

Abstract
Background and Purpose
Generation of cGMP via NO‐sensitive soluble guanylyl cyclase (sGC) has been implicated in the regulation of renal functions. Chronic kidney disease (CKD) is associated with decreased NO bioavailability, increased oxidative stress and oxidation of sGC to its haem‐free form, apo‐sGC. Apo‐sGC cannot be activated by NO, resulting in impaired cGMP signalling that is associated with chronic kidney disease progression. We hypothesised that sGC activators, which activate apo‐sGC independently of NO, increase renal cGMP production under conditions of oxidative stress, thereby improving renal blood flow (RBF) and kidney function.
Experimental Approach
Two novel sGC activators, runcaciguat and BAY‐543, were tested on murine kidney. We measured cGMP levels in real time in kidney slices of cGMP sensor mice, vasodilation of pre‐constricted glomerular arterioles and RBF in isolated perfused kidneys. Experiments were performed at baseline conditions, under L‐NAME‐induced NO deficiency, and in the presence of oxidative stress induced by ODQ.
Key Results
Mouse glomeruli showed NO‐induced cGMP increases. Under baseline conditions, sGC activator did not alter glomerular cGMP concentration or NO‐induced cGMP generation. In the presence of ODQ, NO‐induced glomerular cGMP signals were markedly reduced, whereas sGC activator induced strong cGMP increases. L‐NAME and ODQ pretreated isolated glomerular arterioles were strongly dilated by sGC activator. sGC activator also increased cGMP and RBF in ODQ‐perfused kidneys.
Conclusion and Implication
sGC activators increase glomerular cGMP, dilate glomerular arterioles and improve RBF under disease‐relevant oxidative stress conditions. Therefore, sGC activators represent a promising class of drugs for chronic kidney disease treatment.
LINKED ARTICLES
This article is part of a themed issue on cGMP Signalling in Cell Growth and Survival. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v179.11/issuetoc
Keywords: cGMP imaging, glomerular arterioles, GC, NO, renal blood flow, sGC activators, vasodilation
Abbreviations
- Ang II
angiotensin II
- ANP
atrial natriuretic peptide
- CFP
cyan fluorescent protein
- DEA/NO
diethylamine NONOate
- L‐NAME
N‐nitroarginine methyl ester
- ODQ
1H‐[1,2,4]oxadiazolo[4,3‐a]quinoxalin‐1‐one
- R (cGMP)
relative CFP/YFP ratio change of cGMP sensor
- RBF
renal blood flow
- sGC
soluble guanylyl cyclase
- SGLT2i
sodium‐glucose‐cotransporter 2 inhibitors
- SNAP
S‐nitroso‐N‐acetylpenicillamine
- YFP
yellow fluorescent protein
- ZSF1
Zucker diabetic fatty/spontaneously hypertensive heart failure F1 hybrid rat
What is already known
Impaired NO/sGC/cGMP signalling is associated with vasoconstriction, hypertension and progression of chronic kidney disease.
What does this study add
sGC activators increase the glomerular cGMP concentration and dilate glomerular arterioles.
sGC activators improve renal blood flow under oxidative stress conditions.
What is the clinical significance
sGC activators represent a promising new class of drugs for treating chronic kidney disease.
1. INTRODUCTION
Chronic hypertension leads to end‐organ damage, which especially affects blood vessels and kidneys. End‐organ damage goes along with gradual increase in proteinuria, impaired kidney function including reduced glomerular filtration rate (GFR) and the development of chronic kidney disease (CKD) (Griffin, 2017). Persisting chronic kidney disease often results in end‐stage renal disease and ultimately the need for a kidney transplant (Brenner et al., 2001; Coresh, 2017). Currently, the first‐line management of chronic kidney disease consists of inhibiting the renin‐angiotensin‐aldosterone system, which is known to play a pivotal role in chronic kidney disease development (Siragy & Carey, 2010). Although therapies inducing reduction of systemic blood pressure (BP) have shown beneficial effects on kidney function in patients with renal disease, these effects are not fully maintained in chronic therapy (Breyer & Susztak, 2016) or in patients with type‐2‐diabetes and advanced end‐stage renal disease (Schievink et al., 2016). Recently, sodium‐glucose‐cotransporter 2 (SGLT2) inhibitors which are approved for the treatment of type‐2‐diabetes, have been demonstrated to prevent GFR decline in chronic kidney disease patients (Lin et al., 2019; Muskiet et al., 2017). However, despite reduction of cardiovascular risk by SGLT2 inhibitors in chronic kidney disease patients (Briasoulis et al., 2018; Vallon & Thomson, 2017), an initial GFR decrease under SGLT2 inhibitors was reported (Sugiyama et al., 2020). Thus, there is still a need for new chronic kidney disease therapies with a different molecular mode of action, which can overcome limitations of currently used drugs.
The cGMP signalling system is a central regulator of cardiovascular homeostasis and has great potential as a target for new effective pharmacological therapies. Recent studies illustrated that genetic variants in components of this pathway significantly influence BP and the risk of cardiovascular and renal disease (Emdin et al., 2018; Erdmann et al., 2013; International Consortium for Blood Pressure, Ehret et al., 2011; Maass et al., 2015). The soluble guanylyl cyclase (sGC) generates cGMP upon activation by NO. An impairment of the NO/sGC/cGMP pathway and a decline in cGMP contributes to the development vascular dysfunction. Enhanced NO signalling also was associated with a higher GFR as determined via cystatin C and creatinine (Krishnan et al., 2018; Ott et al., 2012). Genetic variants of sGC and cGMP‐dependent protein kinase type I, a downstream effector of sGC, are causally associated with altered vascular structure and remodelling, and sGC gain of function is associated with a higher GFR and lower risk for chronic kidney disease (Emdin et al., 2018). Therefore, sGC‐enhancing drugs might be beneficial for treating chronic kidney disease patients.
Recent studies indicated that oxidative stress, which is triggered by comorbidities in chronic kidney disease, like hypertension, diabetes or obesity, could be one of the drivers of kidney function decline in chronic kidney disease (Coppolino et al., 2018; Samarghandian et al., 2017; Sinha & Dabla, 2015; Su et al., 2019). Oxidative stress is leading to sGC oxidation and ultimately the loss of its haem group, resulting in the formation of haem‐free apo‐sGC. Apo‐sGC can no longer bind and be stimulated by the endogenous ligand NO (Stasch et al., 2015). Thus, oxidative stress disrupts NO/sGC/cGMP signalling and, thereby, counteracts physiological regulation of kidney function by NO. sGC activators are small molecules that activate sGC in a haem‐ and NO‐independent manner and, thus, have the potential to restore sGC/cGMP signalling under conditions of oxidative stress. Runcaciguat is a novel potent and selective sGC activator, which activates apo‐sGC in vitro, ex vivo and in vivo (Hahn, 2018) and has great potential to prevent the decline of kidney function (Benardeau et al., 2020; Hahn et al., 2021). However, the potential kidney‐specific mechanisms behind the renoprotective effects of runcaciguat are not well characterised. Therefore, we aimed to investigate where and under what conditions runcaciguat and its close analogue BAY‐543 (Rühle et al., 2020) induce cGMP production in the kidney and how these sGC activators influence the diameter of isolated glomerular arterioles as well as renal blood flow (RBF) in isolated perfused whole kidney preparations.
Our data show that sGC activators increase the glomerular cGMP concentration under oxidative stress conditions. Moreover, sGC activators induce dilation of pre‐constricted glomerular afferent and efferent arterioles in the absence of NO and presence of oxidative stress and they significantly improve RBF under oxidative stress conditions. These findings show that sGC activators have direct effects on the renal vasculature and counteract the oxidative stress‐induced decline in RBF. Thus, sGC activators could represent a new therapeutic approach for the treatment of chronic kidney disease patients.
2. METHODS
2.1. Materials
The sGC activator runcaciguat (BAY 1101042) (Hahn, 2018; Hahn et al., 2021) and its analogue BAY‐543 (Rühle et al., 2020) were synthesised by Bayer AG (Wuppertal, Germany) and dissolved in DMSO (Sigma‐Aldrich, Darmstadt, Germany). Structurally, runcaciguat and BAY‐543 are closely related, differing only in one side group (Figure S1). In the previous studies cited above, their EC50 to activate purified sGC (11 nM for both compounds) and their ability to dilate pre‐contracted rabbit arteries ex vivo (IC50 of 199 and 75 nM for runcaciguat and BAY‐543, respectively) were found to be very similar.
The sGC oxidant 1H‐[1,2,4]oxadiazolo[4,3‐a]quinoxaline‐1‐one (ODQ) was from Axxora (Ann Arbor, MI, USA; real‐time cGMP imaging) or Sigma‐Aldrich (all other experiments). Atrial natriuretic peptide (ANP) was from Tocris (Minneapolis, MN, USA), angiotensin II (Ang II) and N‐nitroarginine methyl ester (L‐NAME) from Sigma‐Aldrich, and the L‐ and N‐type calcium channel blocker cilnidipine from Cayman Chemical (Ann Arbor, MI, USA). The NO donor diethylamine NONOate (DEA/NO) was from Axxora. (DEA/NO) was stored in 100 mM stock solutions in NaOH (10 mM) and diluted in perfusion buffer immediately before application. Due to its short half‐life time (≈16 min at room temperature and pH 7.4), DEA/NO is well‐suited to induce fast, transient cGMP increases during real‐time cGMP imaging experiments. For analysis of isolated glomerular arterioles and perfused kidneys, the NO‐releasing compound S‐nitroso‐N‐acetylpenicillamine (SNAP; Cayman Chemical) was preferred as it has slower kinetics of NO release (several hours, varies between tissues) as compared with DEA/NO which ensures sustained continuous NO release at 37°C in these experiments.
