

Abstract
Astrocytes are glial cells that support neurological function in the central nervous system (CNS), in part, by providing structural support for neuronal synapses and blood vessels, participating in electrical and chemical transmission, and providing trophic support via soluble factors. Dysregulation of astrocyte function contributes to neurological decline in CNS diseases. Neurological diseases are highly heterogeneous but share common features of cellular stress including the accumulation of misfolded proteins. Endoplasmic reticulum (ER) stress has been reported in nearly all neurological and neurodegenerative diseases. ER stress occurs when there is an accumulation of misfolded proteins in the ER lumen and the protein folding demand of the ER is overwhelmed. ER stress initiates the unfolded protein response (UPR) to restore homeostasis by abating protein translation and, if the cell is irreparably damaged, initiating apoptosis. Although protein aggregation and misfolding in neurological disease has been well described, cell‐specific contributions of ER stress and the UPR in physiological and disease states are poorly understood. Recent work has revealed a role for active UPR signaling that may drive astrocytes toward a maladaptive phenotype in various model systems. In response to ER stress, astrocytes produce inflammatory mediators, have reduced trophic support, and can transmit ER stress to other cells. This review will discuss the current known contributions and consequences of activated UPR signaling in astrocytes.
Keywords: astrocytes, cell signaling, endoplasmic reticulum, glia, protein folding, translation
Main Points
Astrocytes experience endoplasmic reticulum (ER) stress during neuroinflammatory and neurodegenerative diseases.
ER stress activates the unfolded protein response (UPR) in astrocytes and drives the production of inflammatory molecules including IL‐6, CCL2, CCL20, CCL3 CXCL1, CXCL10, Lcn2, VEGF, and C3.
Astrocytes under ER stress can induce UPR and inflammatory signaling in neighboring cells.
Activated UPR in astrocytes may contribute to neurotoxicity and/or reduced neuronal and synaptic support.
1. INTRODUCTION
1.1. Endoplasmic reticulum stress and the unfolded protein response
Secreted and membrane bound proteins are translated and processed in the endoplasmic reticulum (ER). Within the ER, proteins mature by folding into the proper tertiary and quaternary structure and acquire necessary post‐translational modifications. The ER is also critical for membrane lipid production and for the regulation of intracellular Ca2+ (Schwarz & Blower, 2016). Often, proteins within the ER fail to fold into the correct form. Fortunately, the cell has intrinsic quality control mechanisms that eliminate misfolded proteins, such as chaperone‐mediated folding (Kim et al., 2013) and ER associated degradation (ERAD) (Christianson & Ye, 2014; Hampton, 2002). However, when these control mechanisms are overwhelmed, misfolded proteins accumulate in the ER lumen. The aberrant accumulation of misfolded proteins and concomitant induction of ER stress has been observed in many diseases and cell types (Oakes & Papa, 2015; Schröder & Kaufman, 2005). ER stress occurs when a cell can no longer keep up with the demand to fold proteins due to the number of misfolded proteins in the ER lumen. ER stress initiates a highly conserved adaptive mechanism called the unfolded protein response (UPR). The intracellular signaling stimulated by ER stress is aimed at restoring homeostasis; however, if the stress is not alleviated, prolonged ER stress can drive cell death and inflammation which may contribute to pathology (Oakes & Papa, 2015).
ER stress can occur transiently in physiological conditions when there is an increased demand for protein secretion, or in pathogenic states where ER stress occurs due to genetic mutations, oxidative stress, ischemia, or other maladaptive cellular states. Physiological ER stress has been best characterized in cells harboring high secretory capacities such as pancreatic β cells and antibody‐producing B cells (Brozzi et al., 2016; Fonseca et al., 2011; Gao et al., 2012; Iwakoshi et al., 2003; Lipson et al., 2008; Ma & Hendershot, 2004; Zhang et al., 2005). Although UPR activation is necessary to maintain homeostasis and clearly plays a role in homeostatic processes, tight regulation of the UPR is paramount for maintaining cellular health. Persistent activation of the UPR is reported in multiple diseases, including diabetes, cancer, and neurodegeneration (Clarke et al., 2014; Hetz & Mollereau, 2014; Scheuner & Kaufman, 2008).
There are three known proteins which sense the accumulation of misfolded proteins and transmit distinct signals to the cytosol and nucleus to modify transcriptional and translational programs to cope with ER stress. These trans‐ER membrane proteins are inositol requiring enzyme 1 (IRE1), protein kinase R‐like ER kinase (PERK) and activating transcription factor (ATF) 6. These enzymes are maintained in their inactive state through interaction with the ER‐resident protein chaperone glucose regulated protein (GRP) 78 (also known as binding immunoglobulin protein [BiP]) (Bertolotti et al., 2000). GRP78 binds broadly to hydrophobic residues that are exposed by misfolded proteins (Flynn et al., 1991). Excess misfolded proteins recruit GRP78 away from the luminal domains of PERK, IRE1, and ATF6 allowing activation (Bertolotti et al., 2000; Shen et al., 2002). PERK and IRE1 can also directly interact with misfolded proteins which contributes to its activation via a ligand‐receptor type interaction (Gardner & Walter, 2011; Karagöz et al., 2017; Wang et al., 2016, 2018; Zhou et al., 2006). Figure 1 provides an overview of the UPR signal transducing molecules.
FIGURE 1.
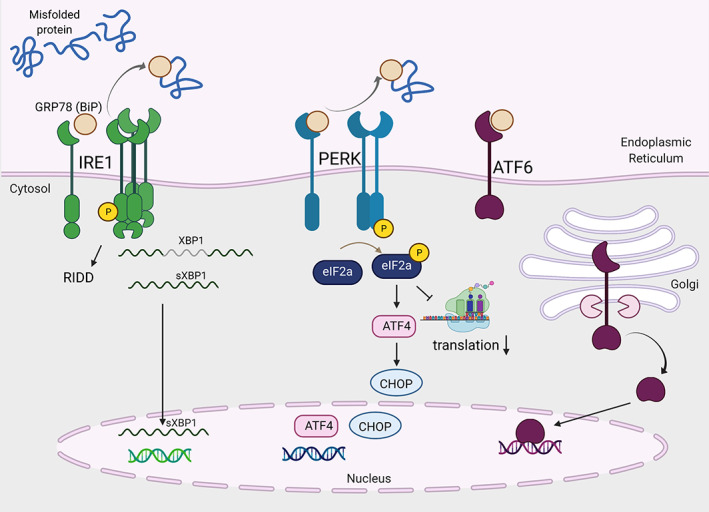
The canonical unfolded protein response (UPR) is activated by three trans‐endoplasmic reticulum (ER) membrane sensors: IRE1, PERK, and ATF6. Figure was created in Biorender
IRE1 is the most evolutionarily conserved UPR initiator and contains both kinase and endoribonuclease domains. Following release of GRP78, IRE1 oligomerizes in the ER membrane facilitating trans‐autophosphorylation of IRE1 which increases RNase activity (Lee et al., 2008; Ron & Hubbard, 2008; Sidrauski & Walter, 1997). IRE1 then splices the mRNA of x‐box‐binding protein (XBP1) to remove a small stop codon‐containing intron which allows translation of the functional transcription factor leading to expression of genes encoding molecular chaperones and ERAD. Further, the activation of IRE1 kinase can promote stress signaling pathways such as c‐Jun N‐terminal kinase (JNK) and nuclear factor kappa‐light‐chain‐enhancer of activated B cells (NF‐κB) (Calfon et al., 2002; Urano et al., 2000; Zha et al., 2015). Additionally, the RNase activity of IRE1 mediates regulated IRE1‐dependent decay (RIDD) in which a subset of ER‐targeted mRNAs are degraded (Hollien & Weissman, 2006; Moore & Hollien, 2015). Collectively, IRE1 drives XBP‐1‐dependent gene expression that includes ER chaperones and, through RNA degradation, reduces nascent polypeptide entry into the ER to reduce the folding demand (Hollien & Weissman, 2006; Lee et al., 2003).
PERK is a trans‐ER membrane serine/threonine kinase which is activated by misfolded proteins in the ER lumen. Following release of GRP78, PERK dimerizes and trans‐ and auto‐phosphorylates to increase its kinase activity (Bertolotti et al., 2000; Harding et al., 1999). PERK phosphorylates the eukaryotic initiation factor (eIF) 2α which leads to binding and inhibition of the guanine nucleotide exchange factor (GEF) eIF2B. This prevents formation of the complex needed to load the 43S ribosome with methionine, thus preventing translation initiation (Harding et al., 1999; Jackson et al., 2010). Under these conditions, some proteins are selectively translated. For example, activating transcription factor (ATF) 4 is translated when eIF2α is phosphorylated. ATF4 translation can lead to expression of CHOP (encoded by the gene ddit3). In many cases, CHOP acts as a proapoptotic factor. Overall, PERK activation reduces the protein load on the ER, and if mechanisms fail to restore homeostasis, initiate cell death.
ATF6 is a transmembrane glycoprotein that is a member of the basic leucine‐zipper proteins (bZIP) transcription factor family. Upon the accumulation of misfolded proteins and disassociation of GRP78, ATF6 localizes to the Golgi apparatus where is cleaved by site‐1 and site‐2 proteases (Ye et al., 2000), revealing a nuclear localization sequence. Subsequently, ATF6 translocates to the nucleus and binds promoter sequences to increase gene expression of ER protein chaperones and UPR regulators to increase folding capacity of the ER (Haze et al., 1999).
Overall, IRE1, PERK, and ATF6 are activated in response to the accumulation of misfolded proteins within the ER lumen to promote efficient protein folding through the upregulation of protein chaperones and by reducing the folding burden on the ER by eliminating influx of mRNA and polypeptides. If these mechanisms are insufficient, persistent UPR activation will promote apoptosis to eliminate the irreparably damaged cell.
1.2. UPR and astrocytes
The UPR is activated transiently to restore homeostasis, however, chronic UPR activation has been implicated in central nervous system (CNS) diseases including, but not limited to, Alzheimer's disease (AD), multiple sclerosis (MS), traumatic brain injury (TBI), stroke, prion disease, Parkinson's disease (PD), and Huntington's disease (HD) (Alberdi et al., 2013; Bell et al., 2016; Chen et al., 2016; Clayton & Popko, 2016; Duran‐Aniotz et al., 2017; Halliday et al., 2017; Halliday & Mallucci, 2014; Hoozemans et al., 2012; Leitman et al., 2013; Ma et al., 2013; Moreno et al., 2012, 2013; Smith et al., 2020; Sun et al., 2015; Torres et al., 2010). Many of these CNS disorders include components of misfolded or aggregated proteins. However, the UPR has been primarily studied in neurons and oligodendrocytes (recently reviewed in [Clayton & Popko, 2016; Lin & Stone, 2020; Martinez et al., 2018]).
Astrocytes comprise a large portion of the CNS and are vital for proper neuronal survival and function (Aoki et al., 2001; Brenner et al., 2001). Historically, astrocytes were considered a homogenous population that primarily played a role in structural support to the CNS, however, technical advances and meticulous experimentation have shown that astrocytes are a heterogeneous and dynamic population of CNS‐resident cells, playing important roles in both homeostasis and disease (Batiuk et al., 2020; Clarke et al., 2021; Khakh & Deneen, 2019). For example, astrocytes support synapse formation and function through both physical interactions and secreted molecules (Clarke & Barres, 2013). Astrocytes play a role in synaptic pruning during development, which is essential for proper neural development (Stevens et al., 2007). Astrocytes also support synaptic function by regulating ion homeostasis (Ca2+, Cl−, K+), water transport, and neurotransmitter reuptake and recycling (Sofroniew & Vinters, 2010; Verkhratsky & Nedergaard, 2018).
