

Abstract
A cytochrome c heme protein was recently engineered to catalyze the formation of carbon–silicon bonds via carbene insertion into Si–H bonds, a reaction that was not previously known to be catalyzed by a protein. High chemoselectivity toward C–Si bond formation over competing C–N bond formation was achieved, although this trait was not screened for during directed evolution. Using computational and experimental tools, we now establish that activity and chemoselectivity are modulated by conformational dynamics of a protein loop that covers the substrate access to the iron–carbene active species. Mutagenesis of residues computationally predicted to control the loop conformation altered the protein’s chemoselectivity from preferred silylation to preferred amination of a substrate containing both N–H and Si–H functionalities. We demonstrate that information on protein structure and conformational dynamics, combined with knowledge of mechanism, leads to understanding of how non-natural and selective chemical transformations can be introduced into the biological world.
Graphical Abstract
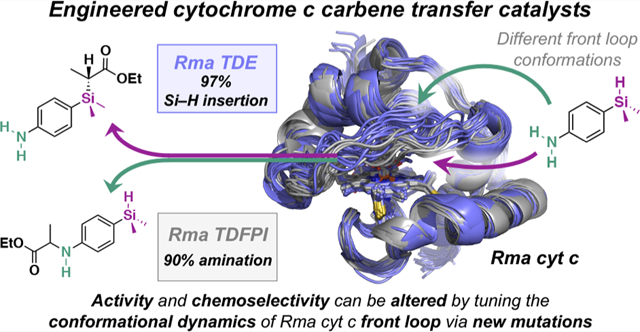
INTRODUCTION
The Arnold lab has expanded the catalytic repertoire of enzymes toward new-to-nature chemistries and recently created a carbon–silicon bond-forming enzyme1 by directed evolution of Rhodothermus marinus cytochrome c (Rma cyt c), whose native function is electron transport.2 The laboratory-evolved protein (Rma cyt c V75T M100D M103E variant, Rma TDE) is capable of catalyzing carbene insertions into Si–H bonds to form new carbon–silicon bonds (Scheme 1). Rma cyt c has also been evolved to efficiently catalyze carbene insertions into B–H bonds in a selective manner,3,4 high-lighting the versatility and evolvability of this protein scaffold toward unnatural carbene transfer reactions. Although the catalytic activities of these novel enzymes are established in previous work,1,3–5 how and why this particular protein scaffold leads to new biocatalysts that efficiently and selectively catalyze these new-to-nature reactions remains unknown. Here, we investigate the reaction mechanism and use our mechanistic studies to help explain how certain mutations alter and significantly enhance enzyme reactivity and selectivity.
Scheme 1a.
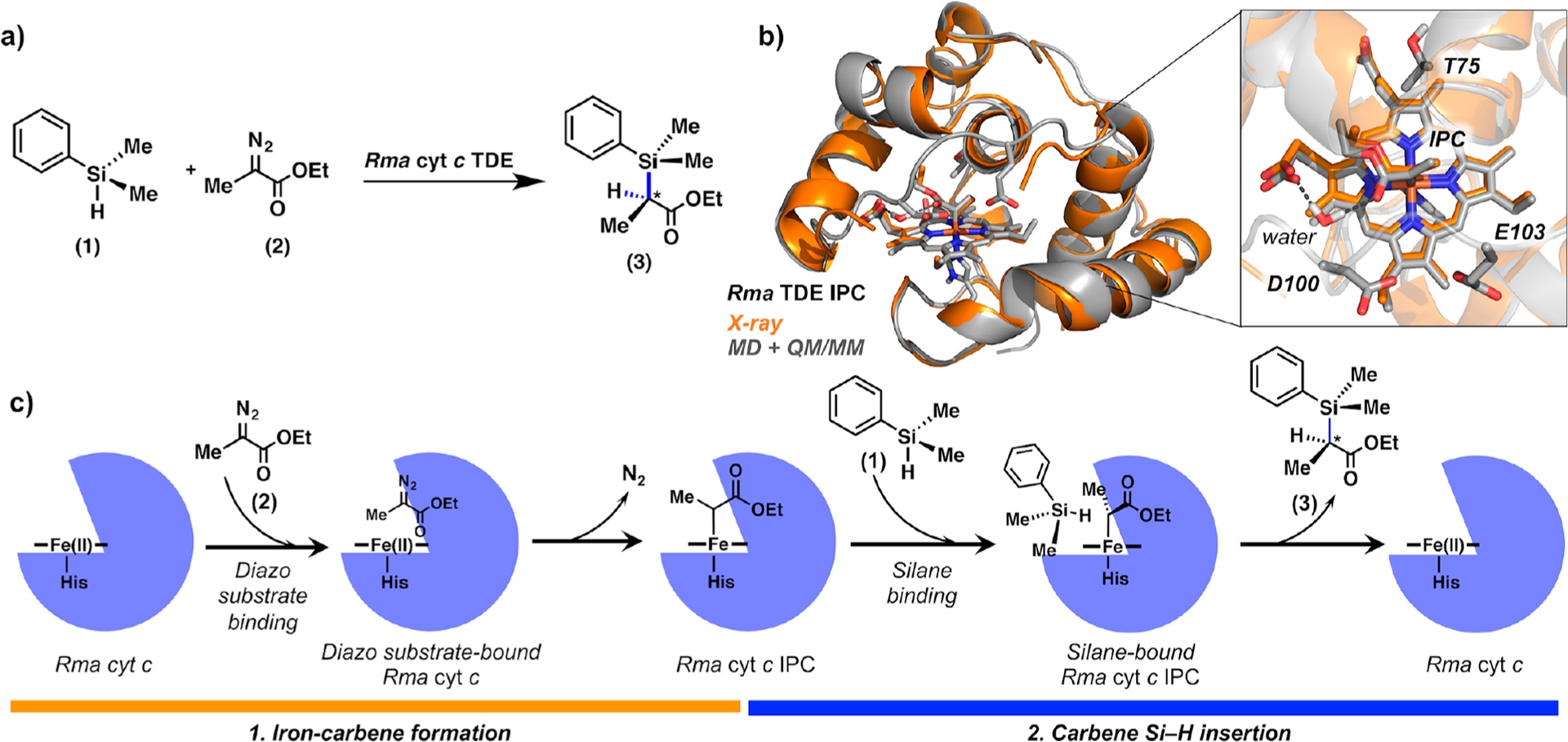
a(a) Stereoselective carbon–silicon bond formation catalyzed by Rma cytochrome c V75T M100D M103E (Rma TDE). Rma TDE was evolved to catalyze carbene insertions into Si–H bonds using silane 1 and ethyl 2-diazopropanoate (Me-EDA, 2) as substrates. (b) Overlay of the X-ray crystal structure of carbene-bound Rma TDE (1.29 Å; PDB code 6CUN; in orange) and the computationally modeled (MD + QM/MM-optimized, in gray) structure from our previous work.7 Iron porphyrin carbene (IPC), mutated residues, and computationally characterized bridging water molecule are shown in stick format. (c) Schematic description of the proposed mechanism for Rma cytochrome c-catalyzed carbon–silicon bond formation via (1) iron–carbene formation followed by (2) carbene Si–H insertion.