2.2. Animals
In the present study, we used mice, because they are amenable to genetic modifications and murine kidneys represent a good model to analyse renal function in mammals. All animal procedures were performed in accordance with the Directive 2010/63/EU, the German Tierschutz‐Versuchstierverordnung and the local authorities in Tübingen, Berlin and Regensburg. Animal studies are reported in compliance with the ARRIVE guidelines (Percie du Sert et al., 2020) and with the recommendations made by the British Journal of Pharmacology (Lilley et al., 2020). Mice were housed in groups of up to eight animals in open type III cages at 22°C and 50%–60% humidity in a 12 h light/12 h dark cycle with access to standard rodent chow (ssniff; Soest, Germany) and tap water ad libitum.
For real‐time cGMP imaging, 8‐ to 18‐week‐old R26‐CAG‐cGi500(L1) mice (Thunemann et al., 2013) of either sex on a C57BL/6N genetic background were used. Genotyping of these animals was performed by PCR analysis of ear biopsy DNA using the following primers: P1 (CTCTGCTGCCTCCTGGCTTCT), P2 (CGAGGCGGATCACAAGCAATA) and P3 (TCAATGGGCGGGGGTCGTT). These primers amplify a 330 base pair fragment of the wildtype allele (P1 and P2) and a 250 base pair fragment of the transgene (P1 and P3). These mice were killed with CO2 followed by cervical dislocation.
Other experiments were performed with C57BL/6N mice (RRID:IMSR_CRL:027; Charles River, Wilmington, MA, USA) at an age of 12–16 weeks. Only male mice were used for these studies to exclude sex‐specific variation of arteriole diameters and RBF. If not stated otherwise, experimental animals were killed by cervical dislocation.
2.3. Preparation of kidney slices
For the preparation of acute tissue slices, kidneys were dissected from R26‐CAG‐cGi500(L1) mice, and the renal capsule was carefully removed in ice‐cold carbogen‐gassed Ringer buffer (127.0 mM NaCl, 2.5 mM KCl, 0.5 mM MgCl2, 1.1 mM CaCl2, 1.1 mM NaH2PO4, 26.0 mM NaHCO3, 20.0 mM d‐glucose). The pH was adjusted by continuous gassing with carbogen. Kidneys were sectioned with a vibratome (VT1200, Leica, Buffalo Grove, IL, USA) to a thickness of 700 μm. Whole kidneys and slices were incubated in ice‐cold carbogen‐gassed Ringer buffer for up to 8 h until real‐time cGMP imaging was performed as described below.
2.4. Real‐time cGMP imaging
Förster resonance energy transfer (FRET)/cGMP imaging was performed as described previously (Thunemann et al., 2013). The set‐up consisted of an upright Examiner.Z1 microscope (Zeiss, Oberkochen, Germany), a Yokogawa CSU‐X1 spinning disc confocal scanner (Yokogawa Denki, Musashino, Japan), three diode lasers (445, 488 and 561 nm), three water immersion objectives (W N‐ACHROMAT 10/0.3, W Plan‐APOCHROMAT 20/1.0 DIC [UV] VIS‐IR, W Plan‐APOCHROMAT 40/1.0 DIC VIS‐IR; all from Zeiss) and one air objective (EC Plan‐NEOFLUAR 2.5/0.085; Zeiss). Yellow fluorescent protein (YFP) fluorescence of the cGMP sensor cGi500 was detected with a CCD camera (Spot Pursuit, Diagnostic Instruments, Sterling Heights, MI, USA) through a 525/50 nm emission filter after excitation with the 488 nm laser. For FRET‐based imaging, the donor fluorophore, cyan fluorescent protein (CFP), was excited with the 445 nm laser, and a Dual‐View beam splitter (Photometrics, Tucson, AZ, USA) with 505 nm dichroic mirror, 470/24 nm and 535/30 nm emission filters was used for simultaneous acquisition of CFP and YFP emission. Signals were recorded with an electron‐multiplying charged‐coupled device (EM‐CCD) camera (QuantEM 512SC, Photometrics) at a frame rate of 0.2 Hz and an exposure time of 0.2 s. The system was controlled by VisiView 4.0.0.12 (Visitron Systems, Puchheim, Germany). Kidney slices were continuously superfused with carbogen‐gassed Ringer buffer with or without drugs at a flow rate of 1 ml/min at room temperature. The custom‐built superfusion system consisted of an fast protein liquid chromatography pump (Pharmacia P‐500, GE Healthcare, Chicago, IL, USA), fast protein liquid chromatography injection valves (Pharmacia V‐7, GE Healthcare), a magnetic platform (Warner Instruments, Hamden, CT, USA), a superfusion chamber (RC‐26, Warner Instruments), a mesh‐assisted Slice Hold‐Down (SHD‐26H/10, Warner Instruments) and sample loops of different sizes (7 ml for ODQ; 2 ml for other drugs). To ensure that drug exposure was comparable between different tissue slices, the same drug volumes were applied for the same time span. A vacuum pump with adjustable vacuum (Laboport N86, KNF Neuberger, Hamburg, Germany) was connected to the system to constantly remove excess buffer.
2.5. Image acquisition and post‐processing
Online image acquisition was performed with VisiView 4.0.0.12 (Visitron Systems, Puchheim, Germany), and offline post‐processing and analysis were performed with Fiji software (RRID:SCR_002285) (Rueden et al., 2017; Schindelin et al., 2012). Images were aligned in x/y dimension with the Fiji plugin MultiStackReg v1.45 (RRID:SCR_016098). For further evaluation, Excel (RRID:SCR_016137; Office 16; Microsoft, Redmond, WA, USA) and Origin 2019 (RRID:SCR_014212; OriginLab, Northampton, MA, USA) were used. CFP and YFP emission were used to calculate the CFP/YFP ratio. The relative CFP/YFP ratio change (black traces in the respective graphs; referred to as R (cGMP)), which correlates with the cGMP concentration change, was obtained by normalisation to the baseline recorded for ~3 min at the beginning of each experiment. This normalisation is necessary to account for variations in the basal fluorescence intensity between the preparations. For peak evaluation, R (cGMP) traces were smoothed according to the Savitzky–Golay filtering method (smoothing window = 30 points) and the Peak Analyzer tool of Origin was used to calculate the AUC for each signal.
2.6. Dissection and perfusion of glomerular arterioles
Kidneys were removed and sliced along the corticomedullary axis. Afferent and efferent arterioles were prepared according to procedures detailed by Liu and colleagues (Liu et al., 2012). In short, afferent and efferent arterioles with attached glomeruli were isolated and transferred into a chamber assembled on the stage of an inverted microscope. Arterioles were perfused using a system of pipettes, which allowed to hold and perfuse the vessels. Both, afferent and efferent arterioles were perfused from the free end, so orthograde in case of afferent arterioles and retrograde in case of efferent arterioles. Perfusion pressure was 100 mmHg for afferent arterioles and 40 mmHg for efferent arterioles, and the perfusion rates were in physiological ranges. DMEM (DMEM/F‐12, Gibco, Darmstadt, Germany) containing 0.1% BSA (Carl Roth, Karlsruhe, Germany) was used during vessel dissection. The same solution was present in the experimental chamber, whereas the DMEM perfusion solution which was applied to the arteriolar lumen contained 1.0% BSA.
2.7. Protocols for perfusion of glomerular arterioles
Arterioles were allowed to acclimatise for 10 min after establishing the perfusion. Viability was tested by short‐term application of KCl (100 mM). Only vessels which showed full and sustained constriction were used for subsequent experiments. Arterioles were treated for 15 min with the non‐specific NOS inhibitor L‐NAME to induce NO deficiency, or for 10 min with the sGC‐oxidising substance ODQ and vehicle (DMSO), respectively. Ang II, which belongs to the strongest vasoconstrictors particularly in the renal vasculature (Patzak et al., 2001), was applied to pre‐constrict the vessels. Here, Ang II was applied in ascending concentrations (2 min for each concentration). After application of the highest Ang II concentration, cumulative concentrations of runcaciguat or of the known vasodilators NO (in the form of SNAP; sGC‐dependent) and cilnidipine (sGC‐independent) were administered to the Ang II‐constricted arteries for 2 min each. In the ‘L‐NAME + vehicle’ group, DMSO was applied instead of runcaciguat in corresponding concentrations. To analyse the time course of runcaciguat‐induced vasodilation, arterioles pretreated with L‐NAME and pre‐constricted with Ang II were subjected to a prolonged 10 min treatment with runcaciguat or vehicle.
2.8. Measurement of arteriole diameters
Perfused arterioles were continuously displayed on the computer screen using a video camera and the respective software (Moticam 2.0, Motic Asia, Kowloon, Hong Kong). Luminal diameters served for the estimation of arteriolar tone and reactivity. For the assessment of concentration–response relationships, pictures were recorded at a frame rate of 1 Hz. Then, the vessel diameter in five pictures was averaged in steady state conditions for each concentration effect to limit the influence of movement artefacts. For long‐term investigations of runcaciguat action (10 min), pictures were recorded at a frame rate of 0.1 Hz. Either way, the arteriolar diameters were measured using ImageJ2 (RRID:SCR_003070) (Rueden et al., 2017). For analysis of the Ang II concentration–response relationship, arteriolar diameters were normalised to the initial diameter after L‐NAME/ODQ/vehicle and before Ang II application. For evaluation of the dilation of pre‐constricted arterioles induced by runcaciguat, SNAP and cilnidipine, diameter changes were normalised to the total constriction induced by L‐NAME/ODQ/vehicle + Ang II. Both normalisation procedures are typically applied in vessel physiology to limit the influence of intra‐ and inter‐individual variations of arteriole diameters and thereby focus the evaluation on pharmacological effects. The part of the vessel with the strongest response was chosen for analysis.