In addition to their supportive role, astrocytes respond to insult and injury, can promote neurotoxicity, and direct CNS inflammation by promoting microglial activation and leukocyte trafficking. Inflammation, particularly proinflammatory cytokines such as interleukin (IL)‐6, IL‐1, tumor necrosis factor (TNF)‐α, and the complement system, play an important role in neurological diseases and are associated with worsened neurological outcomes (Becher et al., 2017; Glass et al., 2010; Han et al., 2021; Heneka et al., 2014; Skaper et al., 2018). Astrocytes are key directors of inflammation within the CNS. It is well established that astrocytes undergo transcriptional and phenotypical changes in response to injury, called astrogliosis (Sofroniew & Vinters, 2010). During astrogliosis, astrocytes are more proliferative, glial fibrillary acidic protein (GFAP) expression increases, signaling molecules and cytokines are upregulated, the extracellular matrix remodels, and changes in ability of astrocytes to properly regulate synapses and the blood brain barrier (BBB) occur. Reactive astrocytes can have differential roles depending on the injury or disease. For example, reactive astrocytes worsen AD, but during ischemia or spinal cord injury, reactive astrocytes promote overall neural recovery (Bao et al., 2012; Choudhury & Ding, 2016; Faulkner et al., 2004; Hamby & Sofroniew, 2010; Li et al., 2014; Reichenbach et al., 2019; Shimada et al., 2011; Sofroniew, 2009; Voskuhl et al., 2009).
Perturbations in astrocyte function are now implicated in many neurological diseases including PD, AD, ischemic stroke, epilepsy, and ALS (Sofroniew & Vinters, 2010). This highlights the importance of astrocytes in maintaining and directing neurological function. However, the mechanisms by which cellular stressors initiate astrocyte dysfunction that contributes to disease are not well understood. Recently, single cell RNA sequencing (scRNAseq) revealed that subpopulations of astrocytes that are expanded during experimental autoimmune encephalomyelitis (EAE) have increased UPR signaling, suggesting that the UPR is associated with EAE (Wheeler et al., 2020). Additionally, overexpression of spliced XBP1 in astrocyte‐like glial cells in Caenorhabditis elegans extends life span (Frakes et al., 2020). Astrocytes express all the initiating sensors of the UPR (IRE1, PERK, and ATF6) and express the ER stress sensitive molecule, old astrocyte specifically induced substance (OASIS). Further, astrocytes are largely resistant to aberrant ER stress‐induced cell death (Meares et al., 2014). GRP78 is important in protecting astrocytes as shown by overexpression in the in vitro stroke model of oxygen glucose deprivation (Ouyang et al., 2011).
Astrocytes have been demonstrated to express many cytokines, chemokines, and reactive species that contribute to the inflammatory environment of the CNS (Farina et al., 2007). Table 1 describes molecules that have been demonstrated to be induced by ER stress in astrocytes. Further, the UPR has been explicitly linked to initiating inflammation in other cells types. For example, UPR activation augments inflammatory responses stimulated by the bacterial cell wall component lipopolysaccharide (LPS) and can directly drive activation of the acute phase response (Martinon & Glimcher, 2011; Zhang et al., 2006). Taken together, accumulating evidence suggests that chronic ER stress and UPR signaling in astrocytes may play a pathological role in neurological disease.
TABLE 1.
Endoplasmic reticulum (ER) stress‐induced changes in astrocytes
| Factor | Increased (↑), decreased (↓), no change (—) | Method | Reference |
|---|---|---|---|
| Cytokines | |||
| IL‐1α/β | — | ELISA | (Meares et al., 2014) |
| IL‐6 | ↑ | ELISA and qPCR | (Guthrie et al., 2016; Meares et al., 2014; Sims & Meares, 2019) |
| IFN‐γ | — | ELISA | (Meares et al., 2014) |
| TNF‐α | — | ELISA | (Guthrie et al., 2016; Meares et al., 2014) |
| MCSF | ↑ | ELISA | (Meares et al., 2014) |
| LIF | ↑ | ELISA and qPCR | (Sanchez et al., 2019) |
| IL‐11 | ↑/— | qPCR/ELISA | (Sanchez et al., 2019) |
| OSM | ↑/— | qPCR/ELISA | (Guthrie et al., 2016; Sanchez et al., 2019; Sims & Meares, 2019) |
| Chemokines | |||
| CXCL1 | ↑ | ELISA and qPCR | (Meares et al., 2014; Sims & Meares, 2019) |
| CXCL9 | — | ELISA | (Meares et al., 2014) |
| CXCL10 | ↑ | ELISA and qPCR | (Meares et al., 2014; Smith et al., 2020) |
| CCL2 | ↑ | ELISA | (Guthrie et al., 2016; Meares et al., 2014; Sims & Meares, 2019) |
| CCL3 | ↑ | ELISA | (Meares et al., 2014) |
| CCL4 | ↑ | ELISA | (Meares et al., 2014) |
| CCL5 | — | ELISA | (Meares et al., 2014) |
| CCL11 | ↑ | ELISA | (Meares et al., 2014) |
| CCL20 | ↑ | qPCR | (Guthrie et al., 2016; Meares et al., 2014) |
| Growth factors/ECM proteins | |||
| VEGF | ↑ | ELISA | (Meares et al., 2014) |
| CNTF | ↓ | ELISA, qPCR | (Sanchez et al., 2019) |
| BDNF | — | RNAseq | (Sims & Meares, 2019) |
| GDNF | — | RNAseq | (Sims & Meares, 2019) |
| Collagen | ↓ | LC/MS | (Smith et al., 2020) |
| Fibronectin | ↓ | LC/MS | (Smith et al., 2020) |
| Glypican‐4 | ↓ | LC/MS | (Smith et al., 2020) |
| Complement | |||
| C1qa | ↓ | RNAseq | (Sims & Meares, 2019) |
| C3 | ↑ | qPCR/RNA scope | (Smith et al., 2020) |
| Serping1 | ↓ | qPCR | (Smith et al., 2020) |
| Gliosis markers | |||
| Vimentin | ↑ | qPCR | (Smith et al., 2020) |
| Lcn2 | ↑ | qPCR | (Smith et al., 2020) |
| Serpina3n | ↓ | qPCR | (Smith et al., 2020) |
| CD109 | ↓ | qPCR | (Smith et al., 2020) |
| Stress related | |||
| GADD45α | ↑ | qPCR | (Sims & Meares, 2019) |
| TRIB3 | ↑ | qPCR | (Guthrie et al., 2016; Sims & Meares, 2019) |
| ERO1B | ↑ | qPCR | (Sims & Meares, 2019) |
| ATF6 | ↑ | qPCR | (Sims & Meares, 2019) |
| ATF4 | ↑ | qPCR/immunoblot | (Sims & Meares, 2019; Smith et al., 2020) |
| CHOP (ddit3) | ↑ | Immunoblot | (Sims & Meares, 2019) |
| PERK | ↑ | qPCR | (Sims & Meares, 2019) |
| GADD34 | ↑ | Immunoblot | (Smith et al., 2020) |
| Lipoproteins | |||
| Apolipoprotein E | ↓ | LC/MS | (Smith et al., 2020) |
| Low density lipoprotein in receptor related protein 1 | ↓ | LC/MS | (Smith et al., 2020) |
2. IRE1 IN ASTROCYTES
IRE1 signaling has been linked to cell death and inflammation in the CNS. Evidence of active IRE1 signaling has been reported in post‐mortem human tissue in clinically confirmed cases of AD, HD, and glioma in addition to many mouse in vivo and in vitro disease models. In an immortalized astrocytic cell line, SVGA, cells infected with HIV‐1 require IRE1 signaling to activate JNK and activator protein (AP)‐1 to induce cell death (Shah et al., 2016). Further, nitric oxide (NO) has also been demonstrated to activate IRE1‐dependent signaling in human glioma cell lines. Treating human astrocytoma (CRT‐MG) cells with an NO donor and the ER stress inducer, thapsigargin, increased apoptosis that coincided with IRE1 nuclease activity, IRE1/TRAF2 complex formation, and p‐JNK1/2 levels, implying that treatment of NO subsequently activates the IRE1‐α/TRAF2/JNK pathway. IRE1 knockdown confirmed that intracellular NO affects IRE1‐dependent phosphorylation of CREB in human glioma cells (Kim et al., 2010). Together, this suggests pathogenic stimuli (viral infection and reactive nitrogen species) can activate the IRE1 arm of the UPR and contribute to cell death in in vitro astrocytoma cell lines.
In vivo evidence suggests that IRE1 signaling in astrocytes is associated with the neurodegenerative diseases AD and MS. In brain tissue from AD patients, phosphorylated IRE1 is increased and correlates with disease severity based on Braak Staging, a pathology‐based characterization of AD (Braak et al., 2003; Braak & Braak, 1991; Duran‐Aniotz et al., 2017). To investigate the role of IRE1 in a mouse model of AD, IRE1 was deleted in the nervous system using Nestin‐cre and crossed with the 5XFAD genetic model of AD. Genetic deletion of the RNase domain of IRE1 significantly reduced amyloid deposition and astrocyte activation. Further, deficiency of IRE1 signaling improved synaptic function and long‐term potentiation, suggesting restored memory and learning capacity of the mice. This led to the amelioration of disease hallmarks including Aβ1‐42 production, amyloid plaque deposition, and cognitive deficits. Additionally, deletion of IRE1 reduced astrogliosis, based on GFAP staining, in the 5XFAD hippocampus. In this case, attenuation of gliosis may be through a direct effect on astrocytes or due to reduced overall disease burden (Duran‐Aniotz et al., 2017). Further AD studies are needed to delineate the astrocyte‐specific contributions of IRE1 signaling.
In a large pharmacogenetic screen to identify signaling pathways involved in pathogenic neuroinflammation in MS, astrocytes were stimulated in vitro with TNF‐α and IL‐1β, two cytokines known to be associated with the pathogenesis of EAE and MS. Here, Wheeler et. al determined that IRE1 is phosphorylated and XBP1 was spliced, suggesting activation of IRE1 signaling during astrocyte‐mediated neuroinflammation. To confirm this in vivo, this study used cell‐specific lentiviral delivery of short hairpin (sh)—RNA targeting the gene that encodes for IRE1 (ern1) to knockdown expression in astrocytes during active EAE, which reduced disease severity. These studies demonstrated that abrogating expression of IRE1 under control of the astrocyte selective GFAP promoter ameliorated EAE disease course and reduced inflammatory mediators produced by astrocytes (Wheeler et al., 2019). This suggests that IRE1 signaling in astrocytes is pathogenic in the murine EAE model of MS.
These studies, collectively, imply that IRE1 signaling in astrocytes can be activated by various stimuli and that activated IRE1 can integrate with many signaling pathways that promote inflammation or cell death.
3. PERK IN ASTROCYTES
Activated PERK signaling has been reported in a variety of neurological diseases including AD, MS, prion disease, neurotropic viral infection, and ALS (Hoozemans et al., 2012; Ito et al., 2009; Moreno et al., 2013; Nijholt et al., 2012). Using immunocytochemistry to analyze brain tissue of human MS samples, the UPR proteins GRP78, XBP‐1, and CHOP were increased in acute MS lesions (Mháille et al., 2008). In models of prion disease, neuronal cell lines were infected with PrP, the misfolded protein associated with prion disease. Prion infected neurons were more susceptible to cell death, and targeting PERK signaling in in vivo models of prion disease is protective (Moreno et al., 2012, 2013; Torres et al., 2010). Some reports show that prolonged expression of CHOP is pro‐apoptotic, but this has not directly been demonstrated in primary astrocytes or in vivo models (Kawahara et al., 2001; Oyadomari et al., 2001). However, the Venezuelan equine encephalomyelitis virus (VEEV) induces apoptosis of the astrocyte‐like glioblastoma cell line (U87Mg) through CHOP expression that is activated by PERK (Baer et al., 2016). PERK knockdown in primary astrocytes reduces viral load of VEEV, but there is no difference in viral load between U87Mg cells with or without PERK expression (Dahal et al., 2021).
