

SUMMARY
DNA methylation is an important epigenetic mark. In plants, de novo DNA methylation occurs mainly through the RNA‐directed DNA methylation (RdDM) pathway. Researchers have previously inferred that a flowering regulator, MULTICOPY SUPPRESSOR OF IRA1 4 (MSI4)/FVE, is involved in non‐CG methylation at several RdDM targets, suggesting a role of FVE in RdDM. However, whether and how FVE affects RdDM genome‐wide is not known. Here, we report that FVE is required for DNA methylation at thousands of RdDM target regions. In addition, dysfunction of FVE significantly reduces 24‐nucleotide siRNA accumulation that is dependent on factors downstream in the RdDM pathway. By using chromatin immunoprecipitation and sequencing (ChIP‐seq), we show that FVE directly binds to FVE‐dependent 24‐nucleotide siRNA cluster regions. Our results also indicate that FVE may function in RdDM by physically interacting with RDM15, a downstream factor in the RdDM pathway. Our study has therefore revealed that FVE, by associating with RDM15, directly regulates DNA methylation and siRNA accumulation at a subset of RdDM targets.
Keywords: DNA methylation, RNA‐directed DNA methylation, small RNAs, WD40, FVE/MSI4
Significance Statement
The RNA‐directed DNA methylation (RdDM) pathway is one of the most important DNA methylation pathways in plants. In this manuscript, we show that FVE is required for DNA methylation and for 24‐nucleotide siRNA accumulation at a subset of RdDM targets. Moreover, FVE physically interacts with RDM15 and shares DNA methylation targets with RDM15 in the genome. Our results support the notion that FVE functions in RdDM, particularly in RNA polymerase V‐mediated downstream step.
INTRODUCTION
DNA methylation is a conserved epigenetic modification that is critical for gene expression and genomic stability (Law et al., 2010; Zhang et al., 2018). DNA methylation occurs exclusively in the CG context in mammals. In contrast, cytosines in plants are methylated in three contexts: CG, CHG, and CHH (where H represents A, T, or C) (Lister et al., 2008; Zhang et al., 2006). The maintenance of DNA methylation in CG, CHG, and CHH contexts depends on different DNA methyltransferases. CG methylation is maintained by DNA METHYLTRANSFERASE 1 (MET1), the plant ortholog of mammalian DNA (cytosine‐5)‐METHYLTRANSFERASE 1 (DNMT1) (Finnegan et al., 1996; Kankel et al., 2003). The plant‐specific CHROMOMETHYLASE 3 (CMT3), which can bind to H3K9me2, catalyzes CHG methylation (Du et al., 2012; Lindroth et al., 2001). CHH methylation is maintained by CHROMOMETHYLASE 2 (CMT2) and DOMAINS REARRANGED METHYLASE 2 (DRM2) (Cao and Jacobsen, 2002; Liu and Lang, 2020; Long et al., 2019).
DNA methylation can be either maintained or established de novo. In Arabidopsis, de novo DNA methylation is mediated by a process known as RNA‐directed DNA methylation (RdDM), which involves the biogenesis of small interfering RNAs (siRNAs) and siRNA‐guided DNA methylation (Matzke and Mosher, 2014). A number of accessory factors in the RdDM pathway have been revealed. RNA polymerase IV (Pol IV), for example, initiates the production of siRNAs by generating single‐stranded RNAs, which are then converted into double‐stranded RNAs (dsRNAs) by RNA‐dependent RNA polymerase 2 (RDR2) (Haag et al., 2012; Xie et al., 2004). DICER‐LIKE PROTEIN 3 (DCL3) cleaves dsRNAs into 24‐nucleotide (nt) siRNAs, which are subsequently loaded into the ARGONAUTE (AGO) protein, mainly AGO4 and AGO6 (Duan et al., 2015; Zilberman et al., 2004). AGO‐loaded siRNAs are paired with scaffold RNAs transcribed by Pol V (Haag and Pikaard, 2011). Finally, DNA methyltransferase DRM2 is recruited and catalyzes de novo DNA methylation of cytosine in all sequence contexts (Zhang et al., 2018).
Pol IV and Pol V are plant‐specific RNA polymerases that are derived from Pol II and that function in RdDM (Pikaard et al., 2012). The precise recruitment of Pol IV and Pol V to RdDM targets greatly affects the pattern of DNA methylation in the genome. The recruitment of Pol IV relies on SAWADEE HOMEODOMAIN HOMOLOG 1 (SHH1) (Law et al., 2013), and the interaction between Pol IV and CLASSY1 (CLSY1) is required for Pol IV‐dependent RNA production (Zhou et al., 2018). Production of scaffold RNAs by Pol V requires the chromatin‐remodeling DDR complex, which contains DEFECTIVE IN RNA‐DIRECTED DNA METHYLATION 1 (DRD1), DEFECTIVE IN MERISTEM SILENCING 3 (DMS3), and RDM1 (Kanno et al., 2004; Law et al., 2010). In addition, SUPPRESSOR OF VARIEGATION 3‐9 HOMOLOG PROTEIN 2 (SUVH2) and SUVH9 function in the recruitment of Pol V by interacting with the DDR complex (Johnson et al., 2014; Liu et al., 2014). However, these known regulatory factors can only explain the recruitment of Pol IV and Pol V at a subset of RdDM targets. The mechanism underlying Pol IV and Pol V recruitment to other RdDM target is incompletely understood.
Flowering transition is a vital process that is precisely regulated by environmental factors and endogenous components in the so‐called autonomous pathway (Simpson and Dean, 2002). FLOWERING LOCUS C (FLC), which encodes a flowering repressor, is downregulated by genes in the autonomous pathway by RNA processing and epigenetic regulation (Simpson, 2004; Wu et al., 2020). MSI4/FVE, a WD40 domain‐containing protein, is homologous with the mammalian RETINOBLASTOMA‐ASSOCIATED PROTEINs RbAp46/48 (Qian and Lee, 1995). Like its mammalian homologs RbAp46/48, FVE can induce changes in histone modification at the FLC locus by associating with the HISTONE DEACETYLASE 6 (HDA6) complex, leading to transcriptional silencing of FLC (Ausin et al., 2004; Gu et al., 2011). FVE is also involved in POLYCOMB REPRESSIVE COMPLEX 2 (PRC2), which modulates the enrichment of H3K27 trimethylation (H3K27me3) on FLC and the downstream target FLOWERING LOCUS T (FT) (Pazhouhandeh et al., 2011). Dysfunction of FVE causes a late‐flowering phenotype in Arabidopsis (Kim et al., 2004). In addition to the various effects of FVE on histone modification noted earlier in the Introduction, loss‐of‐function of FVE causes reduction of DNA methylation levels at several RdDM target loci including AtMu1 (DNA transposon), AtSN1 (retrotransposon), FWA, and IG/LINE (intergenic transcripts) (Baurle and Dean, 2008; Gu et al., 2011). However, how FVE modulates DNA methylation in the RdDM pathway remains poorly understood.
To explore the function of FVE in the RdDM pathway, we used next‐generation sequencing in order to characterize the DNA methylome and 24‐nt siRNAs profiles in plants with FVE mutation. Our results suggest that FVE is required for RdDM at thousands of regions genome‐wide. Analysis of 24‐nt siRNA profiles of the fve mutant indicated that FVE has significant effects on Pol V‐dependent but not Pol IV‐dependent 24‐nt siRNA accumulation, suggesting a role of FVE downstream in the RdDM pathway. A chromatin immunoprecipitation and sequencing (ChIP‐seq) assay revealed that FVE proteins directly bind to the FVE‐dependent 24‐nt siRNA‐targeted loci. In addition, FVE physically interacted with an RdDM downstream effector, RNA‐DIRECTED DNA METHYLATION 15 (RDM15), and the analysis of DNA methylomes of rdm15 and fve mutants showed that RDM15 and FVE have shared DNA methylation targets in the genome, suggesting that these two proteins may function together in RdDM. Our results demonstrate that FVE is required for siRNA biogenesis and de novo DNA methylation at a subset of RdDM targets in Arabidopsis.