We employed experimental and computational tools to investigate the mechanism of Rma TDE-catalyzed carbene Si–H insertion (Scheme 1a); our results are similar to those reported by Fasan and Zhang for a similar myoglobin-catalyzed carbene Si–H insertion.6 We then investigate how the reaction is affected by the Rma TDE protein scaffold and mutations introduced by directed evolution. We propose that the conformational dynamics of a single loop near the heme play a key role in determining the activity and chemoselectivity of the enzyme. Finally, we use our findings to guide enzyme engineering to switch silylation–amination chemoselectivity and demonstrate that this altered selectivity is mediated by new and distinct conformational dynamics of the loop.
RESULTS AND DISCUSSION
1. Characterization of the Reaction Mechanism of Rma cyt c-Catalyzed Si–H Carbene Insertions.
Heme protein-catalyzed carbene insertion reactions are proposed to occur through the formation of a reactive iron porphyrin carbene (IPC) intermediate, followed by transfer of the carbene to a second substrate such as an olefin or an amine N–H bond (Scheme 1).7–10 Previous studies of iron porphyrin-catalyzed carbene transfer reactions have suggested that three distinct IPC electronic states could be involved in these reactions: the closed-shell singlet (CSS), open-shell singlet (OSS), and triplet (T) states.8–13 The protein structure, particularly the amino acid ligand that coordinates the iron porphyrin, has been shown to influence the relative stabilities of various electronic states.7
To characterize the mechanism of carbene insertion into Si–H bonds catalyzed by Rma cytochrome c M100D V75T M103E (Rma TDE), we performed density functional theory (DFT) calculations using a simple truncated model (imidazole-ligated iron porphyrin) and hybrid quantum mechanics/molecular mechanics (QM/MM) calculations (see Supporting Information methods, section IIb). In our previous work, MD simulations revealed the presence of a bridging water molecule that establishes persistent H-bond interactions with the heme carboxylate group and the carbonyl group of the carbene once the IPC is formed (Scheme 1b).7 This interaction helps maintain the carbene in a single conformation and controls the stereoselectivity of the carbene transfer reaction. In order to further characterize the impact of this water molecule on the reaction mechanism, we explicitly considered it in the QM/MM calculations which account for the effect of the protein environment surrounding the histidine-ligated iron porphyrin. We considered the closed-shell singlet (CSS), open-shell singlet (OSS), and triplet (T) electronic states along the reaction pathway (see Supporting Information methods, section IIa).
The first step of the reaction (Scheme 1c) involves iron–carbene formation from the ethyl 2-diazopropanoate substrate (Me-EDA, 2) and loss of N2. Both DFT and QM/MM calculations showed that the closed-shell singlet transition state (TS) is lowest in energy, while the open-shell singlet TS and triplet TS are higher in energy (Figure 1a and Figure S4a). This differs from earlier studies showing the open-shell singlet is favored with a thiolate ligand.11 In the second reaction step, C–Si bond formation, DFT and QM/MM calculations showed that the closed-shell singlet transition state is lowest in energy. Intrinsic reaction coordinate (IRC) calculations corresponding to the lowest-energy QM/MM transition state (TDE-TS2-CSS) show that the enzymatic Si–H insertion is concerted but highly asynchronous, consisting of partial hydride-transfer from the silane to the carbene carbon, followed by C–Si bond formation (Figure 1c). In contrast, the higher in energy triplet transition state (TDE-TS2-T) favors a radical stepwise pathway (Figures S4–S6). These results are consistent with those recently reported by Fasan and Zhang on myoglobin (Mb)-catalyzed carbene insertions into Si–H bonds, where they used truncated model DFT calculations and radical trap experiments to conclude that the Mb-catalyzed carbene insertion into Si–H bonds takes place through a concerted mechanism involving a closed-shell singlet transition state.6
Figure 1.
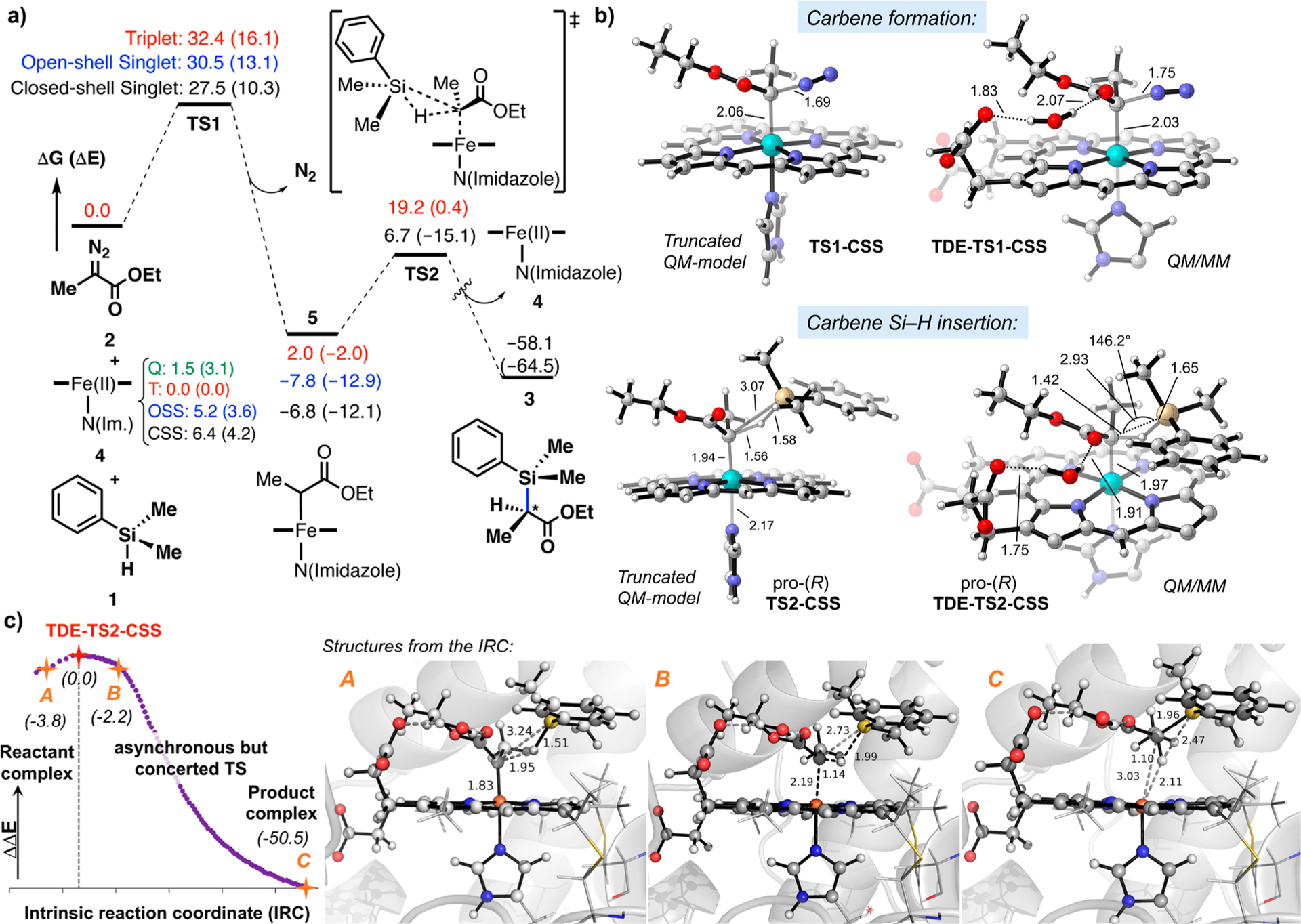
(a) Computed reaction pathway for carbene Si–H insertion reaction between PhMe2SiH 1 and Me-EDA 2 catalyzed by a model imidazole-ligated iron porphyrin (truncated DFT model). Results with three iron electronic states are given. The calculated intrinsic electronic and Gibbs free energies are given in kcal·mol−1. Gibbs free energies do not consider the formation of substrate–substrate or enzyme–substrate reactant complexes, thus neglecting the stabilizing interactions that overcome the unfavorable −TΔS term of a bimolecular reaction (usually about 12 kcal· mol−1 in the free energies). (b) Optimized geometries for the lowest in energy TS1-CSS and TS2-CSS obtained from truncated QM and QM/MM models (only atoms included in the QM region are shown for clarity). (c) Intrinsic reaction coordinate (IRC) calculation for the lowest energy Si–H carbene insertion transition state (TDE-TS2-CSS) calculated using QM/MM (atoms included in the QM region are shown in ball and stick format). Relative electronic energies are given in kcal·mol−1. Key distances and angles are shown in Å and degrees.
