: 10.1126/science.277.5327.805 [DOI] [PubMed] [Google Scholar]
- 10.Langkau N, Martin N, Brandt R, et al. TSC1 and TSC2 mutations in tuberous sclerosis, the associated phenotypes and a model to explain observed TSC1/ TSC2 frequency ratios. Eur J Pediatr. 2002;161(7):393–402. doi: 10.1007/s00431-001-0903-7 [DOI] [PubMed] [Google Scholar]
- 11.Huang J, Dibble CC, Matsuzaki M, Manning BD. The TSC1- TSC2 complex is required for proper activation of mTOR complex 2. Mol Cell Biol. 2008;28(12):4104–4115. doi: 10.1128/MCB.00289-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garami A, Zwartkruis FJ, Nobukuni T, et al. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell. 2003;11(6):1457–1466. doi: 10.1016/S1097-2765(03)00220-X [DOI] [PubMed] [Google Scholar]
- 13.Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10(1):151–162. doi: 10.1016/S1097-2765(02)00568-3 [DOI] [PubMed] [Google Scholar]
- 14.Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc Natl Acad Sci USA. 2004;101(37):13489–13494. doi: 10.1073/pnas.0405659101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jozwiak J, Jozwiak S, Grzela T, Lazarczyk M. Positive and negative regulation of TSC2 activity and its effects on downstream effectors of the mTOR pathway. Neuromolecular Med. 2005;7(4):287–296. doi: 10.1385/NMM:7:4:287 [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Gao X, Saucedo LJ, Ru B, Edgar BA, Pan D. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat Cell Biol. 2003;5(6):578–581. doi: 10.1038/ncb999 [DOI] [PubMed] [Google Scholar]
- 17.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–293. doi: 10.1016/j.cell.2012.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jacinto E, Loewith R, Schmidt A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6(11):1122–1128. doi: 10.1038/ncb1183 [DOI] [PubMed] [Google Scholar]
- 19.Jones AC, Shyamsundar MM, Thomas MW, et al. Comprehensive mutation analysis of TSC1 and TSC2-and phenotypic correlations in 150 families with tuberous sclerosis. Am J Hum Genet. 1999;64(5):1305–1315. doi: 10.1086/302381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Slegtenhorst M, Verhoef S, Tempelaars A, et al. Mutational spectrum of the TSC1 gene in a cohort of 225 tuberous sclerosis complex patients: no evidence for genotype-phenotype correlation. J Med Genet. 1999;36(4):285–289. [PMC free article] [PubMed] [Google Scholar]
- 21.Kozlowski P, Roberts P, Dabora S, et al. Identification of 54 large deletions/duplications in TSC1 and TSC2 using MLPA, and genotype-phenotype correlations. Hum Genet. 2007;121(3–4):389–400. doi: 10.1007/s00439-006-0308-9 [DOI] [PubMed] [Google Scholar]
- 22.Dabora SL, Franz DN, Ashwal S, et al. Multicenter Phase 2 trial of sirolimus for tuberous sclerosis: kidney angiomyolipomas and other tumors regress and VEGF-D levels decrease. PLoS One. 2011;6(9):e23379. doi: 10.1371/journal.pone.0023379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davies DM, de Vries PJ, Johnson SR, et al. Sirolimus therapy for angiomyolipoma in tuberous sclerosis and sporadic lymphangioleiomyomatosis: a phase 2 trial. Clin Cancer Res. 2011;17(12):4071–4081. doi: 10.1158/1078-0432.CCR-11-0445 [DOI] [PubMed] [Google Scholar]
- 24.McCormack FX, Inoue Y, Moss J, et al. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. New Engl J Med. 2011;364(17):1595–1606. doi: 10.1056/NEJMoa1100391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sancak O, Nellist M, Goedbloed M, et al. Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype–phenotype correlations and comparison of diagnostic DNA techniques in Tuberous Sclerosis Complex. Eur J Hum Genet. 2005;13(6):731–741. doi: 10.1038/sj.ejhg.5201402 [DOI] [PubMed] [Google Scholar]