RESULTS
FVE regulates DNA methylation at RdDM targets
Previous studies have shown that the autonomous pathway component FVE affects methylation levels at several RdDM targets including AtMu1, AtSN1, FWA, and IG/LINE (Gu et al., 2011; Veley and Michaels, 2008). To study how FVE functions in DNA methylation, we used the fve mutants fve‐3 and fve‐4 in the following analysis. To determine whether FVE also regulates DNA methylation at other RdDM loci, we assessed the DNA methylation level at several known RdDM targets using DNA methylation‐sensitive PCR or an individual bisulfite sequencing assay in the Col‐0 wild type (WT) and fve‐3, fve‐4, nrpd1‐3, and nrpe1‐11 mutants. NRPD1 and NRPE1 are the largest subunits exclusive to Pol IV and Pol V, respectively (Zhang et al., 2018). In the fve‐3, fve‐4, nrpd1‐3, and nrpe1‐11 mutants, we found reduced DNA methylation levels at all tested loci compared to the WT (Figure S1). To determine the genome‐wide effect of FVE on DNA methylation, we performed whole‐genome bisulfite sequencing with 14‐day‐old seedlings of fve‐4 and WT. We found that, like nrpd1‐3 and nrpe1‐11, fve‐4 had lower CHG and CHH methylation levels in the transposable element (TE) regions and gene flanking regions than the WT, although the reduction in fve‐4 was less than that in nrpd1‐3 and nrpe1‐11 (Figure S2a). Our results suggest that FVE may broadly affect RdDM in the genome.
To further characterize the role of FVE in the RdDM pathway, we identified differentially methylated regions (DMRs) in fve‐4 relative to the WT using the R package ‘methylkit’ (see the Experimental Procedures section). A total of 5013 hypo‐DMRs and 387 hyper‐DMRs were identified in fve‐4 relative to the WT (Table S1). With the same method, 9218 hypo‐DMRs in nrpd1‐3 and 8859 hypo‐DMRs in nrpe1‐11 were identified. The distribution of hypo‐DMRs in the genome revealed FVE prominently regulates DNA methylation in TEs, i.e., 57% of the hypo‐DMRs in fve‐4 were distributed in TE regions (Figure 1a). This distribution pattern was similar to that of nrpd1‐3 and nrpe1‐11 hypo‐DMRs (Figure 1a). A detailed examination of the influence of FVE on TEs also revealed that FVE resembles NRPD1 and NRPE1 in affecting DNA methylation of TEs in different sizes (Figure 1b). These results demonstrate that FVE resembles NRPD1 and NRPE1 in affecting DNA methylation in TEs.
Figure 1.
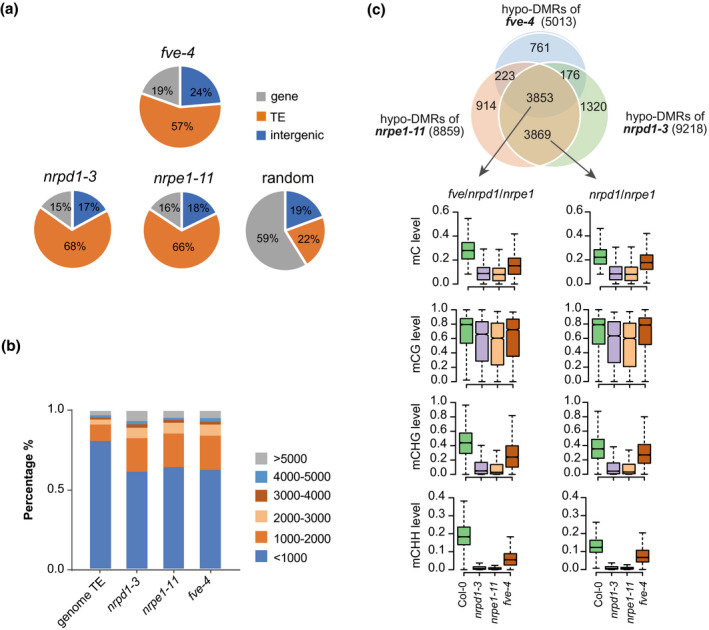
Effects of FVE on DNA methylation levels at the RdDM target loci.
(a) Pie charts showing the distribution of hypo‐DMRs in fve‐4, nrpd1‐3, nrpe1‐11, and random selected regions in wild type.
(b) Length distributions of TEs in the genome and in different groups of hypo‐DMR regions in nrpd1‐3, nrpe1‐11, and fve‐4.
(c) Venn diagrams showing the overlap among hypo‐DMRs in nrpd1‐3, nrpe1‐11, and fve‐4 versus Col‐0. The fve/nrpd1/nrpe1 hypo‐DMRs refer to 3853 overlapping DMRs in fve‐4, nrpd1‐3, and nrpe1‐11. The nrpd1/nrpe1 hypo‐DMRs refer to 3869 DMRs that only overlapped between nrpd1‐3 and nrpe1‐11. The numbers of hypo‐DMRs for each mutant are shown in parentheses. The box plots show mC, mCG, mCHG, and mCHH (where H represents A, C, or T) levels of different subgroups of hypo‐DMRs in Col‐0, nrpd1‐3, nrpe1‐11, and fve‐4.
We found that fve‐4 hypo‐DMRs highly (76.9%) overlapped with nrpe1‐11 hypo‐DMRs and nrpd1‐3 hypo‐DMRs (Figure 1c). Among the DMRs shared between fve, nrpd1, and nrpe1 (Figure 1c), DNA methylation, especially CHG and CHH methylation, was significantly decreased in fve‐4 compared to the WT, although the reduction was less in fve‐4 than in nrpd1‐3 and nrpe1‐11. Among the DMRs shared between nrpd1 and nrpe1 (Figure 1c), we also observed reductions in CHG and CHH methylation in fve‐4, suggesting that these regions may also be FVE targets but did not pass the cutoff for identification of DMRs. The same analysis was also performed on the methylome dataset of fve‐3 (Figure S2b), and the results were similar to those obtained for fve‐4. These results demonstrate that FVE affects RdDM in a genome‐wide manner.
FVE is required for the accumulation of 24‐nt siRNAs
De novo DNA methylation is triggered by siRNAs (Zhang et al., 2018), and we carried out small RNA sequencing in fve‐4 and the WT. The total amount of 24‐nt siRNA was 30% lower in the fve‐4 mutants (Figure S3a). To determine whether FVE affects the abundance of RdDM‐dependent 24‐nt siRNAs, we compared them to the abundances of 24‐nt siRNAs in nrpd1‐3 and nrpe1‐11 mutants. Using ShortStack (Law et al., 2013), we identified 12 646 24‐nt siRNA clusters that were lost in the nrpd1‐3 mutant compared to the WT (Table S2), defined as Pol IV‐dependent siRNA clusters, among which 4819 Pol V‐dependent 24‐nt siRNA clusters were identified (Table S2). In addition, we identified 3040 Pol IV‐dependent 24‐nt siRNA clusters that were lost in the fve‐4 mutant (Table S2), defined as FVE‐dependent 24‐nt siRNA clusters. FVE‐dependent 24‐nt siRNA clusters corresponded to 24% of Pol IV‐dependent 24‐nt siRNA clusters and to >45% of Pol V‐dependent 24‐nt siRNA clusters (Figure 2a). FVE‐dependent siRNA levels were all lower in fve‐4, nrpd1‐3, and nrpe1‐11 compared to the WT (Figure 2b). These results revealed a role of FVE in the accumulation of RdDM‐dependent 24‐nt siRNAs.
Figure 2.
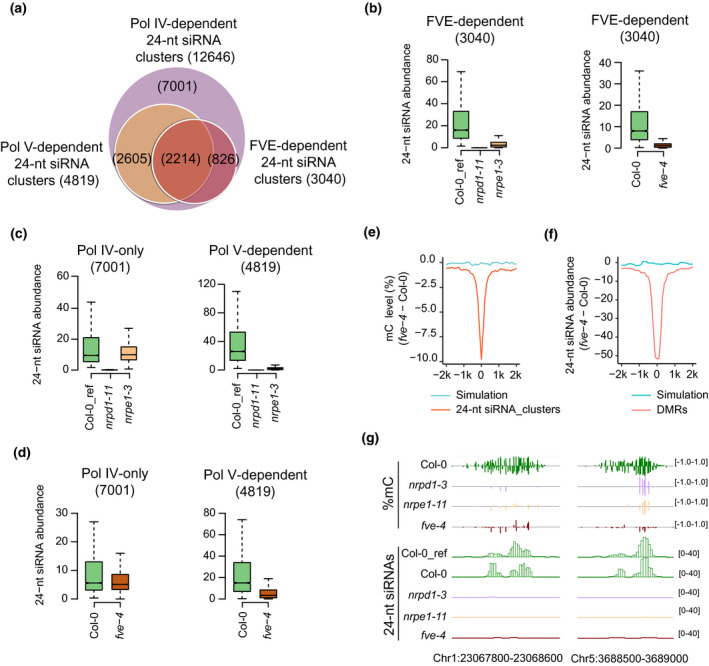
Effects of FVE on 24‐nt siRNA accumulation.