Our earlier Hammett analysis1 showed that electron-donating substitutions on the aryl group of the silane increase the initial rate of the reaction, which indicates that the Si–H insertion step is rate-determining in Rma TDE-catalyzed reaction. To further interrogate the reaction mechanism and rate-determining step (RDS) experimentally, the initial-rate primary kinetic isotope effect (KIE) of the Rma TDE-catalyzed reaction was determined using dimethyl(phenyl)silane (PhMe2Si-H) and dimethyl(phenyl)silane-d (PhMe2Si-D). These KIE experiments reflect the competition between PhMe2Si-H vs PhMe2Si-D for the carbene intermediate. The KIE value obtained for Rma TDE was 2.1 ± 0.2, indicating that Si–H bond cleavage is rate-determining in this particular biocatalytic system (see Figure S1). The experimental initial-rate KIE for Rma TDE is comparable to that computed for TDE-TS2-CSS (1.74, see Table S1). Similarly, a KIE value of 1.19 was recently reported for Mb(H64V,V68A)-catalyzed reaction between ethyl α-diazoacetate (EDA) and silane 1.6 The larger KIE measured for the Rma TDE-catalyzed reaction might indicate that it corresponds to a later TS on the concerted hydride transfer and C–Si bond-forming reaction coordinate, as compared to the Mb(H64V,V68A)-catalyzed reaction. This is supported by the TDE-TS2-CSS QM/MM optimized geometry, where the C–Si and C–H bonds being formed (2.93 and 1.42 Å, respectively, Figure 1b) are shorter (i.e., more product-like) than those in the optimized enzyme-free QM model TS2-CSS structure (3.07 and 1.56 Å, respectively, Figure 1b) and the QM model TS reported by Fasan and Zhang (3.1 and 1.7 Å, respectively).6 The occurrence of a TS in Rma TDE in which the hydride is more transferred to the carbene is attributed to the presence of the water molecule that is H-bonded to the carbene carbonyl group. This hydrogen bond interaction, characteristic of the Rma TDE enzyme, increases the electron withdrawing character of the ester group conjugated to the carbene C atom, which in turn enhances the electrophilicity of the carbene. Consequently, the hydride transfer occurring during the asynchronous but concerted process is favored (Figure 1c).
In addition to the initial-rate KIE value, the end-point KIE value was also measured for Rma TDE and determined to be 1.34 ± 0.08 (see Figure S1). This is similar to the end-point KIE values reported for Rh-, Ir-, and Cu-catalyzed reactions (1.29 and 1.5 for Rh, 1.6 for Ir, 1.08 for Cu), where the C–Si bond formation step is believed to be both rate-limiting and concerted.14–16 Overall, our experimental results suggest that C–Si bond formation step is rate-limiting in the Rma TDE-catalyzed carbene Si–H insertion reaction, while our computations indicate that this carbene insertion step follows a concerted, asynchronous mechanism.
2. Understanding How Directed Evolution and Silane Binding Affect the Catalytic Efficiency of Rma TDE-Catalyzed C–Si Bond Formation.
Mutations V75T, M100D, and M103E, introduced to wild-type Rma cyt c during direction evolution, improve the total turnover number of this enzyme for C–Si bond formation.1 Previously, we showed that these mutations played a role in (1) generating space near the catalytic iron–heme cofactor and therefore creating a new enzyme active site in Rma TDE and (2) facilitating the access of silane substrate 1 to the active site of carbene-bound Rma TDE by modulating the conformation and dynamics of a loop that covers the access to the heme pocket (“front loop” residues 98–103) (Figure 2a,b).7 We hypothesized that the protein front loop conformation and dynamics might also modulate silane binding in a catalytically competent pose and thus contribute to the rate-determining step of the reaction (Scheme 1).
Figure 2.
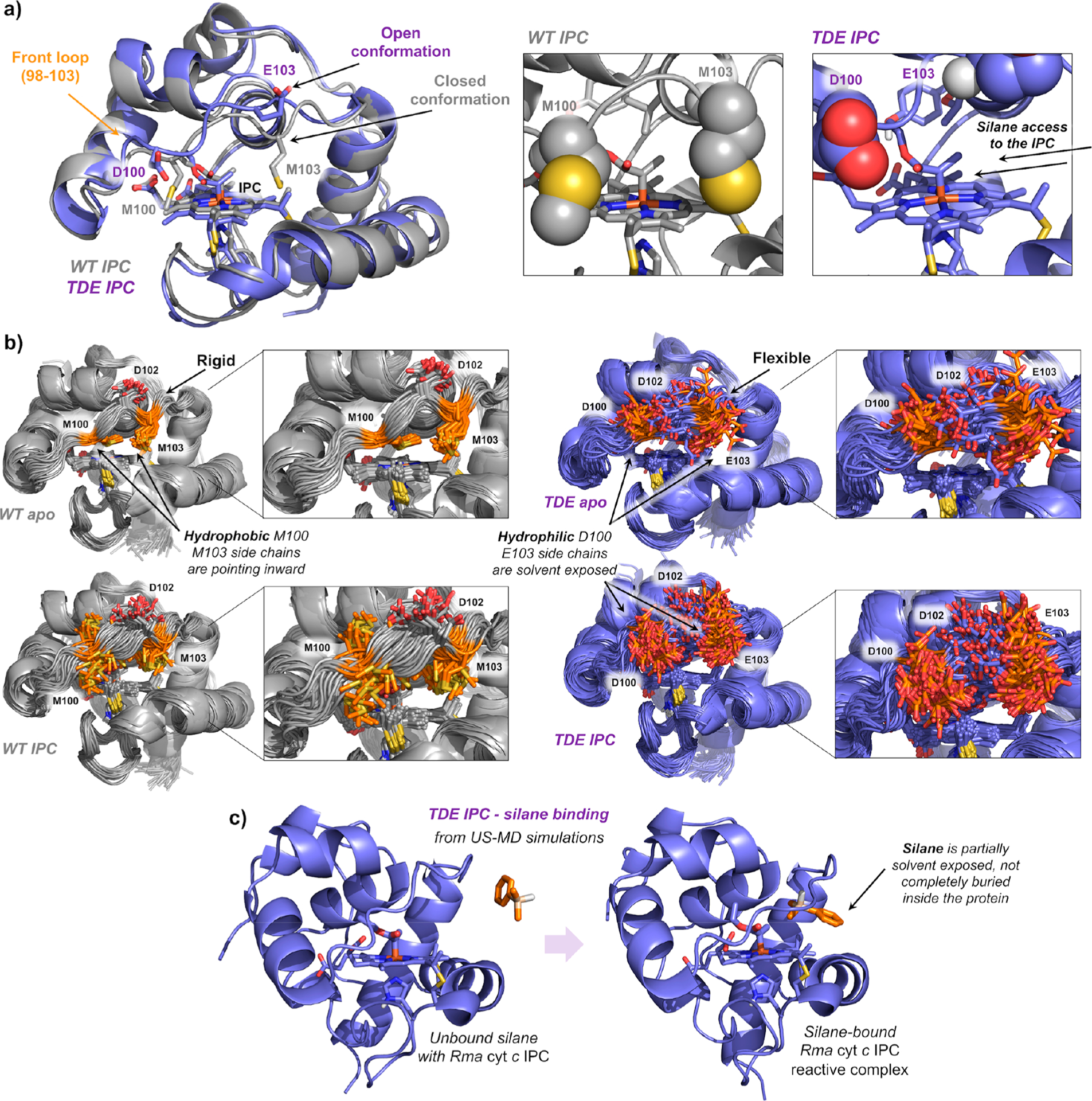
(a) Representative snapshots obtained from MD simulations of IPC bound in wild-type Rma cyt c (gray) and the Rma TDE variant (purple). Front loop M100/D100 and M103/E103 residues are represented in space filling format. Hydrophobic methionines (M100 and M103) close the substrate channel in wild-type Rma cyt c, but mutations M100D and M103E introduce hydrophilic residues that are exposed to the solvent in Rma TDE along the MD simulations, allowing an easier access of the silane substrate to interact with the IPC. V75T creates new key H-bond with Y71 that induces a more hydrophobic environment for the IPC intermediate. (b) Overlay of 50 snapshots (every 10 ns) obtained from 500 ns MD simulations for wild-type Rma cyt c and Rma TDE variants in its apo and IPC-bound states. Residues mutated in the front loop (original M100 and M103 in WT, and D100 and E103 in TDE variant) are shown in orange. Insets highlight the flexibility acquired by the front loop due to newly introduced mutations (going from “WT apo” to “TDE apo”) and the formation of the IPC (going from “WT apo” and “TDE apo” to “WT IPC” and “TDE IPC”, respectively). Corresponding RMSF analysis are reported in Figure S8. (c) Representative snapshots of silane binding to Rma TDE IPC in a catalytically relevant pose obtained from umbrella sampling MD simulations.
