

Abstract
The human genome contains 11 APOBEC (apolipoprotein B mRNA editing catalytic polypeptide‐like) cytidine deaminases classified into four families. These proteins function mainly in innate antiviral immunity and can also restrict endogenous retrotransposable element multiplication. The present study focuses on APOBEC3C (A3C), a member of the APOBEC3 subfamily. Some APOBEC3 proteins use their enzymatic activity on genomic DNA, inducing mutations and DNA damage, while other members facilitate DNA repair. Our results show that A3C is highly expressed in cells treated with DNA‐damaging agents. Its expression is regulated by p53. Depletion of A3C slightly decreases proliferation and does not affect DNA repair via homologous recombination or nonhomologous end joining. The A3C interactomes obtained from control cells and cells exposed to the genotoxin etoposide indicated that A3C is a nucleolar protein. This was confirmed by the detection of either endogenous or ectopic A3C in nucleoli. Interestingly, we show that A3C is excluded from areas of DNA breaks in live cells. Our data also indicate that the C‐terminal part of A3C is responsible for its nucleolar localization and exclusion from DNA damage sites.
Keywords: APOBEC3C, cancer biology, DNA repair, nucleolus, p53
APOBEC3C, via its C‐terminal domain, is targeted to nucleoli and actively excluded from DNA nuclear lesion sites. Its expression is stimulated by genotoxins in a p53‐dependent manner, but APOBEC3C is expendable for double‐stranded DNA repair.
Abbreviations
- A3C
APOBEC3C
- APOBEC
apolipoprotein B mRNA editing catalytic polypeptide‐like
- ATM
ataxia telangiectasia mutated
- BrdU
5‐bromo‐2′‐deoxyuridine
- CDK4
cyclin‐dependent kinase 4
- CMV
cytomegalovirus
- DRGFP
direct repeat green fluorescent protein
- GO
gene ontology
- HR
homologous recombination
- NHEJ
nonhomologous end joining
- NLS
nuclear localization signal
- NoD
nucleolar localization sequence Detector
- NPM‐1
nucleophosmin
- PFA
paraformaldehyde
- PI
propidium iodide
- ssDNA
single stranded DNA
Introduction
Cancer has become the leading cause of death in developed countries [1], and it is the second leading cause of death in the world [2]. Most chemotherapy drugs currently used in the clinic are nonspecific DNA‐damaging agents [3, 4, 5]. The present study started from the observation that APOBEC3C (A3C) expression is strongly increased upon treating osteosarcoma cells with DNA‐damaging chemotherapy drugs. The genes encoding the apolipoprotein B mRNA editing catalytic polypeptide‐like 3 (APOBEC3) family are located in a cluster on chromosome 22 that arose through duplication [6]. The family consists of seven members (A–D and F–H). They possess either one (APOBEC3A, APOBEC3C, and APOBEC3H) or two cytidine deaminase domains (APOBEC3B, APOBEC3D, APOBEC3F, and APOBEC3G). These proteins have an intrinsic ability to bind RNA and single stranded DNA (ssDNA) [7]. Their major function is apparently to restrict lentiviral infection and endogenous retro‐element integration. They achieve this by inducing, via their cytidine deaminase activity, C to T mutations in the ssDNA intermediates produced during reverse transcription [8, 9]. These proteins bind ssDNA with relative sequence specificity [10]. In recent years APOBEC3 proteins have come into focus from the perspective of cancer. Driven by the advancements in DNA sequencing, investigators created a catalog of somatic mutations in breast cancer cells, and observed a phenomenon of localized C to T hypermutation that the authors termed kataegis. They hypothesized that AID/APOBEC proteins could be responsible for kataegis [11]. Subsequent studies showed that this was the case, and aberrant overexpression of several A3 members has been implicated in cancer cell genome hypermutation [12, 13, 14, 15]. In fact, A3A overexpression can trigger the DNA damage response, which leads to cell cycle arrest [16]. A3 deamination of genomic DNA preferentially takes place on the lagging strand during DNA replication [17]. Increasing the mutational burden of cancer cells leads to increased heterogeneity, and potential generation of treatment resistant clones, which is the reason this is still an active area of research. Unexpectedly, A3G was shown to promote DNA repair in a cytidine deaminase‐dependent manner, and thereby aiding lymphoma cell resistance to radiation treatment [18]. A3G and its activity are required for double‐stranded DNA break repair, although the exact mechanism is not elucidated [19]. These observations open the door to the possibility that other A3 members are involved in DNA repair and could be potential therapeutic targets in cancer cells.
This study focuses on A3C, which was implicated in restriction of HIV‐1 and other lentiviruses [20, 21], hepatitis B virus [22], and endogenous retroelements [23]. However, A3C has not been studied in the context of cancer. Since both pro‐repair and pro‐mutagenic functions have been reported for members of this protein family and since no study has focused on A3C so far, we aimed here to: (a) investigate if A3C is involved in DNA repair; (b) provide information about A3C expression regulation, localization, and interacting partners. Our results indicate that A3C is highly expressed in cells treated with various DNA‐damaging agents, especially those inducing double‐strand breaks. Endogenous, but not exogenous, A3C expression is regulated by p53, both in untreated conditions and when exposed to DNA break‐inducing agents. Pull‐down experiments coupled to mass spectrometry indicated that A3C interacts mostly with RNA‐binding and nucleolar proteins and to some DNA repair proteins. The tropism of A3C for the nucleolus was further validated using live cell microscopy and purified nucleoli. Interestingly, we found that A3C is excluded from areas of damaged DNA. Lastly, we show that the C‐terminal part of A3C is responsible for its nucleolar localization and exclusion from areas of damaged DNA.
Results
This study was driven by our observation that human osteosarcoma (U2OS) cells increase their A3C expression when treated with DNA‐damaging drugs, the largest class of anticancer agents currently used in the clinic [3]. Initially, we recorded this increase in response to cisplatin and etoposide (Fig. 1A) and then also upon cell exposure to a broader array of single‐ and double‐strand DNA break‐inducing agents (Fig. 1B). The A3C protein level increase was between 4‐ and 6‐fold above basal levels, depending on the type of drug used (Fig. 1B). Next, we focused on factors that could modulate A3C expression. APOBEC3 family members can be controlled by the p53 transcription factor [24, 25]. Upon double‐strand DNA breaks, p53 is stabilized via phosphorylation at serine 15 by the ataxia telangiectasia mutated (ATM) kinase [26]. We blocked ATM activity using the small molecule inhibitor KU60019 [27] and observed that this inhibited etoposide‐induced endogenous A3C increase (Fig. 1C; also compare lanes 2 and 3 in Fig. 2). This was also the case in the HCT116 colorectal cancer cell line (Fig. 1D). Etoposide‐induced A3C expression was almost completely blocked when p53 was silenced (Fig. 1E). To elucidate whether phosphorylation of p53 at serine 15 is required for A3C induction, we took advantage of the LNZ‐308 glioblastoma cell line that does not express p53. We transfected LNZ‐308 cells with wild‐type p53, or its S15A mutant, and we observed that both constructs were able to increase A3C levels in these cells (Fig. 1F), indicating that S15 phosphorylation of p53 is not necessary for induction of A3C levels.
Fig. 1.