Further, evidence of activated PERK signaling in astrocytes has been reported in neuropathological studies of human AD and PD brains (Hoozemans et al., 2012; Nijholt et al., 2011). Additional studies have established that PERK is phosphorylated in glial cells in brains from tauopathy‐associated dementias (Nijholt et al., 2012). In a 2014 study, Devi and Ohno examined the role of a hemizygous PERK knockout crossed to the genetic AD model, 5XFAD. Genetic PERK ablation reduces phosphorylated eIF2α and ATF4. PERK haploinsufficiency in 5XFAD mice partially rescued memory loss in a behavioral fear conditioning model. These cognitive improvements coincided with a reduction in amyloid‐β plaque burden in hippocampal and cortical regions of 5XFAD mice. Importantly, the maladaptive effects of PERK signaling were specific to onset of AD; there were no measured cognitive changes in unaffected PERK+/− mice compared to control animals (Devi & Ohno, 2014).
In sporadic ALS and in the transgenic ALS mouse model that expresses mutant superoxide dismutase (SOD)G93A, immunohistochemistry staining of spinal cords demonstrated that many astrocytes, along with other cell types, expressed CHOP, suggesting that PERK signaling is activated in astrocytes in ALS (Ito et al., 2009). Another study modeling ALS in mice demonstrated that astrocytes are activated, as quantified by GFAP immunofluorescence staining. Here, mice expressing wild type human SOD, which has been reported to spontaneously aggregate and model spontaneous ALS, were exposed to the pharmacological inhibitor of N‐linked glycosylation (tunicamycin) to induce UPR activation, which was shown to increase SOD1 aggregation. Importantly, wild type littermates did not have a significant increase in GFAP staining upon tunicamycin treatment (Medinas et al., 2018). This suggests that SOD aggregation and UPR activation enhance GFAP expression, which is associated with a reactive astrocyte phenotype, in a murine ALS model.
PERK is also active in models of acute brain injury. In the TBI model of controlled closed cortical impact, PERK is phosphorylated and colocalizes with GFAP, a marker of reactive astrocytes. In vitro analyses identified that the calcineurin isoform β (CNβ) can interact with PERK to drive its oligomerization and downstream activation of the UPR, independent of CNβ phosphatase activity. Here, the authors found that CNβ loss is detrimental in TBI and photothrombotic stroke models. In stroke, this is associated with reduced phosphorylation of eIF2α and increased GFAP expression. Additionally, overexpression of CNβ attenuated astrocyte toxicity from oxidative and hypoxic insults through a PERK dependent mechanism (Chen et al., 2016). This suggests a potential role for PERK signaling in promoting astrocyte survival after acute injury. These findings are consistent with other reports using the blast injury model of TBI in which GFAP expression is induced upon injury and is reduced when treated with the phosphatase inhibitor salubrinal to maintain eIF2α phosphorylation (Logsdon et al., 2016). Salubrinal reduced impulsive‐like behavior induced by repeated blast injury, suggesting that prolonging eIF2α phosphorylation in acute injury models may be protective (Logsdon et al., 2016). Similarly, p‐eIF2α, GRP78, and CHOP expression is increased post TBI and these markers of ER stress colocalize with GFAP‐positive astrocytes and multiple other cell types. Here, salubrinal improved motor function and spatial defects as tested by the Morris Water Maze post‐TBI (Wang et al., 2019). Although these studies did not directly define functional mechanisms in astrocytes, they provide evidence of reactive astrocyte activation and ER stress during acute neuronal injury. Importantly, while enhancing or maintaining eIF2α phosphorylation immediately after injury provides protection, reversing the p‐eIF2α‐mediated translational block using the eIF2B agonist ISRIB during the chronic phase (4 weeks post injury) improves cognitive function (Chou et al., 2017). These studies indicate that PERK signaling, and potentially other eIF2α kinases, have differential effects during the acute and chronic phases following cerebral injury. The role of PERK signaling in astrocytes in either phase is unknown and warrants additional astrocyte‐specific loss‐ or gain‐of‐function studies.
Additionally, vanishing white matter disease (VWM) demonstrates the importance of downstream PERK signaling in astrocytes. VWM is a leukoencephalopathy in which dysfunctional astrocytes are thought to drive pathogenesis (Bugiani et al., 2018). VWM is caused by an autosomal recessive mutation in eIF2B, which reduce function and cause prolonged suppression of protein translation in response to stimuli that promote eIF2α phosphorylation (Dooves et al., 2016; Moon & Parker, 2018). This highlights a role for phosphorylated eIF2α‐driven translational repression in preserving astrocyte homeostasis and directly links signaling components downstream of PERK to neurological disease.
To date, multiple reports link UPR‐dependent PERK signaling in astrocytes to inflammatory gene expression and/or neurotoxicity (Guthrie et al., 2016; Meares et al., 2014; Smith et al., 2020; Sprenkle et al., 2017). ER stress‐inducing pharmacological agents thapsigargin and tunicamycin promote phosphorylation of eIF2α in primary murine astrocytes (Guthrie et al., 2016; Sims & Meares, 2019; Smith et al., 2020; Sprenkle et al., 2019). A 2014 study demonstrated that gene expression of inflammatory markers (IL‐6, CCL2), astrocyte markers (GFAP, OASIS), and ER stress‐related genes (GRP78, CHOP, PERK, ATF4) are upregulated throughout the course of EAE in brain and spinal cord tissue. Downstream markers of PERK activation such as phosphorylation of eIF2α and CHOP expression are exhibited in thapsigargin‐treated astrocytes concomitantly with upregulation of IL‐6, CCL2, and CCL20. Additionally, ER stress augmented IL‐6 expression induced by IL‐6 or oncostatin M (OSM) in a PERK‐dependent fashion. This suggests that astrocytes may contribute to the UPR and inflammatory response seen in CNS tissue during EAE. It is important to note that these inflammatory proteins are induced at the protein level even under conditions of phosphorylated eIF2α, which functions to attenuate translation, demonstrating that these proteins are translated during UPR activation (Meares et al., 2014).
PERK is an important driver of inflammatory gene expression in astrocytes in response to ER stress. A partial (heterozygous) or complete (homozygous) genetic loss of PERK in primary astrocytes was associated with a lower astrocyte‐driven expression of as IL‐6, CCL2, and CCL20 analyzed by qPCR or ELISA. Further, primary astrocytes treated with thapsigargin and a PERK inhibitor, GSK2606414, reduced production of cytokines and chemokines measured by ELISA. This demonstrates that PERK activation contributes to both transcriptional and translational activation of inflammatory mediators in astrocytes (Guthrie et al., 2016). Therefore, unresolved UPR activation may contribute to prolonged, aberrant inflammatory activation via PERK signaling that may contribute to the non‐resolving nature of neurological diseases.
Cytokines such as IL‐6, which is driven by PERK activation in astrocytes, rely on Janus kinase (JAK)—Signal transducer and activator of transcription (STAT) signaling to exert their effects. JAK–STAT signaling has been directly linked to astrocyte‐driven pathology in neurodegeneration. STAT3 activation occurs in astrocytes in response to acute injury and is required for astrocytes to form glial scars and take on a reactive astrocyte phenotype (astrogliosis) (Wanner et al., 2013). Astrocytes in models of AD, HD, and MS also express higher levels of phosphorylated STAT3 (Ben Haim et al., 2015). Additionally, JAK inhibition ameliorates disease progression in rodent models of PD and MS (Liu et al., 2014; Qin et al., 2016).
PERK signaling activates downstream signaling in a JAK1 dependent mechanism, and inhibiting JAK1 kinase activity reduced ER stress‐induced inflammatory gene expression. Importantly, JAK1 inhibition does not impact all ER stress‐induced gene expression. Further, it has been shown that PERK activates JAK1 to drive a subset of gene expression that is distinct from those induced by the JAK/STAT activating cytokine OSM (Sims & Meares, 2019). This demonstrates that UPR signaling modulates inflammatory responses in a manner distinct from traditional inflammatory signaling. Taken together, this evidence suggests that PERK and JAK–STAT signaling in neurodegenerative disease models may promote aberrant inflammation. Targeting PERK signaling in astrocytes may be a mechanism to selectively attenuate immune responses in neurological diseases.
Targeting the UPR to selectively attenuate inflammation is supported by work in other cell types. For example, UPR signaling in macrophages activates proinflammatory cytokine signaling via the IRE1 pathway. Here, ER stress activates the nucleotide‐binding oligomerization domain‐containing protein (NOD) 1/2 and sXBP1 in an IRE1 dependent manner. Contrary to macrophages, PERK drives IL‐6 expression in astrocytes. This highlights that the UPR regulates inflammation using distinct mechanisms in different cell types (Keestra‐Gounder et al., 2016; Martinon et al., 2010). ER stress‐induced IL‐6 production in astrocytes differs from macrophages in that it requires PERK and JAK1 but is independent of IRE1 and nuclear factor‐κB (NF‐κB) (Guthrie et al., 2016; Meares et al., 2014). Additionally, endothelial cells produce IL‐6 in response to ER stress, but here, this IL‐6 expression is dependent on both ATF4 and sXBP1 (Gargalovic et al., 2006). ER stress induced IL‐6 expression in astrocytes does not rely on ATF4 signaling, as demonstrated using siRNA‐mediated knockdown of ATF4 in primary astrocyte cultures (Sims & Meares, 2019). This illustrates the need for more careful investigations regarding the nuances of UPR signaling in various cell types. For example, the UPR in the CNS literature focuses heavily on neurons and oligodendrocytes, however, these findings may not apply to astrocytes. Although astrocytes induce IL‐6 and other inflammatory molecules in a PERK‐dependent fashion, this is not the case for other IL‐6 family members. Importantly, ciliary neurotrophic factor (CNTF) is downregulated upon ER stress induction in cultured astrocytes (Sanchez et al., 2019). This suggests trophic support from astrocytes can be restricted by the UPR. Indeed, it has been shown that ER stressed astrocytes lose trophic support for neuronal synapse formation (Smith et al., 2020).
Collectively, multiple studies have demonstrated that PERK signaling promotes an astrocyte‐driven inflammatory response. Although inflammation provides a beneficial and restorative role, chronic inflammation is thought to contribute to neurological disease. PERK signaling in astrocytes may be a target to selectively attenuate damaging inflammation while retaining beneficial inflammatory signaling in the CNS. Further studies and conditional deletion of PERK and downstream signaling components in astrocytes are needed to solidify the role in disease models.
4. ATF6 AND OASIS IN ASTROCYTES
ATF6 and OASIS (CREB3L1) are bZIP transcription factors similarly activated in response to ER stress. OASIS is a molecule primarily expressed in astrocytes in the CNS. Upon activation, it is transported to the Golgi apparatus, is cleaved, and the N‐terminal domain promotes expression of ERAD‐associated genes (Kondo et al., 2005). ATF6 is activated (cleaved) in embryonal astrocytes during differentiation suggesting a role for ATF6 in astrocyte development (Saito et al., 2012). OASIS is also important for astrocyte differentiation. In mice lacking OASIS, astrocyte development was impaired. OASIS was shown to bind the promoter of glial cells missing transcription factor 1 (Gcm1) and promote Gcm1 expression. Gcm1 may regulate GFAP promoter methylation allowing transcriptional activation. The reduced expression of Gcm1 in OASIS−/− mice may, in part, underlie the reduced astrocyte differentiation (Saito et al., 2012). To date, few studies have been performed examining the role of ATF6 in astrocytes during disease states. In a murine model of ischemic stroke, middle cerebral artery occlusion (MCAO), ATF6 knockout mice exhibited reduced infarct area as analyzed by the metabolic stain triphenyl tetrazolium chloride (TTC). Concomitantly, ATF6α knockout mice had reduced STAT3 activation and expression of GFAP in the ischemic area of the brain 3 days post MCAO as measured by immunoblotting (Yoshikawa et al., 2015). This study suggests that ATF6 is protective during ischemia via an astrocyte‐dependent mechanism.