Using umbrella sampling MD (US-MD) simulations (see Supporting Information methods, section IIc), we compared the silane binding to carbene-bound wild-type Rma cyt c (WT IPC) and carbene-bound Rma TDE (TDE IPC) in a reactive conformation (Figure S7). In both cases, the protein front loop must change conformation to accommodate the incoming silane substrate, and the silane substrate is only loosely bound by both protein complexes. The silane substrate is highly solvent exposed and not completely surrounded by the protein residues (Figure 2c). Nevertheless, the silane binding affinity in the TDE IPC reactive intermediate estimated from US-MD simulations is ~2 kcal·mol−1 more favorable than the silane-bound WT IPC. MD simulations show that the front loop is more flexible in TDE IPC than in WT IPC (Figure 2b and Figure S7), which allows the front loop to adopt conformations that better favor silane binding (Figure S8, residues 98–103). This increased flexibility in the front loop may be due to the hydrophilicity of the D100 and E103 side chains in TDE IPC, which are solvent exposed and are pointing outward the active site in contrast to M100 and M103, facilitating the silane approach to the reactive carbene (see Figure 2a,b).
From kinetics experiments, we have determined that Rma TDE has a 7-fold increase in initial rate of carbene Si–H insertion relative to the wild-type protein.1 According to transition state theory (TST), a 7-fold increase in rate represents an energy difference of ~1.2 kcal·mol−1 between the RDS barriers for wild-type Rma cyt c and Rma TDE. This experimentally estimated energy difference (ΔΔG‡RDS) of 1.2 kcal·mol−1 agrees with the computationally estimated energy difference for silane binding in a catalytically competent pose (Figure S7). Taken together, these results describe how silane binding in a reactive conformation differs between Rma cyt c and Rma TDE and indicate that changes in silane binding were key to improving the catalytic activity of Rma cyt c toward carbene Si–H insertions.
US-MD simulations also showed that the preferred conformation of the carbene in the enzyme active site, which we characterized in our previous work,7 is not altered as the silane approaches the enzyme. This observation, together with the concerted nature of the Si–H carbene insertion TS we characterized, supports our previous hypothesis of how the cytochrome c protein controls stereoselectivity: the stereo-chemical outcome of C–Si bond formation is solely determined by how the silane substrate approaches the IPC intermediate in the enzyme active site (Figures 2a and S7).
3. How Rma TDE Controls Chemoselectivity: C–Si vs C–N Bond Formation.
In addition to catalyzing the formation of C–Si bonds, carbene transfer biocatalysts can catalyze formation of other C–X bonds (X = C, N, O, S, and B).3,4,17–25 In synthetic chemistry, metal carbenes are known to be reactive toward a broad array of functional groups, most notably unsaturated bonds (e.g., olefins and alkynes) and nucleophiles, as well as a variety of X–H bonds.26–32 However, such reactivity also makes chemoselectivity difficult to control.21,33,34 Because chemoselective carbene transfer catalysts would be useful for chemical synthesis, we sought to elucidate the principles that govern carbene transfer chemoselectivity. Previously, we discovered that Rma TDE differentiates reactions at Si–H and N–H bonds with 97% selectivity for silylation, even though selection pressure for chemoselectivity was not applied during the directed evolution of this enzyme (where silane 1 was used for activity screening).1 This preference for Si–H functionalization differs from the intrinsic reactivities of a theoretical protein-free reaction (see below) and is the opposite of what is observed in related carbene transfer competition experiments conducted with a myoglobin enzyme.21 Interestingly, wild-type Rma cyt c functionalizes N–H and Si–H bonds with approximately equal preference, which means that the mutations in Rma TDE somehow enhance chemoselectivity for Si–C bond formation.1
The mechanisms of imidazole-ligated iron porphyrin-catalyzed carbene transfers to N–H and Si–H bonds were compared for the reaction of p-dimethylsilylaniline (Figure 3a,b). N–H functionalization can occur through a concerted N–H insertion mechanism akin to that discussed earlier for Si–H insertion (Figure 3c) or through nucleophilic attack by the nitrogen lone pair followed by subsequent hydrogen shift from the ylide (Figure 3b).11,19 By use of DFT calculations to model the reaction between imidazole-ligated IPC and para-dimethylsilylaniline 6, the preferred amination pathway was determined to be N-nucleophilic attack followed by ylide rearrangement, in agreement with Shaik’s previous mechanistic proposal.11 This transition state for nucleophilic amination is lower in energy than that for concerted Si–H insertion (ΔΔG‡ = 3.5 kcal·mol−1, Figure 3a–c); thus, in the absence of the protein scaffold, amination is preferred over Si–H carbene insertion. In addition, silylation and amination have geometrically distinct transition states, as the required substrate approaches to the carbene carbon are different in these reactions (Figure 3a,b).
Figure 3.