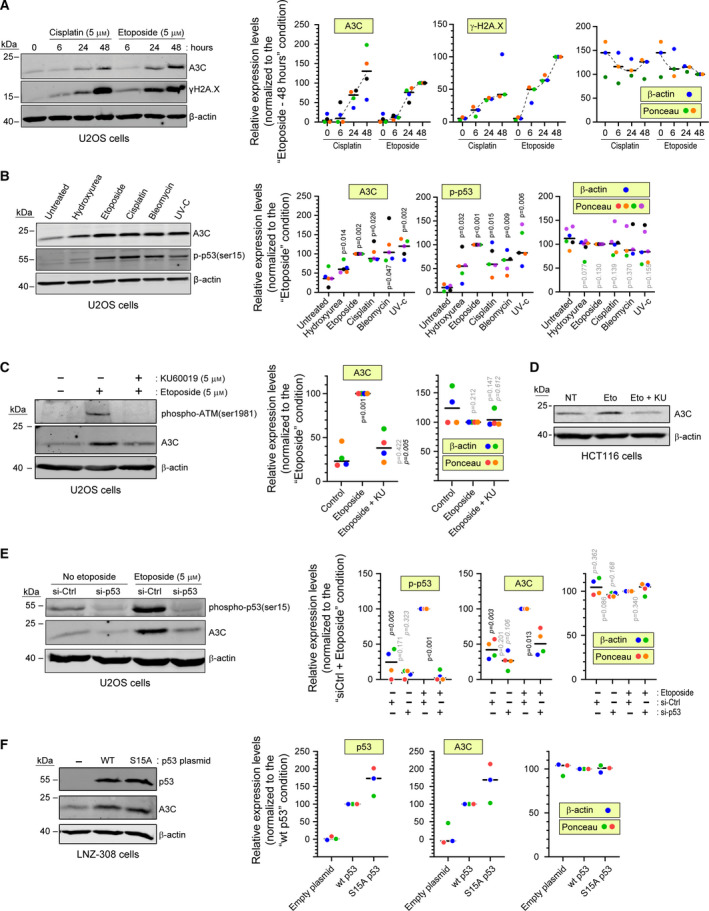
Genotoxic stress exposure induces A3C expression in a p53‐dependent manner. (A) U2OS cells were left untreated or treated with either 5 µm cisplatin or 5 µm etoposide. The cells were lysed at the indicated time points. (B) Cells were left untreated or treated with either 5 mm hydroxyurea, 5 µm etoposide, 5 µm cisplatin, 5 µg·mL−1 bleomycin, or 20 J·m−2 UV‐C and then incubated for 48 h and lysed. (C) U2OS cells were treated with the indicated combinations of etoposide and KU60019 for 48 h. (D) Western blot showing A3C levels in HCT116 cells that were left untreated (NT) or stimulated 48 h with 5 µm etoposide alone (Eto) or in combination with 5 µm KU60019 (Eto+KU). (E) U2OS cells were transfected with a control siRNA or a siRNA targeting p53. Forty‐eight hours later, the cells were treated or not with etoposide for an additional 48‐h period. (F) LNZ‐308 cells were transfected for 48 h with the indicated p53 plasmids. In all panels, expression of the indicated proteins was assessed by western blotting. Quantitation of the western blot signals, performed using ImageJ, is shown on the right hand side of the panels. Loading controls were based on β‐actin or Ponceau staining quantitation. Individual experiments are color coded. The black horizontal bars correspond to the median. The P‐values were obtained using paired t tests. Shaded P values are those > 0.05. In panel B, P values correspond to the comparison with the untreated condition. In panel C, nonitalicized P values correspond to the comparison with the control condition, while the italicized P values correspond to the comparison with the ‘Etoposide’ condition. In panel E, nonitalicized p values correspond to the comparison with the ‘si‐Ctrl‘ condition for a given etoposide concentration, while the italicized P values correspond to the comparison with the ‘Etoposide’ condition for a given siRNA.
Fig. 2.
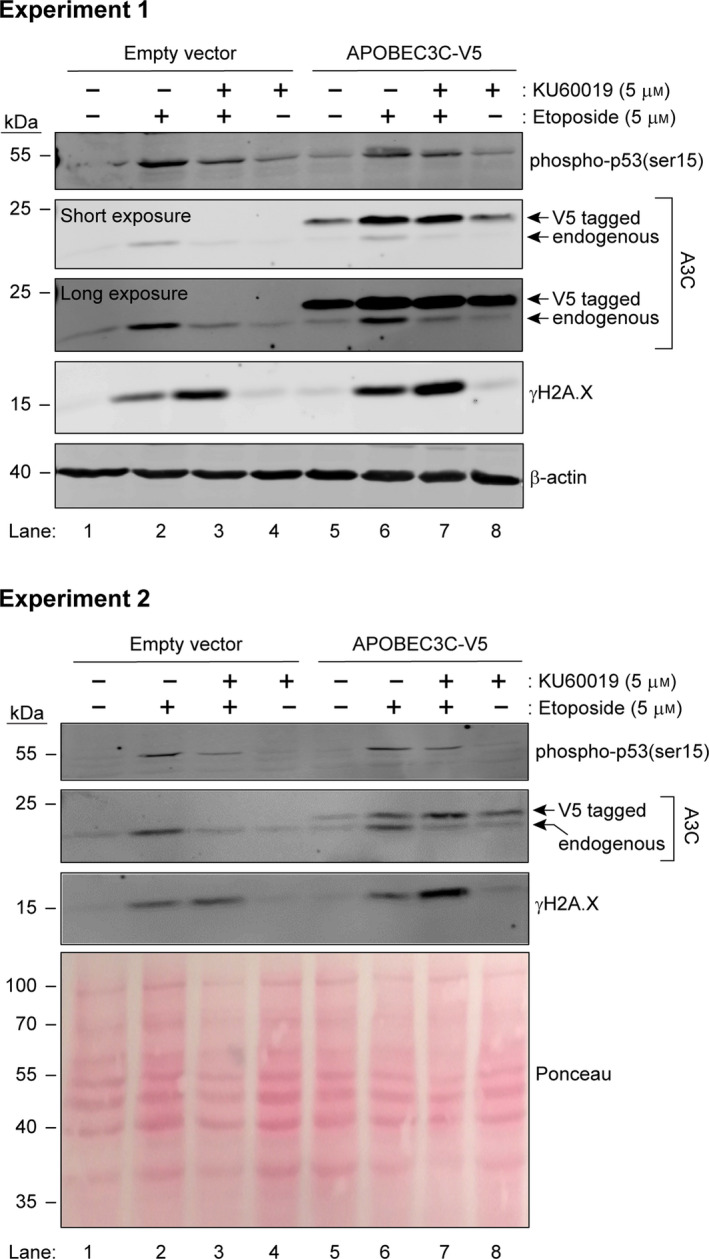
Regulation of A3C expression at the transcriptional and post‐transcriptional levels. U2OS cells were transfected with empty pcDNA3 or pcDNA3 encoding a V5‐tagged version of A3C. Twenty‐four hours later, the cells were treated with etoposide and KU60019, alone or in combination, for an additional 48‐h period. Expression of the indicated proteins was assessed by western blotting. Two independent experiments are shown.
We uncovered an additional, ATM‐ and p53‐independent, mechanism controlling A3C levels when we monitored the expression of ectopically expressed A3C. This was achieved using a pcDNA3 eukaryotic mammalian expression vector. The expression of the constructs cloned into pcDNA3 is driven by the cytomegalovirus (CMV) promoter, which is not known to be positively modulated by p53 [28]. Our experiments showed that levels of ectopic A3C were also increased upon etoposide treatment (Fig. 2, compare lane 5 with lane 6). This increase was not blocked by ATM inhibition (Fig. 2, compare lane 6 with lane 7), in contrast to what was observed with the endogenous protein. These results indicate that A3C levels are controlled at the transcriptional level by p53 in an ATM‐dependent manner and by an uncharacterized post‐transcriptional mechanism.
As APOBEC3 proteins are part of the innate antiviral response [8], we wondered if exposure to interferons would increase A3C levels. We found however that treating U2OS cells with α, β, or γ interferons did not modulate the levels of endogenous A3C (Fig. 3).
Fig. 3.
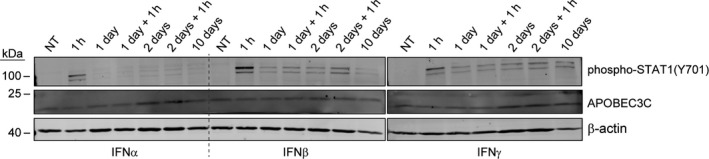
Interferons do not modulate A3C levels. U2OS cells were treated with 100 ng·mL−1 α, β, or γ interferons for the indicated periods. The lanes labeled ‘+ 1 h’ represent conditions in which fresh interferon was added one hour before harvesting. For the ‘10 days’ condition, fresh interferon was added every other day. Expression of the indicated proteins was assessed by western blotting.