- 26.Dabora SL, Jozwiak S, Franz DN, et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am J Hum Genet. 2001;68(1):64–80. doi: 10.1086/316951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frassanito P, Noya C, Tamburrino G. Current trends in the management of subependymal giant cell astrocytomas in tuberous sclerosis. Childs Nerv Syst. 2020;36(10):2527–2536. doi: 10.1007/s00381-020-04889-9 [DOI] [PubMed] [Google Scholar]
- 28.Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355(13):1345–1356. doi: 10.1056/NEJMra055323 [DOI] [PubMed] [Google Scholar]
- 29.Franz DN, Belousova E, Sparagana S, et al. Long-term use of everolimus in patients with tuberous sclerosis complex: final results from the EXIST-1 study. PLoS One. 2016;11(6):e0158476. doi: 10.1371/journal.pone.0158476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sasongko TH, Ismail NF, Zabidi-Hussin Z. Rapamycin and rapalogs for tuberous sclerosis complex. Cochrane Database Syst Rev. 2016;7(7):Cd011272. doi: 10.1002/14651858.CD011272.pub2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Curatolo P, Bjornvold M, Dill PE, et al. The role of mTOR inhibitors in the treatment of patients with tuberous sclerosis complex: evidence-based and expert opinions. Drugs. 2016;76(5):551–565. doi: 10.1007/s40265-016-0552-9 [DOI] [PubMed] [Google Scholar]
- 32.MacKeigan JP, Krueger DA. Differentiating the mTOR inhibitors everolimus and sirolimus in the treatment of tuberous sclerosis complex. Neuro Oncol. 2015;17(12):1550–1559. doi: 10.1093/neuonc/nov152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Franz DN, Belousova E, Sparagana S, et al. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2013;381(9861):125–132. doi: 10.1016/S0140-6736(12)61134-9 [DOI] [PubMed] [Google Scholar]
- 34.Franz DN, Capal JK. mTOR inhibitors in the pharmacologic management of tuberous sclerosis complex and their potential role in other rare neurodevelopmental disorders. Orphanet J Rare Dis. 2017;12(1):51. doi: 10.1186/s13023-017-0596-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bissler JJ, Kingswood JC, Radzikowska E, et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet. 2013;381(9869):817–824. doi: 10.1016/S0140-6736(12)61767-X [DOI] [PubMed] [Google Scholar]
- 36.Gupta N, Henske EP. Pulmonary manifestations in tuberous sclerosis complex. Am J Med Genet C Semin Med Genet. 2018;178(3):326–337. doi: 10.1002/ajmg.c.31638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bissler JJ, McCormack FX, Young LR, et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. 2008;358(2):140–151. doi: 10.1056/NEJMoa063564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sadowski K, Kotulska K, Jozwiak S. Management of side effects of mTOR inhibitors in tuberous sclerosis patients. Pharmacol Rep. 2016;68(3):536–542. doi: 10.1016/j.pharep.2016.01.005 [DOI] [PubMed] [Google Scholar]
- 39.Wataya-Kaneda M, Uemura M, Fujita K, et al. Tuberous sclerosis complex: recent advances in manifestations and therapy. Int J Urol. 2017;24(9):681–691. doi: 10.1111/iju.13390 [DOI] [PubMed] [Google Scholar]
- 40.Ebrahimi-Fakhari D, Meyer S, Vogt T, Pfohler C, Muller CSL. Dermatological manifestations of tuberous sclerosis complex (TSC). J Dtsch Dermatol Ges. 2017;15(7):695–700. [DOI] [PubMed] [Google Scholar]
- 41.Wheless JW, Almoazen H. A novel topical rapamycin cream for the treatment of facial angiofibromas in tuberous sclerosis complex. J Child Neurol. 2013;28(7):933–936. doi: 10.1177/0883073813488664 [DOI] [PubMed] [Google Scholar]