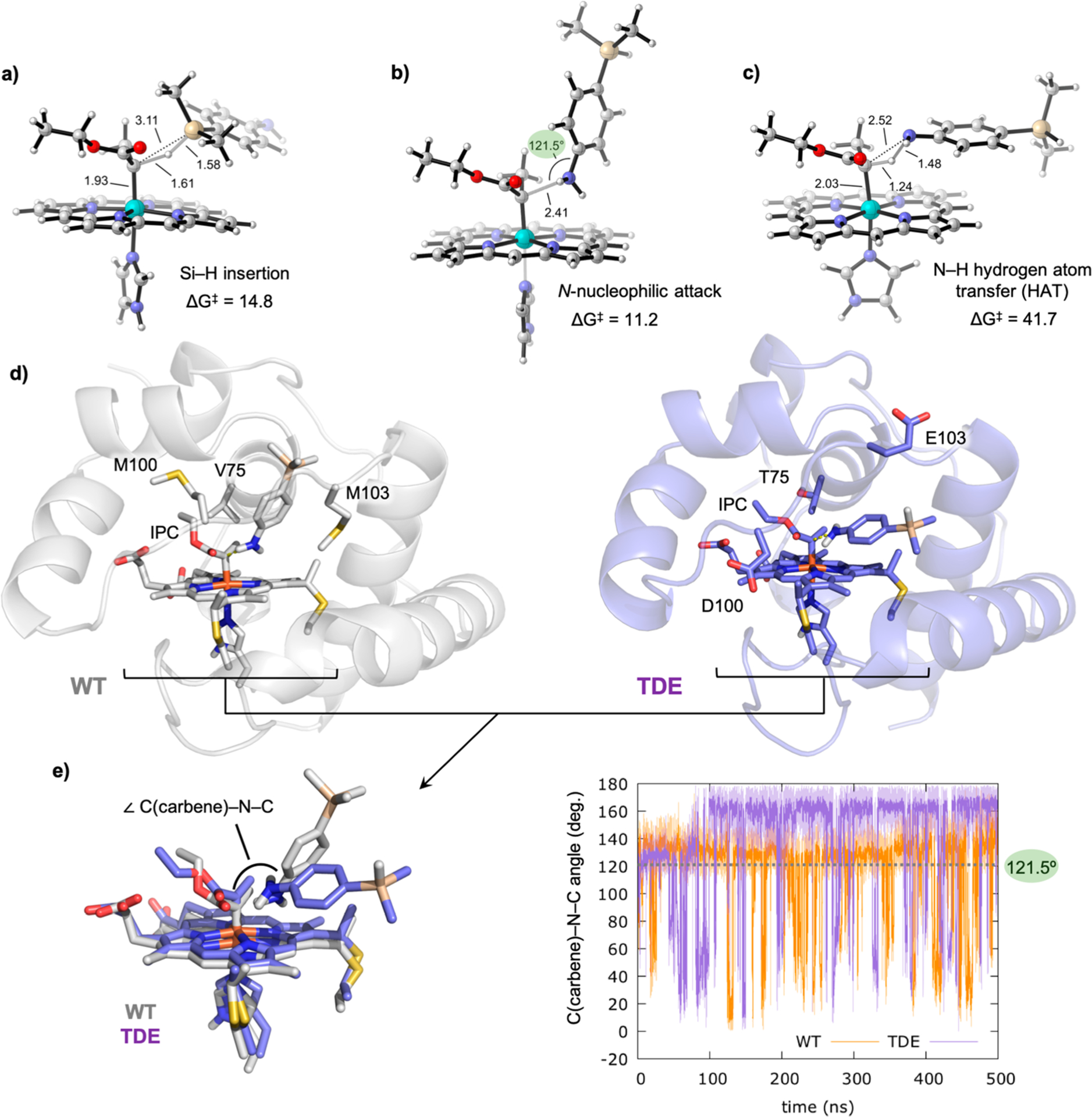
DFT optimized transition states using p-dimethylsilylaniline 6 as substrate: (a) Si–H carbene insertion; (b) N-nucleophilic attack to the electrophilic IPC carbon atom; (c) direct N–H carbene insertion. Gibbs free energies are estimated from lowest in energy OSS IPC 5 and are given in kcal·mol−1. Key distances and angles are given in Å and degrees, respectively. Different orientations of the substrate toward the carbene are required. (d) Analysis of the substrate binding pose and near attack conformations through constrained MD simulations (N(silane)–C(carbene) distance constrained along 500 ns MD trajectories). Representative snapshots of constrained MD simulations (constrained N–C distance to 2.5 Å, see Supporting Information methods section IIc for details) in the wild-type and TDE IPC Rma enzyme active sites are shown. (e) C(carbene)–N(silane)–C(silane) attack angles explored along the constrained MD simulations in wild-type (orange graph) and TDE (purple graph) IPCs. The ideal C(carbene)–N(silane)–C(silane) attack angle value for an effective N-nucleophilic attack is 121.5°, determined from DFT optimized TS.
We investigated how chemoselectivity might be controlled in the enzyme active site of Rma cyt c. Using carbene-bound wild-type Rma cyt c and Rma TDE as starting structures, we docked p-dimethylsilylaniline 6 into both enzyme active sites, with conformation and orientation of 6 resembling how this substrate attacks the carbene in the DFT-optimized N-nucleophilic attack TS (Figure 3b). The docked structures then served as starting points for MD simulations, in which the N(substrate 6)–C(carbene) distance was kept constrained during the entire simulation (see Supporting Information methods, section IIc). These constrained MD simulations showed that in wild-type Rma cyt c, 6 preferentially explores a near attack conformation that resembles the N-nucleophilic attack TS, in which the measured ∠C(carbene)–N(substrate 6)–C(substrate 6) angle is 121.5° (Figure 3d,e). This is in contrast to the Rma TDE system, where 6 prefers a binding pose that resembles the near attack conformation for the energetically disfavored DFT optimized N–H hydrogen abstraction TS (ΔG‡ = 41.7 kcal·mol−1, Figure 3c) with an enlarged ∠C(carbene)–N(substrate 6)–C(substrate 6) angle (Figure 3e) rather than the corresponding N-nucleophilic attack TS. This change in the preferred near attack conformation of the substrate is due to the new conformation explored by the protein front loop in Rma TDE, which is induced by mutations as previously described (Figure 3b and Figure 2a). Taken together, DFT calculations and MD simulations suggest that Rma TDE prevents amination by disfavoring the N-nucleophilic attack transition state through front loop conformation and dynamics that, at the same time, favor silylation.
4. From Mechanistic Understanding to New Protein Function: Switching the Chemoselectivity of Rma TDE from Silylation to Amination.
The development of novel carbene transfer enzymes has largely been driven by repurposing existing heme proteins and evolving these proteins for improved activity for a specific reaction.1,3,4,17–21,23,24,35 Fundamentally, the process of repurposing and evolving enzymes for novel reactivity is an alteration of an enzyme’s selectivity, taking advantage of their intrinsic catalytic promiscuity.36 Understanding the factors that determine enzyme selectivity may therefore facilitate the development of new carbene transfer biocatalysts for chemical synthesis. In order to further tailor these enzymes for new catalytic applications, we sought to engineer and characterize an enzyme variant with inverted chemoselectivity, favoring amination over silylation.