Next, we focused on the identification of A3C‐interacting partners. We performed anti‐V5 pull‐down experiments in cells transfected with empty pcDNA3 or with pcDNA3 encoding V5‐tagged A3C. These experiments were performed using untreated cells and cells stimulated with etoposide for 48 h. Mass spectrometry results of pull‐down samples showed that the majority of A3C‐interacting partners were proteins involved in RNA processing and ribosome biogenesis (Fig. 4A, Table S1). The sets of A3C‐interacting proteins were similar between control and etoposide‐treated cells (Table S1). However, since etoposide increases ectopic A3C expression (see Figs 2 and 4D, ‘Input’ lanes), the pull‐down of A3C in etoposide‐treated samples was more efficient (Fig. 3C, ‘1/40 of V5‐IP’ lanes), generating a higher signal to noise ratio (Table S1). Therefore, we used the data obtained from the etoposide‐treated cells for the gene ontology (GO) analysis (Fig. 4B). The top GO terms enriched in the list of A3C‐interacting partners were related to RNA processing and biogenesis, and to DNA repair. In the context of the known functions of APOBEC family members, the RNA‐binding and DNA repair binding GO terms were of particular interest. We decided to validate one interacting partner from each of these categories. We checked whether DDX17, an RNA helicase [29], and PARP1, a protein involved in DNA repair [30], are detected in V5 immunoprecipitates from control and A3C‐expressing cells, treated or not with etoposide. Fig. 4C shows that there was indeed interaction between A3C and endogenous DDX17, and that this interaction was more prominent in cells treated with etoposide, as expected from the ability of this genotoxin to increase ectopic A3C expression. Similar results were obtained for PARP1, although the interaction was clear only in etoposide‐treated cells. The efficiency of A3C expression and pull‐down is shown in Fig. 4D.
Fig. 4.
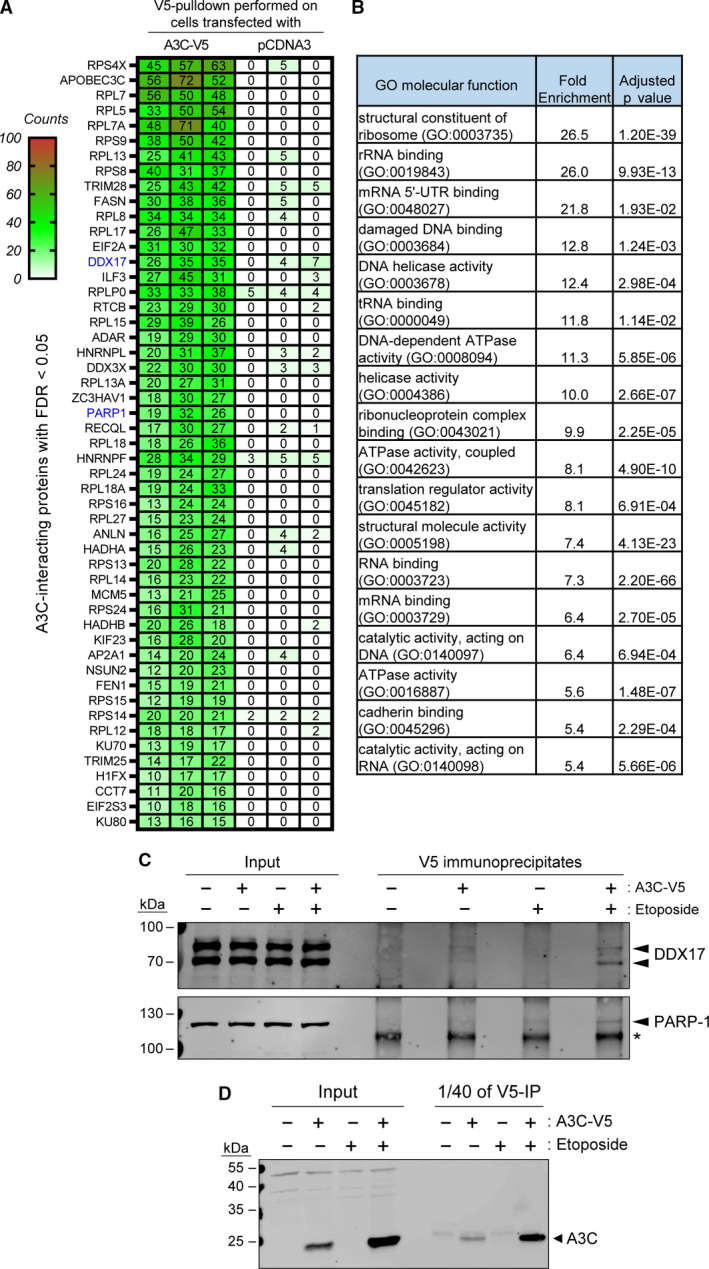
A3C interactome. (A) Heatmap showing the 50 most represented proteins found in A3C pull‐down samples (with the corresponding values obtained in the control pull‐down). Each column represents one individual experiment. Numbers represent the peptide fragments that were detected as unique for each protein (exclusive spectral counts). Only proteins with FDR < 0.05 are shown. Proteins are ordered by largest to smallest differences between A3C and control pull‐down detected peptides. The proteins the names of which are labeled in blue are those that are tested in panel C. (B) GO analysis of the 190 proteins significantly detected as A3C‐interacting partners. The table only lists the GO terms that are enriched 5‐fold or more. (C) U2OS cells were transfected with empty pcDNA3 or pcDNA3 encoding a V5‐tagged version of A3C. Twenty‐four hours later, the cells were treated or not with 25 µm etoposide for an additional 48‐h period. The V5 immunoprecipitates were analyzed by western blotting using DDX17 and PARP1 antibodies. The location of these proteins is indicated by black arrowheads. The asterisk denotes a nonspecific band. (D) A3C levels in the samples analyzed by immunoprecipitation in panel C.
As mentioned above, some A3 family members have been shown to be required for DNA repair, while others have been reported to be detrimental to mammalian genome integrity. No study has focused so far on the role of A3C in DNA repair. The results in Fig. 1A,B indicate that the DNA double‐strand break‐inducing drug etoposide is among the most potent A3C inducer among the compounds we tested. Therefore, we employed GFP‐based reporters that measure the efficiency of double‐strand DNA break repair. The two main pathways that mammalian cells use for repairing double‐strand DNA breaks are homologous recombination (HR) and nonhomologous end joining (NHEJ) [3]. The direct repeat green fluorescent protein (DRGFP) reporter developed by Maria Jasin was used to assess DNA repair via the HR pathway [31]. NHEJ efficiency can be measured using the EJ5GFP reporter developed by Jeremy Stark [32] (Fig. 5A). Upon A3C knockdown, we did not observe any difference in the cells' ability to repair DNA breaks as measured by the aforementioned reporters (Fig. 5B,C). A3C depletion did not seem to affect the extent of DNA damage (measured using the γH2A.X mark) induced by etoposide (Fig. 5D). The specificity of the siRNA pool targeting A3C is shown in Fig. 5E. To further assess the impact of A3C on how cells respond to DNA damage, we created two independent CRISPR/Cas9‐mediated A3C knockout cell lines (Fig. 6A) and tested their sensitivity to etoposide. The knockout lines appeared to grow less than the control cells but were otherwise as sensitive to etoposide as the wild‐type cells (Fig. 6B).
Fig. 5.