OASIS activation has also been linked, in astrocytes, to AD disease mechanisms. Apolipoprotein E (ApoE) is a protein involved in catabolizing triglycerides and the ApoE4 allele is strongly associated with the development of AD, although causal associations between ApoE4 and AD are not fully known, reviewed in Belloy et al. (2019). Primary astrocytes expressing mutant APOE, to model human ApoE4, exhibit reduced ApoE expression and increased UPR activation, including cleavage of OASIS and genes downstream of the IRE1 and PERK pathways. This suggests that ApoE can induce cleavage of OASIS in astrocytes and activate the UPR in astrocytes and promote neuronal toxicity (Zhong et al., 2009). Collectively, there are limited studies on the role of ATF6 and OASIS, however, these studies demonstrate that activation must be well‐regulated for proper astrocyte function.
4.1. Non‐cell autonomous effects of ER stressed astrocytes
In a 2017 study, Sprenkle et al was the first to describe that astrocytes can transmit ER stress to other cell types. A phenomenon that was previously described in cancer cells and termed transmissible ER stress (TERS) (Mahadevan et al., 2012). This suggested that UPR activation in astrocytes can induce UPR signaling in neighboring cells. In this study, astrocyte conditioned media (ACM) collected from astrocytes treated with the ER stress‐inducing agent thapsigargin or tunicamycin was transferred to HT‐22 hippocampal neuronal cells. The cells that were exposed to thapsigargin treated ACM exhibited higher gene expression and protein levels of GRP78, spliced XBP1, and CHOP, indicating that astrocytes secrete a soluble factor that stimulates an ER stress response. Further, this study showed that neurons experiencing ER stress also secrete a molecule that induces ER stress in cultures of neurons, astrocytes, and microglia (Sprenkle et al., 2019). This study identified that UPR activation can be transmitted between cells of the nervous system. These studies are consistent with previous work that demonstrated that ER stress is also transmissible between cancer or myocardial cells and macrophages, which also respond to ER stress by producing inflammatory molecules, albeit these mechanisms are distinct from those identified in astrocytes (Mahadevan et al., 2011; Sanchez et al., 2019; Zhang et al., 2017). ER stressed astrocytes, through a PERK‐dependent process, also increase microglial expression of IL‐1β and IL‐6 (Meares et al., 2014). Independently of PERK, ER stressed astrocytes reduce microglial expression of arginase, CD206 and insulin like growth factor 1 (Guthrie et al., 2016). Together, these data indicate that in response to ER stress, astrocytes can shift microglia to an inflammatory phenotype. Additionally, Wheeler and colleagues demonstrated that XBP1 knockdown in astrocytes decreases the number of monocytes that traffic to the CNS during EAE. Macrophages that trafficked to the CNS during EAE in the GFAP‐driven XBP1 knockdown had reduced expression of inflammatory genes involved in IL‐6 signaling, NF‐κB signaling, and chemokine signaling. Similarly, microglia in the astrocyte specific XBP1 knockdown had reduced proinflammatory gene expression in comparison to EAE animals with XBP1 expression astrocytes (Wheeler et al., 2019).
ACM from healthy astrocytes is known to support synaptogenesis (Allen et al., 2012; Baldwin & Eroglu, 2017; Hughes et al., 2010). To determine if UPR activation impacts the ability of astrocytes to support synapses, Mallucci and colleagues collected ACM from thapsigargin‐treated astrocytes. By immunostaining pre and post synaptic terminals, ACM from UPR activated astrocytes was shown to reduce synaptogenesis. Further, inhibiting PERK pharmacologically restored the ability of ACM to promote synapse formation, suggesting that UPR activation via PERK inhibits astrocyte‐mediated neurotrophic functions. Further, this study tested if targeting PERK‐eIF2α signaling in vivo could be neuroprotective. Using mice that over express prion protein (PrP) and succumb to prion infection. Astrocyte specific lentiviral overexpression of GADD34, an eIF2α‐specific phosphatase, was markedly protective in prion‐infected mice. GADD34 overexpression (to reduce PERK signaling) in astrocytes prevented neurodegeneration in the hippocampus, had an increased number of pyramidal neurons, reduced astrocyte reactivity based on morphology and GFAP staining, and extended the life span of these mice in comparison to control PrP animals. This study shows both in vitro and in vivo that UPR activation via the PERK pathway alters the transcriptome and secreted molecules of astrocytes and this is linked to a reduction in neuronal synapse formation (Smith et al., 2020). These studies expand upon and corroborate the previous findings that PERK inhibition is protective in prion infection (Halliday et al., 2017; Halliday & Mallucci, 2014; Moreno et al., 2012, 2013). Further, this suggests that UPR‐activated astrocytes have pathogenic roles in prion infection and identifies PERK signaling as a central driver in this process.
Consistent with the notion that astrocytes have a significant role in directing the milieu of the inflammatory environment in the CNS, viral infections have also shown to induce the UPR in astrocytes, leading to pathogenic non‐cell‐autonomous astrocyte dependent pathology. The HIV protein Tat has been shown to induce ER stress in astrocytes leading to GFAP‐dependent neurotoxicity (Fan & He, 2016). Inflammation and expression of the human endogenous retrovirus protein, syncytin‐1, promote ER stress in astrocytes in MS (Deslauriers et al., 2011). This study demonstrated that ER stress proteins were upregulated in MS patient brains, along with the human endogenous retrovirus protein (HERV) syncytin‐1. Syncytin‐1 induces splicing of XBP1 and leads to downstream inflammation. These mechanisms were confirmed by transfecting primary human fetal astrocytes with syncinctin‐1. This induced splicing of XBP1, indicating that the IRE1 pathway is activated. Further, Nos2 was concomitantly upregulated and contributed to oligodendrocyte toxicity in the EAE model. Together, this suggests that IRE1 signaling is stimulated by the HERV protein syncytin‐1 to initiate a sXBP1‐dependent nitric oxide and neuroinflammatory response.
Additionally, Zika virus has been shown to activate the UPR in astrocytes. ZIKV infection of astrocytes caused an over expression of UPR‐related genes BiP, XBP1, CHOP, and growth arrest and DNA damage‐inducible protein (GADD) 34, Under these conditions, cell viability was decreased, RNA metabolism genes and micro‐RNAs were downregulated, however, astrocyte‐derived soluble factors glial cell line‐derived neurotrophic factor (GDNF) and neuronal growth factor (NGF) were upregulated, highlighting that some molecules were still being translated under ER stress conditions (Kozak et al., 2017). However, these results are associated with UPR activation, and direct evidence for the non‐cell autonomous action of UPR signaling in astrocytes still requires investigation. These results lay the groundwork for further studies examining the role of ZIKV and other neurotrophic viral infections in astrocytes.
In summary, astrocytes play a critical role at directing the overall CNS environment due to their close physical and trophic connection to other CNS cells as well as blood vessels. UPR activation is emerging as an important process by which astrocytes influence the survival, activation and function of other CNS resident and infiltrating cells.
5. DISCUSSION AND PERSPECTIVES
The UPR has been studied in a multitude of disease states and cell types, however, the astrocyte‐specific roles of the UPR is a relatively new field. ER stress has been primarily characterized in the CNS focusing on neurons and oligodendrocytes (Hetz & Saxena, 2017; Sprenkle et al., 2017). As more studies are performed, it is evident that UPR activation has diverse roles in each CNS cell type. A working model is proposed in Figure 2. For example, EAE is ameliorated by PERK activation in oligodendrocytes, but PERK knockdown in astrocytes had no effect on the development of EAE (Lin et al., 2007; Wheeler et al., 2019). Although the UPR is known to activate pathways that have been associated with apoptosis, there is little evidence that UPR signaling in astrocytes induces cell death. Instead, UPR‐activated astrocytes are posited to be in a unique position to contribute to the inflammatory environment of the CNS because astrocytes are the most populous glial cell, can be neurotoxic, and direct CNS inflammation by promoting microglial activation and leukocyte trafficking (Clarke et al., 2018; Liddelow et al., 2017; Meares et al., 2012). Inflammatory and reactive astrocytes are attributed to neurotoxicity in many disease models. Understanding how astrocytes are fine‐tuned to produce these neurotoxic responses is of vital importance; neuronal loss cannot be overcome and leads to motor and cognitive decline. Here, we provide evidence that UPR signaling could substantially contribute to astrocyte‐driven neuronal dysfunction. To gain complete understanding of the UPR in the CNS, more advanced cell‐specific studies must be completed. Gain and loss of function studies in vivo in astrocyte conditional models are underway and will provide a wealth of information on the functional role of ER stress signaling in astrocytes.
FIGURE 2.
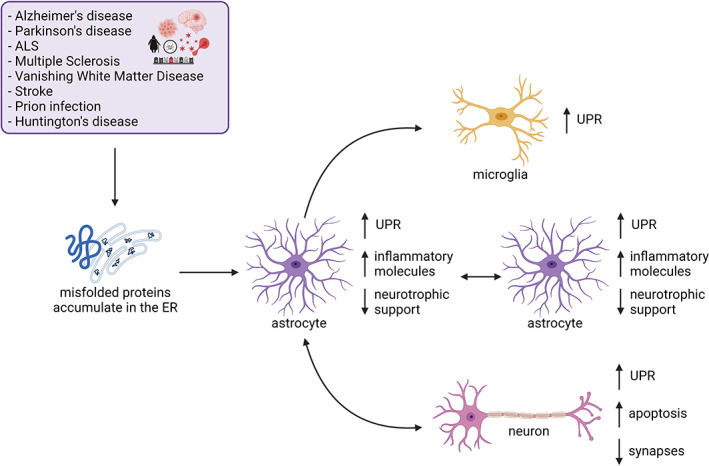
Proposed mechanisms by which endoplasmic reticulum stress signaling in astrocytes impacts the overall central nervous system (CNS) environment. Here, we propose a model where astrocytes experience endoplasmic reticulum stress during disease states, activating the unfolded protein response (UPR). UPR‐related molecules are upregulated in astrocytes, concomitant with inflammatory genes. Further, UPR‐activated astrocytes have reduced capacity to support neuronal synapses and activated UPR in astrocytes is associated with increased synaptic loss and lower numbers of neurons during disease. Further, astrocytes have the capacity to “transmit” endoplasmic reticulum (ER) stress to other neuronal cell types including other astrocytes, microglia, and neurons. Figure was created in Biorender
Targeting UPR signaling in astrocytes has been tested in select models of neurological diseases. For example, IRE1 knockdown in astrocytes is protective in the EAE model of MS (Wheeler et al., 2019). Therefore, IRE1 signaling may be pathogenic in neurological diseases that are associated with excessive neuroinflammation. Astrocyte‐specific overexpression of GADD34, an eIF2α‐specific phosphatase, is protective in prion disease (Smith et al., 2020), consistent with studies using small molecules to target PERK signaling (Moreno et al., 2013). These studies suggest a pathogenic role for PERK signaling in astrocytes in diseases driven by protein misfolding. Together, with in vitro evidence that PERK signaling in astrocytes promotes inflammation and that ER stress is transmissible (Guthrie et al., 2016; Meares et al., 2014; Sanchez et al., 2019; Sims & Meares, 2019; Smith et al., 2020; Sprenkle et al., 2017), these studies suggest that UPR signaling in astrocytes may profoundly impact multiple neurological diseases. Additional studies using astrocyte‐selective Cre drivers such GFAP‐Cre (Garcia et al., 2004; Gregorian et al., 2009) and ALDH1L1‐CreERT2 (Srinivasan et al., 2016) to target specific UPR components in additional disease models of neural injury and neurodegeneration will ultimately define ER stress signaling in astrocytes and its impact on disease.
Further, recent studies have established that substantial regional heterogeneity of astrocytes exists (Matias et al., 2019). Currently, astrocyte specific UPR signaling has not been characterized or compared across regional locations of the CNS. The location and composition of misfolded proteins is known to vary among each neurodegenerative disease. Therefore, this prompts the possibility that UPR signaling in astrocytes can greatly differ based on location, disease state or stage.