2.9. Preparation and analysis of isolated perfused whole kidneys
Mice were anaesthetised with an intraperitoneal injection of xylazine hydrochloride (10 mg·kg−1, Serumwerk Bernburg, Bernburg, Germany) and ketamine HCl (100 mg·kg−1; betapharm Arzneimittel, Augsburg, Germany) and placed on a warmed table. Isolation and perfusion of mouse kidneys followed a published method (Schweda et al., 2003). In short, the abdominal cavity was opened by a midline incision, and the aorta was clamped distal to the right renal artery. The mesenteric artery was ligated and a metal perfusion cannula (0.8 mm outer diameter) was inserted into the abdominal aorta. Then, the aorta was ligated proximal to the right renal artery and perfusion was started in situ with an initial flow rate of 1 ml·min−1. The right kidney was excised, placed in a tempered humid chamber and perfused at constant pressure (100 mmHg). Finally, the renal vein was cannulated (1.5 mm outer diameter polypropylene catheter). The venous effluent was drained outside the humid chamber and collected for determination of venous blood flow. The basic perfusion medium supplied from a 37°C‐tempered 200 ml‐reservoir consisted of a modified Krebs–Henseleit solution containing amino acids (10 ml·L−1 Aminoplasmal B. Braun 10%), 8.7 mM d‐glucose, 0.3 mM pyruvate, 2.0 mM l‐lactate, 1.0 mM ketoglutarate, 1.0 mM l‐malate and 6.0 mM urea. The perfusate was supplemented with 60 g·L−1 BSA, 10 mU·L−1 vasopressin 8‐lysine and freshly washed human red blood cells (10% haematocrit). Ampicillin (30 mg·L−1) and flucloxacillin (30 mg·L−1) were added to inhibit possible bacterial growth in the medium. To improve the functional preservation of the preparation, the perfusate was continuously dialysed against a 10‐fold volume of the same composition, but lacking erythrocytes and BSA. For oxygenation of the perfusion medium, the dialysate was gassed with 94% O2/6% CO2.
2.10. Determination of renal blood flow and cGMP secretion
Perfusate flow from kidneys isolated as described above was calculated by collection and gravimetric determination of the venous effluent. After establishing a constant perfusion pressure (100 mmHg), perfusate flow rates stabilised within 12–15 min. Stock solutions of the indicated drugs were added to the perfusate. For evaluation, the amount of venous effluent was normalised to the perfusate flow under control conditions or after application of 30 μM ODQ as indicated in the respective graphs to exclude differences in baseline perfusion between the preparations. For determination of renal cGMP production, venous effluent was collected over a period of 1 min during four intervals along the study and its cGMP concentration was determined after acetylation of samples using a cGMP enzyme immunoassay Kit (Cat# 581021, Cayman Chemical, Hamburg, Germany). The cGMP secretion was calculated by multiplying cGMP concentration and perfusate flow.
2.11. Data and statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). Variances in group sizes within individual comparisons are due to exclusion of defective preparations and yields, which were lower than expected. Data are presented as mean ± SD or mean ± SEM as specified in the figure legends. P values <0.05 were considered significant. Randomisation and blinding were not applicable because the experimental setups demanded application of substances in distinct succession. Instead, analysis and evaluation were performed uniformly and were based exclusively on objective parameters.
Statistical analysis was performed with Origin 2019 (OriginLab, Northampton, MA, USA; FRET/cGMP imaging) or GraphPad PRISM 8 (RRID:SCR_002798; GraphPad Software, San Diego, CA, USA; analysis of isolated perfused kidneys). For not normally distributed data sets, statistical differences were analysed non‐parametrically by Mann–Whitney U‐test. In case of normally distributed data, statistical differences were analysed parametrically by Student's t‐test (equal variances) or Welch's t‐test (unequal variances). The DEA/NO concentration–response curve was calculated with the sigmoidal ‘DoseResp’ fitting function of origin.
Time‐ and concentration‐dependent differences of diameter changes of arterioles were statistically assessed with the free software R (RRID:SCR_001905; version 3.6.3) (R Core Team, 2020) using the Brunner test (Brunner & Langer, 1999). Sample sizes subjected to statistical analysis were at least five animals per group (n = 5), with n = number of independent values. This test is a non‐parametric counterpart of the two‐factorial ANOVA and tests the main hypothesis of a global difference between two groups. It is appropriate for the comparison of serial repeated measurements without normal distribution.
2.12. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in the IUPHAR/BPS Guide to PHARMACOLOGY http://www.guidetopharmacology.org and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander et al., 2019).
3. RESULTS
3.1. Renal glomeruli express a functional NO/cGMP signalling pathway
To measure the spatiotemporal cGMP dynamics in the kidney, we used transgenic cGMP sensor mice expressing the FRET‐based cGMP indicator cGi500 (Russwurm et al., 2007; Thunemann et al., 2013). Specifically, we used the R26‐CAG‐cGi500(L1) mouse line that expresses the cGMP sensor globally in all tissues. By FRET imaging of live kidney slices ex vivo, cGMP signals were recorded in real time in response to the endogenous sGC ligand NO and in response to the sGC activator BAY‐543 alone and in combination with NO. The sGC‐oxidising agent ODQ was used to mimic chronic kidney disease‐related oxidative stress conditions. Slices were kept in carbogen‐gassed Ringer buffer during preparation and measurements to keep the tissue vital.
Visual inspection of the sensor‐derived fluorescence confirmed that the cGMP sensor was broadly expressed in the kidney of cGMP sensor mice (Figure 1a). Note that the yellow fluorescence seen in the photomicrographs indicates expression of the sensor protein but not the cGMP concentration. The latter is determined by ratiometric analysis of the sensor's CFP and YFP fluorescence as described in Section 2.5. Individual glomeruli were selected as regions of interest and several glomeruli were averaged to quantify the cGMP concentration in the absence and presence of various drugs.
FIGURE 1.
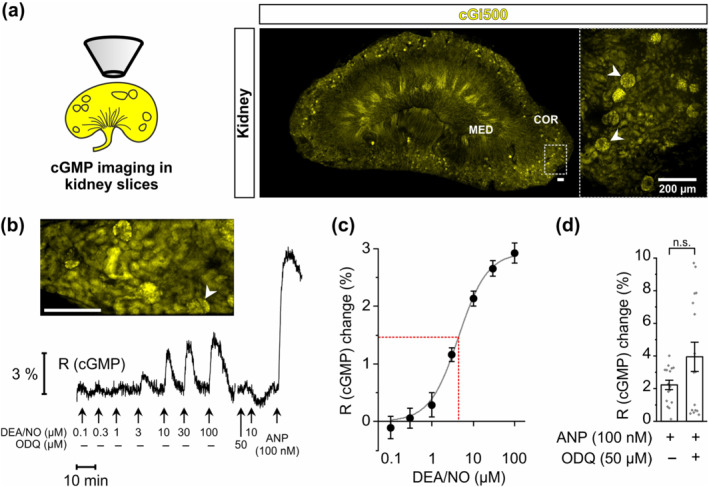
NO‐induced cGMP generation in real time in glomeruli of kidney slices. FRET‐based cGMP imaging was performed with acute kidney slices from R26‐CAG‐cGi500(L1) mice. (a) Schematic representation and representative images of kidney slices. Yellow colour represents yellow fluorescent protein (YFP) fluorescence of the cGMP sensor cGi500 in renal cells. White arrowheads point towards some glomeruli. Dashed rectangle indicates the magnified region shown to the right. (b) Representative real‐time FRET/cGMP measurement of a kidney slice. During recording, increasing concentrations of diethylamine NONOate (DEA/NO) or 100 nM atrial natriuretic peptide (ANP) were applied to the slice for 2 min each. Before the second last stimulation with 10 μM DEA/NO, slices were pre‐incubated for 5 min with ODQ (50 μM). Black trace represents the cyan fluorescent protein (CFP)/YFP ratio R, which indicates cGMP concentration changes. The white arrowhead points towards the glomerulus measured in this experiment. (c) Concentration‐response curve (solid grey line) based on the relative R (cGMP) changes induced by increasing concentrations of DEA/NO (n = 19). The dashed red line indicates EC50. (d) Statistical analysis was performed with the relative R (cGMP) changes induced by ANP (100 nM) in the presence and absence of ODQ (n = 16). Data represent mean ± SEM. COR, renal cortex; MED, renal medulla. Scale bars, 200 μm
First, we tested the capacity of renal glomeruli to generate cGMP in response to the NO‐releasing compound DEA/NO. DEA/NO concentration‐dependently increased the cGMP concentration in the glomeruli of cGMP sensor mice with an EC50 of 4.4 ± 0.1 μM (Figure 1b,c). Application of ODQ (50 μM) slightly reduced the basal cGMP production and, as expected, abolished NO‐induced cGMP generation (Figure 1b). As a control, atrial natriuretic peptide (ANP), which increases cGMP via simulation of the particulate guanylyl cyclase A, was applied to the kidney slices at the end of each experiment. ANP (100 nM) potently increased glomerular cGMP levels independent of ODQ application (Figure 1b,d), indicating that sGC‐independent cGMP generation was not affected by ODQ and that the tissue was still vital at the end of each experiment. Together, these data demonstrated the presence of a functional and oxidation‐sensitive NO/sGC/cGMP signalling pathway in the glomeruli of murine kidneys. In subsequent FRET imaging experiments, 10 μM DEA/NO was used as a concentration that induces non‐saturated cGMP responses.
3.2. The sGC activator BAY‐543 increases glomerular cGMP under oxidative stress
Next, we analysed modulation of glomerular sGC/cGMP signalling by the sGC activator BAY‐543, a close analogue of runcaciguat. As expected from our previous results (Figure 1b,c), under baseline conditions (i.e. in the absence of ODQ), application of DEA/NO alone reproducibly induced an increase of cGMP in glomeruli (Figure 2a,c). In the absence of ODQ, application of BAY‐543 did not alter the cGMP concentration and did not affect NO‐induced cGMP signals. Interestingly, in the presence of ODQ, BAY‐543 (10 μM) increased the cGMP concentration to a similar extent as 10 μM DEA/NO (Figure 2b,c). Under the same oxidative stress conditions, that is, in the presence of ODQ, NO no longer increase cGMP (Figure 2b,c), consistent with the findings reported in Figure 1b. Altogether, our data shown in Figures 1 and 2 indicated that the sGC activator BAY‐543 stimulates cGMP production in mouse glomeruli under oxidative stress conditions, which have impaired NO‐induced cGMP signalling.
FIGURE 2.