(a) Venn diagrams showing the overlap among FVE‐, Pol IV‐, and Pol V‐dependent 24‐nt siRNA clusters. The numbers of 24‐nt siRNA are shown in parentheses.
(b) Box plots showing abundance of FVE‐dependent 24‐nt siRNAs in Col‐0 wild type and nrpd1‐3, nrpe1‐11, and fve‐4 mutants. Statistical analysis was performed using Wilcoxon tests. A list of P‐values is provided in Table S4.
(c) Box plots showing abundance of 24‐nt siRNAs in Col‐0 wild type and nrpd1‐3 and nrpe1‐11 mutants in the Pol IV‐only (left) and Pol V‐dependent (right) 24‐nt siRNA cluster regions. Statistical analysis was performed using Wilcoxon tests. A list of P‐values is provided in Table S4.
(d) Box plots showing abundance of 24‐nt siRNAs in Col‐0 wild type and fve‐4 in the Pol IV‐only (left) and Pol V‐dependent (right) 24‐nt siRNA cluster regions. Statistical analysis was performed using Wilcoxon tests. A list of P‐values is provided in Table S4.
(e) Changes of DNA methylation levels in fve‐4 relative to Col‐0 wild type at FVE‐dependent 24‐nt siRNA cluster regions.
(f) Changes of 24‐nt siRNA levels in fve‐4 relative to Col‐0 wild type at fve‐4 hypo‐DMRs.
(g) Genome browser views of DNA methylation levels and 24‐nt siRNA accumulation in Col‐0, fve‐4, nrpd1‐3, and nrpe1‐11 at selected RdDM loci.
Biogenesis of 24‐nt siRNAs and siRNA‐guided DNA methylation are the two main steps in the RdDM pathway (Zhang et al., 2018). The RdDM‐dependent 24‐nt siRNAs can be divided into two subclasses: upstream 24‐nt siRNAs, which are only affected in upstream RdDM mutants, such as nrpd1, and downstream 24‐nt siRNAs, which are also affected in downstream RdDM mutants, such as nrpe1 (Law et al., 2013). The Pol IV‐only 24‐nt siRNAs, which represent upstream siRNAs, were substantially lost in nrpd1‐3 but not in nrpe1‐11, whereas Pol V‐dependent siRNAs were lost in both nrpd1‐3 and nrpe1‐11 (Figure 2c; Figure S3b). In addition, dysfunction of FVE weakly reduced the number of expressed upstream 24‐nt siRNAs, but strongly reduced the number of expressed downstream 24‐nt siRNAs (Figure 2d; Figure S3c). Moreover, more than 70% (2214/3040) of the FVE‐dependent 24‐nt siRNA clusters overlapped with Pol V‐dependent 24‐nt siRNA clusters (Figure 2a). The genomic distribution of FVE‐dependent siRNA clusters resembled that of Pol V‐ but not of Pol IV‐dependent siRNA clusters (Figure S3d). In addition, we found that the levels of Pol V‐dependent transcripts, including IGN6, IGN20, and IGN26 (Wierzbicki et al., 2012; Wierzbicki et al., 2008), were all decreased in fve‐4 compared to WT (Figure S3e). All of these results suggest that FVE plays a role in the accumulation of downstream siRNAs in the RdDM pathway, suggesting that FVE functions in a downstream step of RdDM.
To further understand the relationship between the siRNAs and DNA methylation in the fve‐4 mutant, we examined changes in DNA methylation at FVE‐dependent siRNA clusters. The DNA methylation level, including mCG, mCHG, and mCHH, was decreased in regions of FVE‐dependent siRNA clusters (Figure 2e; Figure S3f). The siRNA enrichment at hypo‐DMRs in the fve‐4 mutant was also analyzed. The siRNA levels were clearly lower at hypo‐DMRs in the fve‐4 mutant than in the WT (Figure 2f). On the other hand, we found that some fve‐4 hypo‐DMRs are not associated with the change of 24‐nt siRNA accumulation in fve‐4 (Figure S3g), indicating that like NRPE1 (Dou et al., 2013; Mosher et al., 2008), FVE can regulate DNA methylation at some genomic regions where 24‐nt siRNAs are not affected by FVE. The DNA methylation levels and 24‐nt siRNA levels at several representative fve‐4 hypo‐DMRs are shown in Figure 2(g).
FVE is enriched at FVE‐dependent RdDM targets
Because FVE is known to regulate FLC gene expression (Kim et al., 2004), we determined whether FVE affects the transcript levels of genes involved in RdDM. We found that the transcript levels of currently known RdDM genes were not significantly downregulated in fve‐4 (Figure S4a). To determine whether FVE directly binds to target loci, we used ChIP‐seq to assess FVE enrichment at target loci. Transgenic lines expressing FVE‐3xFLAG or FVE‐3xMYC driven by the FVE native promoter in the fve‐4 mutant were generated. The late‐flowering phenotype of the fve‐4 mutant was rescued in FVE‐3xFLAG and FVE‐3xMYC transgenic lines, and the expression of FVE proteins was measured (Figure S4b,c). ChIP‐seq of 14‐day‐old seedlings of FVE‐3xFLAG transgenic lines revealed that FVE was enriched at FVE‐dependent 24‐nt siRNA cluster regions relative to Pol IV‐only siRNA cluster regions (Figure 3a). This observation was consistent with our finding that FVE regulates downstream but not upstream 24‐nt siRNA accumulation in the RdDM pathway. Consistent with the latter finding, FVE‐enriched regions were found to coincide with 24‐nt siRNA losses and a reduction in DNA methylation in fve (Figure 3b). Together, these findings indicate that FVE directly functions in RdDM.
Figure 3.
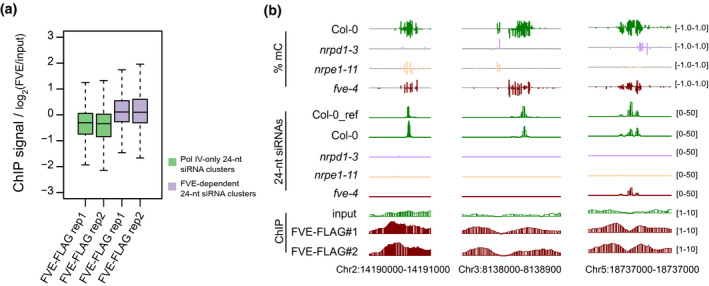
Enrichment of FVE at 24‐nt siRNA target loci.
(a) Box plots showing the enrichment signals of FVE at FVE‐dependent 24‐nt siRNA cluster regions and Pol IV‐only 24‐nt siRNA cluster regions. Two independent biological replicates of FVE‐3xFLAG ChIP‐seq data are shown.
(b) Genome browser views of DNA methylation levels, 24‐nt siRNA accumulation, and ChIP signals of FVE‐3xFLAG at selected loci in the genome. Two replicates of FVE‐3xFLAG are displayed.
FVE physically interacts with RDM15, a known RdDM downstream factor
FVE contains six WD40 repeat domains, which provide a platform for the interaction with other proteins (Jain and Pandey, 2018). To reveal how FVE functions in RdDM, we carried out immunoprecipitation assays followed by mass spectrometry (IP‐MS) with FVE‐3xFLAG/fve‐4 and FVE‐3xMYC/fve‐4 seedlings. RDM15, a known downstream RdDM factor (Niu et al., 2021), was copurified by FVE in both transgenic lines, indicating that FVE is associated with RDM15 in vivo (Figure 4a). To verify the interaction between FVE and RDM15, we performed a split‐luciferase complementation (split‐LUC) imaging assay, and once again detected an interaction between FVE and RDM15 (Figure 4b). The interaction was also validated by yeast two‐hybrid assays (Figure 4c). These results are consistent with the inference that FVE is associated with RDM15 in vivo.