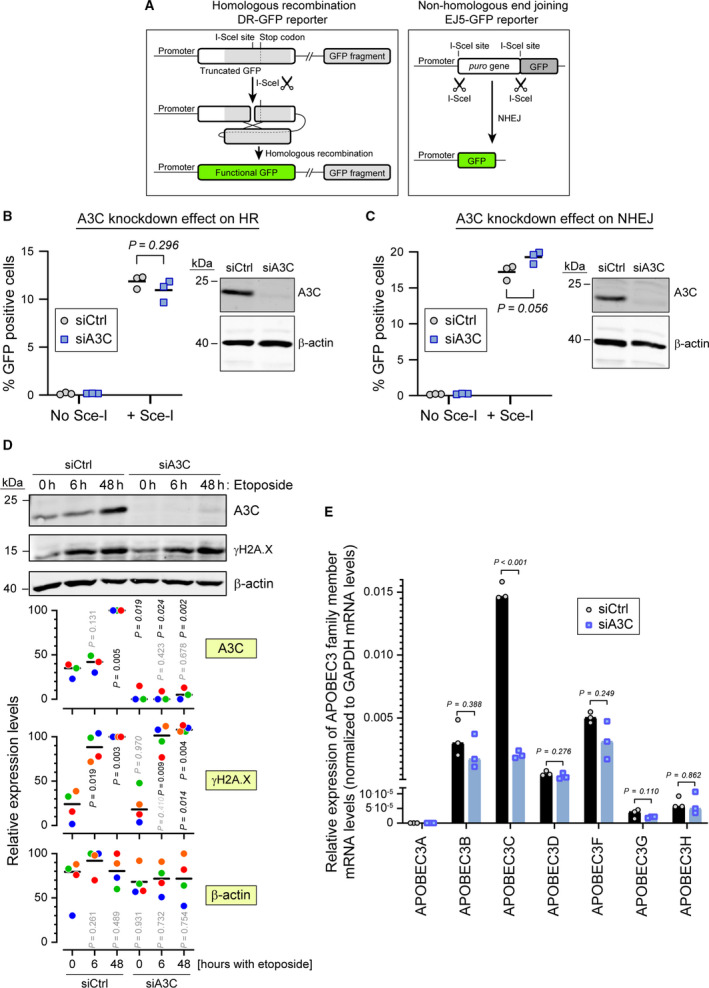
A3C is not required for efficient double‐strand DNA break repair. (A) Schematic representation (not to scale) of the DRGFP reporter (left) and EJ5GFP reporter (right). The DRGFP construct bears a cDNA encoding the full‐length GFP that contains an I‐SceI restriction site disrupting the reading frame and leading to a premature stop codon. This cDNA is followed by a 5′‐ and 3′‐truncated GFP fragment that does not contain the I‐SceI restriction site. When cells expressing this reporter are transduced with a lentivirus encoding the I‐SceI enzyme, the reporter is cleaved at the I‐SceI site. The ensuing DNA repair by HR removes the termination codon in the full‐length cDNA allowing for the production of a functional GFP (green). The EJ5GFP bears a puromycin resistance gene flanked by two I‐SceI restriction sites. This gene is followed by a GFP‐encoding cDNA. In cells expressing this reporter, removal of the puromycin resistance gene is achieved by transduction with an I‐SceI encoding lentivirus. The ensuing NHEJ‐mediated repair brings the promoter and the GFP cDNA close together, allowing for GFP production. (B, C) U2OS‐DRGFP (panel B) or U2OS‐EF5GFP (panel C) cells were transfected with either control siRNA or siRNA targeting A3C for 48 h and then transduced or not with a lentivirus carrying the ISce‐I endonuclease. Forty‐eight hours later the percentage of GFP positive cells was quantified by flow cytometry. Statistical analysis: unpaired Student’s t test. The right side of each panel shows the efficiency of A3C knockdown as assessed by western blotting. (D) Wild‐type U2OS cells were transfected with control siRNA (siCtrl) or siRNAs targeting A3C (siA3C) for 72 h, and then treated with 5 µm etoposide for the indicated time periods. Cell lysates were used for western blotting, with the indicated antibodies. The quantitation shown below the western blots are normalized to the ‘siCtrl, 48 h‘ values for A3C and γH2A.X and to the maximum values for β‐actin. Data points with the same color are derived from the same independent experiment. The black horizontal bars correspond to the median. The p‐values were obtained through paired t tests. For A3C and γH2A.X: nonitalicized and italicized values were those obtained by comparison with the corresponding ‘0 h‘ condition and between the ‘siCtrl‘ and ‘siA3C’ conditions at a given time point, respectively. For β‐actin, the p‐values were obtained by comparison with the ‘siCtrl, 0 h‘ condition. Shaded P values are those > 0.05. (E) A3C in U2OS cells was knocked down using a pool of 30 different siRNAs (siA3C). As a negative control, the cells were also treated with a nontargeting control siRNA pool (siCtrl). The specificity of the knock down was assessed by quantitating the mRNA levels of APOBEC3 family members.
Fig. 6.
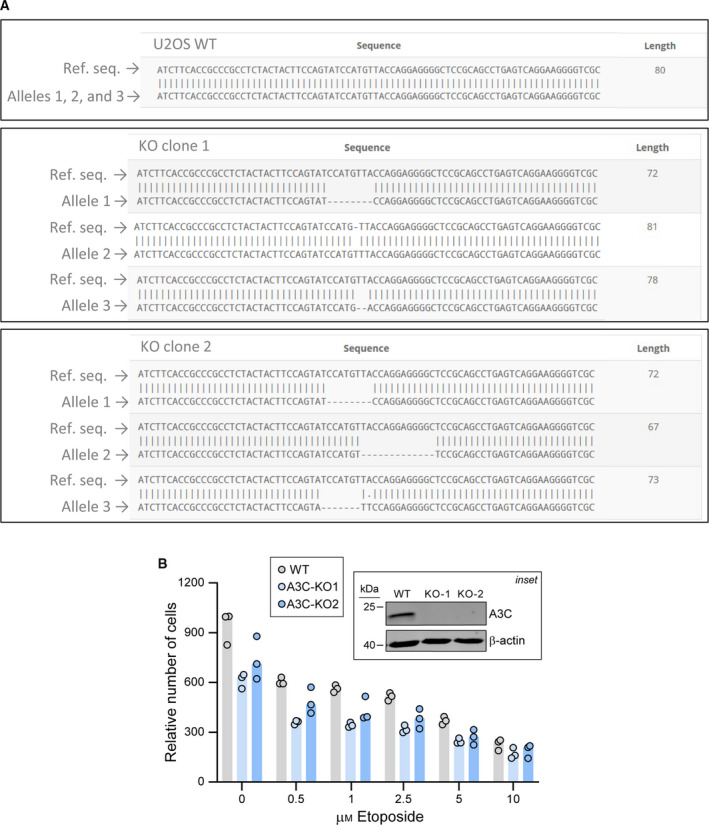
CRISPR/Cas9‐mediated A3C gene disruption and its impact on etoposide sensitivity. (A) A3C gene sequencing in wild‐type (WT) and two CRISPR/Cas9‐generated A3C knockout U2OS cells (KO1 and KO2). The presence of frameshift mutations located in the vicinity of the sgRNA target site was detected in the three A3C alleles of the knockout clones. (B) Wild‐type (WT) and A3C knockout (KO) U2OS cells were seeded in 12‐well plates and treated with increasing concentrations of etoposide for 72 h. The relative number of cells was determined by Presto‐Blue assay. Inset: the presence of A3C protein in these cells was evaluated by western blotting. The cells were stimulated with 5 μm etoposide for 48 h to boost A3C expression.
Our data indicate that U2OS cells lacking A3C proliferate less than control cells (Fig. 6B, first three bars). We confirmed this proliferation defect using siRNA‐mediated A3C knockdown (Fig. 7A). We decided to characterize in more details the impact of A3C depletion on the cell cycle. We employed DNA content staining by propidium iodide (PI) coupled with a 5‐bromo‐2′‐deoxyuridine (BrdU) incorporation assay to obtain a sharp resolution of the cell cycle phases. BrdU is a thymidine analog that is incorporated into newly synthetized DNA and therefore labels cells in the S‐phase of the cell cycle. Upon A3C depletion in U2OS cells, we observed a decrease in the percentage of cells in S‐phase and an increase in the percentage of cells in the G1 phase of the cell cycle (Fig. 7B,C). This phenotype was also observed when A3C was knocked down in HCT116 cells (Fig. 7D–F).
Fig. 7.
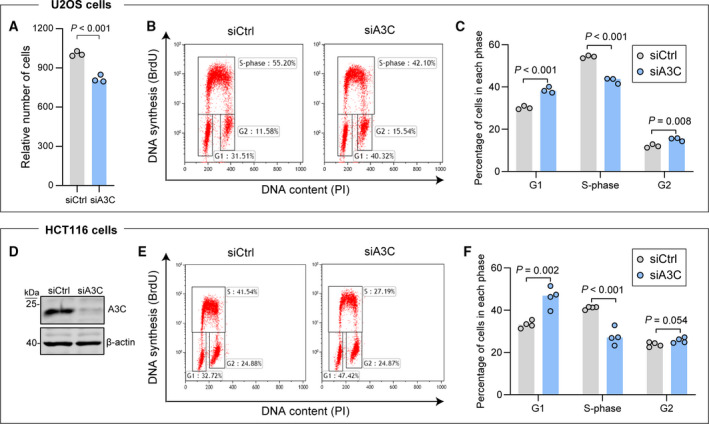
A3C depletion reduces cell proliferation. (A) U2OS cells were transfected with either control siRNA (siCtrl) or siRNAs targeting A3C (siA3C) for 48 h. The cells were then trypsinized and plated at a density of 4∙104 cells·well−1 (12‐well plates). Seventy‐two hours later, the relative cell number was quantitated using a Presto‐Blue assay. (B) Representative cell cycle profiles of control and A3C knockdown cells. (C) Quantitation of cell cycle distribution in control and A3C‐depleted U2OS cells. (D) Western blot depicting the efficiency of A3C knockdown in HCT116 cells. (E, F) FACS analysis of cell cycle profiles in control and A3C knockdown HCT116 cells (representative profiles in panel E, and quantitation in panel F). Statistical analysis: panels C and F, two tailed unpaired t tests (P values corrected for multiple comparisons with the Benjamini–Hochberg method).