We investigated the engineering of Rma TDE to favor the amination product of p-dimethylsilylaniline (7) instead of the competing silylation product (8), which Rma TDE prefers. We hypothesized that this altered selectivity could be achieved by modulating the conformational dynamics of the front loop. To test this, we introduced selectivity-altering mutations to three selected positions to modify the front loop conformation, guided by MD simulations. Taking Rma TDE as parent protein, the amino acid residues chosen for site-saturation mutagenesis were E103, M99, and N80 (Figure 4). E103 (on the front loop) was selected because it was previously shown to affect the chemoselectivity.1 Residues M99 (on the front loop) and N80 (on the D helix of Rma cyt c) were selected because our MD simulations suggested that these residues might play an important role in determining front loop conformation by establishing different hydrogen bonding interactions between their main and side chains (Figure S9 and Figure S10).
Figure 4.
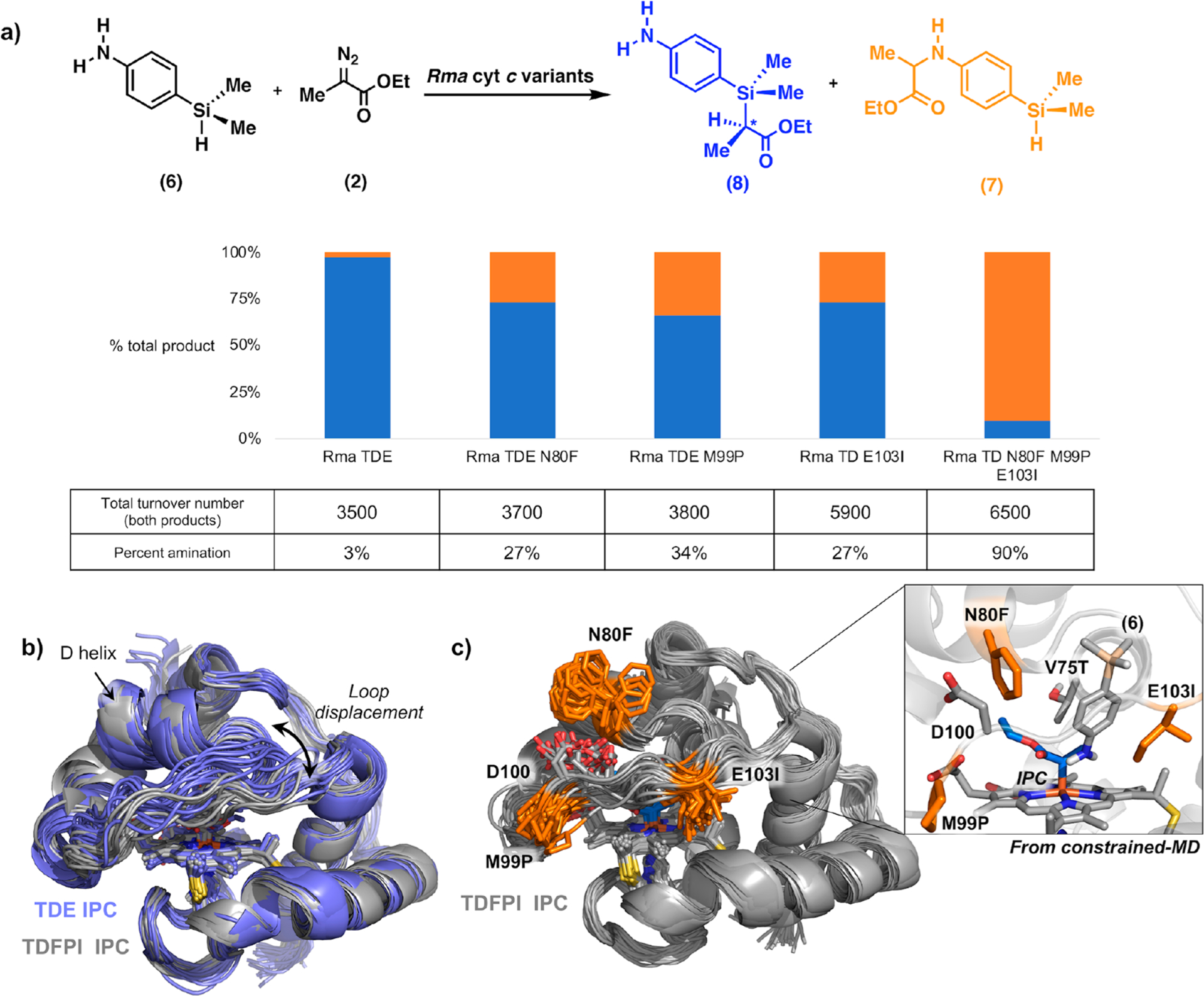
(a) Site saturation mutagenesis of Rma TDE for chemoselective amination over Si–H carbene insertion using p-dimethylsilylaniline 6 and diazo compound 2. Reactions were performed with heat-treated cell lysates with 1 μM Rma cyt c protein, 10 mM 6, 10 mM 2, and 10 mM sodium dithionite in M9-N buffer. Chemoselectivity was measured as the percent of amination product over total product formed and was determined by gas chromatography. Total turnover number (TTN) is defined as the combined concentration of 7 and 8, over protein concentration. See Supporting Information for full description of the experimental procedures. (b) Overlay of different snapshots obtained from 500 ns MD trajectories of Rma TDE (purple) and Rma TDFPI (gray) IPC-bound complexes. The most important structural changes correspond to the preferred “top-open” conformation adopted by the front loop (residues 98–103) in Rma TDFPI as compared to the parent Rma TDE variant. (c) Overlay of snapshots obtained from 500 ns MD trajectory for Rma TDFPI IPC-bound complex. The mutations N80F, M99P, and E103I are highlighted in orange. Inset: Snapshot from constrained-MD simulation of Rma TDFPI IPC-bound complex with substrate 6, showing how substrate 6 prefers to adopt a binding pose that favors amination via N-nucleophilic attack.
We generated site-saturation mutagenesis libraries at each of the three sites and screened the variants for chemoselectivity, as quantified by the product ratios of carbene transfer to an amine substrate (p-isopropylaniline) and a silane substrate (dimethylphenylsilane, 1) in a single competition reaction. Variants displaying improved selectivity for the amination reaction were then evaluated for their chemoselectivity on p-dimethylsilylaniline (6). The most amination-selective variants from each library were determined to be E103I, M99P, and N80F, which produced the amination product as 27–34% of the total product, as compared to 3% for the parent Rma TDE (Figure 4a). Although only mutations at E103 had been previously found to alter the enzyme chemoselectivity,1 our results showed that mutations at each of these three positions have a similar effect on it. This demonstrates that mutations that influence front loop conformational dynamics may alter the chemoselectivity of the carbene insertion. When we combined these three mutations in a single protein (Rma TD N80F M99P E103I, abbreviated as Rma TDFPI), the resulting enzyme produces the amination product as 90% of the total product, suggesting that the front loop now occupies a new and distinct conformation that selectively favors amination over silylation. Rma TDFPI also exhibited ~1.9-fold increased turnover with respect to the parent Rma TDE variant, mainly attributed to E103I mutation (Figure 4b).