Figure 4.
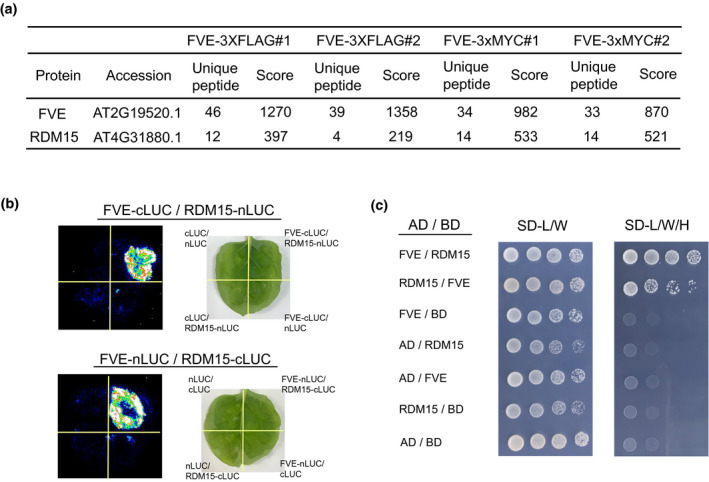
FVE physically interacts with RDM15.
(a) The RDM15 protein was detected by LC‐MS/MS following immunoprecipitation of FLAG‐tagged FVE and MYC‐tagged FVE. Two biological replicates are shown for each tagged FVE purification here.
(b) Analysis of the interactions between FVE and RDM15 by split‐luciferase complementation assays in tobacco (Nicotiana benthamiana) leaves.
(c) Analysis of the interactions between FVE and RDM15 by yeast two‐hybrid assay.
FVE and RDM15 cooperatively affect DNA methylation at some RdDM target loci
Because RDM15 is an RdDM factor and regulates DNA methylation at a subset of RdDM target loci (Niu et al., 2021), we determined whether FVE and RDM15 affect DNA methylation at the same genomic regions. Published methylome data of rdm15 mutants were used in this analysis. We found that, relative to the WT, fve‐4 hypo‐DMRs were also hypomethylated in rdm15 (Figure 5a). This reduction of DNA methylation in rdm15 mutants occurred at CG, CHG, and CHH contexts (Figure 5a). We further analyzed the methylomes of rdm15 mutants, and identified 1354 hypo‐DMRs in rdm15 using the same ‘methylkit’ method as described earlier. About 75% (1019/1354) of the rdm15 hypo‐DMRs overlapped with DMRs shared between fve, nrpd1, and nrpe1 (Figure 5b). The large overlapping rate supported the inference that FVE and RDM15 may function together in vivo to regulate RdDM. Examples of shared hypo‐DMRs in rdm15 and fve‐4 are provided in Figure 5(c).
Figure 5.
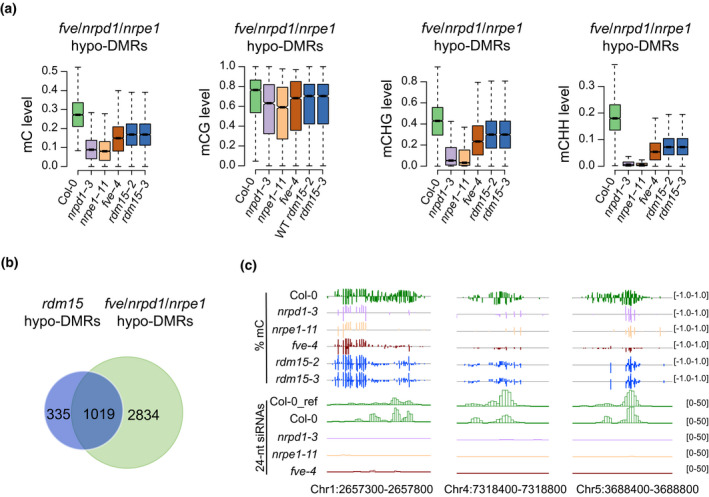
FVE and RDM15 cooperatively regulate DNA methylation at some RdDM target loci.
(a) Box plots showing mC, mCG, mCHG, and mCHH levels of fve/nrpd1/nrpe1 hypo‐DMR regions in Col‐0 wild type and the nrpd1‐3, nrpe1‐11, fve‐4, rdm15‐2, and rdm15‐3 mutants.
(b) Venn diagrams showing the overlap between hypo‐DMRs of rdm15 (rdm15 versus Col‐0) and fve/nrpd1/nrpe1 hypo‐DMRs.
(c) Genome browser views of DNA methylation levels and 24‐nt siRNA accumulation at several loci in different genotypes.
DISCUSSION
Previous studies have shown that the flowering regulators FVE, FPA, and FCA determine DNA methylation levels at several RdDM target loci (Baurle and Dean, 2008; Baurle et al., 2007; Gu et al., 2011). However, genome‐wide DNA methylation at RdDM loci was not observed in fpa or fca single mutants (Stroud et al., 2013). In the current study, we characterized the methylome of fve‐4 and found thousands of hypo‐DMRs in fve‐4 versus the WT (Col‐0), demonstrating that FVE has global effects on DNA methylation (Figure 1; Figure S3d). A comparison of hypo‐DMRs among fve‐4, nrpd1‐3, and nrpe1‐11 suggested that the majority of FVE‐dependent hypo‐DMRs overlap with nrpd1‐3 and nrpe1‐11 hypo‐DMRs (Figure 1c). These results have therefore indicated the important involvement of FVE in RdDM.
In RdDM, DNA methylation occurs at loci that are targeted by siRNAs (Zhang et al., 2018). In the current study, we found that FVE regulates 24‐nt siRNA accumulation, especially the accumulation of downstream RdDM siRNAs (Figure 2). ChIP‐seq of FVE‐3xFLAG transgenic lines revealed that FVE was enriched at FVE‐dependent 24‐nt siRNA cluster regions rather than Pol IV‐only 24‐nt siRNA cluster regions, indicated that FVE directly functions downstream of the RdDM pathway (Figure 3a). A previous study reported that FVE was copurified by DRM2, a downstream factor of RdDM (Zhong et al., 2014), and a most recent study reported that FVE can interact with SUVH9, a factor involved in association of Pol V with chromatin (Zhou et al., 2021). Consistently, we found that FVE physically interacts with RDM15, a downstream factor of RdDM, and that rdm15 and fve‐4 mostly share the same hypo‐DMRs (Figures 4 and 5). These results suggest that FVE and RDM15 may work together to regulate RdDM. The current study therefore increases our understanding of the RdDM pathway in Arabidopsis. Several other RdDM components, such as PKL, were also copurified by FVE (Table S3). In the future, it will be interesting to study how PKL is involved in FVE‐dependent RdDM.
FVE is required for transcriptional repression of the floral regulator FLC (Kim et al., 2004; Michaels and Amasino, 1999). FVE can interact with histone deacetylase HDA6 to facilitate histone deacetylation at FLC (Gu et al., 2011). HDA6 functions in both RdDM and maintenance of DNA methylation (Blevins et al., 2014). To examine whether FLC repression is related to FVE‐ or HDA6‐dependent DNA methylation, we used a genome browser to determine the DNA methylation states at the FLC locus. The ChIP‐seq signals of FLAG‐tagged FVE were detected in the promoter regions of FLC (Figure S5a). However, the methylation levels along the FLC locus were nearly unchanged in fve‐4 or hda6 compared to the WT (Col‐0), and this was also true for nrpd1‐3 and nrpe1‐11 (Figure S5a). Our results are consistent with the previous results indicating that FLC repression is not mediated by changes in DNA methylation in the FLC locus (Jean Finnegan et al., 2005). In particular, loss‐of‐function of the RdDM components NRPD1 or NRPE1 in Arabidopsis did not result in late‐flowering phenotypes (Figure S5b). Our results therefore suggest that FVE repression of FLC expression is independent of its function in RdDM.
In summary, we found that FVE directly affects genome‐wide 24‐nt siRNA accumulation and DNA methylation in Arabidopsis, and FVE may function in RdDM by physically interacting with RDM15, a known RdDM component. We also showed that the function of FVE in flowering is independent of its function in RdDM.