We did not record changes in the distribution between the different cell cycle phases in A3C knockout U2OS cells compared to control cells (Fig. 8A,B). Yet, U2OS cells lacking A3C grow slower (Fig. 5E) and their DNA synthesis rate is decreased compared to wild‐type cells (Fig. 8C). Possibly, adaptation following A3C gene disruption has permitted A3C knockout cells to recover a normal pattern of cell cycle phase distribution despite the remaining defects in DNA synthesis and proliferation. Adaptation and compensation effects following gene knockout have been reported earlier, explaining some of the phenotypic differences observed between acute knockdown and permanent knockout of a gene [33].
Fig. 8.
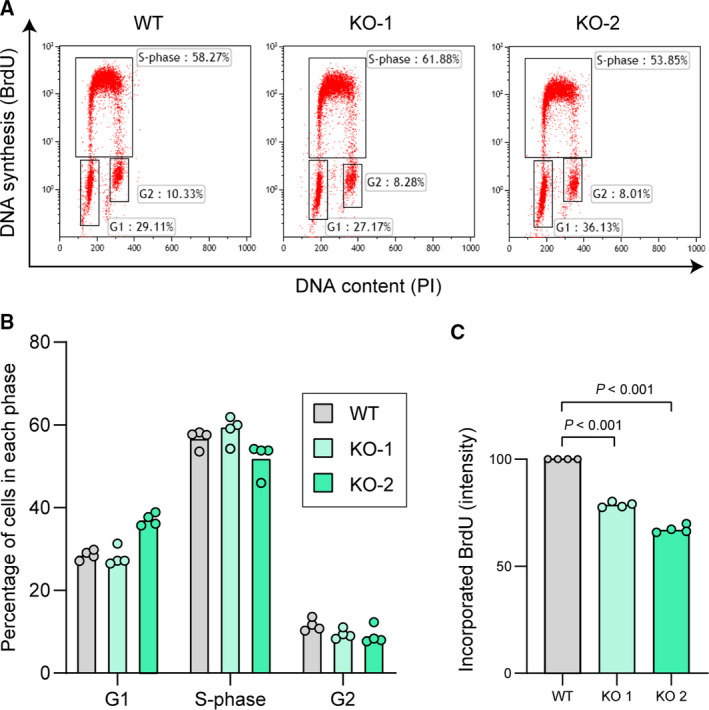
Effect of permanent A3C loss on cell proliferation. (A‐B) Representative cell cycle profiles (panel A) and the corresponding cell cycle distribution quantitation (panel B) of wild‐type U2OS cells (WT) and two A3C knockout (KO) U2OS cell lines. (C) DNA synthesis rate (amount of BrdU incorporated in a 25‐min pulse) in the cells shown in panels A–B. Graphs indicate the median and the individual experimental values normalized to the wild‐type cells. Statistical analysis: unpaired Student’s t tests.
Our mass spectrometry results suggest that A3C is present in the nucleolus. Several of the top A3C‐interacting partners have been observed in nucleoli, such as RPL7 [34], DDX5 [35], and DHX30 [36]. A3C also seems to interact with the prototypic nucleolus marker Nucleolin (Table S1). Previous studies that investigated the subcellular location of GFP‐APOBEC3 proteins have not found GFP‐A3C in nucleoli [37]. Of note, in the aforementioned study, the authors used GFP‐A3C‐expressing cells that were fixed with paraformaldehyde (PFA). Although this is a very popular fixation protocol, it has been shown to sometimes affect the subcellular localization of proteins [38, 39]. Therefore, we generated A3C expression constructs that were N‐terminally tagged with GFP, and analyzed these cells both live and after PFA fixation. In live cells, GFP‐A3C was detected in the cytoplasm and nucleoli, with occasional nuclear staining. However, after PFA fixation, A3C was no longer detected in nucleoli and its nuclear staining considerably increased (Fig. 9A). This shows that fixation induces artefactual A3C distribution in cells. Consequently, the remaining microscopy data we are presenting were generated from live cells.
Fig. 9.
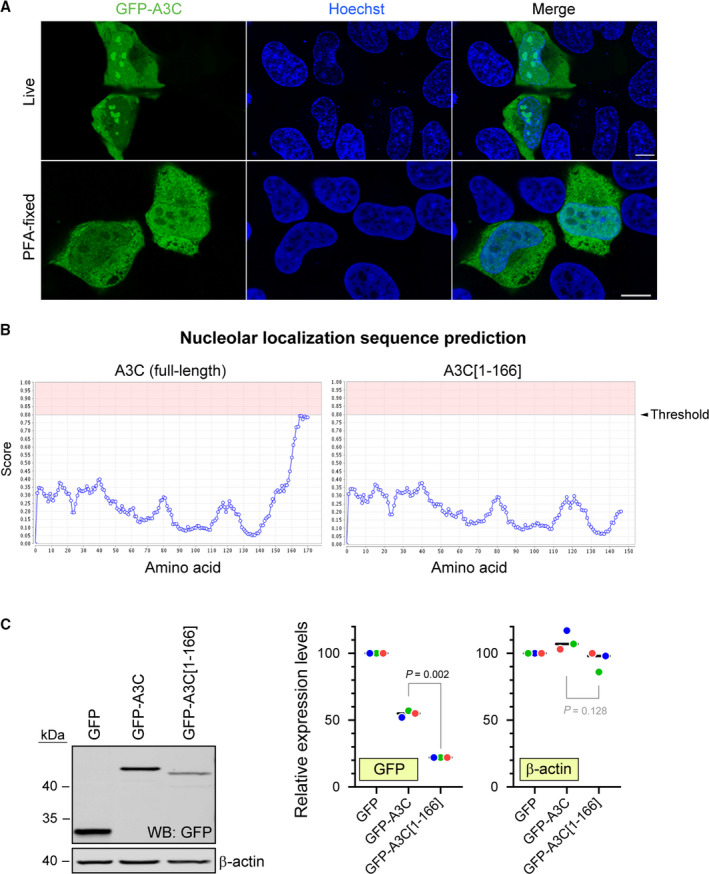
Effect of cell fixation on A3C subcellular location and role of the nucleolar localization sequence of A3C on its expression. (A) Top row: U2OS cells were transfected with GFP‐APOBEC3C and 24 h later were analyzed using a confocal microscope. Bottom row: cells were transfected as above and 24 h later the cells were fixed in PFA, permeabilized, and stained with an anti‐GFP antibody. Nuclei were counterstained with Hoechst 33342. (B) Nucleolar localization sequence prediction in full‐length A3C (left) and A3C[1‐166] (right). (C) Western blot depicting the expression levels of GFP constructs in U2OS cells transfected for 24 h with the plasmids encoding the indicated constructs. The quantitation shown on the right is normalized to the ‘GFP’ values. Data points with the same color are derived from the same independent experiment. The black horizontal bars correspond to the median. The P‐values were obtained through paired t tests. Shaded P values are those > 0.05.