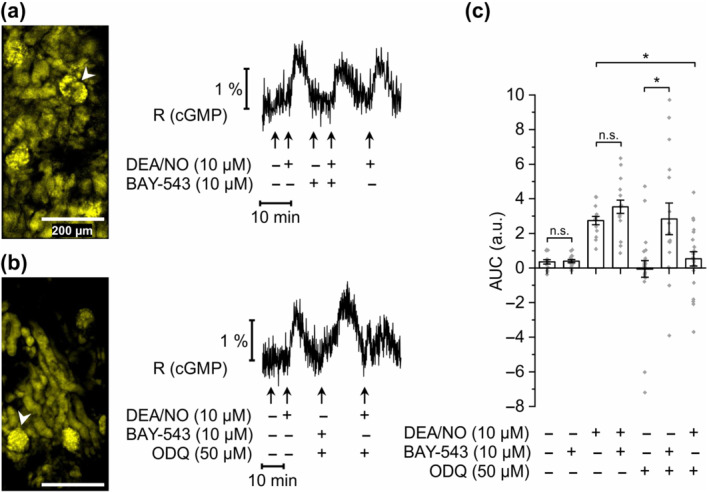
sGC activator‐induced cGMP generation in glomeruli of ODQ‐treated kidney slices. FRET‐based cGMP imaging was performed in glomeruli of kidney slices from R26‐CAG‐cGi500(L1) mice. (a) Representative measurement of a glomerulus without ODQ application. During the recording, diethylamine NONOate (DEA/NO; 10 μM), BAY‐543 (10 μM), or a combination of both substances was applied to the slices for 2 min. Black trace represents the cyan fluorescent protein/yellow fluorescent protein (CFP/YFP) ratio R, which indicates cGMP concentration changes. White arrowhead points towards the glomerulus represented in the measurement shown to the right. (b) Representative measurement of a glomerulus with ODQ application. DEA/NO (10 μM), BAY‐543 (10 μM), or a combination of both substances was applied with or without 5 min pretreatment with ODQ (50 μM). (c) Statistical analysis was performed with the AUC of the signals. Note that negative AUC values in the presence of ODQ might be due to reduced endogenous cGMP generation compared with baseline conditions. Data represent mean ± SEM (n = 14, 16, 14, 16, 24, 16 and 21 from left to right). Scale bars, 200 μm. *P < .05; n.s., not significant
3.3. Runcaciguat induces dilation of renal arterioles
To further explore the physiological effects of cGMP production induced by sGC activators on kidney function, we assessed their effect on the diameters of glomerular vessels in ex vivo experiments. Afferent and efferent glomerular arterioles were isolated from mouse kidneys, mounted on pipettes and perfused. The diameter of afferent and efferent arterioles were measured under resting conditions and in the presence of vasoactive agents. For both afferent and efferent arterioles, three groups of vessels were randomly assigned as ‘L‐NAME + runcaciguat’, ‘ODQ + runcaciguat’ and ‘L‐NAME + vehicle’, which correspond to the groups used for pretreatment with L‐NAME or ODQ and subsequent application of runcaciguat or vehicle. The perfusion conditions were strictly identical in these three groups. In one set of experiments, vessels were pretreated with the NOS inhibitor L‐NAME (10−4 M, 15 min) to induce NO deficiency. Then, increasing concentrations of Ang II were added on top of L‐NAME to pre‐constrict the vessels, followed by vehicle or runcaciguat (‘L‐NAME + vehicle’ or ‘L‐NAME + runcaciguat’ group; for an example of the experimental protocol, see Figure 3a,b). Another set of experiments was performed in a similar manner, but vessels were pre‐incubated with ODQ (10−5 M, 10 min) to mimic oxidative stress and oxidise the haem group of sGC (‘ODQ + runcaciguat’ group). Ang II concentration‐dependently decreased the vessel diameter by approximately 60% of the initial diameter at concentrations between 10−8 M and 10−6 M in both afferent (Figure 3c) and efferent arterioles (Figure 3d). The absolute afferent and efferent arteriole diameters under resting conditions as well as before and after Ang II application were similar throughout the three different groups (Table 1). In addition, superimposition of the Ang II concentration–response curves produced from afferent (Figure 3c) and efferent arterioles (Figure 3d) before application of vehicle or runcaciguat showed similar sensitivity of the vessel preparations to Ang II. Together, these results indicated a good reproducibility of the measurements and similar initial conditions for the subsequent application of vehicle or runcaciguat.
FIGURE 3.
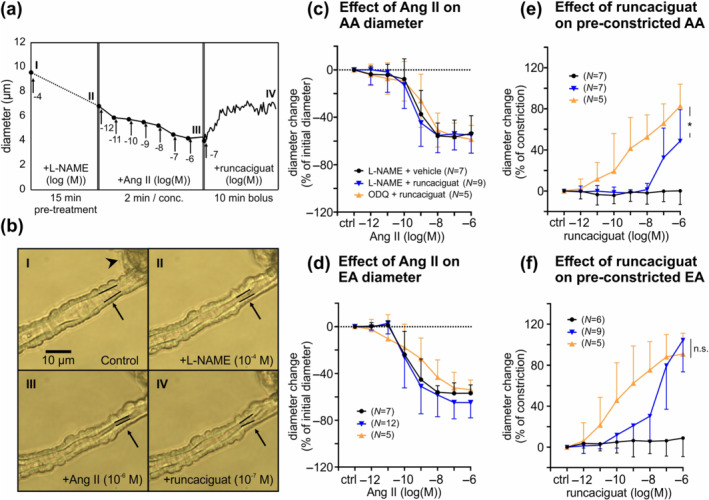
Runcaciguat‐induced dilation of angiotensin II (Ang II) pre‐constricted glomerular arterioles pretreated with L‐NAME or ODQ. Diameter changes of afferent (AA) and efferent arterioles (EA) upon application of various substances were monitored. (a) Representative sequence of an afferent arteriole diameter measurement: Endogenous NO generation was inhibited with L‐NAME (10−4 M) and then the afferent arteriole was pre‐constricted with incremental concentrations of Ang II (10−12 to 10−6 M). The diameter change of the pre‐constricted afferent arteriole in response to runcaciguat (10−7 M, bolus application) was followed. (b) Images illustrate the monitored vessel during each step of the sequence as indicated by roman numbers. Black arrows point towards the segment subjected to quantitative evaluation. The inner vessel wall in this segment is highlighted with black lines. Black arrowhead points towards the attached glomerulus. (c, d) Resting afferent and efferent arterioles were pretreated with L‐NAME (‘L‐NAME + vehicle’ and ‘L‐NAME + runcaciguat’ groups) or ODQ (10−5 M; ‘ODQ + runcaciguat’ group) and then pre‐constricted with incremental concentrations of Ang II in presence of L‐NAME or ODQ. Values are given as percent change of the initial diameter after L‐NAME or ODQ pretreatment and before Ang II application. (e, f) Pre‐constricted afferent and efferent arterioles were treated with cumulative concentrations of runcaciguat or vehicle (DMSO). Values are given as percent of the maximal constriction induced by Ang II. Data represent mean ± SD. In panels (c)–(f), n values are indicated in parentheses. The colour code given in panels (d)–(f) corresponds to the experimental groups indicated in panel (c). scale bar, 10 μm. *P < 0.05
TABLE 1.
Diameters of glomerular arterioles at different time points during the experiment
| Afferent arteriole diameter (μm) | Efferent arteriole diameter (μm) | |||||
|---|---|---|---|---|---|---|
| I | II | III | I | II | III | |
| L‐NAME + vehicle | 7.7 ± 2.8 (n = 7) | 7.3 ± 3.0 (n = 7) | 3.3 ± 1.7 (n = 7) | 7.7 ± 1.4 (n = 7) | 6.7 ± 1.6 (n = 7) | 3.4 ± 1.7 (n = 7) |
| L‐NAME + runcaciguat | 9.1 ± 1.6 (n = 9) | 8.6 ± 2.1 (n = 9) | 3.2 ± 1.0 (n = 9) | 7.5 ± 1.2 (n = 12) | 7.0 ± 1.5 (n = 12) | 2.6 ± 2.0 (n = 12) |
| ODQ + runcaciguat | 7.9 ± 1.4 (n = 5) | 6.7 ± 0.7 (n = 5) | 2.8 ± 0.8 (n = 5) | 6.6 ± 1.1 (n = 5) | 6.2 ± 2.5 (n = 5) | 2.9 ± 1.1 (n = 5) |
Note: Afferent and efferent arterioles were pretreated with L‐NAME (10−4 M; ‘L‐NAME + vehicle’ and ‘L‐NAME + runcaciguat’ groups) or ODQ (10−5 M; ‘ODQ + runcaciguat’ group) and pre‐constricted with incremental concentrations of Ang II (10−12–10−6 M). Shown are absolute diameters of afferent and efferent arterioles under resting conditions (I), after application of L‐NAME or ODQ (II) and after addition of the highest Ang II concentration (III). An example for the procedure is shown in Figure 3a. Data represent mean ± SD. n values are indicated in parentheses.
Addition of increasing concentrations of runcaciguat (10−12 M to 10−6 M) to the pre‐constricted afferent (Figure 3e) or efferent arterioles (Figure 3f) for 2 min induced strong vasodilation. ODQ pretreated afferent arterioles were significantly more sensitive to runcaciguat than L‐NAME pretreated afferent arterioles (Figure 3e,). ODQ pretreated efferent arterioles also showed a trend towards increased sensitivity to runcaciguat as compared with L‐NAME pretreated efferent arterioles, but the difference did not reach statistical significance (Figure 3f). The dilating effect of runcaciguat on L‐NAME pretreated vessels started at significantly lower concentrations in efferent arterioles than in afferent arterioles but was not significantly different between ODQ pretreated efferent and afferent arterioles (Figure 3e,f). These observations suggested a greater sensitivity for sGC activator of efferent over afferent arterioles under NO deficiency. The highest applied dose of runcaciguat (10−6 M) induced significantly stronger dilation of L‐NAME pretreated efferent than afferent arterioles (efferent: 110%, afferent: 49%, Figure 3e,f). Maximal dilatations did not differ significantly in vessels pretreated with ODQ (efferent: 91%, afferent: 83%, Figure 3e,f). The dilation induced by runcaciguat (10−6 M) in the presence of ODQ was comparable with the maximal dilation that could be achieved by application of the NO donor SNAP or the L‐ and N‐type calcium channel blocker cilnidipine (Figure S2). Further experiments showed that SNAP‐induced but not cilnidipine‐induced dilation was efficiently blocked by ODQ (Figure S2). Together, these results indicated that runcaciguat is a potent vasodilator of glomerular arterioles under conditions of NO deficiency and oxidative stress.