EXPERIMENTAL PROCEDURES
Plant materials and growth conditions
All plant materials used in this study were of the Columbia‐0 (Col) ecotype, and the mutants used have been previously described. fve‐3 and fve‐4 have point mutations, and nrpd1‐3 (SALK_128428) and nrpe1‐11 (SALK_029919) are T‐DNA insertion mutants. All seeds were stratified for 2 days at 4°C before they were sown on 1/2 Murashige and Skoog (MS) plates containing 1% sucrose and 0.7% agar. Fourteen‐day‐old seedlings in plates or plants in soil were grown at 22°C under long‐day conditions (16 h light and 8 h dark).
Plasmid and transgenic plant construction
To construct the FVE‐FLAG or FVE‐MYC vector, the genome region containing FVE genes with the 2‐kb upstream region (as the native promoter region) was amplified from Col‐0 DNA using the primers listed in Table S5. The fragments were then cloned into the pCAMBIA1305‐3xFLAG or pCAMBIA1305‐3xMYC vector. The constructed vectors were transformed into fve‐4 mutant plants. Transformants (T0) were selected on 1/2 MS plates containing 20 μg ml−1 hygromycin B (Roche, Basel, Switzerland). Putative transformants (T1) were transferred to soil and screened for the presence of FVE proteins using Western blot. Only homozygous plants containing the FVE transgene were transferred to soil for seed collection in subsequent generations.
Locus‐specific DNA methylation analysis
For DNA methylation‐sensitive restriction endonuclease digestion followed by PCR (Chop‐PCR), genomic DNA was extracted from 14‐day‐old seedlings or leaves using the cetyl trimethyl ammonium bromide (CTAB) method (Rowland and Nguyen, 1993). One microgram of genomic DNA was digested overnight with methylation‐sensitive restriction endonucleases HpaII and HaeIII (NEB, Ipswich, MA, USA) and then used to amplify the indicated regions by PCR using primers flanking the endonuclease recognition sites. Loss of DNA methylation was reflected by a loss of or a decrease in PCR products. Five RdDM loci were selected using the published methylomes of nrpd1‐3 and nrpe1‐11 (Zhang et al., 2013). The primers used for Chop‐PCR are listed in Table S5.
For individual‐locus bisulfite sequencing, genomic DNA was extracted using the DNeasy Plant Mini Kit (Qiagen, Venlo, The Netherlands). The extracted DNA was then treated with the BisulFlash DNA Modification Kit (Epigentek, Farmingdale, NY, USA) according to the manufacturer’s instructions. The bisulfite‐treated DNA was used as the PCR template with primers designed for specific target regions (Table S5). The resulting PCR fragments were cloned into the pMD18‐T vector (Takara, Kusatsu, Japan), and recombinant plasmids were then transformed into Escherichia coli Top10 competent cells. At least 10 single clones were sequenced per amplicon, and the sequencing data were analyzed using Kismeth (Gruntman et al., 2008).
Whole‐genome bisulfite sequencing and data analysis
Genomic DNA was extracted from 14‐day‐old seedlings with the DNeasy Plant Maxi Kit (Qiagen, Venlo, The Netherlands) and then used for library construction using Illumina’s standard DNA methylation analysis protocol and the NEB Ultra II DNA Library Prep Kit. The samples were sequenced with the Illumina HiSeq2500 sequencing platform at the Genomics Core Facility of the Shanghai Centre for Plant Stress Biology, Chinese Academy of Sciences.
For data analysis, reads containing adapters and low‐quality reads (q < 20) were trimmed using cutadapt (Martin, 2011) and Trimmomatic (Bolger et al., 2014), respectively, and clean reads that were shorter than 45 nt were discarded. Clean reads were then mapped to the Arabidopsis thaliana TAIR 10 genome using BSMAP (Xi and Li, 2009) with default parameters. When the methratio.py script was used to extract the methylation ratio from BSMAP mapping results, the option ‐r was used to remove potential PCR duplicates.
DMRs were identified using the R package ‘methylkit’ (Akalin et al., 2012) with a 500‐bp window and a 500‐bp step size, a differential methylation statistic (q) cutoff value of 0.01, and an absolute methylation percentage change cutoff value of 8. The whole‐genome bisulfite sequencing data in Figure S2(b) were downloaded from the NCBI GEO database with accession number GSE39901. The whole‐genome bisulfite sequencing data for rdm15 in Figure 5 were downloaded from the NCBI GEO database with accession number GSE154302.
Small RNA sequencing and data analysis
Genomic small RNA extracted from 14‐day‐old Col‐0 and fve‐4 seedlings was used to construct libraries, which were then sequenced. Small RNA sequence data of Col‐0 (marked as Col‐0_ref), nrpe1‐11, and nrpd1‐3 downloaded from the NCBI GEO database with accession number GSE44209 were also used. For sequencing data of small RNAs, adapters were trimmed using cutadapt (v1.8.3), low‐quality tags were trimmed by Trimmomatic (Bolger et al., 2014), and reads longer than 15 nt were retained for further analyses. The small RNA‐seq data were analyzed according to Zhou et al. (2018). The trimmed siRNA reads were mapped to the Arabidopsis genome TAIR 10 using ShortStack (v3.8.5) (Johnson et al., 2016) with either the multi‐mapping mode (‐‐mmap f) or the no‐multi‐mapping, none mode (‐‐mmap n). A custom script was then used to retain only perfectly matching reads and reads with a single mismatch at their 3′ terminus. The siRNA reads passing this custom filter were then used to call siRNA clusters using ShortStack with the ‐‐mincov 20, pad 100, ‐‐dicermin 21, and ‐‐dicermax 24 options. Then, the 24‐nt siRNA clusters that were identified in Col‐0 and absent in nrpd1‐3 were defined as NRPD1‐dependent siRNA clusters. The abundance of 24‐nt siRNAs at DMRs was calculated by counting 24‐nt siRNA reads, which were normalized to per 100 million base pair values. In Figure S3(b,c), the Pol IV‐only and Pol V‐dependent 24‐nt siRNA clusters were defined according to the reference (Law et al., 2013). In Figure S3(b), all of the siRNA data were downloaded from the NCBI GEO database with accession number GSE45368. SiRNA data for Col‐0 and fve‐4 in Figure S3(c) were sequenced for this project.
ChIP‐seq and data analysis
Adapters and 3′‐ends of reads with sufficiently low quality scores (q < 20) were trimmed, and clean reads that were shorter than 45 nt were discarded. Clean paired‐end reads were mapped to the TAIR 10 genome of A. thaliana with Bowtie2 (version 2.3.4.1) with default parameters (Langmead and Salzberg, 2012), and samtools (Li et al., 2009) was used to remove potential PCR duplicates. Only uniquely aligned reads were retained for the downstream analysis. To generate the relative ChIP‐seq signal in 24‐nt siRNA clusters, the depth number in each region was summed. The relative signal (y‐axis) in each bin was defined as follows: log2{n (histone modification) × N (input) / [N (histone modification) × n (input)]}, where n represents the sum of the depths of the corresponding library in each bin and N is the number of mapped reads of the corresponding library.
Immunoprecipitation and LC‐MS/MS analysis
Fourteen‐day‐old seedlings of two FVE transgenic lines were used. Dynabeads (10003D, Invitrogen, Carlsbad, CA, USA) conjugated with FLAG antibody (F1804, Sigma, St Louis, MO, USA) or MYC antibody (Abmart, Shanghai, China) were applied for immunoprecipitation. Affinity purification was performed as previously described (Law et al., 2010). Pull‐down protein samples were subjected to LC‐MS/MS analysis as previously described (Lang et al., 2015).
Split‐luciferase complementary assays
Split‐LUC assays were performed using Agrobacterium tumefaciens GV3101 carrying different constructs in 4‐week‐old tobacco leaves. The coding sequences of the FVE and RDM15 proteins were cloned into pCAMBIA‐cLUC and pCAMBIA‐nLUC vectors, respectively. Luciferase activity was detected with a luminescence imaging system at 48 h post‐infiltration (Lang et al., 2015).
Yeast two‐hybrid assays
Full‐length coding sequences of FVE and RDM15 proteins were cloned into pGADT7 and/or pGBKT7 vectors to generate activation domain (AD) and binding domain (BD) constructs. The AD and BD constructs were then cotransformed into yeast strain AH109. Associations were screened on synthetic dropout (SD) medium lacking tryptophan (Trp), leucine (Leu), and histidine (His).