The observation that A3C sometimes localizes to the nucleus and that it seems to accumulate in the nucleolus prompted us to investigate the potential presence of nuclear/nucleolar localization signals in A3C. Using the online tool NLS‐Mapper [40] to analyze the A3C coding sequence (NM_014508.3, 190 amino acids), we found a strong nuclear localization signal (NLS) sequence in the C‐terminal part of A3C. This NLS sequence (‘R179LLKRRLRES188’) presented a NLS‐Mapper score of 7/10 (10 being the strongest). In addition, using the Nucleolar localization sequence Detector (NoD) [41], we found that the C‐terminal part of A3C also presents a sequence that is at the threshold of being recognized as a nucleolar localization signal (Fig. 9B). Since both the potential nuclear and nucleolar localization sequences were detected in the C‐terminal part of A3C, we decided to create a C‐terminally truncated A3C mutant lacking the last 24 amino acids. This mutant was referred to as A3C[1‐166]. The expression of these constructs was tested by western blotting (Fig. 9C). The A3C[1‐166] mutant was expressed at considerably lower levels than its full‐length counterpart.
Using a fluorescent version of the nucleolar marker nucleophosmin (NPM‐1), we obtained additional evidence that full‐length A3C localizes to nucleoli in U2OS cells (Fig. 10A). In contrast, the A3C[1‐166] mutant was excluded from nucleoli but not from the nucleus (Fig. 10B). Similar results were obtained in HCT116 cells (Fig. 10C). These data indicate that the C‐terminal part of A3C is responsible for its nucleolar, but not nuclear, localization.
Fig. 10.
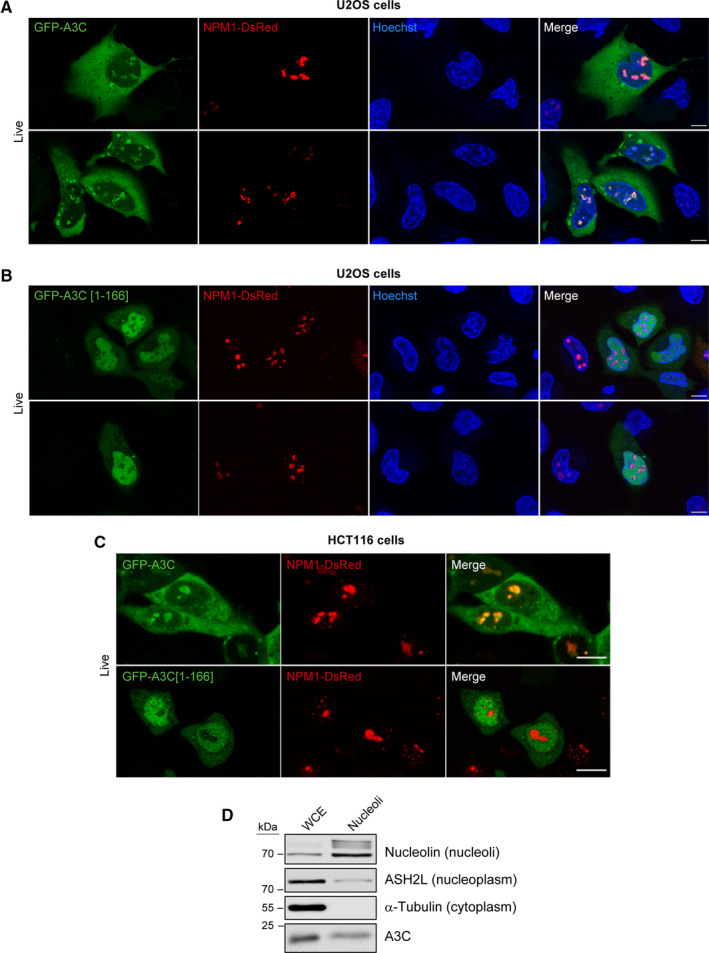
The C‐terminal end of A3C is required for its nucleolar location. (A, B) U2OS cells were co‐transfected with hNPM1‐DsRed.cmv and either GFP‐hAPOBEC3C‐V5 (panel A) or GFP‐hAPOBEC3C[1‐166]‐V5 (panel B). The cells were analyzed 24 h post‐transfection using a confocal microscope. Nuclei were stained with Hoechst 33342. (C) As panels A and B but using HCT116 cells instead of U2OS cells. (D) Western blot indicating the presence of A3C in whole cell extracts (WCE) and purified nucleoli from HCT116 cells. Scale bars in panels A to C, 10 µm.
Our data pointing toward A3C nucleolar localization were gathered in conditions of ectopic A3C expression. Ectopic expression of proteins to supra‐physiological levels could alter their subcellular localization. As we could not rely on immunocytochemistry to investigate the endogenous A3C location due to fixation affecting its distribution, we employed a nucleoli purification protocol. In our hands, HCT116 cells were much more amenable to nucleoli purification than U2OS cells. Fig. 10D shows that endogenous A3C is detected in the nucleoli of HCT116 extracts. Endogenous A3C therefore also localizes to nucleoli.
As A3C expression is induced in cells treated with DNA‐damaging agents and because some interacting partners of A3C are involved in DNA repair (PARP1, RECQL; see Fig. 4A), we decided to investigate the potential recruitment of A3C to DNA damage sites in live cells. We used a DNA double‐strand break reporter consisting of a truncated 53BP1 protein fused to a mApple fluorescent tag [42]. 53BP1 is an integral component of mammalian DNA damage signaling and repair pathways. It is swiftly recruited to DNA damage sites and plays a role in DNA repair pathway choice, promoting NHEJ [43]. We co‐transfected U2OS cells with GFP‐A3C and Apple‐53BP1trunc and imaged live cells using confocal microscopy (Fig. 11A,B). In untreated conditions, the Apple‐53BP1trunc signal was detected throughout the nucleus. GFP‐A3C localized to the cytoplasm and nucleolus, as described above. After 5 h of etoposide treatment, Apple‐53BP1trunc formed foci at DNA lesion sites. GFP‐A3C, but not the truncated GFP‐A3C[1‐166] version, was excluded from these areas (Fig. 11A,B). Similar results were obtained in HCT116 cells (Fig. 11C).
Fig. 11.
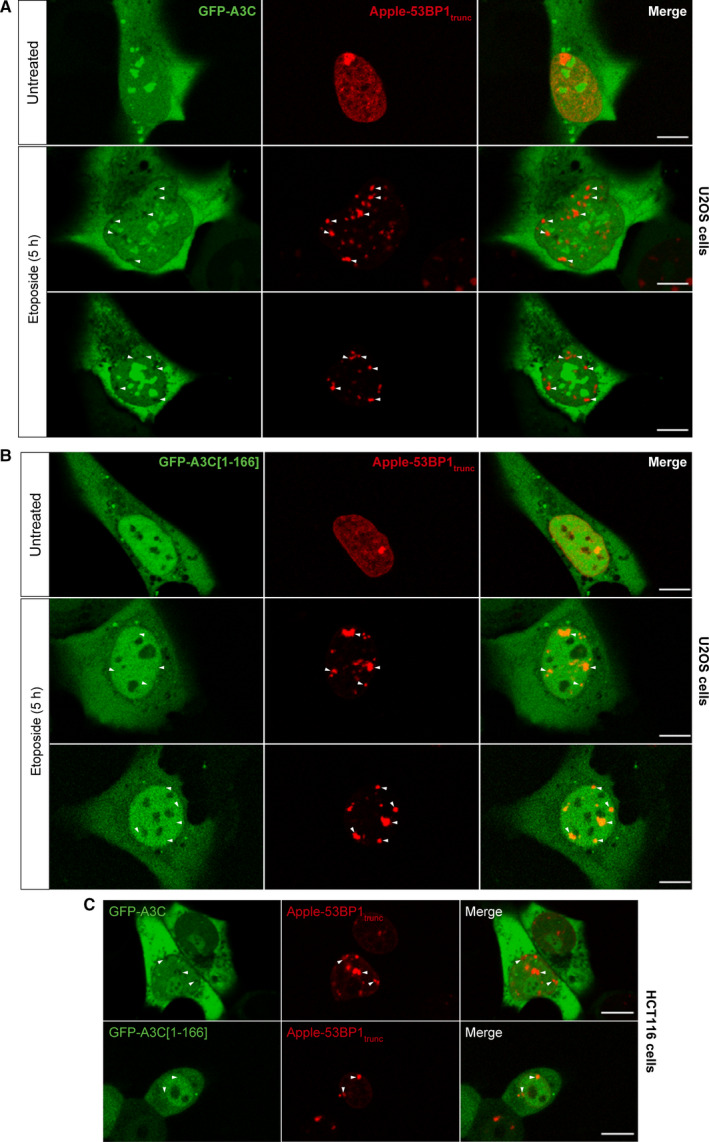
A3C is excluded from DNA damage sites. (A) Confocal images of live U2OS cells co‐transfected with mApple‐h53BP1[1220‐1709].lti and GFP‐hAPOBEC3C‐V5. Twenty‐four hours post‐transfection, the cells were left either untreated (top rows) or treated with 1 µm etoposide for 5 h (second and third rows). White arrowheads indicate 53BP1 foci formation upon accumulation at DNA double‐strand breaks. (B) Same procedure as in panel A, except that the cells were transfected with GFP‐hAPOBEC3C[1‐166]‐V5. Scale bars, 10 µm. (C) As panels A and B but using HCT116 cells instead of U2OS cells. The micrographs represent live cells analyzed by confocal microscopy. Scale bars: 10 µm.