To further evaluate the time course of sGC activator action, we evaluated how afferent and efferent arteriole diameters changed during prolonged 10 min bolus applications of runcaciguat (10−7 M). In these experiments, arterioles were pretreated with L‐NAME (10−4 M) and pre‐constricted with Ang II (10−12–10−6 M, not shown). Again, the diameters before application of runcaciguat or vehicle were similar (data not shown). The maximum vasodilatation achieved 10 min after application of runcaciguat was 50% of constriction in afferent arterioles (Figure 4a) and 60% in efferent arterioles (Figure 4b). Maximal vasodilation and kinetics of diameter changes were not significantly different between afferent and efferent arterioles.
FIGURE 4.
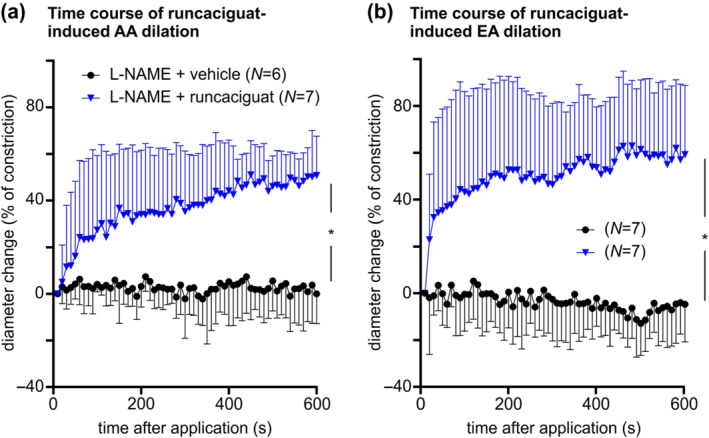
Time course of runcaciguat‐induced dilation of angiotensin II (Ang II) pre‐constricted glomerular arterioles. Glomerular arterioles were pretreated with L‐NAME (10−4 M) and pre‐constricted with Ang II (10−12 to 10−6 M), as shown before. Then, runcaciguat (10−7 M; ‘L‐NAME + runcaciguat’) or DMSO (‘L‐NAME + vehicle’) were applied and diameter changes of (a) afferent arterioles and (b) efferent arteriole were monitored over 10 min. Data represent mean ± SD. n values are indicated in parentheses. The colour code given in panel (b) corresponds to the experimental groups indicated in panel (a). *P < 0.05
Overall, these data indicated a reduction in renal resistance triggered by the sGC activator runcaciguat under conditions of oxidative stress and NO deficiency. At the tested concentration range, runcaciguat induced dilation of afferent and efferent arterioles. The vasodilating effect of runcaciguat was stronger after pretreatment with ODQ than with L‐NAME, suggesting that runcaciguat activates the sGC more potently under conditions of oxidative stress.
3.4. Runcaciguat increases cGMP and improves RBF in perfused mouse kidneys
To complement the cGMP imaging data generated in kidney slices (Figures 1 and 2) and vascular reactivity measurements with isolated glomerular arterioles (Figures 3 and 4), we went on to analyse RBF and cGMP production under close‐to‐native conditions in whole kidneys. To do so, mouse kidneys were isolated and perfused at constant pressure (100 mmHg) as described previously (Schweda et al., 2003). Once perfusion of the kidney had stabilised (i.e. after 20 min perfusion), ODQ was infused to mimic NO deficiency and oxidative stress as observed in chronic kidney disease patients (Elshamaa et al., 2011; Martens & Edwards, 2011; Stasch et al., 2015). ODQ (30 μM) strongly reduced RBF to approximately 50% of control (Figure 5a). In line with this finding, ODQ also reduced the cGMP content in the venous effluent as compared with baseline conditions (Figure 5b). Reduction of RBF by ODQ plateaued roughly 10 min after the start of infusion. Under these oxidative stress conditions, runcaciguat (10 μM) increased the cGMP content from 1.21 ± 0.6 to 13.1 ± 0.7 pmol·min−1 per g kidney (Figure 5b). At the same time, runcaciguat dose‐dependently increased RBF to a level above the baseline recorded in the absence of ODQ at the beginning of the experiment (Figure 5a). After this increase in RBF by runcaciguat, addition of the NO donor SNAP did not significantly change cGMP production (13.1 ± 0.7 versus 15.3 ± 1.2 pmol·min−1 per g kidney) (Figure 5b) and did not further change RBF (Figure 5a). In addition, we analysed the effects of SNAP on RBF and cGMP secretion under basal versus oxidative stress conditions (Figure 5c,d). Under basal conditions, SNAP potently increased renal perfusion and cGMP content, while it failed to do so in the presence of ODQ. In summary, these data showed that in the presence of ODQ mimicking chronic kidney disease‐related oxidative stress conditions, runcaciguat increases the renal cGMP concentration and RBF in perfused mouse kidneys, whereas NO is no longer able to do so under these conditions.
FIGURE 5.
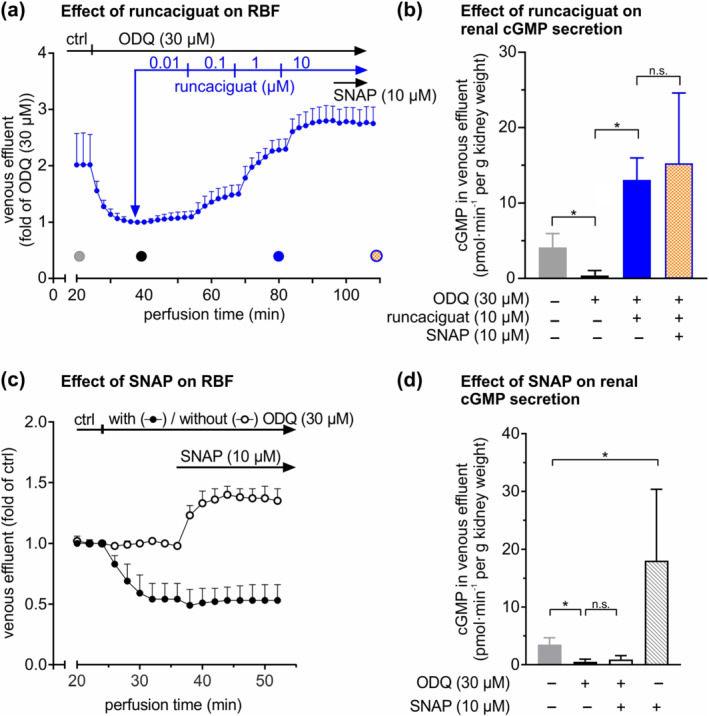
Renal blood flow (RBF) and cGMP secretion in perfused mouse kidneys. Kidneys were isolated and perfused at constant pressure (100 mmHg). (a) RBF was analysed by measuring the amount of venous effluent. After stabilization of perfusion (i.e. after 20 min), ODQ (30 μM) followed by incremental concentrations of runcaciguat (0.01, 0.1, 1 and 10 μM) and S‐nitroso‐N‐acetylpenicillamine (SNAP; 10 μM) were applied sequentially, while the venous effluent was continuously quantified. Filled circles indicate time points at which the cGMP concentration in the venous effluent was determined (n = 6). (b) The cGMP concentration in the venous effluent was determined via enzyme immunoassay and multiplied by the perfusate flow to determine cGMP secretion as a measure of intrarenal cGMP formation. (c, d) Similarly, the effects of SNAP (10 μM) in the absence or presence of ODQ (30 μM) on RBF (c) and on renal cGMP secretion (d) were determined (n = 5). Data represent mean ± SD. *P < 0.05; n.s., not significant; ctrl, control
4. DISCUSSION
This study showed that sGC activators increase the glomerular cGMP concentration, induce dilation of pre‐constricted glomerular arterioles and improve RBF under conditions of NO deficiency and oxidative stress. It is known that oxidation of sGC from its native to the haem‐free form, apo‐sGC, prevents binding of NO and thereby shuts down the NO/sGC/cGMP signalling pathway (Stasch et al., 2015). This leads to reduced production of cGMP, which is important for the regulation of body homeostasis and kidney function (Krishnan et al., 2018; Wang‐Rosenke et al., 2008). As sGC activators act in an NO‐ and haem‐independent manner, these compounds might be able to restore cGMP signalling under pathophysiological conditions involving oxidative stress. Because chronic kidney disease is associated with oxidative stress (Matsuda & Shimomura, 2013; Moon & Won, 2017) and oxidation of sGC might be a critical driver of chronic kidney disease, sGC activators represent a promising new option for chronic kidney disease treatment. However, the mode of action of sGC activators in renal tissue and specifically their impact on renal blood vessels is poorly understood.