- 42.Lin YT, Yu CL, Tu YK, Chi CC. Efficacy and safety of topical mechanistic target of rapamycin inhibitors for facial angiofibromas in patients with tuberous sclerosis complex: a systematic review and network meta-analysis. Biomedicines. 2022;10(4):826. doi: 10.3390/biomedicines10040826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rowley SA, O’Callaghan FJ, Osborne JP. Ophthalmic manifestations of tuberous sclerosis: a population based study. Brit J Ophthalmol. 2001;85(4):420–423. doi: 10.1136/bjo.85.4.420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kwiatkowska J, Waldoch A, Meyer-Szary J, Potaz P, Grzybiak M. Cardiac tumors in children: a 20-year review of clinical presentation, diagnostics and treatment. Adv Clin Exp Med. 2017;26(2):319–326. doi: 10.17219/acem/62121 [DOI] [PubMed] [Google Scholar]
- 45.Dahdah N. Everolimus for the treatment of tuberous sclerosis complex–related cardiac rhabdomyomas in pediatric patients. J Pediatr. 2017;190:21–26.e7. doi: 10.1016/j.jpeds.2017.06.076 [DOI] [PubMed] [Google Scholar]
- 46.Sugalska M, Tomik A, Józwiak S, Werner B. Treatment of cardiac rhabdomyomas with mTOR inhibitors in children with tuberous sclerosis complex – a systematic review. Int J Environ Res Public Health. 2021;18(9):4907. doi: 10.3390/ijerph18094907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stelmaszewski EV, Parente DB, Farina A, et al. Everolimus for cardiac rhabdomyomas in children with tuberous sclerosis. The ORACLE study protocol (everOlimus for caRdiac rhAbdomyomas in tuberous sCLErosis): a randomised, multicentre, placebo-controlled, double-blind Phase II trial. Cardiol Young. 2020;30(3):337–345. doi: 10.1017/S1047951119003147 [DOI] [PubMed] [Google Scholar]
- 48.Nabbout R, Belousova E, Benedik MP, et al. Historical patterns of diagnosis, treatments, and outcome of epilepsy associated with tuberous sclerosis complex: results from TOSCA registry. Front Neurol. 2021;12:697467. doi: 10.3389/fneur.2021.697467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Calcagnotto ME, Paredes MF, Tihan T, Barbaro NM, Baraban SC. Dysfunction of synaptic inhibition in epilepsy associated with focal cortical dysplasia. J Neurosci. 2005;25(42):9649–9657. doi: 10.1523/JNEUROSCI.2687-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guerrini R, Carrozzo R. Epileptogenic brain malformations: clinical presentation, malformative patterns and indications for genetic testing. Seizure. 2001;10(7):532–543. doi: 10.1053/seiz.2001.0650 [DOI] [PubMed] [Google Scholar]
- 51.Leo A, Constanti A, Coppola A, Citraro R, De Sarro G, Russo E. Chapter 8 – mTOR signaling in epilepsy and epileptogenesis: preclinical and clinical studies. Maiese, Kenneth. In: Molecules to Medicine with mTOR. Boston: Academic Press; 2016:123–142. [Google Scholar]
- 52.Orlova KA, Crino PB. The tuberous sclerosis complex. Ann New York Acad Sci. 2010;1184:87–105. doi: 10.1111/j.1749-6632.2009.05117.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chu-Shore CJ, Major P, Camposano S, Muzykewicz D, Thiele EA. The natural history of epilepsy in tuberous sclerosis complex. Epilepsia. 2010;51(7):1236–1241. doi: 10.1111/j.1528-1167.2009.02474.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.DiMario FJ. Brain abnormalities in tuberous sclerosis complex. J Child Neurol. 2004;19(9):650–657. doi: 10.1177/08830738040190090401 [DOI] [PubMed] [Google Scholar]
- 55.Chan JA, Zhang H, Roberts PS, et al. Pathogenesis of tuberous sclerosis subependymal giant cell astrocytomas: biallelic inactivation of TSC1 or TSC2 leads to mTOR activation. J Neuropathol Exp Neurol. 2004;63(12):1236–1242. doi: 10.1093/jnen/63.12.1236 [DOI] [PubMed] [Google Scholar]