A computational model for carbene-bound Rma TDFPI was then generated and used to perform MD simulations to understand the effects of the new mutations. Our simulations revealed a significant change in front loop conformation in Rma TDFPI compared to Rma TDE (Figure 4b). Notably, the M99P and E103I mutations work synergistically to disfavor the silylation transition state by changing the conformational preference of the front loop (M99P) and blocking the preferred binding pose required for the silane to approach the IPC (E103I) (Figure 4c). The new active site found in Rma TDFPI has a substrate access tunnel between the D helix and the protein front loop that guides substrate 6 to approach the carbene-bound heme from the top, a trajectory that favors a near attack, amination conformation via an N-nucleophilic attack transition state (Figure 4c). This trajectory is also favored by the N80F mutation: due to steric repulsion between the phenylalanine side chain and the protein front loop, the front loop is now distanced from the D helix where N80F resides. Finally, by docking substrate 6 into carbene-bound Rma TDFPI with an amination-like conformation and constraining the N(substrate 6)–C(carbene) distance (2.5 Å), constrained MD simulations were used to analyze the catalytically competent binding poses of substrate 6 in Rma TDFPI. These simulations showed that the binding pose of substrate 6 is modulated by the new loop conformation and hydrophobic interactions between E103I and the aromatic ring of 6, which stabilizes the substrate in a near attack conformation that leads to the N-nucleophilic attack TS (Figure 4c and Figure S11). These results demonstrate that the conformational dynamics of the front loop play a fundamental role in determining the reactivity profile of Rma cyt c-derived carbene transfer catalysts and that these dynamics are easily altered through mutagenesis, generating enzymes with altered selectivity.
CONCLUSIONS
The catalytic versatility and selectivity observed in engineered carbene transfer enzymes have led to growing interest in developing these systems for chemical catalysis. Understanding the catalytic mechanism of these engineered enzymes and how the protein scaffold influences reaction outcomes offers fundamental insights into how non-natural enzymes function and how we might design new ones. Here, we have characterized, through computation and experiment, how laboratory-evolved Rma cyt c variants catalyze the formation of carbon–silicon bonds. We determined that Rma TDE catalyzes carbene insertion into Si–H bonds through a concerted, nonradical mechanism and that Si–C bond formation corresponds to the rate-limiting step. We also found that specific H-bond interactions in the Rma TDE active site increase the electrophilicity of the carbene as compared to other enzymatic systems. Binding organosilane to the enzyme in a catalytically competent pose requires unfavorable displacement of the protein front loop, rendering C–Si bond formation the rate-determining step of the overall reaction. Directed evolution transformed Rma TDE into a faster C–Si bond-forming enzyme by making silane binding in a catalytic conformation more favorable in Rma TDE than in the wild-type protein; this in turn reduces the rate-determining energy barrier of carbene Si–H insertion. The mutations in Rma TDE also confer chemoselectivity to the enzyme by stabilizing a substrate binding pose that favors silylation over amination, through modulation of the protein front loop conformational dynamics. Finally, taking advantage of the mechanistic details uncovered in this study and guided by computational modeling, we introduced mutations that ultimately switched the enzyme chemoselectivity from silylation to amination by favoring a new conformation of the protein front loop. These results demonstrate how protein conformational dynamics and knowledge of reaction mechanism can guide the evolution of new enzymatic reactions, offering a framework for future mechanism-guided engineering of useful and novel carbine transfer enzymes.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health, National Institute for General Medical Sciences Grant GM-124480 (to K.N.H.); the National Science Foundation Division of Molecular and Cellular Biosciences Grant MCB-2016137 and the Division of Chemical, Bioengineering, Environmental, and Transport Systems Grant CBET-1937902 (to F.H.A.); the Spanish Ministry of Science and Innovation MICINN (Grant PID2019-111300GA-I00 to M.G.-B and Grants CTQ2015-70524-R and RYC-2013-14706 to G.J.-O.). M.G.-B. thanks the Ramón Areces Foundation for a postdoctoral fellowship, the Spanish MINECO for a Juan de la Cierva—Incorporación fellowship (Grant IJCI-2017-33411), and the Generalitat de Catalunya AGAUR for a Beatriu de Pinós H2020 MSCA-Cofund 2018-BP-00204 project. R.D.L. is supported by NIH National Research Service Award Training Grant 5 T32 GM07616. Computational resources were provided by the University of California, Los Angeles Institute for Digital Research and Education and the Extreme Science and Engineering Discovery Environment, which is supported by National Science Foundation Grant OCI-1053575. We thank Dr. K. Chen and Prof. X. Huang for helpful discussions and comments.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c02146.
Detailed description of experimental and computational methods and procedures, spectroscopic data for all new compounds, supplementary figures and tables, and Cartesian coordinates of DFT optimized structures (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.1c02146
The authors declare no competing financial interest.
Contributor Information
Marc Garcia-Borràs, Department of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, United States; Institut de Química Computacional i Catàlisi (IQCC) and Departament de Química, Universitat de Girona, 17003 Girona, Spain.
S. B. Jennifer Kan, Division of Chemistry and Chemical Engineering 210-41, California Institute of Technology, Pasadena, California 91125, United States.
Russell D. Lewis, Division of Biology and Bioengineering, California Institute of Technology, Pasadena, California 91125, United States.
Allison Tang, Division of Chemistry and Chemical Engineering 210-41, California Institute of Technology, Pasadena, California 91125, United States.
Gonzalo Jimenez-Osés, CIC bioGUNE, 48170 Derio, Spain.
Frances H. Arnold, Division of Biology and Bioengineering and Division of Chemistry and Chemical Engineering 210-41, California Institute of Technology, Pasadena, California 91125, United States.
K. N. Houk, Department of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, United States.
REFERENCES
- (1).Kan SBJ; Lewis RD; Chen K; Arnold FH Directed evolution of cytochrome c for carbon–silicon bond formation: Bringing silicon to life. Science 2016, 354 (6315), 1048–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Stelter M; Melo AMP; Pereira MM; Gomes CM; Hreggvidsson GO; Hjorleifsdottir S; Saraiva LM; Teixeira M; Archer M A Novel Type of Monoheme Cytochrome c: Biochemical and Structural Characterization at 1.23 Å Resolution of Rhodothermus marinus Cytochrome c. Biochemistry 2008, 47 (46), 11953–11963. [DOI] [PubMed] [Google Scholar]