To explore the spatiotemporal dynamics of NO‐ and sGC activator‐induced cGMP signals in the kidney, we performed real‐time cGMP imaging in kidney slices of transgenic cGMP sensor mice. To our knowledge, this is the first demonstration of cGMP signals in live kidney tissue. We showed that NO and sGC activator increase the cGMP concentration in glomeruli in the absence and presence of oxidative stress, respectively. Furthermore, runcaciguat induced dilation of glomerular arterioles and improved RBF under disease‐relevant conditions of oxidative stress and NO depletion. Although the present study did not experimentally distinguish between specific renal cell types, the combined results strongly suggest that runcaciguat acts on apo‐sGC in vascular smooth muscle cells of glomerular arterioles, leading to an increase of cGMP, activation of cGMP‐dependent protein kinase type I and vasodilation. This model is consistent with the cellular distribution of sGC in the kidney. Using a highly specific antibody against the β1 subunit of sGC, expression of the protein was detected in renal vascular cells, including glomerular arterioles (Theilig et al., 2001). Expression data, however, do not show the functionality of sGC in a specific compartment. It is well known that enzyme activity is regulated by post‐translational mechanisms including, in the case of sGC, protein oxidation. Our cGMP imaging data link sGC expression in glomeruli with respective enzyme activity in the absence and presence of oxidative stress. Because the R26‐CAG‐cGi500(L1) mice used in this study express the cGMP sensor in all cell types, we cannot exclude the possibility that cGMP signals measured in our FRET imaging experiments were also derived from non‐vascular smooth muscle cells that express sGC, such as interstitial fibroblasts (Theilig et al., 2001). However, it is unlikely that these cells were involved in sGC activator‐induced dilation of renal arterioles. To analyse which specific renal cell types are capable of generating cGMP in response to NO and sGC activators, additional experiments could be performed using transgenic mice with cell type‐specific expression of the cGMP sensor (Thunemann et al., 2013). Furthermore, it would be interesting to analyse the effects of sGC activators on cGMP signals in chronic kidney disease kidneys.
Real‐time cGMP imaging illustrated that BAY‐543 induces cGMP generation in glomeruli specifically in the presence of oxidative stress. This finding complements previous in vitro experiments showing that apo‐sGC has a higher sensitivity for sGC activators than native sGC (Schmidt et al., 2009). To further dissect the mechanism of sGC activator action on kidney function, we investigated the effects of runcaciguat on the diameter of murine glomerular afferent and efferent arteries. It is known that pre‐glomerular resistance and glomerular haemodynamics contribute to the control of the murine GFR (Patzak et al., 2004). Under pathological conditions, such as hypertension and type‐2‐diabetes, an increase in vascular resistance provokes a reduction of vascular diameter and arterial remodelling in the kidney, leading to impaired vascular reactivity and kidney perfusion (Polichnowski et al., 2013; Touyz et al., 2018). In the present study, we showed that runcaciguat improves vascular reactivity of pre‐constricted murine glomerular afferent and efferent arterioles under conditions of NO deficiency and oxidative stress. Runcaciguat induced a concentration‐dependent relaxation of pre‐constricted glomerular arterioles with an apparent higher sensitivity for ODQ over L‐NAME pretreated arterioles. This finding is consistent with a predominant action of sGC activator under oxidative stress as measured in our cGMP imaging experiments with kidney slices. Interestingly, the stronger action of runcaciguat in efferent compared with afferent arterioles indicate an influence on the GFR. This must be investigated in further experiments, for example, via cannulation of the ureter during experiments on isolated perfused kidneys.
To validate the cGMP imaging and vascular reactivity data obtained in kidney slices and isolated arterioles in a more physiological setting, we evaluated the effects of runcaciguat in perfused whole mouse kidneys. In order to mimic chronic kidney disease‐related oxidative stress, ODQ was infused. ODQ rapidly induced stable reduction of kidney perfusion, which could be reversed by runcaciguat in a concentration‐dependent manner. In parallel, ODQ induced a significant reduction of the renal cGMP content, which was restored by runcaciguat. At the same time, the improvement of kidney perfusion and cGMP secretion by the NO donor SNAP was completely abolished by ODQ. These data indicate that in contrast to NO, runcaciguat increases cGMP levels and RBF under oxidative stress conditions. Together with the experiments in isolated renal arterioles, these findings strongly suggest a beneficial effect of sGC activators on the perfusion of diseased kidneys by dilating glomerular arterioles under conditions of NO deficiency and oxidative stress. Our study was performed with mouse kidneys, but it is likely that sGC activators have similar effects on human kidney perfusion. Indeed, a recent study reported that the sGC activator BAY 60–2770 relaxes human intra‐renal arteries (Frees et al., 2020), indicating that the murine kidney is a suitable model for preclinical studies.
Within the scope of this study, the effect of sGC activators on the renal vasculature was analysed in detail. In addition to their vasodilative effects, sGC activators may also be renoprotective via antifibrotic, antiproliferative and anti‐inflammatory effects on vascular and non‐vascular compartments of the kidney. In this context, it is interesting to note that the sGC activator BI 703704 (Boustany‐Kari et al., 2016) and the sGC stimulator praliciguat (Liu et al., 2020) inhibited the progression of diabetic nephropathy in obese ZSF1 rats in doses that did not alter BP, perhaps via suppression of inflammation and apoptosis in tubular cells (Liu et al., 2020). sGC activators may inhibit glomerular remodelling also via direct effects on vascular smooth muscle cells independent of vasodilation. It is well known that NO/sGC/cGMP signalling in vascular smooth muscle cells regulates vascular plasticity and remodelling during diseases like atherosclerosis (Lehners et al., 2018).
In summary, our results indicate that sGC activators increase cGMP production in vascular smooth muscle cells in glomerular arterioles and, thereby, improve kidney perfusion under disease‐relevant conditions of oxidative stress and NO depletion. The unique selectivity of sGC activators for oxidised apo‐sGC has great potential to limit off‐target effects on healthy tissue, where sGC is mainly present in its native reduced form (Sharkovska et al., 2010). Most of the comorbidities in chronic kidney disease are associated with increased oxidative stress burden, leading to endothelial dysfunction with low NO production as well as NO‐unresponsive apo‐sGC. It is an intriguing concept that sGC activators could overcome the pathophysiological blockade of the endogenous NO/sGC/cGMP in renal tissue. Thus, sGC activators like runcaciguat could provide a novel and effective chronic kidney disease treatment.
AUTHOR CONTRIBUTIONS
T.S., F.S., A.P., R.F. and A.B. designed the project. D.S., M.Z.X., T.S., F.S. and S.F. acquired and analysed the data. T.S., M.G.H., F.E. and P.S. provided novel sGC activators. D.S., J.R.K., A.P., P.S., R.F. and A.B. wrote, and all authors edited and approved the manuscript.
CONFLICT OF INTERESTS
T.S., M.G.H., I.M., J.R.K., F.E. and P.S. are employees of Bayer AG. A.B. has been an employee of Bayer AG and is now employed at Novo Nordisk A/S. D.S., M.Z.X., F.S., S.F., A.P. and R.F. have received a restricted research grant from Bayer AG to conduct experiments.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the British Journal of Pharmacology guidelines for Design and Analysis and Animal Experimentation, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1. Structure and activity of the soluble guanylyl cyclase (sGC) activator runcaciguat and its analogue BAY‐543. Structurally, both sGC activators are similar. In previous in vitro and ex vivo experiments (references included on the bottom left), they activated sGC, enhanced cGMP production, and dilated pre‐contracted arterioles with similar potency. MEC, minimal effective concentration.
Figure S2. Analysis of maximal vasodilation achieved with sGC‐dependent and sGC‐independent agents. Diameter changes of afferent glomerular arterioles (AA) upon application of various substances were monitored. (a, b) Resting AA were pre‐treated with ODQ (10−5 M) or vehicle as indicated and then pre‐constricted with Ang II (10−6 M) in the presence of ODQ or vehicle. Values are given as percent change of the initial diameter after ODQ or vehicle pre‐treatment and before Ang II application. (c, d) Pre‐constricted AA were treated with cumulative concentrations of SNAP (c) or the L‐ and N‐type calcium channel blocker cilnidipine (d). Values are given as percent of the maximal constriction induced by Ang II. Data represent mean ± SD. n values are indicated in parentheses. The colour code given in panels c‐d corresponds to the experimental groups indicated in panels a‐b. *P < .05.
ACKNOWLEDGMENTS
The authors like to thank Markus Wolters for his help with initial cGMP imaging of kidney slices, Hana Cernecka for her support in refining the IPK graphs and Maria T. Kristina Zaldivia for reading the manuscript. R.F. and S.F. received grants from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – FOR 2060 projects FE 438/5‐2 and FE 438/6‐2, Projektnummer 374031971 ‐ TRR 240, and Projektnummer 335549539 ‐ GRK 2381.
Stehle, D. , Xu, M. Z. , Schomber, T. , Hahn, M. G. , Schweda, F. , Feil, S. , Kraehling, J. R. , Eitner, F. , Patzak, A. , Sandner, P. , Feil, R. , & Bénardeau, A. (2022). Novel soluble guanylyl cyclase activators increase glomerular cGMP, induce vasodilation and improve blood flow in the murine kidney. British Journal of Pharmacology, 179(11), 2476–2489. 10.1111/bph.15586
Funding information Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – FOR 2060 projects, Grant/Award Numbers: FE 438/5‐2, FE 438/6‐2, 374031971 ‐ TRR 240, 335549539 ‐ GRK 2381
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request. Some data may not be made available because of privacy or ethical restrictions. The authors state that no material from other sources than the presented experiments was used. Citation was indicated according to British Journal of Pharmacology guidelines.