Critically, limited therapies and cures exist for most neurological diseases. Therefore, it is logical to assume that identifying novel therapeutic targets to regulate disease‐associated signaling cascades is vital for the design of effective treatments. Considering the well‐established association of protein aggregation and accumulation in neurological disorders and the recent advances in astrocyte biology, understanding how astrocytes experiencing ER stress influence the CNS environment may be a critical link in understanding signaling pathways that contribute to neurological dysfunction. ER stress has been linked to neurotoxicity in models of neurodegeneration. However, neurotoxicity may be the result of multiple unconfirmed mechanisms including: (1) initiation of apoptosis by the UPR in neurons, (2) loss of supportive function in other CNS cells, such as glia, (3) UPR signaling in glial cells have a gain of neurotoxic function, or (4) a combination of these possibilities. Altogether, an accumulation of evidence suggests that the duration, cell type, and inflammatory environment of the CNS dictates the consequences of active UPR signaling.
ACKNOWLEDGMENTS
The authors would like to thank past and present members of the Meares lab at West Virginia University for their support and thoughtful conversations about the content of this literature review. We would also like to acknowledge Biorender, which was used to create our figures. This work was supported by the National Institute of Neurological Disorders and Stroke.
Sims, S. G. , Cisney, R. N. , Lipscomb, M. M. , & Meares, G. P. (2022). The role of endoplasmic reticulum stress in astrocytes. Glia, 70(1), 5–19. 10.1002/glia.24082
Funding information National Institute of Neurological Disorders and Stroke, Grant/Award Numbers: 1F31NS113482, 5R01NS099304
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- Alberdi, E. , Wyssenbach, A. , Alberdi, M. , Sanchez‐Gomez, M. V. , Cavaliere, F. , Rodriguez, J. J. , Verkhratsky, A. , & Matute, C. (2013). Ca(2+)‐dependent endoplasmic reticulum stress correlates with astrogliosis in oligomeric amyloid beta‐treated astrocytes and in a model of Alzheimer's disease. Aging Cell, 12(2), 292–302 10.1111/acel.12054 [DOI] [PubMed] [Google Scholar]
- Allen, N. J. , Bennett, M. L. , Foo, L. C. , Wang, G. X. , Chakraborty, C. , Smith, S. J. , & Barres, B. A. (2012). Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature, 486(7403), 410–414 10.1038/nature11059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki, Y. , Haginoya, K. , Munakata, M. , Yokoyama, H. , Nishio, T. , Togashi, N. , Ito, T. , Suzuki, Y. , Kure, S. , Iinuma, K. , Brenner, M. , & Matsubara, Y. (2001). A novel mutation in glial fibrillary acidic protein gene in a patient with Alexander disease. Neuroscience Letters, 312(2), 71–74 10.1016/s0304-3940(01)02139-5(01)02139‐5 [DOI] [PubMed] [Google Scholar]
- Baer, A. , Lundberg, L. , Swales, D. , Waybright, N. , Pinkham, C. , Dinman, J. D. , Jacobs, J. L. , & Kehn‐Hall, K. (2016). Venezuelan equine encephalitis virus induces apoptosis through the unfolded protein response activation of EGR1. Journal of Virology, 90(7), 3558–3572 10.1128/JVI.02827-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin, K. T. , & Eroglu, C. (2017). Molecular mechanisms of astrocyte‐induced synaptogenesis. Current Opinion in Neurobiology, 45, 113–120 10.1016/j.conb.2017.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao, Y. , Qin, L. , Kim, E. , Bhosle, S. , Guo, H. , Febbraio, M. , Haskew‐Layton, R. E. , Ratan, R. , & Cho, S. (2012). CD36 is involved in astrocyte activation and astroglial scar formation. Journal of Cerebral Blood Flow and Metabolism, 32(8), 1567–1577 10.1038/jcbfm.2012.52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batiuk, M. Y. , Martirosyan, A. , Wahis, J. , de Vin, F. , Marneffe, C. , Kusserow, C. , Koeppen, J. , Viana, J. F. , Oliveira, J. F. , Voet, T. , Ponting, C. P. , Belgard, T. G. , & Holt, M. G. (2020). Identification of region‐specific astrocyte subtypes at single cell resolution. Nature Communications, 11(1), 1220 10.1038/s41467-019-14198-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becher, B. , Spath, S. , & Goverman, J. (2017). Cytokine networks in neuroinflammation. Nature Reviews. Immunology, 17(1), 49–59 10.1038/nri.2016.123 [DOI] [PubMed] [Google Scholar]
- Bell, M. C. , Meier, S. E. , Ingram, A. L. , & Abisambra, J. F. (2016). PERK‐opathies: An endoplasmic reticulum stress mechanism underlying neurodegeneration. Current Alzheimer Research, 13(2), 150–163 10.2174/1567205013666151218145431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belloy, M. E. , Napolioni, V. , & Greicius, M. D. (2019). A quarter century of APOE and Alzheimer's disease: Progress to date and the path forward. Neuron, 101(5), 820–838 10.1016/j.neuron.2019.01.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Haim, L. , Ceyzériat, K. , Carrillo‐de Sauvage, M. A. , Aubry, F. , Auregan, G. , Guillermier, M. , Ruiz, M. , Petit, F. , Houitte, D. , Faivre, E. , Vandesquille, M. , Aron‐Badin, R. , Dhenain, M. , Déglon, N. , Hantraye, P. , Brouillet, E. , Bonvento, G. , & Escartin, C. (2015). The JAK/STAT3 pathway is a common inducer of astrocyte reactivity in Alzheimer's and Huntington's diseases. The Journal of Neuroscience, 35(6), 2817–2829 10.1523/JNEUROSCI.3516-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolotti, A. , Zhang, Y. , Hendershot, L. , Harding, H. , & Ron, D. (2000). Dynamic interaction of BiP and ER stress transducers in the unfolded‐protein response. Nature Cell Biology, 2(6), 326–332. [DOI] [PubMed] [Google Scholar]
- Braak, H. , & Braak, E. (1991). Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathologica, 82(4), 239–259 10.1007/bf00308809 [DOI] [PubMed] [Google Scholar]
- Braak, H. , Del Tredici, K. , Rub, U. , de Vos, R. A. , Jansen Steur, E. N. , & Braak, E. (2003). Staging of brain pathology related to sporadic Parkinson's disease. Neurobiology of Aging, 24(2), 197–211. 10.1016/s0197-4580(02)00065-9 [DOI] [PubMed] [Google Scholar]
- Brenner, M. , Johnson, A. B. , Boespflug‐Tanguy, O. , Rodriguez, D. , Goldman, J. E. , & Messing, A. (2001). Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nature Genetics, 27(1), 117–120 10.1038/83679 [DOI] [PubMed] [Google Scholar]
- Brozzi, F. , Gerlo, S. , Grieco, F. A. , Juusola, M. , Balhuizen, A. , Lievens, S. , Gysemans, C. , Bugliani, M. , Mathieu, C. , Marchetti, P. , Tavernier, J. , & Eizirik, D. L. (2016). Ubiquitin D regulates IRE1alpha/c‐Jun N‐terminal kinase (JNK) protein‐dependent apoptosis in pancreatic beta cells. The Journal of Biological Chemistry, 291(23), 12040–12056 10.1074/jbc.M115.704619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugiani, M. , Vuong, C. , Breur, M. , & van der Knaap, M. S. (2018). Vanishing white matter: a leukodystrophy due to astrocytic dysfunction. Brain Pathology, 28(3), 408–421 10.1111/bpa.12606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calfon, M. , Zeng, H. , Urano, F. , Till, J. H. , Hubbard, S. R. , Harding, H. P. , Clark, S. G. , & Ron, D. (2002). IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP‐1 mRNA. Nature, 415(6867), 92–96 10.1038/415092a [DOI] [PubMed] [Google Scholar]
- Chen, Y. , Holstein, D. M. , Aime, S. , Bollo, M. , & Lechleiter, J. D. (2016). Calcineurin beta protects brain after injury by activating the unfolded protein response. Neurobiology of Disease, 94, 139–156 10.1016/j.nbd.2016.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou, A. , Krukowski, K. , Jopson, T. , Zhu, P. J. , Costa‐Mattioli, M. , Walter, P. , & Rosi, S. (2017). Inhibition of the integrated stress response reverses cognitive deficits after traumatic brain injury. Proceedings of the National Academy of Sciences of the United States of America, 114(31), E6420–E6426 10.1073/pnas.1707661114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury, G. R. , & Ding, S. (2016). Reactive astrocytes and therapeutic potential in focal ischemic stroke. Neurobiology of Disease, 85, 234–244 10.1016/j.nbd.2015.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson, J. C. , & Ye, Y. (2014). Cleaning up in the endoplasmic reticulum: Ubiquitin in charge. Nature Structural & Molecular Biology, 21(4), 325–335 10.1038/nsmb.2793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, B. E. , Taha, D. M. , Tyzack, G. E. , & Patani, R. (2021). Regionally encoded functional heterogeneity of astrocytes in health and disease: A perspective. GLIA, 69(1), 20–27 10.1002/glia.23877 [DOI] [PubMed] [Google Scholar]
- Clarke, H. J. , Chambers, J. E. , Liniker, E. , & Marciniak, S. J. (2014). Endoplasmic reticulum stress in malignancy. Cancer Cell, 25(5), 563–573 10.1016/j.ccr.2014.03.015 [DOI] [PubMed] [Google Scholar]
- Clarke, L. E. , & Barres, B. A. (2013). Emerging roles of astrocytes in neural circuit development. Nature Reviews. Neuroscience, 14(5), 311–321 10.1038/nrn3484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, L. E. , Liddelow, S. A. , Chakraborty, C. , Münch, A. E. , Heiman, M. , & Barres, B. A. (2018). Normal aging induces A1‐like astrocyte reactivity. Proceedings of the National Academy of Sciences of the United States of America, 115(8), E1896–E1905 10.1073/pnas.1800165115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton, B. L. L. , & Popko, B. (2016). Endoplasmic reticulum stress and the unfolded protein response in disorders of myelinating glia. Brain Research, 1648(Pt B), 594–602 10.1016/j.brainres.2016.03.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahal, B. , Lehman, C. W. , Akhrymuk, I. , Bracci, N. R. , Panny, L. , Barrera, M. D. , Bhalla, N. , Jacobs, J. L. , Dinman, J. D. , & Kehn‐Hall, K. (2021). PERK is critical for alphavirus nonstructural protein translation. Viruses, 13(5), 892. 10.3390/v13050892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deslauriers, A. M. , Afkhami‐Goli, A. , Paul, A. M. , Bhat, R. K. , Acharjee, S. , Ellestad, K. K. , Noorbakhsh, F. , Michalak, M. , & Power, C. (2011). Neuroinflammation and endoplasmic reticulum stress are coregulated by crocin to prevent demyelination and neurodegeneration. Journal of Immunology, 187(9), 4788–4799 10.4049/jimmunol.1004111 [DOI] [PubMed] [Google Scholar]
- Devi, L. , & Ohno, M. (2014). PERK mediates eIF2alpha phosphorylation responsible for BACE1 elevation, CREB dysfunction and neurodegeneration in a mouse model of Alzheimer's disease. Neurobiology of Aging, 35(10), 2272–2281 10.1016/j.neurobiolaging.2014.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooves, S. , Bugiani, M. , Postma, N. L. , Polder, E. , Land, N. , Horan, S. T. , van Deijk, A. L. , van de Kreeke, A. , Jacobs, G. , Vuong, C. , Klooster, J. , Kamermans, M. , Wortel, J. , Loos, M. , Wisse, L. E. , Scheper, G. C. , Abbink, T. E. , Heine, V. M. , & van der Knaap, M. S. (2016). Astrocytes are central in the pathomechanisms of vanishing white matter. The Journal of Clinical Investigation, 126(4), 1512–1524 10.1172/JCI83908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran‐Aniotz, C. , Cornejo, V. H. , Espinoza, S. , Ardiles, A. O. , Medinas, D. B. , Salazar, C. , Foley, A. , Gajardo, I. , Thielen, P. , Iwawaki, T. , Scheper, W. , Soto, C. , Palacios, A. G. , Hoozemans, J. J. M. , & Hetz, C. (2017). IRE1 signaling exacerbates Alzheimer's disease pathogenesis. Acta Neuropathologica, 134(3), 489–506 10.1007/s00401-017-1694-x [DOI] [PubMed] [Google Scholar]