- (3).Kan SBJ; Huang X; Gumulya Y; Chen K; Arnold FH Genetically programmed chiral organoborane synthesis. Nature 2017, 552, 132–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Huang X; Garcia-Borràs M; Miao K; Kan SBJ; Zutshi A; Houk KN; Arnold FH A Biocatalytic Platform for Synthesis of Chiral α-Trifluoromethylated Organoborons. ACS Cent. Sci 2019, 5 (2), 270–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Chen K; Huang X; Zhang S-Q; Zhou AZ; Kan SBJ; Hong X; Arnold FH Engineered Cytochrome c-Catalyzed Lactone-Carbene B–H Insertion. Synlett 2019, 30 (04), 378–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Khade RL; Chandgude AL; Fasan R; Zhang Y Mechanistic Investigation of Biocatalytic Heme Carbenoid Si–H Insertions. ChemCatChem 2019, 11 (13), 3101–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Lewis RD; Garcia-Borràs M; Chalkley MJ; Buller AR; Houk KN; Kan SBJ; Arnold FH Catalytic iron-carbene intermediate revealed in a cytochrome c carbene transferase. Proc. Natl. Acad. Sci. U. S. A 2018, 115 (28), 7308–7313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hayashi T; Tinzl M; Mori T; Krengel U; Proppe J; Soetbeer J; Klose D; Jeschke G; Reiher M; Hilvert D Capture and characterization of a reactive haem–carbenoid complex in an artificial metalloenzyme. Nat. Catal 2018, 1 (8), 578–584. [Google Scholar]
- (9).Khade RL; Fan W; Ling Y; Yang L; Oldfield E; Zhang Y Iron Porphyrin Carbenes as Catalytic Intermediates: Structures, Mössbauer and NMR Spectroscopic Properties, and Bonding. Angew. Chem., Int. Ed 2014, 53 (29), 7574–7578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Khade RL; Zhang Y Catalytic and Biocatalytic Iron Porphyrin Carbene Formation: Effects of Binding Mode, Carbene Substituent, Porphyrin Substituent, and Protein Axial Ligand. J. Am. Chem. Soc 2015, 137 (24), 7560–7563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Sharon DA; Mallick D; Wang B; Shaik S Computation Sheds Insight into Iron Porphyrin Carbenes’ Electronic Structure, Formation, and N–H Insertion Reactivity. J. Am. Chem. Soc 2016, 138 (30), 9597–9610. [DOI] [PubMed] [Google Scholar]
- (12).Khade RL; Zhang Y C–H Insertions by Iron Porphyrin Carbene: Basic Mechanism and Origin of Substrate Selectivity. Chem. - Eur. J 2017, 23 (70), 17654–17658. [DOI] [PubMed] [Google Scholar]
- (13).Wei Y; Tinoco A; Steck V; Fasan R; Zhang Y Cyclopropanations via Heme Carbenes: Basic Mechanism and Effects of Carbene Substituent, Protein Axial Ligand, and Porphyrin Substitution. J. Am. Chem. Soc 2018, 140 (5), 1649–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Yasutomi Y; Suematsu H; Katsuki T Iridium(III)-Catalyzed Enantioselective Si–H Bond Insertion and Formation of an Enantioenriched Silicon Center. J. Am. Chem. Soc 2010, 132 (13), 4510–4511. [DOI] [PubMed] [Google Scholar]
- (15).Dakin LA; Ong PC; Panek JS; Staples RJ; Stavropoulos P Speciation and Mechanistic Studies of Chiral Copper(I) Schiff Base Precursors Mediating Asymmetric Carbenoid Insertion Reactions of Diazoacetates into the Si–H Bond of Silanes. Organometallics 2000, 19 (15), 2896–2908. [Google Scholar]
- (16).Chen D; Zhu D-X; Xu M-H Rhodium(I)-Catalyzed Highly Enantioselective Insertion of Carbenoid into Si–H: Efficient Access to Functional Chiral Silanes. J. Am. Chem. Soc 2016, 138 (5), 1498–1501. [DOI] [PubMed] [Google Scholar]
- (17).Tyagi V; Bonn RB; Fasan R Intermolecular carbene S–H insertion catalysed by engineered myoglobin-based catalysts. Chem. Sci 2015, 6 (4), 2488–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Sreenilayam G; Fasan R Myoglobin-catalyzed intermolecular carbene N–H insertion with arylamine substrates. Chem. Commun 2015, 51 (8), 1532–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Wang ZJ; Peck NE; Renata H; Arnold FH Cytochrome P450-catalyzed insertion of carbenoids into N–H bonds. Chem. Sci 2014, 5 (2), 598–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Zhang RK; Chen K; Huang X; Wohlschlager L; Renata H; Arnold FH Enzymatic assembly of carbon–carbon bonds via iron-catalysed sp3 C–H functionalization. Nature 2019, 565 (7737), 67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Moore EJ; Steck V; Bajaj P; Fasan R Chemoselective Cyclopropanation over Carbene Y–H Insertion Catalyzed by an Engineered Carbene Transferase. J. Org. Chem 2018, 83 (14), 7480–7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Yang H; Swartz AM; Park HJ; Srivastava P; Ellis-Guardiola K; Upp DM; Lee G; Belsare K; Gu Y; Zhang C; Moellering RE; Lewis JC Evolving artificial metalloenzymes via random mutagenesis. Nat. Chem 2018, 10, 318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Key HM; Dydio P; Clark DS; Hartwig JF Abiological catalysis by artificial haem proteins containing noble metals in place of iron. Nature 2016, 534, 534–537. [DOI] [PubMed] [Google Scholar]
- (24).Dydio P; Key HM; Hayashi H; Clark DS; Hartwig JF Chemoselective, Enzymatic C–H Bond Amination Catalyzed by a Cytochrome P450 Containing an Ir(Me)-PIX Cofactor. J. Am. Chem. Soc 2017, 139 (5), 1750–1753. [DOI] [PubMed] [Google Scholar]
- (25).Srivastava P; Yang H; Ellis-Guardiola K; Lewis JC Engineering a dirhodium artificial metalloenzyme for selective olefin cyclopropanation. Nat. Commun 2015, 6, 7789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Doyle MP Catalytic methods for metal carbene transformations. Chem. Rev 1986, 86 (5), 919–939. [DOI] [PubMed] [Google Scholar]
- (27).Ford A; Miel H; Ring A; Slattery CN; Maguire AR; McKervey MA Modern Organic Synthesis with α-Diazocarbonyl Compounds. Chem. Rev 2015, 115 (18), 9981–10080. [DOI] [PubMed] [Google Scholar]
- (28).Miller DJ; Moody CJ Synthetic applications of the O-H insertion reactions of carbenes and carbenoids derived from diazocarbonyl and related diazo compounds. Tetrahedron 1995, 51 (40), 10811–10843. [Google Scholar]
- (29).Doyle MP; Forbes DC Recent Advances in Asymmetric Catalytic Metal Carbene Transformations. Chem. Rev 1998, 98 (2), 911–936. [DOI] [PubMed] [Google Scholar]
- (30).Davies HML; Morton D Guiding principles for site selective and stereoselective intermolecular C–H functionalization by donor/acceptor rhodium carbenes. Chem. Soc. Rev 2011, 40 (4), 1857–1869. [DOI] [PubMed] [Google Scholar]
- (31).Zhu S-F; Zhou Q-L Transition-Metal-Catalyzed Enantioselective Heteroatom–Hydrogen Bond Insertion Reactions. Acc. Chem. Res 2012, 45 (8), 1365–1377. [DOI] [PubMed] [Google Scholar]
- (32).Keipour H; Carreras V; Ollevier T Recent progress in the catalytic carbene insertion reactions into the silicon–hydrogen bond. Org. Biomol. Chem 2017, 15 (26), 5441–5456. [DOI] [PubMed] [Google Scholar]
- (33).Padwa A; Austin DJ; Hornbuckle SF; Semones MA; Doyle MP; Protopopova MN Control of chemoselectivity in catalytic carbenoid reactions. Dirhodium(II) ligand effects on relative reactivities. J. Am. Chem. Soc 1992, 114 (5), 1874–1876. [Google Scholar]
- (34).Padwa A; Austin DJ; Price AT; Semones MA; Doyle MP; Protopopova MN; Winchester WR; Tran A Ligand effects on dirhodium(II) carbene reactivities. Highly effective switching between competitive carbenoid transformations. J. Am. Chem. Soc 1993, 115 (19), 8669–8680. [Google Scholar]
- (35).Jeschek M; Panke S; Ward TR Artificial Metalloenzymes on the Verge of New-to-Nature Metabolism. Trends Biotechnol. 2018, 36 (1), 60–72. [DOI] [PubMed] [Google Scholar]
- (36).Renata H; Wang ZJ; Arnold FH Expanding the Enzyme Universe: Accessing Non-Natural Reactions by Mechanism-Guided Directed Evolution. Angew. Chem., Int. Ed 2015, 54 (11), 3351–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.