REFERENCES
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , Armstrong, J. F. , Faccenda, E. , Harding, S. D. , Pawson, A. J. , & Sharman, J. L. (2019). The Concise Guide to PHARMACOLOGY 2019/20: Catalytic receptors. British Journal of Pharmacology, 176(Suppl 1), S247–S296. 10.1111/bph.14751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benardeau, A. , Hahn, M. G. , Gerisch, M. , Meyer, M. , Menshykau, D. , Hetzel, T. , Schlender, J. , Hartmann, E. , Schomber, T. , Patzak, A. , Schweda, F. , Kraehling, J. R. , Sandner, P. , & Eitner, F. (2020). The novel soluble guanylyl cyclase (sGC) activator runcaciguat induced renal vasodilation and attenuated kidney damage in a rat CKD model. Journal of the American Society of Nephrology, 31(Suppl), PO0600, 232. [Google Scholar]
- Boustany‐Kari, C. M. , Harrison, P. C. , Chen, H. , Lincoln, K. A. , Qian, H. S. , Clifford, H. , Wang, H. , Zhang, X. , Gueneva‐Boucheva, K. , Bosanac, T. , Wong, D. , Fryer, R. M. , Richman, J. G. , Sarko, C. , & Pullen, S. S. (2016). A soluble guanylate cyclase activator inhibits the progression of diabetic nephropathy in the ZSF1 rat. The Journal of Pharmacology and Experimental Therapeutics, 356(3), 712–719. 10.1124/jpet.115.230706 [DOI] [PubMed] [Google Scholar]
- Brenner, B. M. , Cooper, M. E. , de Zeeuw, D. , Keane, W. F. , Mitch, W. E. , Parving, H. H. , Remuzzi, G. , Snapinn, S. M. , Zhang, Z. , & Shahinfar, S. (2001). Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. The New England Journal of Medicine, 345(12), 861–869. 10.1056/NEJMoa011161 [DOI] [PubMed] [Google Scholar]
- Breyer, M. D. , & Susztak, K. (2016). Developing treatments for chronic kidney disease in the 21st century. Seminars in Nephrology, 36(6), 436–447. 10.1016/j.semnephrol.2016.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briasoulis, A. , Al Dhaybi, O. , & Bakris, G. L. (2018). SGLT2 inhibitors and mechanisms of hypertension. Current Cardiology Reports, 20(1), 1–7. 10.1007/s11886-018-0943-5 [DOI] [PubMed] [Google Scholar]
- Brunner, E. , & Langer, F. (1999). Nichtparametrische analyse longitudinaler Daten. München, Wien: R. Oldenbourg Verlag. [Google Scholar]
- Coppolino, G. , Leonardi, G. , Andreucci, M. , & Bolignano, D. (2018). Oxidative stress and kidney function: A brief update. Current Pharmaceutical Design, 24(40), 4794–4799. 10.2174/1381612825666190112165206 [DOI] [PubMed] [Google Scholar]
- R Core Team . (2020). R: a language and environment for statistical computing. Available from https://www.R-project.org (accessed 24 November 2020)
- Coresh, J. (2017). Update on the burden of CKD. Journal of the American Society of Nephrology, 28(4), 1020–1022. 10.1681/ASN.2016121374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , Hoyer, D. , Insel, P. A. , Izzo, A. A. , Ji, Y. , MacEwan, D. J. , Sobey, C. G. , Stanford, S. C. , Teixeira, M. M. , Wonnacott, S. , & Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175(7), 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elshamaa, M. F. , Sabry, S. , Badr, A. , El‐Ahmady, M. , Elghoroury, E. A. , Thabet, E. H. , Kandil, D. , & Kamel, S. (2011). Endothelial nitric oxide synthase gene intron4 VNTR polymorphism in patients with chronic kidney disease. Blood Coagulation & Fibrinolysis, 22(6), 487–492. 10.1097/MBC.0b013e328346ef71 [DOI] [PubMed] [Google Scholar]
- Emdin, C. A. , Khera, A. V. , Klarin, D. , Natarajan, P. , Zekavat, S. M. , Nomura, A. , Haas, M. , Aragam, K. , Ardissino, D. , Wilson, J. G. , Schunkert, H. , McPherson, R. , Watkins, H. , Elosua, R. , Bown, M. J. , Samani, N. J. , Baber, U. , Erdmann, J. , Gormley, P. , … Kathiresan, S. (2018). Phenotypic consequences of a genetic predisposition to enhanced nitric oxide signaling. Circulation, 137(3), 222–232. 10.1161/CIRCULATIONAHA.117.028021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdmann, J. , Stark, K. , Esslinger, U. B. , Rumpf, P. M. , Koesling, D. , de Wit, C. , Kaiser, F. J. , Braunholz, D. , Medack, A. , Fischer, M. , & Zimmermann, M. E. (2013). Dysfunctional nitric oxide signalling increases risk of myocardial infarction. Nature, 504(7480), 432–436. 10.1038/nature12722 [DOI] [PubMed] [Google Scholar]
- Frees, A. , Assersen, K. B. , Jensen, M. , Hansen, P. B. L. , Vanhoutte, P. M. , Madsen, K. , Federlein, A. , Lund, L. , Toft, A. , & Jensen, B. L. (2020). Natriuretic peptides relax human intrarenal arteries through natriuretic peptide receptor type—A recapitulated by soluble guanylyl cyclase agonists. Acta Physiologica (Oxford, England), 231, e13565. 10.1111/apha.13565 [DOI] [PubMed] [Google Scholar]
- Griffin, K. A. (2017). Hypertensive kidney injury and the progression of chronic kidney disease. Hypertension, 70(4), 687–694. 10.1161/HYPERTENSIONAHA.117.08314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn, M. G. (2018). BAY‐1101042: A potent NO‐ and heme‐independent sGC activator suitable for oral dosing. Drugs of the Future, 43(10), 783–792. [Google Scholar]
- Hahn, M. G. , Lampe, T. , Sheikh, S. E. , Griebenow, N. , Woltering, E. , Schlemmer, K.‐H. , Dietz, L. , Gerisch, M. , Wunder, F. , Becker‐Pelster, E. M. , Mondritzki, T. , Tinel, H. , Knorr, A. , Kern, A. , Lang, D. , Hueser, J. , Schomber, T. , Benardeau, A. , Eitner, F. , … Stasch, J. P. (2021). Discovery of the soluble guanylate cyclase activator runcaciguat (BAY 1101042). Journal of Medicinal Chemistry, 64(9), 5323–5344. 10.1021/acs.jmedchem.0c02154 [DOI] [PubMed] [Google Scholar]
- International Consortium for Blood Pressure Genome‐Wide Association Studies , Ehret, G. B. , Munroe, P. B. , Rice, K. M. , Bochud, M. , Johnson, A. D. , Smith, A. V. , Psaty, B. M. , Abecasis, G. R. , Chakravarti, A. , Elliott, P. , van Duijn, C. M. , Newton‐Cheh, C. , Levy, D. , Caulfield, M. J. , & Johnson, T. (2011). Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature, 478(7367), 103–109. 10.1038/nature10405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan, S. M. , Kraehling, J. R. , Eitner, F. , Benardeau, A. , & Sandner, P. (2018). The impact of the nitric oxide (NO)/soluble guanylyl cyclase (sGC) signaling cascade on kidney health and disease: A preclinical perspective. International Journal of Molecular Sciences, 19(6), 1–18. 1712. 10.3390/ijms19061712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehners, M. , Dobrowinski, H. , Feil, S. , & Feil, R. (2018). cGMP signaling and vascular smooth muscle cell plasticity. Journal of Cardiovascular Development and Disease, 5(2), 20, 1–18. 10.3390/jcdd5020020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley, E. , Stanford, S. C. , Kendall, D. E. , Alexander, S. P. , Cirino, G. , Docherty, J. R. , George, C. H. , Insel, P. A. , Izzo, A. A. , Ji, Y. , Panettieri, R. A. , Sobey, C. G. , Stefanska, B. , Stephens, G. , Teixeira, M. , & Ahluwalia, A. (2020). ARRIVE 2.0 and the British Journal of Pharmacology: Updated guidance for 2020. British Journal of Pharmacology, 177(16), 3611–3616. 10.1111/bph.15178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y. H. , Huang, Y. Y. , Hsieh, S. H. , Sun, J. H. , Chen, S. T. , & Lin, C. H. (2019). Renal and glucose‐lowering effects of empagliflozin and dapagliflozin in different chronic kidney disease stages. Frontiers in Endocrinology, 22(10), 1–11, 820. 10.3389/fendo.2019.00820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, G. , Shea, C. M. , Jones, J. E. , Price, G. M. , Warren, W. , Lonie, E. , Yan, S. , Currie, M. G. , Profy, A. T. , Masferrer, J. L. , & Zimmer, D. P. (2020). Praliciguat inhibits progression of diabetic nephropathy in ZSF1 rats and suppresses inflammation and apoptosis in human renal proximal tubular cells. American Journal of Physiology. Renal Physiology, 319(4), F697–F711. 10.1152/ajprenal.00003.2020 [DOI] [PubMed] [Google Scholar]
- Liu, Z. Z. , Viegas, V. U. , Perlewitz, A. , Lai, E. Y. , Persson, P. B. , Patzak, A. , & Sendeski, M. M. (2012). Iodinated contrast media differentially affect afferent and efferent arteriolar tone and reactivity in mice: A possible explanation for reduced glomerular filtration rate. Radiology, 265(3), 762–771. 10.1148/radiol.12120044 [DOI] [PubMed] [Google Scholar]
- Maass, P. G. , Aydin, A. , Luft, F. C. , Schachterle, C. , Weise, A. , Stricker, S. , Lindschau, C. , Vaegler, M. , Qadri, F. , Toka, H. R. , Schulz, H. , & Bahring, S. (2015). PDE3A mutations cause autosomal dominant hypertension with brachydactyly. Nature Genetics, 47(6), 647–653. 10.1038/ng.3302 [DOI] [PubMed] [Google Scholar]
- Martens, C. R. , & Edwards, D. G. (2011). Peripheral vascular dysfunction in chronic kidney disease. Cardiology Research and Practice, 2011, 267257–267259. 10.4061/2011/267257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda, M. , & Shimomura, I. (2013). Increased oxidative stress in obesity: Implications for metabolic syndrome, diabetes, hypertension, dyslipidemia, atherosclerosis, and cancer. Obesity Research & Clinical Practice, 7(5), e330–e341. 10.1016/j.orcp.2013.05.004 [DOI] [PubMed] [Google Scholar]
- Moon, J. S. , & Won, K. C. (2017). Oxidative stress: Link between hypertension and diabetes. The Korean Journal of Internal Medicine, 32(3), 439–441. 10.3904/kjim.2017.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muskiet, M. H. A. , Heerspink, H. J. L. , & van Raalte, D. H. (2017). SGLT2 inhibition: A new era in renoprotective medicine? The Lancet Diabetes and Endocrinology, 5(8), 569–571. 10.1016/S2213-8587(17)30222-X [DOI] [PubMed] [Google Scholar]