- Fan, Y. , & He, J. J. (2016). HIV‐1 tat induces unfolded protein response and endoplasmic reticulum stress in astrocytes and causes neurotoxicity through glial fibrillary acidic protein (GFAP) activation and aggregation. The Journal of Biological Chemistry, 291(43), 22819–22829 10.1074/jbc.M116.731828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina, C. , Aloisi, F. , & Meinl, E. (2007). Astrocytes are active players in cerebral innate immunity. Trends in Immunology, 28(3), 138–145 10.1016/j.it.2007.01.005 [DOI] [PubMed] [Google Scholar]
- Faulkner, J. R. , Herrmann, J. E. , Woo, M. J. , Tansey, K. E. , Doan, N. B. , & Sofroniew, M. V. (2004). Reactive astrocytes protect tissue and preserve function after spinal cord injury. The Journal of Neuroscience, 24(9), 2143–2155 10.1523/JNEUROSCI.3547-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn, G. C. , Pohl, J. , Flocco, M. T. , & Rothman, J. E. (1991). Peptide‐binding specificity of the molecular chaperone BiP. Nature, 353(6346), 726–730 10.1038/353726a0 [DOI] [PubMed] [Google Scholar]
- Fonseca, S. G. , Gromada, J. , & Urano, F. (2011). Endoplasmic reticulum stress and pancreatic beta‐cell death. Trends in Endocrinology and Metabolism, 22(7), 266–274 10.1016/j.tem.2011.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frakes, A. E. , Metcalf, M. G. , Tronnes, S. U. , Bar‐Ziv, R. , Durieux, J. , Gildea, H. K. , Kandahari, N. , Monshietehadi, S. , & Dillin, A. (2020). Four glial cells regulate ER stress resistance and longevity via neuropeptide signaling in C. elegans . Science, 367(6476), 436–440 10.1126/science.aaz6896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, Y. , Sartori, D. J. , Li, C. , Yu, Q. C. , Kushner, J. A. , Simon, M. C. , & Diehl, J. A. (2012). PERK is required in the adult pancreas and is essential for maintenance of glucose homeostasis. Molecular and Cellular Biology, 32(24), 5129–5139 10.1128/MCB.01009-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia, A. D. , Doan, N. B. , Imura, T. , Bush, T. G. , & Sofroniew, M. V. (2004). GFAP‐expressing progenitors are the principal source of constitutive neurogenesis in adult mouse forebrain. Nature Neuroscience, 7(11), 1233–1241 10.1038/nn1340 [DOI] [PubMed] [Google Scholar]
- Gardner, B. M. , & Walter, P. (2011). Unfolded proteins are Ire1‐activating ligands that directly induce the unfolded protein response. Science, 333(6051), 1891–1894 10.1126/science.1209126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gargalovic, P. S. , Gharavi, N. M. , Clark, M. J. , Pagnon, J. , Yang, W.‐P. , He, A. , Truong, A. , Baruch‐Oren, T. , Berliner, J. A. , Kirchgessner, T. G. , & Lusis, A. J. (2006). The unfolded protein response is an important regulator of inflammatory genes in endothelial cells. Arteriosclerosis, Thrombosis, and Vascular Biology, 26(11), 2490–2496 10.1161/01.ATV.0000242903.41158.a1 [DOI] [PubMed] [Google Scholar]
- Glass, C. K. , Saijo, K. , Winner, B. , Marchetto, M. C. , & Gage, F. H. (2010). Mechanisms underlying inflammation in neurodegeneration. Cell, 140(6), 918–934 10.1016/j.cell.2010.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorian, C. , Nakashima, J. , Le Belle, J. , Ohab, J. , Kim, R. , Liu, A. , Smith, K. B. , Groszer, M. , Garcia, A. D. , Sofroniew, M. V. , Carmichael, S. T. , Kornblum, H. I. , Liu, X. , & Wu, H. (2009). Pten deletion in adult neural stem/progenitor cells enhances constitutive neurogenesis. The Journal of Neuroscience, 29(6), 1874–1886 10.1523/JNEUROSCI.3095-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie, L. N. , Abiraman, K. , Plyler, E. S. , Sprenkle, N. T. , Gibson, S. A. , McFarland, B. C. , Rajbhandari, R. , Rowse, A. L. , Benveniste, E. N. , & Meares, G. P. (2016). Attenuation of PKR‐like ER kinase (PERK) signaling selectively controls endoplasmic reticulum stress‐induced inflammation without compromising immunological responses. The Journal of Biological Chemistry, 291(30), 15830–15840 10.1074/jbc.M116.738021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday, M. , Hughes, D. , & Mallucci, G. R. (2017). Fine‐tuning PERK signaling for neuroprotection. Journal of Neurochemistry, 142, 812–826. 10.1111/jnc.14112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday, M. , & Mallucci, G. R. (2014). Targeting the unfolded protein response in neurodegeneration: A new approach to therapy. Neuropharmacology, 76Pt A, 169–174 10.1016/j.neuropharm.2013.08.034 [DOI] [PubMed] [Google Scholar]
- Hamby, M. E. , & Sofroniew, M. V. (2010). Reactive astrocytes as therapeutic targets for CNS disorders. Neurotherapeutics, 7(4), 494–506 10.1016/j.nurt.2010.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton, R. Y. (2002). ER‐associated degradation in protein quality control and cellular regulation. Current Opinion in Cell Biology, 14(4), 476–482 10.1016/s0955-0674(02)00358-7(02)00358‐7 [DOI] [PubMed] [Google Scholar]
- Han, R. T. , Kim, R. D. , Molofsky, A. V. , & Liddelow, S. A. (2021). Astrocyte‐immune cell interactions in physiology and pathology. Immunity, 54(2), 211–224 10.1016/j.immuni.2021.01.013 [DOI] [PubMed] [Google Scholar]
- Harding, H. , Zhang, Y. , & Ron, D. (1999). Protein translation and folding are coupled by an endoplasmic‐reticulum‐resident kinase. Nature, 397(6716), 271–274. [DOI] [PubMed] [Google Scholar]
- Haze, K. , Yoshida, H. , Yanagi, H. , Yura, T. , & Mori, K. (1999). Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Molecular Biology of the Cell, 10(11), 3787–3799 10.1091/mbc.10.11.3787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka, M. T. , Kummer, M. P. , & Latz, E. (2014). Innate immune activation in neurodegenerative disease. Nature Reviews. Immunology, 14(7), 463–477 10.1038/nri3705 [DOI] [PubMed] [Google Scholar]
- Hetz, C. , & Mollereau, B. (2014). Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nature Reviews. Neuroscience, 15(4), 233–249 10.1038/nrn3689 [DOI] [PubMed] [Google Scholar]
- Hetz, C. , & Saxena, S. (2017). ER stress and the unfolded protein response in neurodegeneration. Nature Reviews. Neurology, 13(8), 477–491 10.1038/nrneurol.2017.99 [DOI] [PubMed] [Google Scholar]
- Hollien, J. , & Weissman, J. S. (2006). Decay of endoplasmic reticulum‐localized mRNAs during the unfolded protein response. Science, 313(5783), 104–107 10.1126/science.1129631 [DOI] [PubMed] [Google Scholar]
- Hoozemans, J. J. , van Haastert, E. S. , Nijholt, D. A. , Rozemuller, A. J. , & Scheper, W. (2012). Activation of the unfolded protein response is an early event in Alzheimer's and Parkinson's disease. Neurodegenerative Diseases, 10(1–4), 212–215 10.1159/000334536 [DOI] [PubMed] [Google Scholar]
- Hughes, E. G. , Elmariah, S. B. , & Balice‐Gordon, R. J. (2010). Astrocyte secreted proteins selectively increase hippocampal GABAergic axon length, branching, and synaptogenesis. Molecular and Cellular Neurosciences, 43(1), 136–145 10.1016/j.mcn.2009.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito, Y. , Yamada, M. , Tanaka, H. , Aida, K. , Tsuruma, K. , Shimazawa, M. , Hozumi, I. , Inuzuka, T. , Takahashi, H. , & Hara, H. (2009). Involvement of CHOP, an ER‐stress apoptotic mediator, in both human sporadic ALS and ALS model mice. Neurobiology of Disease, 36(3), 470–476 10.1016/j.nbd.2009.08.013 [DOI] [PubMed] [Google Scholar]
- Iwakoshi, N. N. , Lee, A. H. , Vallabhajosyula, P. , Otipoby, K. L. , Rajewsky, K. , & Glimcher, L. H. (2003). Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP‐1. Nature Immunology, 4(4), 321–329 10.1038/ni907 [DOI] [PubMed] [Google Scholar]
- Jackson, R. J. , Hellen, C. U. T. , & Pestova, T. V. (2010). The mechanism of eukaryotic translation initiation and principles of its regulation. Nature Reviews. Molecular Cell Biology, 11(2), 113–127 10.1038/nrm2838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagöz, G. E. , Acosta‐Alvear, D. , Nguyen, H. T. , Lee, C. P. , Chu, F. , & Walter, P. (2017). An unfolded protein‐induced conformational switch activates mammalian IRE1. eLife, 6, e30700, 10.7554/eLife.30700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara, K. , Oyadomari, S. , Gotoh, T. , Kohsaka, S. , Nakayama, H. , & Mori, M. (2001). Induction of CHOP and apoptosis by nitric oxide in p53‐deficient microglial cells. FEBS Letters, 506(2), 135–139 10.1016/s0014-5793(01)02898-8(01)02898‐8 [DOI] [PubMed] [Google Scholar]
- Keestra‐Gounder, A. M. , Byndloss, M. X. , Seyffert, N. , Young, B. M. , Chávez‐Arroyo, A. , Tsai, A. Y. , Cevallos, S. A. , Winter, M. G. , Pham, O. H. , Tiffany, C. R. , de Jong, M. F. , Kerrinnes, T. , Ravindran, R. , Luciw, P. A. , McSorley, S. J. , Bäumler, A. J. , & Tsolis, R. M. (2016). NOD1 and NOD2 signalling links ER stress with inflammation. Nature, 532(7599), 394–397 10.1038/nature17631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakh, B. S. , & Deneen, B. (2019). The emerging nature of astrocyte diversity. Annual Review of Neuroscience, 42, 187–207 10.1146/annurev-neuro-070918-050443 [DOI] [PubMed] [Google Scholar]
- Kim, Y. E. , Hipp, M. S. , Bracher, A. , Hayer‐Hartl, M. , & Hartl, F. U. (2013). Molecular chaperone functions in protein folding and proteostasis. Annual Review of Biochemistry, 82, 323–355 10.1146/annurev-biochem-060208-092442 [DOI] [PubMed] [Google Scholar]
- Kim, Y. H. , Joo, H. S. , & Kim, D. S. (2010). Nitric oxide induction of IRE1‐alpha‐dependent CREB phosphorylation in human glioma cells. Nitric Oxide, 23(2), 112–120 10.1016/j.niox.2010.04.009 [DOI] [PubMed] [Google Scholar]
- Kondo, S. , Murakami, T. , Tatsumi, K. , Ogata, M. , Kanemoto, S. , Otori, K. , Iseki, K. , Wanaka, A. , & Imaizumi, K. (2005). OASIS, a CREB/ATF‐family member, modulates UPR signalling in astrocytes. Nature Cell Biology, 7(2), 186–194 10.1038/ncb1213 [DOI] [PubMed] [Google Scholar]