Discussion
As cancer is trending to become the prevalent cause of death worldwide [1], and one of the largest classes of anticancer drugs is represented by DNA‐damaging agents [4], understanding how cancer cells react to chemotherapy drugs represents valuable information. The present study highlights the fact that upon exposure to genotoxic stress, osteosarcoma and colorectal cancer cells increase the expression of the A3C protein, which localizes mostly to the cytoplasm, and accumulates in the nucleoli. This expression is controlled by the p53 master transcription factor as it is the case for other APOBEC3 family members [25]. Additionally we show that phosphorylation of p53 at serine 15 is not needed for p53 to induce A3C expression, despite the fact that this modification was proposed to be crucial for p53 transcriptional activity [44].
Conflicting data have been reported concerning the subcellular location of A3C. In fixed HeLa and LLC‐Mk2 cells, A3C was not found in nucleoli [37, 45]. An in‐depth proteomic analysis of nucleolar components in HeLa cells did not detect A3C as a resident nucleolar protein [34]. In contrast, He et al. have reported that A3C is localized exclusively in nucleolin‐positive structures in the nucleus of fixed HeLa cells [46]. We show here that in live U2OS and HCT116 cells the highest densities of A3C are found within nucleoli. A3C is also clearly present in the cytoplasm but almost not detected in non‐nucleolar nuclear regions. Fixation completely disrupts this pattern as we saw that A3C in fixed U2OS cells is excluded from nucleoli and redistributes to non‐nucleolar nuclear regions. The cytoplasmic location appears unaffected by fixation. Our work therefore indicates that the conflicting data concerning A3C location in cells likely result from the methods used to visualize A3C in cells, fixation generally inducing artefactual A3C subcellular redistribution.
We provide evidence that the C‐terminal part of A3C is responsible for its nucleolar localization. Indeed, an A3C construct lacking the last 24 amino acids was no longer found in nucleoli but accumulated mostly in the remaining parts of the nucleus. A different member of the APOBEC3 family, A3H, has also been shown to accumulate in nucleoli [47]. Nucleoli are membrane‐less nuclear structures and represent the main site of ribosomal RNA synthesis and also ribosome assembly. Additionally, this organelle has been reported to contribute to DNA repair [48, 49], telomere maintenance [50, 51] and various other cellular processes [52]. There is also substantial evidence that viral pathogens, including influenza [53] and coronaviruses [54], use the nucleolar components for the assembly of ribo‐nucleo‐protein (RNP) particles. Other viruses use nucleoli as the main site of replication [55]. Interestingly, A3C has been shown to restrict the replication of human coronaviruses [45], which could explain its localization in this cellular compartment.
To our knowledge, this present work is the first to provide the full interactome of A3C. These mass spectrometry data indicate that A3C interacts mostly with nucleolar and RNA‐binding proteins. Some interaction between A3C and the LINE1 retrotransposon‐encoded ORFp1 protein was detected, consistent with a previous documentation of this interaction [56]. We did not find evidence of an interaction between A3C and TRIB3, a known interacting partner of endogenous A3A [57]. On the other hand, A3C bound to DHX9, a partner and modulator of A3B antiviral activity [58]. Although A3B was shown to associate with cyclin‐dependent kinase 4 (CDK4) and to modulate the nuclear import of cyclin D1 [59], which could be relevant for our observation of a decreased proliferation upon A3C depletion, we did not detect interaction between A3C and CDK4. We did however, detect some interaction with CDK9 and its partner cyclin T1.
The A3C interactome also revealed that A3C interacts with proteins that are recruited to DNA breaks such as PARP1 and RECQL. As a result, we checked if A3C itself is localized to DNA damage sites and found that it is actually excluded from these areas. Moreover, we observed that the C‐terminally truncated mutant of A3C is not excluded from regions of DNA damage. We did not observe any toxicity while expressing the truncated A3C mutant, indicating that, even though it is not excluded from damage sites, it does not interfere with repair of endogenous damage or with DNA replication. However, the mutant was expressed at considerably lower levels than the full‐length A3C, indicating a potential issue with protein stability.
Interestingly, PARP1 has been shown to accumulate in the nucleolus under normal conditions and to relocate to other nuclear regions in response to genotoxic stress exposure [60, 61]. Future studies are required to determine in which cellular compartment the interactions between A3C and PARP1 or RECQL occur. It is currently unknown why the expression of most A3 increases upon DNA damage. Due to the fact that these proteins are over‐expressed at such long times postgenotoxin exposure (48 h), we hypothesize that these are secondary effects of DNA damage, with potential implications in viral infection, where A3 proteins exert their main functions.
To our knowledge, we provide the first report of an exclusion of an APOBEC3 family member from areas of active DNA repair. From an evolutionary standpoint, it makes sense that mammalian cells display active exclusion of mutator proteins, such as cytidine deaminases, form areas of DNA damage. Considering that substrates of A3 proteins are ssDNA and that stretches of ssDNA are intermediary molecules in both ssDNA repair via nucleotide excision repair [62] and double‐strand break repair via HR [63], A3C exclusion from damage sites presumably occurs to protect the host cell genome integrity.
Our knockout and knockdown approaches indicate that A3C does not play a major role in DNA repair, in contrast to what was shown for A3G [18, 19]. However, we show that A3C depletion leads to a slight impairment in cell proliferation. Acute silencing of A3C using siRNAs decreases the percentage of cells in S‐phase, while permanent A3C depletion using CRISPR‐mediated knockout leads to a slower rate of DNA synthesis. The different manners by which A3C knockdown and A3C knockout affect cell proliferation may be due to adaptation and compensation effects [64, 65] that had time to take place in cells lacking A3C but not in cells acutely depleted in A3C. Interestingly, knockdown of two different interacting partners of A3C, DDX5 and DDX17, also induces a very similar slight reduction in cell proliferation [66]. The exact mechanism behind the observed reduction in proliferation remains unknown.
Most studies focusing on APOBEC3 family of proteins reported that their substrate is ssDNA [10, 67]. Substrates for two members of this family have been recently extended to RNA though. A3A is implicated in RNA editing in human monocytes in response to hypoxia and interferons [68]. A3G overexpression leads to RNA editing of around 600 genes in HEK 293T cells and these genes are distinct from those edited by A3A [69]. As the nucleolus is a compartment densely populated by RNA and RNA‐binding proteins, future studies investigating the transcriptome of A3C‐expressing cells may uncover if this protein can also edit RNA and whether this is required for its antiviral activity or efficient progression through the cell cycle.
In conclusion, the present study provides basic information regarding the location, expression dynamics, and interaction partners of A3C in osteosarcoma cells and colorectal cancer cells. These data were acquired under normal conditions and following exposure to genotoxins. We provide evidence that A3C is a bona fide nucleolar protein, and we show that the C‐terminal sequence of A3C is required for its nucleolar localization. We also demonstrate that A3C is expendable for double‐stranded DNA repair. Moreover, we show that this protein is excluded from areas of active DNA repair, and this exclusion is also dependent on its C‐terminal sequence. This information may provide the ground for new avenues of research concerning this protein, or other members of the A3 family. Future studies will determine if other members of this family are also excluded from areas of damaged DNA, consolidating this previously undescribed mechanism of mammalian genome safeguarding.
Materials and methods
Cell culture
U2OS (HTB‐96) and HCT116 (CCL‐247) cells were purchased from ATCC (Manassas, VA, USA), and were grown in Dulbecco's modified Eagle's medium (DMEM; Thermo Fisher Scientific, Basel, Switzerland, 10566032) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, 26140079). The cells were grown in a humidified atmosphere with 5% CO2.