Quantitative real‐time PCR
Total RNA was extracted from 14‐day‐old seedlings grown under long‐day conditions. The extracted RNA was treated with RNase‐free DNase to remove genomic DNA. cDNA synthesis was then carried out with the Bioteke RT‐PCR System following the manufacturer’s instructions. The cDNA reaction mixture was diluted three times, and 2 μl was used as template in a 20‐μl PCR reaction with SYBR Green mix (Vazyme, Nanjing, China). Primer sequences are listed in Table S5. Each sample was quantified in triplicate and normalized to the endogenous control ACTIN7 or ACTIN2. Bars indicate the standard deviation of three measurements.
AUTHOR CONTRIBUTIONS
PH, HH, and ZL designed the experiments. PH, XL, PL, and LZ performed the experiments. HH and ZL analyzed the data. PH, HH, PL, WN, ZL, and J‐KZ wrote and revised the manuscript.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
Supporting information
Figure S1. Effects of FVE on DNA methylation levels at RdDM target loci. (a) DNA methylation‐sensitive PCR (Chop‐PCR) assay in Col‐0 wild type and nrpd1‐3, nrpe1‐11, fve‐3, and fve‐4 mutants at selected RdDM target loci. HaeIII and HpaII are methylation‐sensitive restriction endonucleases. Undigested DNA was used as control. Less PCR product suggests lower DNA methylation levels. (b) Individual locus bisulfite sequencing showing DNA methylation levels in Col‐0 wild type and nrpd1‐3, nrpe1‐11, fve‐3, and fve‐4 mutants at RdDM target locus chromosome (Chr) 4:10350860–10351482. CG, CHG, and CHH (where H represents A, T, or C) cytosine contexts are colored as red, blue, and green, respectively. The filled circles represent methylated cytosines while empty circles represent unmethylated cytosines. (c) Analysis of mC, mCG, mCHG, and mCHH levels of Col‐0 wild type and nrpd1‐3, nrpe1‐11, fve‐3, and fve‐4 mutants at Chr4:10350860–10351482.
Figure S2. Analysis of fve‐4 methylome. (a) Average DNA methylation levels of Col‐0 wild type and fve‐4, nrpd1‐3, and nrpe1‐11 mutants in gene regions and TE regions. (b) Box plots showing mC, mCG, mCHG, and mCHH levels of WT, nrpd1‐3, nrpe1‐11, and fve‐3 in two hypo‐DMR regions defined in Figure 1(c).
Figure S3. Influence of FVE on 24‐nt siRNAs and DNA methylation in the genome. (a) Length distribution of the small RNAs in Col‐0 wild type and the fve‐4 mutant. Small RNA counts of each length were calculated and then normalized to 21‐nt siRNAs in each library. (b) Box plots showing 24‐nt siRNA levels in Col‐0 wild type and nrpd1‐3 and nrpe1‐11 mutants at Pol IV‐only and Pol V‐dependent 24‐nt siRNA clusters. (c) Box plots showing 24‐nt siRNA levels in Col‐0 and fve‐4 in Pol IV‐only and Pol V‐dependent 24‐nt siRNA clusters. (d) Circos map showing the distributions of hypo‐DMRs and 24‐nt siRNA clusters in Arabidopsis chromosomes. Genes and TEs are indicated in the outer circles. (e) Relative levels of Pol V‐dependent transcripts in the fve‐4 mutant. The transcript levels were normalized to the endogenous control Actin2. (f) Changes of mCG, mCHG, and mCHH levels in fve‐4 relative to Col‐0 at FVE‐dependent 24‐nt siRNA cluster regions. (g) The siRNA abundance and DNA methylation levels of two groups of fve‐4 hypo‐DMRs. fve‐4 hypo‐DMRs were divided into siRNA‐dependent or ‐independent groups according to the differences of 24‐nt siRNAs between fve‐4 and WT (P < 0.05 and difference < 0). Statistical analysis was performed using Wilcoxon tests. A list of P‐values is provided in Table S4.
Figure S4. Relative transcript levels of RdDM genes in fve‐4 and phenotypes of FVE transgenic lines. (a) Relative transcript levels of genes involved in RdDM in Col‐0 and fve‐4 seedlings as quantified by real‐time PCR. The transcript levels were normalized to the endogenous control Actin7. (b) The late‐flowering phenotype was restored in FLAG‐tagged and MYC‐tagged transgenic lines. Col‐0 wild‐type plants, fve‐4 mutant plants, and FVE‐3xFLAG and FVE‐3xMYC transgenic lines were grown under long‐day conditions (16 h light and 8 h dark, 22°C) for 30 days. (c) Identification of FLAG‐tagged and MYC‐tagged FVE transgenic lines by Western blot.
Figure S5. The function of FVE in flowering is independent of its function in RdDM. (a) Characterization of cytosine methylation and ChIP signals at the FLC region. The locus covers FLC genes and flanking regions within 2k base pairs. (b) Flowering phenotypes of Col‐0, nrpd1‐3, nrpe1‐11, fve‐3, and fve‐4. Plants were grown under long‐day conditions (16 h daylight and 8 h dark, 22°C) for 30 days.
Table S1. List of hypo‐DMRs and hyper‐DMRs identified in fve‐4.
Table S2. List of Pol IV‐, Pol V‐, and FVE‐dependent 24‐nt siRNA clusters.
Table S3. List of copurified proteins by FVE in this study.
Table S4. List of P‐values in this study.
Table S5. List of primers used in this study.
ACKNOWLEDGMENTS
We thank Professor José M. Martínez‐Zapater for kindly providing the fve‐4 seeds. We thank Dr. Kai Tang for bioinformation analysis. We thank Dr. Qingfeng Niu for the RDM15 plasmids. This research work was financially supported by the National Natural Science Foundation of China (NSFC 31900482 to HH) and the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB27040000 to ZL).
DATA AVAILABILITY STATEMENT
The bisulfite sequencing data, siRNA sequencing data, and ChIP sequencing data reported in this paper were deposited at NCBI GEO under accession numbers GSE171805, GSE171813, and GSE171808, respectively.