- Kozak, R. A. , Majer, A. , Biondi, M. J. , Medina, S. J. , Goneau, L. W. , Sajesh, B. V. , Slota, J. A. , Zubach, V. , Severini, A. , Safronetz, D. , Hiebert, S. L. , Beniac, D. R. , Booth, T. F. , Booth, S. A. , & Kobinger, G. P. (2017). MicroRNA and mRNA dysregulation in astrocytes infected with zika virus. Viruses, 9(10), 297. 10.3390/v9100297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, A.‐H. , Iwakoshi, N. N. , & Glimcher, L. H. (2003). XBP‐1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Molecular and Cellular Biology, 23(21), 7448–7459 10.1128/mcb.23.21.7448-7459.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, K. P. K. , Dey, M. , Neculai, D. , Cao, C. , Dever, T. E. , & Sicheri, F. (2008). Structure of the dual enzyme Ire1 reveals the basis for catalysis and regulation in nonconventional RNA splicing. Cell, 132(1), 89–100 10.1016/j.cell.2007.10.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitman, J. , Ulrich Hartl, F. , & Lederkremer, G. Z. (2013). Soluble forms of polyQ‐expanded huntingtin rather than large aggregates cause endoplasmic reticulum stress. Nature Communications, 4, 2753 10.1038/ncomms3753 [DOI] [PubMed] [Google Scholar]
- Li, H. , Zhang, N. , Lin, H. Y. , Yu, Y. , Cai, Q. Y. , Ma, L. , & Ding, S. (2014). Histological, cellular and behavioral assessments of stroke outcomes after photothrombosis‐induced ischemia in adult mice. BMC Neuroscience, 15, 58 10.1186/1471-2202-15-58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow, S. A. , Guttenplan, K. A. , Clarke, L. E. , Bennett, F. C. , Bohlen, C. J. , Schirmer, L. , Bennett, M. L. , Münch, A. E. , Chung, W. S. , Peterson, T. C. , Wilton, D. K. , Frouin, A. , Napier, B. A. , Panicker, N. , Kumar, M. , Buckwalter, M. S. , Rowitch, D. H. , Dawson, V. L. , Dawson, T. M. , … Barres, B. A. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature, 541(7638), 481–487 10.1038/nature21029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, W. , Bailey, S. L. , Ho, H. , Harding, H. P. , Ron, D. , Miller, S. D. , & Popko, B. (2007). The integrated stress response prevents demyelination by protecting oligodendrocytes against immune‐mediated damage. The Journal of Clinical Investigation, 117(2), 448–456 10.1172/JCI29571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, W. , & Stone, S. (2020). Unfolded protein response in myelin disorders. Neural Regeneration Research, 15(4), 636–645 10.4103/1673-5374.266903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipson, K. L. , Ghosh, R. , & Urano, F. (2008). The role of IRE1alpha in the degradation of insulin mRNA in pancreatic beta‐cells. PLoS One, 3(2), e1648 10.1371/journal.pone.0001648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y. , Holdbrooks, A. T. , De Sarno, P. , Rowse, A. L. , Yanagisawa, L. L. , McFarland, B. C. , Harrington, L. E. , Raman, C. , Sabbaj, S. , Benveniste, E. N. , & Qin, H. (2014). Therapeutic efficacy of suppressing the Jak/STAT pathway in multiple models of experimental autoimmune encephalomyelitis. Journal of Immunology, 192(1), 59–72 10.4049/jimmunol.1301513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logsdon, A. F. , Lucke‐Wold, B. P. , Nguyen, L. , Matsumoto, R. R. , Turner, R. C. , Rosen, C. L. , & Huber, J. D. (2016). Salubrinal reduces oxidative stress, neuroinflammation and impulsive‐like behavior in a rodent model of traumatic brain injury. Brain Research, 1643, 140–151 10.1016/j.brainres.2016.04.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, T. , Trinh, M. A. , Wexler, A. J. , Bourbon, C. , Gatti, E. , Pierre, P. , Cavener, D. R. , & Klann, E. (2013). Suppression of eIF2alpha kinases alleviates Alzheimer's disease‐related plasticity and memory deficits. Nature Neuroscience, 16(9), 1299–1305 10.1038/nn.3486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, Y. , & Hendershot, L. M. (2004). ER chaperone functions during normal and stress conditions. Journal of Chemical Neuroanatomy, 28(1–2), 51–65 10.1016/j.jchemneu.2003.08.007 [DOI] [PubMed] [Google Scholar]
- Mahadevan, N. R. , Anufreichik, V. , Rodvold, J. J. , Chiu, K. T. , Sepulveda, H. , & Zanetti, M. (2012). Cell‐extrinsic effects of tumor ER stress imprint myeloid dendritic cells and impair CD8(+) T cell priming. PLoS One, 7(12), e51845 10.1371/journal.pone.0051845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevan, N. R. , Rodvold, J. , Sepulveda, H. , Rossi, S. , Drew, A. F. , & Zanetti, M. (2011). Transmission of endoplasmic reticulum stress and pro‐inflammation from tumor cells to myeloid cells. Proceedings of the National Academy of Sciences of the United States of America, 108(16), 6561–6566 10.1073/pnas.1008942108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez, G. , Khatiwada, S. , Costa‐Mattioli, M. , & Hetz, C. (2018). ER proteostasis control of neuronal physiology and synaptic function. Trends in Neurosciences, 41(9), 610–624 10.1016/j.tins.2018.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon, F. , Chen, X. , Lee, A.‐H. , & Glimcher, L. H. (2010). TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nature Immunology, 11(5), 411–418 10.1038/ni.1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon, F. , & Glimcher, L. H. (2011). Regulation of innate immunity by signaling pathways emerging from the endoplasmic reticulum. Current Opinion in Immunology, 23(1), 35–40 10.1016/j.coi.2010.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matias, I. , Morgado, J. , & Gomes, F. C. A. (2019). Astrocyte heterogeneity: Impact to brain aging and disease. Frontiers in Aging Neuroscience, 11, 59 10.3389/fnagi.2019.00059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meares, G. P. , Liu, Y. , Rajbhandari, R. , Qin, H. , Nozell, S. E. , Mobley, J. A. , Corbett, J. A. , & Benveniste, E. N. (2014). PERK‐dependent activation of JAK1 and STAT3 contributes to endoplasmic reticulum stress‐induced inflammation. Molecular and Cellular Biology, 34(20), 3911–3925 10.1128/MCB.00980-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meares, G. P. , Ma, X. , Qin, H. , & Benveniste, E. N. (2012). Regulation of CCL20 expression in astrocytes by IL‐6 and IL‐17. GLIA, 60(5), 771–781 10.1002/glia.22307 [DOI] [PubMed] [Google Scholar]
- Medinas, D. B. , Rozas, P. , Martinez Traub, F. , Woehlbier, U. , Brown, R. H. , Bosco, D. A. , & Hetz, C. (2018). Endoplasmic reticulum stress leads to accumulation of wild‐type SOD1 aggregates associated with sporadic amyotrophic lateral sclerosis. Proceedings of the National Academy of Sciences of the United States of America, 115(32), 8209–8214 10.1073/pnas.1801109115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mháille, A. N. , McQuaid, S. , Windebank, A. , Cunnea, P. , McMahon, J. , Samali, A. , & FitzGerald, U. (2008). Increased expression of endoplasmic reticulum stress‐related signaling pathway molecules in multiple sclerosis lesions. Journal of Neuropathology and Experimental Neurology, 67(3), 200–211 10.1097/NEN.0b013e318165b239 [DOI] [PubMed] [Google Scholar]
- Moon, S. L. , & Parker, R. (2018). EIF2B2 mutations in vanishing white matter disease hypersuppress translation and delay recovery during the integrated stress response. RNA, 24(6), 841–852 10.1261/rna.066563.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, K. , & Hollien, J. (2015). Ire1‐mediated decay in mammalian cells relies on mRNA sequence, structure, and translational status. Molecular Biology of the Cell, 26(16), 2873–2884 10.1091/mbc.E15-02-0074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno, J. A. , Halliday, M. , Molloy, C. , Radford, H. , Verity, N. , Axten, J. M. , Ortori, C. A. , Willis, A. E. , Fischer, P. M. , Barrett, D. A. , & Mallucci, G. R. (2013). Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion‐infected mice. Science Translational Medicine, 5(206), 206ra138 10.1126/scitranslmed.3006767 [DOI] [PubMed] [Google Scholar]
- Moreno, J. A. , Radford, H. , Peretti, D. , Steinert, J. R. , Verity, N. , Martin, M. G. , Halliday, M. , Morgan, J. , Dinsdale, D. , Ortori, C. A. , Barrett, D. A. , Tsaytler, P. , Bertolotti, A. , Willis, A. E. , Bushell, M. , & Mallucci, G. R. (2012). Sustained translational repression by eIF2alpha‐P mediates prion neurodegeneration. Nature, 485(7399), 507–511 10.1038/nature11058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijholt, D. A. , de Graaf, T. R. , van Haastert, E. S. , Oliveira, A. O. , Berkers, C. R. , Zwart, R. , Ovaa, H. , Baas, F. , Hoozemans, J. J. , & Scheper, W. (2011). Endoplasmic reticulum stress activates autophagy but not the proteasome in neuronal cells: implications for Alzheimer's disease. Cell Death and Differentiation, 18(6), 1071–1081 10.1038/cdd.2010.176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijholt, D. A. , van Haastert, E. S. , Rozemuller, A. J. , Scheper, W. , & Hoozemans, J. J. (2012). The unfolded protein response is associated with early tau pathology in the hippocampus of tauopathies. The Journal of Pathology, 226(5), 693–702 10.1002/path.3969 [DOI] [PubMed] [Google Scholar]
- Oakes, S. A. , & Papa, F. R. (2015). The role of endoplasmic reticulum stress in human pathology. Annual Review of Pathology, 10, 173–194 10.1146/annurev-pathol-012513-104649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang, Y. B. , Xu, L. J. , Emery, J. F. , Lee, A. S. , & Giffard, R. G. (2011). Overexpressing GRP78 influences Ca2+ handling and function of mitochondria in astrocytes after ischemia‐like stress. Mitochondrion, 11(2), 279–286 10.1016/j.mito.2010.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyadomari, S. , Takeda, K. , Takiguchi, M. , Gotoh, T. , Matsumoto, M. , Wada, I. , Akira, S. , Araki, E. , & Mori, M. (2001). Nitric oxide‐induced apoptosis in pancreatic beta cells is mediated by the endoplasmic reticulum stress pathway. Proceedings of the National Academy of Sciences of the United States of America, 98(19), 10845–10850 10.1073/pnas.191207498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, H. , Buckley, J. A. , Li, X. , Liu, Y. , Fox, T. H., 3rd , Meares, G. P. , Yu, H. , Yan, Z. , Harms, A. S. , Li, Y. , Standaert, D. G. , & Benveniste, E. N. (2016). Inhibition of the JAK/STAT pathway protects against alpha‐synuclein‐induced neuroinflammation and dopaminergic neurodegeneration. The Journal of Neuroscience, 36(18), 5144–5159 10.1523/JNEUROSCI.4658-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichenbach, N. , Delekate, A. , Plescher, M. , Schmitt, F. , Krauss, S. , Blank, N. , Halle, A. , & Petzold, G. C. (2019). Inhibition of Stat3‐mediated astrogliosis ameliorates pathology in an Alzheimer's disease model. EMBO Molecular Medicine, 11(2), e9665. 10.15252/emmm.201809665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron, D. , & Hubbard, S. R. (2008). How IRE1 reacts to ER stress. Cell, 132(1), 24–26 10.1016/j.cell.2007.12.017 [DOI] [PubMed] [Google Scholar]
- Saito, A. , Kanemoto, S. , Kawasaki, N. , Asada, R. , Iwamoto, H. , Oki, M. , Miyagi, H. , Izumi, S. , Sanosaka, T. , Nakashima, K. , & Imaizumi, K. (2012). Unfolded protein response, activated by OASIS family transcription factors, promotes astrocyte differentiation. Nature Communications, 3, 967 10.1038/ncomms1971 [DOI] [PubMed] [Google Scholar]