- 56.Baybis M, Yu J, Lee A, et al. mTOR cascade activation distinguishes tubers from focal cortical dysplasia. Ann Neurol. 2004;56(4):478–487. doi: 10.1002/ana.20211 [DOI] [PubMed] [Google Scholar]
- 57.Crino PB, Henske EP. New developments in the neuro-biology of the tuberous sclerosis complex. Neurology. 1999;53(7):1384–1390. doi: 10.1212/WNL.53.7.1384 [DOI] [PubMed] [Google Scholar]
- 58.Joinson C, O’Callaghan FJ, Osborne JP, Martyn C, Harris T, Bolton PF. Learning disability and epilepsy in an epidemiological sample of individuals with tuberous sclerosis complex. Psychol Med. 2003;33(2):335–344. doi: 10.1017/S0033291702007092 [DOI] [PubMed] [Google Scholar]
- 59.Kalantari BN, Salamon N. Neuroimaging of tuberous sclerosis: spectrum of pathologic findings and frontiers in imaging. Am J Roentgenol. 2008;190(5):W304–9. doi: 10.2214/AJR.07.2928 [DOI] [PubMed] [Google Scholar]
- 60.Kotulska K, Borkowska J, Roszkowski M, et al. Surgical treatment of subependymal giant cell astrocytoma in tuberous sclerosis complex patients. Pediatr Neurol. 2014;50(4):307–312. doi: 10.1016/j.pediatrneurol.2013.12.004 [DOI] [PubMed] [Google Scholar]
- 61.Krueger DA, Care MM, Holland K, et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. 2010;363(19):1801–1811. doi: 10.1056/NEJMoa1001671 [DOI] [PubMed] [Google Scholar]
- 62.Kotulska K, Chmielewski D, Borkowska J, et al. Long-term effect of everolimus on epilepsy and growth in children under 3 years of age treated for subependymal giant cell astrocytoma associated with tuberous sclerosis complex. Eur J Paediatr Neurol. 2013;17(5):479–485. doi: 10.1016/j.ejpn.2013.03.002 [DOI] [PubMed] [Google Scholar]
- 63.Moavero R, Cerminara C, Curatolo P. Epilepsy secondary to tuberous sclerosis: lessons learned and current challenges. Childs Nerv Syst. 2010;26(11):1495–1504. doi: 10.1007/s00381-010-1128-8 [DOI] [PubMed] [Google Scholar]
- 64.Specchio N, Pavia GC, de Palma L, et al. Current role of surgery for tuberous sclerosis complex-associated epilepsy. Pediatr Investig. 2022;6(1):16–22. doi: 10.1002/ped4.12312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Krueger DA, Wilfong AA, Holland-Bouley K, et al. Everolimus treatment of refractory epilepsy in tuberous sclerosis complex. Ann Neurol. 2013;74(5):679–687. doi: 10.1002/ana.23960 [DOI] [PubMed] [Google Scholar]
- 66.French JA, Lawson JA, Yapici Z, et al. Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. Lancet. 2016;388(10056):2153–2163. doi: 10.1016/S0140-6736(16)31419-2 [DOI] [PubMed] [Google Scholar]
- 67.Canpolat M, Per H, Gumus H, et al. Rapamycin has a beneficial effect on controlling epilepsy in children with tuberous sclerosis complex: results of 7 children from a cohort of 86. Childs Nerv Syst. 2014;30(2):227–240. doi: 10.1007/s00381-013-2185-6 [DOI] [PubMed] [Google Scholar]
- 68.Cardamone M, Flanagan D, Mowat D, Kennedy SE, Chopra M, Lawson JA. Mammalian target of rapamycin inhibitors for intractable epilepsy and subependymal giant cell astrocytomas in tuberous sclerosis complex. J Pediatr. 2014;164(5):1195–1200. doi: 10.1016/j.jpeds.2013.12.053 [DOI] [PubMed] [Google Scholar]
- 69.Wesseling H, Elgersma Y, Bahn S. A brain proteomic investigation of rapamycin effects in the Tsc1± mouse model. Mol Autism. 2017;8:41. doi: 10.1186/s13229-017-0151-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hwang SK, Lee JH, Yang JE, et al. Everolimus improves neuropsychiatric symptoms in a patient with tuberous sclerosis carrying a novel TSC2 mutation. Mol Brain. 2016;9(1):56. doi: 10.1186/s13041-016-0222-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schneider M, de Vries PJ, Schonig K, Rossner V, Waltereit R. mTOR inhibitor reverses autistic-like social deficit behaviours in adult rats with both Tsc2 haploinsufficiency and develop- mental status epilepticus. Eur Arch Psychiat Clin Neurosci. 2017;267(5):455–463. doi: 10.1007/s00406-016-0703-8 [DOI] [PubMed] [Google Scholar]