References
- Akalin, A. , Kormaksson, M. , Li, S. , Garrett‐Bakelman, F.E. , Figueroa, M.E. , Melnick, A. et al. (2012) methylKit: a comprehensive R package for the analysis of genome‐wide DNA methylation profiles. Genome Biology, 13, R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausin, I. , Alonso‐Blanco, C. , Jarillo, J.A. , Ruiz‐Garcia, L. & Martinez‐Zapater, J.M. (2004) Regulation of flowering time by FVE, a retinoblastoma‐associated protein. Nature Genetics, 36, 162–166. [DOI] [PubMed] [Google Scholar]
- Baurle, I. & Dean, C. (2008) Differential interactions of the autonomous pathway RRM proteins and chromatin regulators in the silencing of Arabidopsis targets. PLoS One, 3(7), e2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baurle, I. , Smith, L. , Baulcombe, D.C. & Dean, C. (2007) Widespread role for the flowering‐time regulators FCA and FPA in RNA‐mediated chromatin silencing. Science, 318, 109–112. [DOI] [PubMed] [Google Scholar]
- Blevins, T. , Pontvianne, F. , Cocklin, R. , Podicheti, R. , Chandrasekhara, C. , Yerneni, S. et al. (2014) A two‐step process for epigenetic inheritance in Arabidopsis. Molecular Cell, 54, 30–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A.M. , Lohse, M. & Usadel, B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, X. & Jacobsen, S.E. (2002) Role of the arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Current Biology, 12, 1138–1144. [DOI] [PubMed] [Google Scholar]
- Dou, K. , Huang, C.F. , Ma, Z.Y. , Zhang, C.J. , Zhou, J.X. , Huang, H.W. et al. (2013) The PRP6‐like splicing factor STA1 is involved in RNA‐directed DNA methylation by facilitating the production of Pol V‐dependent scaffold RNAs. Nucleic Acids Research, 41, 8489–8502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, J. , Zhong, X. , Bernatavichute, Y.V. , Stroud, H. , Feng, S. , Caro, E. et al. (2012) Dual binding of chromomethylase domains to H3K9me2‐containing nucleosomes directs DNA methylation in plants. Cell, 151, 167–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan, C.G. , Zhang, H. , Tang, K. , Zhu, X. , Qian, W. , Hou, Y.J. et al. (2015) Specific but interdependent functions for Arabidopsis AGO4 and AGO6 in RNA‐directed DNA methylation. EMBO Journal, 34, 581–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnegan, E.J. , Peacock, W.J. & Dennis, E.S. (1996) Reduced DNA methylation in Arabidopsis thaliana results in abnormal plant development. Proceedings of the National Academy of Sciences of the United States of America, 93, 8449–8454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruntman, E. , Qi, Y. , Slotkin, R.K. , Roeder, T. , Martienssen, R.A. & Sachidanandam, R. (2008) Kismeth: analyzer of plant methylation states through bisulfite sequencing. BMC Bioinformatics, 9, 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu, X. , Jiang, D. , Yang, W. , Jacob, Y. , Michaels, S.D. & He, Y. (2011) Arabidopsis homologs of retinoblastoma‐associated protein 46/48 associate with a histone deacetylase to act redundantly in chromatin silencing. PLoS Genetics, 7, e1002366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag, J.R. & Pikaard, C.S. (2011) Multisubunit RNA polymerases IV and V: purveyors of non‐coding RNA for plant gene silencing. Nature Reviews Molecular Cell Biology, 12, 483–492. [DOI] [PubMed] [Google Scholar]
- Haag, J.R. , Ream, T.S. , Marasco, M. , Nicora, C.D. , Norbeck, A.D. , Pasa‐Tolic, L. et al. (2012) In vitro transcription activities of Pol IV, Pol V, and RDR2 reveal coupling of Pol IV and RDR2 for dsRNA synthesis in plant RNA silencing. Molecular Cell, 48, 811–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain, B.P. & Pandey, S. (2018) WD40 repeat proteins: signalling scaffold with diverse functions. Protein Journal, 37, 391–406. [DOI] [PubMed] [Google Scholar]
- Jean Finnegan, E. , Kovac, K.A. , Jaligot, E. , Sheldon, C.C. , James Peacock, W. & Dennis, E.S. (2005) The downregulation of FLOWERING LOCUS C (FLC) expression in plants with low levels of DNA methylation and by vernalization occurs by distinct mechanisms. The Plant Journal, 44, 420–432. [DOI] [PubMed] [Google Scholar]
- Johnson, L.M. , Du, J. , Hale, C.J. , Bischof, S. , Feng, S. , Chodavarapu, R.K. et al. (2014) SRA‐ and SET‐domain‐containing proteins link RNA polymerase V occupancy to DNA methylation. Nature, 507, 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, N.R. , Yeoh, J.M. , Coruh, C. & Axtell, M.J. (2016) Improved placement of multi‐mapping small RNAs. G3 Genes|Genomes|Genetics, 6(7), 2103–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kankel, M.W. , Ramsey, D.E. , Stokes, T.L. , Flowers, S.K. , Haag, J.R. , Jeddeloh, J.A. et al. (2003) Arabidopsis MET1 cytosine methyltransferase mutants. Genetics, 163, 1109–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno, T. , Mette, M.F. , Kreil, D.P. , Aufsatz, W. , Matzke, M. & Matzke, A.J. (2004) Involvement of putative SNF2 chromatin remodeling protein'DRD1 in RNA‐directed DNA methylation. Current Biology, 14, 801–805. [DOI] [PubMed] [Google Scholar]
- Kim, H.J. , Hyun, Y. , Park, J.Y. , Park, M.J. , Park, M.K. , Kim, M.D. et al. (2004) A genetic link between cold responses and flowering time through FVE in Arabidopsis thaliana. Nature Genetics, 36, 167–171. [DOI] [PubMed] [Google Scholar]
- Lang, Z. , Lei, M. , Wang, X. , Tang, K. , Miki, D. , Zhang, H. et al. (2015) The methyl‐CpG‐binding protein MBD7 facilitates active DNA demethylation to limit DNA hyper‐methylation and transcriptional gene silencing. Molecular Cell, 57, 971–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B. & Salzberg, S.L. (2012) Fast gapped‐read alignment with Bowtie 2. Nature Methods, 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law, J.A. , Ausin, I. , Johnson, L.M. , Vashisht, A.A. , Zhu, J.K. , Wohlschlegel, J.A. et al. (2010) A protein complex required for polymerase V transcripts and RNA‐ directed DNA methylation in Arabidopsis. Current Biology, 20, 951–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law, J.A. , Du, J. , Hale, C.J. , Feng, S. , Krajewski, K. , Palanca, A.M. et al. (2013) Polymerase IV occupancy at RNA‐directed DNA methylation sites requires SHH1. Nature, 498, 385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindroth, A.M. , Cao, X. , Jackson, J.P. , Zilberman, D. , McCallum, C.M. , Henikoff, S. et al. (2001) Requirement of CHROMOMETHYLASE3 for maintenance of CpXpG methylation. Science, 292, 2077–2080. [DOI] [PubMed] [Google Scholar]
- Lister, R. , O'Malley, R.C. , Tonti‐Filippini, J. , Gregory, B.D. , Berry, C.C. , Millar, A.H. et al. (2008) Highly integrated single‐base resolution maps of the epigenome in Arabidopsis. Cell, 133, 523–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, R. & Lang, Z. (2020) The mechanism and function of active DNA demethylation in plants. Journal of Integrative Plant Biology, 62, 148–159. [DOI] [PubMed] [Google Scholar]
- Liu, Z.W. , Shao, C.R. , Zhang, C.J. , Zhou, J.X. , Zhang, S.W. , Li, L. et al. (2014) The SET domain proteins SUVH2 and SUVH9 are required for Pol V occupancy at RNA‐directed DNA methylation loci. PLoS Genetics, 10, e1003948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long, J.C. , Xia, A.A. , Liu, J.H. , Jing, J.L. , Wang, Y.Z. , Qi, C.Y. et al. (2019) Decrease in DNA methylation 1 (DDM1) is required for the formation of (m) CHH islands in maize. Journal of Integrative Plant Biology, 61, 749–764. [DOI] [PubMed] [Google Scholar]
- Martin, M. (2011) Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet.journal, 17, 10–12. [Google Scholar]
- Matzke, M.A. & Mosher, R.A. (2014) RNA‐directed DNA methylation: an epigenetic pathway of increasing complexity. Nature Reviews Genetics, 15, 394–408. [DOI] [PubMed] [Google Scholar]
- Michaels, S.D. & Amasino, R.M. (1999) FLOWERING LOCUS C encodes a novel MADS domain protein that acts as a repressor of flowering. The Plant Cell, 11, 949–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosher, R.A. , Schwach, F. , Studholme, D. & Baulcombe, D.C. (2008) PolIVb influences RNA‐directed DNA methylation independently of its role in siRNA biogenesis. Proceedings of the National Academy of Sciences of the United States of America, 105, 3145–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu, Q. , Song, Z. , Tang, K. , Chen, L. , Wang, L. , Ban, T. et al. (2021) A histone H3K4me1‐specific binding protein is required for siRNA accumulation and DNA methylation at a subset of loci targeted by RNA‐directed DNA methylation. Nature Communications, 12, 3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazhouhandeh, M. , Molinier, J. , Berr, A. & Genschik, P. (2011) MSI4/FVE interacts with CUL4‐DDB1 and a PRC2‐like complex to control epigenetic regulation of flowering time in Arabidopsis. Proceedings of the National Academy of Sciences of the United States of America, 108, 3430–3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pikaard, C.S. , Haag, J.R. , Pontes, O.M. , Blevins, T. & Cocklin, R. (2012) A transcription fork model for Pol IV and Pol V‐dependent RNA‐directed DNA methylation. Cold Spring Harbor Symposia on Quantitative Biology, 77, 205–212. [DOI] [PubMed] [Google Scholar]