- Sanchez, C. L. , Sims, S. G. , Nowery, J. D. , & Meares, G. P. (2019). Endoplasmic reticulum stress differentially modulates the IL‐6 family of cytokines in murine astrocytes and macrophages. Scientific Reports, 9(1), 14931 10.1038/s41598-019-51481-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuner, D. , & Kaufman, R. J. (2008). The unfolded protein response: A pathway that links insulin demand with beta‐cell failure and diabetes. Endocrine Reviews, 29(3), 317–333 10.1210/er.2007-0039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder, M. , & Kaufman, R. J. (2005). ER stress and the unfolded protein response. Mutation Research, 569(1–2), 29–63 10.1016/j.mrfmmm.2004.06.056 [DOI] [PubMed] [Google Scholar]
- Schwarz, D. S. , & Blower, M. D. (2016). The endoplasmic reticulum: Structure, function and response to cellular signaling. Cellular and Molecular Life Sciences, 73(1), 79–94 10.1007/s00018-015-2052-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah, A. , Vaidya, N. K. , Bhat, H. K. , & Kumar, A. (2016). HIV‐1 gp120 induces type‐1 programmed cell death through ER stress employing IRE1alpha, JNK and AP‐1 pathway. Scientific Reports, 6, 18929 10.1038/srep18929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, J. , Chen, X. , Hendershot, L. , & Prywes, R. (2002). ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Developmental Cell, 3(1), 99–111. [DOI] [PubMed] [Google Scholar]
- Shimada, I. S. , Borders, A. , Aronshtam, A. , & Spees, J. L. (2011). Proliferating reactive astrocytes are regulated by Notch‐1 in the peri‐infarct area after stroke. Stroke, 42(11), 3231–3237 10.1161/STROKEAHA.111.623280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidrauski, C. , & Walter, P. (1997). The transmembrane kinase Ire1p is a site‐specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell, 90(6), 1031–1039 10.1016/s0092-8674(00)80369-4(00)80369‐4 [DOI] [PubMed] [Google Scholar]
- Sims, S. G. , & Meares, G. P. (2019). Janus kinase 1 is required for transcriptional reprograming of murine astrocytes in response to endoplasmic reticulum stress. Frontiers in Cellular Neuroscience, 13, 446 10.3389/fncel.2019.00446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaper, S. D. , Facci, L. , Zusso, M. , & Giusti, P. (2018). An inflammation‐centric view of neurological disease: Beyond the neuron. Frontiers in Cellular Neuroscience, 12, 72 10.3389/fncel.2018.00072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, H. L. , Freeman, O. J. , Butcher, A. J. , Holmqvist, S. , Humoud, I. , Schatzl, T. , Hughes, D. T. , Verity, N. C. , Swinden, D. P. , Hayes, J. , de Weerd, L. , Rowitch, D. H. , Franklin, R. J. M. , & Mallucci, G. R. (2020). Astrocyte unfolded protein response induces a specific reactivity state that causes non‐cell‐autonomous neuronal degeneration. Neuron, 105, 855–866.e5. 10.1016/j.neuron.2019.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew, M. V. (2009). Molecular dissection of reactive astrogliosis and glial scar formation. Trends in Neurosciences, 32(12), 638–647 10.1016/j.tins.2009.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew, M. V. , & Vinters, H. V. (2010). Astrocytes: Biology and pathology. Acta Neuropathologica, 119(1), 7–35 10.1007/s00401-009-0619-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprenkle, N. T. , Lahiri, A. , Simpkins, J. W. , & Meares, G. P. (2019). Endoplasmic reticulum stress is transmissible in vitro between cells of the central nervous system. Journal of Neurochemistry, 148(4), 516–530 10.1111/jnc.14642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprenkle, N. T. , Sims, S. G. , Sánchez, C. L. , & Meares, G. P. (2017). Endoplasmic reticulum stress and inflammation in the central nervous system. Molecular Neurodegeneration, 12(1), 42 10.1186/s13024-017-0183-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan, R. , Lu, T. Y. , Chai, H. , Xu, J. , Huang, B. S. , Golshani, P. , Coppola, G. , & Khakh, B. S. (2016). New transgenic mouse lines for selectively targeting astrocytes and studying calcium signals in astrocyte processes in situ and in vivo. Neuron, 92(6), 1181–1195 10.1016/j.neuron.2016.11.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens, B. , Allen, N. J. , Vazquez, L. E. , Howell, G. R. , Christopherson, K. S. , Nouri, N. , Micheva, K. D. , Mehalow, A. K. , Huberman, A. D. , Stafford, B. , Sher, A. , Litke, A. M. , Lambris, J. D. , Smith, S. J. , John, S. W. , & Barres, B. A. (2007). The classical complement cascade mediates CNS synapse elimination. Cell, 131(6), 1164–1178 10.1016/j.cell.2007.10.036 [DOI] [PubMed] [Google Scholar]
- Sun, S. , Sun, Y. , Ling, S. C. , Ferraiuolo, L. , McAlonis‐Downes, M. , Zou, Y. , Drenner, K. , Wang, Y. , Ditsworth, D. , Tokunaga, S. , Kopelevich, A. , Kaspar, B. K. , Lagier‐Tourenne, C. , & Cleveland, D. W. (2015). Translational profiling identifies a cascade of damage initiated in motor neurons and spreading to glia in mutant SOD1‐mediated ALS. Proceedings of the National Academy of Sciences of the United States of America, 112(50), E6993–E7002 10.1073/pnas.1520639112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres, M. , Castillo, K. , Armisen, R. , Stutzin, A. , Soto, C. , & Hetz, C. (2010). Prion protein misfolding affects calcium homeostasis and sensitizes cells to endoplasmic reticulum stress. PLoS One, 5(12), e15658 10.1371/journal.pone.0015658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urano, F. , Wang, X. , Bertolotti, A. , Zhang, Y. , Chung, P. , Harding, H. P. , & Ron, D. (2000). Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science, 287(5453), 664–666 10.1126/science.287.5453.664 [DOI] [PubMed] [Google Scholar]
- Verkhratsky, A. , & Nedergaard, M. (2018). Physiology of astroglia. Physiological Reviews, 98(1), 239–389 10.1152/physrev.00042.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voskuhl, R. R. , Peterson, R. S. , Song, B. , Ao, Y. , Morales, L. B. , Tiwari‐Woodruff, S. , & Sofroniew, M. V. (2009). Reactive astrocytes form scar‐like perivascular barriers to leukocytes during adaptive immune inflammation of the CNS. The Journal of Neuroscience, 29(37), 11511–11522 10.1523/JNEUROSCI.1514-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, P. , Li, J. , & Sha, B. (2016). The ER stress sensor PERK luminal domain functions as a molecular chaperone to interact with misfolded proteins. Acta Crystallographica Section D: Structural Biology, 72(Pt 12), 1290–1297 10.1107/s2059798316018064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, P. , Li, J. , Tao, J. , & Sha, B. (2018). The luminal domain of the ER stress sensor protein PERK binds misfolded proteins and thereby triggers PERK oligomerization. The Journal of Biological Chemistry, 293(11), 4110–4121 10.1074/jbc.RA117.001294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z. F. , Gao, C. , Chen, W. , Gao, Y. , Wang, H. C. , Meng, Y. , Luo, C. L. , Zhang, M. Y. , Chen, G. , Chen, X. P. , Wang, T. , & Tao, L. Y. (2019). Salubrinal offers neuroprotection through suppressing endoplasmic reticulum stress, autophagy and apoptosis in a mouse traumatic brain injury model. Neurobiology of Learning and Memory, 161, 12–25 10.1016/j.nlm.2019.03.002 [DOI] [PubMed] [Google Scholar]
- Wanner, I. B. , Anderson, M. A. , Song, B. , Levine, J. , Fernandez, A. , Gray‐Thompson, Z. , Ao, Y. , & Sofroniew, M. V. (2013). Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3‐dependent mechanisms after spinal cord injury. The Journal of Neuroscience, 33(31), 12870–12886 10.1523/JNEUROSCI.2121-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler, M. A. , Clark, I. C. , Tjon, E. C. , Li, Z. , Zandee, S. E. J. , Couturier, C. P. , Watson, B. R. , Scalisi, G. , Alkwai, S. , Rothhammer, V. , Rotem, A. , Heyman, J. A. , Thaploo, S. , Sanmarco, L. M. , Ragoussis, J. , Weitz, D. A. , Petrecca, K. , Moffitt, J. R. , Becher, B. , … Quintana, F. J. (2020). MAFG‐driven astrocytes promote CNS inflammation. Nature, 578(7796), 593–599 10.1038/s41586-020-1999-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler, M. A. , Jaronen, M. , Covacu, R. , Zandee, S. E. J. , Scalisi, G. , Rothhammer, V. , Tjon, E. C. , Chao, C. C. , Kenison, J. E. , Blain, M. , Rao, V. T. S. , Hewson, P. , Barroso, A. , Gutierrez‐Vazquez, C. , Prat, A. , Antel, J. P. , Hauser, R. , & Quintana, F. J. (2019). Environmental control of astrocyte pathogenic activities in CNS inflammation. Cell, 176(3), 581–596 e518 10.1016/j.cell.2018.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, J. , Rawson, R. B. , Komuro, R. , Chen, X. , Dave, U. P. , Prywes, R. , Brown, M. S. , & Goldstein, J. L. (2000). ER stress induces cleavage of membrane‐bound ATF6 by the same proteases that process SREBPs. Molecular Cell, 6(6), 1355–1364 10.1016/s1097-2765(00)00133-7 [DOI] [PubMed] [Google Scholar]
- Yoshikawa, A. , Kamide, T. , Hashida, K. , Ta, H. M. , Inahata, Y. , Takarada‐Iemata, M. , Hattori, T. , Mori, K. , Takahashi, R. , Matsuyama, T. , Hayashi, Y. , Kitao, Y. , & Hori, O. (2015). Deletion of Atf6α impairs astroglial activation and enhances neuronal death following brain ischemia in mice. Journal of Neurochemistry, 132(3), 342–353 10.1111/jnc.12981 [DOI] [PubMed] [Google Scholar]
- Zha, X. , Yue, Y. , Dong, N. , & Xiong, S. (2015). Endoplasmic reticulum stress aggravates viral myocarditis by raising inflammation through the IRE1‐associated NF‐kappaB pathway. The Canadian Journal of Cardiology, 31(8), 1032–1040 10.1016/j.cjca.2015.03.003 [DOI] [PubMed] [Google Scholar]
- Zhang, H. , Yue, Y. , Sun, T. , Wu, X. , & Xiong, S. (2017). Transmissible endoplasmic reticulum stress from myocardiocytes to macrophages is pivotal for the pathogenesis of CVB3‐induced viral myocarditis. Scientific Reports, 7, 42162 10.1038/srep42162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, K. , Shen, X. , Wu, J. , Sakaki, K. , Saunders, T. , Rutkowski, D. T. , Back, S. H. , & Kaufman, R. J. (2006). Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell, 124(3), 587–599 10.1016/j.cell.2005.11.040 [DOI] [PubMed] [Google Scholar]
- Zhang, K. , Wong, H. N. , Song, B. , Miller, C. N. , Scheuner, D. , & Kaufman, R. J. (2005). The unfolded protein response sensor IRE1alpha is required at 2 distinct steps in B cell lymphopoiesis. The Journal of Clinical Investigation, 115(2), 268–281 10.1172/JCI21848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, N. , Ramaswamy, G. , & Weisgraber, K. H. (2009). Apolipoprotein E4 domain interaction induces endoplasmic reticulum stress and impairs astrocyte function. The Journal of Biological Chemistry, 284(40), 27273–27280 10.1074/jbc.M109.014464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, J. , Liu, C. Y. , Back, S. H. , Clark, R. L. , Peisach, D. , Xu, Z. , & Kaufman, R. J. (2006). The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proceedings of the National Academy of Sciences of the United States of America, 103(39), 14343–14348 10.1073/pnas.0606480103 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.