- 72.Rosner M, Hengstschlager M. Tuberin binds p27 and negatively regulates its interaction with the SCF component Skp2. J Biol Chem. 2004;279(47):48707–48715. doi: 10.1074/jbc.M405528200 [DOI] [PubMed] [Google Scholar]
- 73.Young LR, Vandyke R, Gulleman PM, et al. Serum vascular endothelial growth factor-D prospectively distinguishes lymphangioleiomyomatosis from other diseases. Chest. 2010;138(3):674–681. doi: 10.1378/chest.10-0573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Karbowniczek M, Astrinidis A, Balsara BR, et al. Recurrent lymphangiomyomatosis after transplantation: genetic analyses reveal a metastatic mechanism. Am J Respir Crit Care Med. 2003;167(7):976–982. doi: 10.1164/rccm.200208-969OC [DOI] [PubMed] [Google Scholar]
- 75.Hatano T, Egawa S. Renal angiomyolipoma with tuberous sclerosis complex: how it differs from sporadic angiomyolipoma in both management and care. Asian J Surg. 2020;43(10):967–972. doi: 10.1016/j.asjsur.2019.12.008 [DOI] [PubMed] [Google Scholar]
- 76.Halpenny D, Snow A, McNeill G, Torreggiani WC. The radiological diagnosis and treatment of renal angiomyolipoma-current status. Clin Radiol. 2010;65(2):99–108. doi: 10.1016/j.crad.2009.09.014 [DOI] [PubMed] [Google Scholar]
- 77.Volpi A, Sala G, Lesma E, et al. Tuberous sclerosis complex: new insights into clinical and therapeutic approach. J Nephrol. 2019;32(3):355–363. doi: 10.1007/s40620-018-0547-6 [DOI] [PubMed] [Google Scholar]
- 78.Feldman ME, Shokat KM. New inhibitors of the PI3KAkt-mTOR pathway: insights into mTOR signaling from a new generation of tor kinase domain inhibitors (TORKinibs). Curr Top Microbiol Immunol. 2010;347(1):241–262. doi: 10.1007/82_2010_64 [DOI] [PubMed] [Google Scholar]
- 79.Sun SY. mTOR kinase inhibitors as potential cancer therapeutic drugs. Cancer Lett. 2013;340(1):1–8. doi: 10.1016/j.canlet.2013.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Karalis V, Bateup HS. Current approaches and future directions for the treatment of mTORopathies. Dev Neurosci. 2021;43(3–4):143–158. doi: 10.1159/000515672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu HJ, Krymskaya VP, Henske EP. Immunotherapy for lymphangioleiomyomatosis and tuberous sclerosis: progress and future directions. Chest. 2019;156(6):1062–1067. doi: 10.1016/j.chest.2019.08.005 [DOI] [PubMed] [Google Scholar]
- 82.Crino PB. The mTOR signalling cascade: paving new roads to cure neurological disease. Nat Rev Neurol. 2016;12(7):379–392. doi: 10.1038/nrneurol.2016.81 [DOI] [PubMed] [Google Scholar]
- 83.Annear NMP, Appleton RE, Bassi Z, et al. Tuberous Sclerosis Complex (TSC): expert recommendations for provision of coordinated care. Front Neurol. 2019;10:1116. doi: 10.3389/fneur.2019.01116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Auvin S, Bissler JJ, Cottin V, et al. A step-wise approach for establishing a multidisciplinary team for the management of tuberous sclerosis complex: a Delphi consensus report. Orphanet J Rare Dis. 2019;14(1):91. doi: 10.1186/s13023-019-1072-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kingswood JC, Bruzzi P, Curatolo P, et al. TOSCA – first international registry to address knowledge gaps in the natural history and management of tuberous sclerosis complex. Orphanet J Rare Dis. 2014;9:182. doi: 10.1186/s13023-014-0182-9 [DOI] [PMC free article] [PubMed] [Google Scholar]