- Ott, I. M. , Alter, M. L. , von Websky, K. , Kretschmer, A. , Tsuprykov, O. , Sharkovska, Y. , Krause‐Relle, K. , Raila, J. , Henze, A. , Stasch, J. P. , & Hocher, B. (2012). Effects of stimulation of soluble guanylate cyclase on diabetic nephropathy in diabetic eNOS knockout mice on top of angiotensin II receptor blockade. PLoS ONE, 7(8), e42623. 10.1371/journal.pone.0042623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patzak, A. , Lai, E. Y. , Mrowka, R. , Steege, A. , Persson, P. B. , & Persson, A. E. (2004). AT1 receptors mediate angiotensin II‐induced release of nitric oxide in afferent arterioles. Kidney International, 66(5), 1949–1958. 10.1111/j.1523-1755.2004.00981.x [DOI] [PubMed] [Google Scholar]
- Patzak, A. , Mrowka, R. , Storch, E. , Hocher, B. , & Persson, P. B. (2001). Interaction of angiotensin II and nitric oxide in isolated perfused afferent arterioles of mice. Journal of the American Society of Nephrology, 12(6), 1122–1127. 10.1681/ASN.V1261122 [DOI] [PubMed] [Google Scholar]
- Percie du Sert, N. , Hurst, V. , Ahluwalia, A. , Alam, S. , Avey, M. T. , Baker, M. , Browne, W. J. , Clark, A. , Cuthill, I. C. , Dirnagl, U. , Emerson, M. , Garner, P. , Holgate, S. T. , Howells, D. W. , Karp, N. A. , Lazic, S. E. , Lidster, K. , MacCallum, C. J. , Macleod, M. , … Würbel, H. (2020). The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biology, 18(7), 1–65, e3000411. 10.1371/journal.pbio.3000410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polichnowski, A. J. , Griffin, K. A. , Long, J. , Williamson, G. A. , & Bidani, A. K. (2013). Blood pressure‐renal blood flow relationships in conscious angiotensin II‐ and phenylephrine‐infused rats. American Journal of Physiology. Renal Physiology, 305(7), F1074–F1084. 10.1152/ajprenal.00111.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueden, C. T. , Schindelin, J. , Hiner, M. C. , DeZonia, B. E. , Walter, A. E. , Arena, E. T. , & Eliceiri, K. W. (2017). ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics, 18(1), 1–26, 529. 10.1186/s12859-017-1934-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rühle, A. , Elgert, C. , Hahn, M. G. , Sandner, P. , & Behrends, S. (2020). Tyrosine 135 of the beta1 subunit as binding site of BAY‐543: Importance of the Y‐x‐S‐x‐R motif for binding and activation by sGC activator drugs. European Journal of Pharmacology, 881, 173203. 10.1016/j.ejphar.2020.173203 [DOI] [PubMed] [Google Scholar]
- Russwurm, M. , Mullershausen, F. , Friebe, A. , Jager, R. , Russwurm, C. , & Koesling, D. (2007). Design of fluorescence resonance energy transfer (FRET)‐based cGMP indicators: A systematic approach. The Biochemical Journal, 407(1), 69–77. 10.1042/BJ20070348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samarghandian, S. , Azimi‐Nezhad, M. , Farkhondeh, T. , & Samini, F. (2017). Anti‐oxidative effects of curcumin on immobilization‐induced oxidative stress in rat brain, liver and kidney. Biomedicine & Pharmacotherapy, 87, 223–229. 10.1016/j.biopha.2016.12.105 [DOI] [PubMed] [Google Scholar]
- Schievink, B. , Kropelin, T. , Mulder, S. , Parving, H. H. , Remuzzi, G. , Dwyer, J. , Vemer, P. , de Zeeuw, D. , & Lambers Heerspink, H. J. (2016). Early renin‐angiotensin system intervention is more beneficial than late intervention in delaying end‐stage renal disease in patients with type 2 diabetes. Diabetes, Obesity & Metabolism, 18(1), 64–71. 10.1111/dom.12583 [DOI] [PubMed] [Google Scholar]
- Schindelin, J. , Arganda‐Carreras, I. , Frise, E. , Kaynig, V. , Longair, M. , Pietzsch, T. , Preibisch, S. , Rueden, C. , Saalfeld, S. , Schmid, B. , Tinevez, J. Y. , White, D. J. , Hartenstein, V. , Eliceiri, K. , Tomancak, P. , & Cardona, A. (2012). Fiji: An open‐source platform for biological‐image analysis. Nature Methods, 9(7), 676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt, H. H. , Schmidt, P. M. , & Stasch, J. P. (2009). NO‐ and haem‐independent soluble guanylate cyclase activators. Handbook of Experimental Pharmacology, 191, 309–339. 10.1007/978-3-540-68964-5_14 [DOI] [PubMed] [Google Scholar]
- Schweda, F. , Wagner, C. , Kramer, B. K. , Schnermann, J. , & Kurtz, A. (2003). Preserved macula densa‐dependent renin secretion in A1 adenosine receptor knockout mice. American Journal of Physiology. Renal Physiology, 284(4), F770–F777. 10.1152/ajprenal.00280.2002 [DOI] [PubMed] [Google Scholar]
- Sharkovska, Y. , Kalk, P. , Lawrenz, B. , Godes, M. , Hoffmann, L. S. , Wellkisch, K. , Geschka, S. , Relle, K. , Hocher, B. , & Stasch, J. P. (2010). Nitric oxide‐independent stimulation of soluble guanylate cyclase reduces organ damage in experimental low‐renin and high‐renin models. Journal of Hypertension, 28(8), 1666–1675. 10.1097/HJH.0b013e32833b558c [DOI] [PubMed] [Google Scholar]
- Sinha, N. , & Dabla, P. K. (2015). Oxidative stress and antioxidants in hypertension‐a current review. Current Hypertension Reviews, 11(2), 132–142. 10.2174/1573402111666150529130922 [DOI] [PubMed] [Google Scholar]
- Siragy, H. M. , & Carey, R. M. (2010). Role of the intrarenal renin‐angiotensin‐aldosterone system in chronic kidney disease. American Journal of Nephrology, 31(6), 541–550. 10.1159/000313363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stasch, J. P. , Schlossmann, J. , & Hocher, B. (2015). Renal effects of soluble guanylate cyclase stimulators and activators: A review of the preclinical evidence. Current Opinion in Pharmacology, 21, 95–104. 10.1016/j.coph.2014.12.014 [DOI] [PubMed] [Google Scholar]
- Su, H. , Wan, C. , Song, A. , Qiu, Y. , Xiong, W. , & Zhang, C. (2019). Oxidative stress and renal fibrosis: Mechanisms and therapies. Advances in Experimental Medicine and Biology, 1165, 585–604. 10.1007/978-981-13-8871-2_29 [DOI] [PubMed] [Google Scholar]
- Sugiyama, S. , Yoshida, A. , Hieshima, K. , Kurinami, N. , Jinnouchi, K. , Tanaka, M. , Suzuki, T. , Miyamoto, F. , Kajiwara, K. , Jinnouchi, T. , & Jinnouchi, H. (2020). Initial acute decline in estimated glomerular filtration rate after sodium‐glucose cotransporter‐2 inhibitor in patients with chronic kidney disease. Journal of Clinical Medical Research, 12(11), 724–733. 10.14740/jocmr4351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theilig, F. , Bostanjoglo, M. , Pavenstadt, H. , Grupp, C. , Holland, G. , Slosarek, I. , Gressner, A. M. , Russwurm, M. , Koesling, D. , & Bachmann, S. (2001). Cellular distribution and function of soluble guanylyl cyclase in rat kidney and liver. Journal of the American Society of Nephrology, 12(11), 2209–2220. 10.1681/ASN.V12112209 [DOI] [PubMed] [Google Scholar]
- Thunemann, M. , Wen, L. , Hillenbrand, M. , Vachaviolos, A. , Feil, S. , Ott, T. , Han, X. , Fukumura, D. , Jain, R. K. , Russwurm, M. , de Wit, C. , & Feil, R. (2013). Transgenic mice for cGMP imaging. Circulation Research, 113(4), 365–371. 10.1161/CIRCRESAHA.113.301063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touyz, R. M. , Alves‐Lopes, R. , Rios, F. J. , Camargo, L. L. , Anagnostopoulou, A. , Arner, A. , & Montezano, A. C. (2018). Vascular smooth muscle contraction in hypertension. Cardiovascular Research, 114(4), 529–539. 10.1093/cvr/cvy023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallon, V. , & Thomson, S. C. (2017). Targeting renal glucose reabsorption to treat hyperglycaemia: The pleiotropic effects of SGLT2 inhibition. Diabetologia, 60(2), 215–225. 10.1007/s00125-016-4157-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang‐Rosenke, Y. , Neumayer, H. H. , & Peters, H. (2008). NO signaling through cGMP in renal tissue fibrosis and beyond: Key pathway and novel therapeutic target. Current Medicinal Chemistry, 15(14), 1396–1406. 10.2174/092986708784567725 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Structure and activity of the soluble guanylyl cyclase (sGC) activator runcaciguat and its analogue BAY‐543. Structurally, both sGC activators are similar. In previous in vitro and ex vivo experiments (references included on the bottom left), they activated sGC, enhanced cGMP production, and dilated pre‐contracted arterioles with similar potency. MEC, minimal effective concentration.
Figure S2. Analysis of maximal vasodilation achieved with sGC‐dependent and sGC‐independent agents. Diameter changes of afferent glomerular arterioles (AA) upon application of various substances were monitored. (a, b) Resting AA were pre‐treated with ODQ (10−5 M) or vehicle as indicated and then pre‐constricted with Ang II (10−6 M) in the presence of ODQ or vehicle. Values are given as percent change of the initial diameter after ODQ or vehicle pre‐treatment and before Ang II application. (c, d) Pre‐constricted AA were treated with cumulative concentrations of SNAP (c) or the L‐ and N‐type calcium channel blocker cilnidipine (d). Values are given as percent of the maximal constriction induced by Ang II. Data represent mean ± SD. n values are indicated in parentheses. The colour code given in panels c‐d corresponds to the experimental groups indicated in panels a‐b. *P < .05.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request. Some data may not be made available because of privacy or ethical restrictions. The authors state that no material from other sources than the presented experiments was used. Citation was indicated according to British Journal of Pharmacology guidelines.