- Qian, Y.W. & Lee, E.Y. (1995) Dual retinoblastoma‐binding proteins with properties related to a negative regulator of ras in yeast. Journal of Biological Chemistry, 270, 25507–25513. [DOI] [PubMed] [Google Scholar]
- Rowland, L.J. & Nguyen, B. (1993) Use of polyethylene glycol for purification of DNA from leaf tissue of woody plants. BioTechniques, 14, 734–736. [PubMed] [Google Scholar]
- Simpson, G.G. (2004) The autonomous pathway: epigenetic and post‐transcriptional gene regulation in the control of Arabidopsis flowering time. Current Opinion in Plant Biology, 7, 570–574. [DOI] [PubMed] [Google Scholar]
- Simpson, G.G. & Dean, C. (2002) Arabidopsis, the Rosetta stone of flowering time? Science, 296, 285–289. [DOI] [PubMed] [Google Scholar]
- Stroud, H. , Greenberg, M.V. , Feng, S. , Bernatavichute, Y.V. & Jacobsen, S.E. (2013) Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell, 152, 352–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veley, K.M. & Michaels, S.D. (2008) Functional redundancy and new roles for genes of the autonomous floral‐promotion pathway. Plant Physiology, 147, 682–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierzbicki, A.T. , Cocklin, R. , Mayampurath, A. , Lister, R. , Rowley, M.J. , Gregory, B.D. et al. (2012) Spatial and functional relationships among Pol V‐associated loci, Pol IV‐dependent siRNAs, and cytosine methylation in the Arabidopsis epigenome. Genes & Development, 26, 1825–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierzbicki, A.T. , Haag, J.R. & Pikaard, C.S. (2008) Noncoding transcription by RNA polymerase Pol IVb/Pol V mediates transcriptional silencing of overlapping and adjacent genes. Cell, 135, 635–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, Z. , Fang, X. , Zhu, D. & Dean, C. (2020) Autonomous pathway: FLOWERING LOCUS C repression through an antisense‐mediated chromatin‐silencing mechanism. Plant Physiology, 182, 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi, Y. & Li, W. (2009) BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics, 10, 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, Z. , Johansen, L.K. , Gustafson, A.M. , Kasschau, K.D. , Lellis, A.D. , Zilberman, D. et al. (2004) Genetic and functional diversification of small RNA pathways in plants. PLoS Biology, 2, e104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H. , Lang, Z. & Zhu, J.K. (2018) Dynamics and function of DNA methylation in plants. Nature Reviews Molecular Cell Biology, 19, 489–506. [DOI] [PubMed] [Google Scholar]
- Zhang, H. , Ma, Z.Y. , Zeng, L. , Tanaka, K. , Zhang, C.J. , Ma, J. et al. (2013) DTF1 is a core component of RNA‐directed DNA methylation and may assist in the recruitment of Pol IV. Proceedings of the National Academy of Sciences of the United States of America, 110, 8290–8295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X. , Yazaki, J. , Sundaresan, A. , Cokus, S. , Chan, S.W. , Chen, H. et al. (2006) Genome‐wide high‐resolution mapping and functional analysis of DNA methylation in arabidopsis. Cell, 126, 1189–1201. [DOI] [PubMed] [Google Scholar]
- Zhong, X. , Du, J. , Hale, C.J. , Gallego‐Bartolome, J. , Feng, S. , Vashisht, A.A. et al. (2014) Molecular mechanism of action of plant DRM de novo DNA methyltransferases. Cell, 157, 1050–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, J.X. , Du, P. , Liu, Z.W. , Feng, C. , Cai, X.W. & He, X.J. (2021) FVE promotes RNA‐directed DNA methylation by facilitating the association of RNA polymerase V with chromatin. The Plant Journal. 10.1111/tpj.15302 [DOI] [PubMed] [Google Scholar]
- Zhou, M. , Palanca, A.M.S. & Law, J.A. (2018) Locus‐specific control of the de novo DNA methylation pathway in Arabidopsis by the CLASSY family. Nature Genetics, 50, 865–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberman, D. , Cao, X. , Johansen, L.K. , Xie, Z. , Carrington, J.C. & Jacobsen, S.E. (2004) Role of Arabidopsis ARGONAUTE4 in RNA‐directed DNA methylation triggered by inverted repeats. Current Biology, 14, 1214–1220. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Effects of FVE on DNA methylation levels at RdDM target loci. (a) DNA methylation‐sensitive PCR (Chop‐PCR) assay in Col‐0 wild type and nrpd1‐3, nrpe1‐11, fve‐3, and fve‐4 mutants at selected RdDM target loci. HaeIII and HpaII are methylation‐sensitive restriction endonucleases. Undigested DNA was used as control. Less PCR product suggests lower DNA methylation levels. (b) Individual locus bisulfite sequencing showing DNA methylation levels in Col‐0 wild type and nrpd1‐3, nrpe1‐11, fve‐3, and fve‐4 mutants at RdDM target locus chromosome (Chr) 4:10350860–10351482. CG, CHG, and CHH (where H represents A, T, or C) cytosine contexts are colored as red, blue, and green, respectively. The filled circles represent methylated cytosines while empty circles represent unmethylated cytosines. (c) Analysis of mC, mCG, mCHG, and mCHH levels of Col‐0 wild type and nrpd1‐3, nrpe1‐11, fve‐3, and fve‐4 mutants at Chr4:10350860–10351482.
Figure S2. Analysis of fve‐4 methylome. (a) Average DNA methylation levels of Col‐0 wild type and fve‐4, nrpd1‐3, and nrpe1‐11 mutants in gene regions and TE regions. (b) Box plots showing mC, mCG, mCHG, and mCHH levels of WT, nrpd1‐3, nrpe1‐11, and fve‐3 in two hypo‐DMR regions defined in Figure 1(c).
Figure S3. Influence of FVE on 24‐nt siRNAs and DNA methylation in the genome. (a) Length distribution of the small RNAs in Col‐0 wild type and the fve‐4 mutant. Small RNA counts of each length were calculated and then normalized to 21‐nt siRNAs in each library. (b) Box plots showing 24‐nt siRNA levels in Col‐0 wild type and nrpd1‐3 and nrpe1‐11 mutants at Pol IV‐only and Pol V‐dependent 24‐nt siRNA clusters. (c) Box plots showing 24‐nt siRNA levels in Col‐0 and fve‐4 in Pol IV‐only and Pol V‐dependent 24‐nt siRNA clusters. (d) Circos map showing the distributions of hypo‐DMRs and 24‐nt siRNA clusters in Arabidopsis chromosomes. Genes and TEs are indicated in the outer circles. (e) Relative levels of Pol V‐dependent transcripts in the fve‐4 mutant. The transcript levels were normalized to the endogenous control Actin2. (f) Changes of mCG, mCHG, and mCHH levels in fve‐4 relative to Col‐0 at FVE‐dependent 24‐nt siRNA cluster regions. (g) The siRNA abundance and DNA methylation levels of two groups of fve‐4 hypo‐DMRs. fve‐4 hypo‐DMRs were divided into siRNA‐dependent or ‐independent groups according to the differences of 24‐nt siRNAs between fve‐4 and WT (P < 0.05 and difference < 0). Statistical analysis was performed using Wilcoxon tests. A list of P‐values is provided in Table S4.
Figure S4. Relative transcript levels of RdDM genes in fve‐4 and phenotypes of FVE transgenic lines. (a) Relative transcript levels of genes involved in RdDM in Col‐0 and fve‐4 seedlings as quantified by real‐time PCR. The transcript levels were normalized to the endogenous control Actin7. (b) The late‐flowering phenotype was restored in FLAG‐tagged and MYC‐tagged transgenic lines. Col‐0 wild‐type plants, fve‐4 mutant plants, and FVE‐3xFLAG and FVE‐3xMYC transgenic lines were grown under long‐day conditions (16 h light and 8 h dark, 22°C) for 30 days. (c) Identification of FLAG‐tagged and MYC‐tagged FVE transgenic lines by Western blot.
Figure S5. The function of FVE in flowering is independent of its function in RdDM. (a) Characterization of cytosine methylation and ChIP signals at the FLC region. The locus covers FLC genes and flanking regions within 2k base pairs. (b) Flowering phenotypes of Col‐0, nrpd1‐3, nrpe1‐11, fve‐3, and fve‐4. Plants were grown under long‐day conditions (16 h daylight and 8 h dark, 22°C) for 30 days.
Table S1. List of hypo‐DMRs and hyper‐DMRs identified in fve‐4.
Table S2. List of Pol IV‐, Pol V‐, and FVE‐dependent 24‐nt siRNA clusters.
Table S3. List of copurified proteins by FVE in this study.
Table S4. List of P‐values in this study.
Table S5. List of primers used in this study.
Data Availability Statement
The bisulfite sequencing data, siRNA sequencing data, and ChIP sequencing data reported in this paper were deposited at NCBI GEO under accession numbers GSE171805, GSE171813, and GSE171808, respectively.