Western blotting
Cells were trypsinized and washed once in ice‐cold PBS. The cells were then lysed using RIPA buffer (1% Triton X‐100, 1% sodium deoxycholate, 0.1% SDS, 20 mm Tris/Cl pH 8.0, 1 mm EDTA, 150 mm NaCl). The lysis buffer was supplemented with complete protease inhibitor (Roche, Basale, Switzerland, 11836145001) and with PhosStop (Roche, 04906837001). Cell lysis was performed for 30 min on ice with light vortexing every 10 min. The samples were sonicated using a Vibra‐Cell 75186 instrument at 40% amplitude, for 5 s and then centrifuged at 15 000 g for 10 min at 4 °C. The supernatant was used for determining the total protein concentration using a BCA kit (Pierce, Witec AG, Sursee, Switzerland, 23225). An equal volume of 2X Laemmli buffer [70] was added to the samples, before boiling them (for 10 min at 95 °C). Thirty micrograms of protein was loaded on SDS/PAGE gels. Transfer was performed on nitrocellulose membranes. Nonfat milk 5% dissolved in TBST (20 mm Tris, 150 mm NaCl, 0.1% Tween‐20) was used for the blocking steps and for antibody incubations. Antibodies were diluted according to the datasheet instructions. For the anti‐APOBEC3C antibody, we observed better performance when using bovine serum albumin (BSA) as blocking and antibody incubation. All antibody incubations were performed overnight at 4 °C with gentle shaking, except beta‐actin, which was incubated for one hour at room temperature. Membranes were washed thrice 20 min with TBST and then incubated with secondary antibodies for one hour at room temperature in the dark. The secondary antibodies were purchased from Invitrogen (A21109, A21057, or A32735; Thermo Fisher Scientific, Waltham, MA, USA). The membranes were washed thrice 20 min with TBST and then analyzed using a LI‐COR Odyssey machine (Bad Homburg, Germany).
Quantitation of the western blot bands was performed using ImageJ. Background values were subtracted in each case. The background values were obtained by quantitating an identical area just above or below the band of interest.
Immunocytochemistry
U2OS cells were plated on 10‐mm glass coverslips placed in 6‐well plates at a 1∙105 cells per well density. The cells were treated and/or transfected as indicated in the figures. At the time of analysis, the cells were washed once with cold PBS and directly fixed for 15 min at room temperature in PFA (2% in PBS). Following PFA fixation, cells were PBS washed and permeabilized in 0.2% Triton X‐100 in PBS, for 10 min at room temperature. The samples were incubated with blocking buffer (DMEM media supplemented with 10% fetal bovine serum). The primary antibody (anti‐GFP) was diluted in blocking buffer and incubated with the samples for one hour at room temperature. The coverslips were washed twice with PBS and the appropriate secondary antibody, diluted in blocking buffer, was added for 1 h at room temperature in the dark. The samples were then incubated in PBS supplemented with 1 µg·mL−1 Hoechst 33342 for 10 min. Three PBS washes were performed and the coverslips were mounted on glass slides using VectaShield (Vector laboratories H‐1000‐10) and sealed with transparent nail polish. The slides were kept in the dark overnight at 4 °C before being analyzed.
Live cell imaging
U2OS and HCT116 cells were seeded on glass bottom 35 mm culture dishes at 1∙105 cells per well and left to adhere overnight. The next day, cells were co‐transfected with GFP‐hAPOBEC3C‐V5 or GFP‐hAPOBEC3C[1‐166]‐V5 (1 μg·plate−1) and, either mApple‐h53BP1[1220‐1709].lti or hNPM1‐DsRed.cmv (1 µg). Ten minutes before the irradiation was performed, Hoechst 33342 (Invitrogen, H3570) was added to the cells (1 µg·mL−1 final concentration), in order to visualize the nuclei. The cells were analyzed using a LSM710 confocal microscope equipped with chamber that maintained the cells in an atmosphere containing 5% CO2, at 37 °C.
Immunoprecipitation
U2OS cells were seeded in 4 individual 10‐cm plates at a 1∙106 cells·plate−1 density and left to adhere overnight. The next day two plates were transfected with 10 μg empty pcDNA3 plasmid and the remaining plates with the same amount of hApobec3c‐V5.dn3. Sixteen hours post‐transfection, the media was changed and one set of plates was treated with 25 μm etoposide and the other set was left untreated. Forty‐eight hours later, the cells were harvested by trypsinization and washed in ice‐cold PBS. Lysis was performed in 1.5 mL of IP‐lysis buffer (50 mm HEPES, 150 mm NaCl, 1 mm EDTA, 1% Triton X‐100, 10% glycerol, 10 μm MgCl2, supplemented with c0mplete protease inhibitor [1 pill per 50 mL]), and with PhosStop (1 pill per 10 mL) for 30 min on ice. The samples were centrifuged at 15 000 g for 10 min at 4 °C, using an Eppendorf 5415R centrifuge equipped with a F45‐24‐11 rotor. The supernatants were moved to new tubes and the protein concentration was determined using a BCA kit (Pierce 23225). A tenth of the lysates were saved as ‘input’ samples. Lysate containing 1 mg of protein were moved to new tubes and 1.5 μg anti‐V5 antibody (Thermo Fisher Scientific, R960) was added to each tube. The tubes were incubated at 4 °C overnight with gently rotation. The next day, 10 µL of prewashed protein G Dynabeads (Thermo Fisher Scientific, 10004D) was added to each tube. The samples were incubated 1 h at 4 °C with slow rotation. The beads were gently washed 5 times with cold IP‐lysis buffer. After the last wash, the beads were resuspended in 40‐µL Laemmli buffer [70] and incubated for 10 min at 95 °C. One 40th of the immunoprecipitated samples, along with the input samples were run on a SDS/PAGE as a quality control. The rest of the immunoprecipitated samples were analyzed by mass spectrometry at the University of Lausanne Protein Analysis Facility (https://www.unil.ch/paf/en/home.html). Three independent experiments were performed.
Mass spectrometry
Protein samples were loaded on a 12% mini polyacrylamide gel and migrated about 2 cm After Coomassie staining, gel lanes between 15 and 300 kDa were excised into 5 pieces, and digested with sequencing‐grade trypsin (Promega, Dübendorf, Switzerland, V5111) as described by Shevchenko et al. [71]. Extracted tryptic peptides were dried and resuspended in 0.05% trifluoroacetic acid, 2% (v/v) acetonitrile for mass spectrometry analyses. Tryptic peptide mixtures were injected on a Dionex RSLC 3000 nanoHPLC system (Dionex, Sunnyvale, CA, USA) interfaced via an EasySpray source to a high‐resolution mass spectrometer QExactive Plus (Thermo Fisher). Peptides were loaded onto a trapping microcolumn Acclaim PepMap100 C18 (20 mm x 100 μm ID, 5 μm, Dionex) before separation on an Easy Spray C18 PepMap column (50 cm x 75 µm ID, 2 µm, 100 Å, Dionex). A gradient from 4% to 76% acetonitrile in 0.1% formic acid was used for peptide separation of each fraction (total time: 65 min). Full MS survey scans were performed at 70 000 resolution. In data‐dependent acquisition controlled by Xcalibur 4.0 software (Thermo Fisher), the 10 most intense multiply charged precursor ions detected in the full MS survey scan were selected for higher energy collision‐induced dissociation (HCD, normalized collision energy NCE = 27%) and analysis in the orbitrap at 17 500 resolution. The window for precursor isolation was of 1.5 m/z units around the precursor and selected fragments were excluded for 60 s from further analysis. MS/MS spectra of raw files were converted by ProteWizard 3.0 (http://proteowizard.sourceforge.net) to mgf format (=text file) and data were analyzed using Mascot 2.6 (Matrix Science, London, UK) as search engine. Mascot was set up to search the Homo sapiens reference proteome database (https://www.uniprot.org/, October 2017 version: 71 785 sequences), and a custom database including usual contaminants (keratins, digestion enzymes, etc.). Trypsin (cleavage at K,R) was used as the enzyme definition, allowing 2 missed cleavages. Mascot was searched with a parent ion tolerance of 10 ppm and a fragment ion mass tolerance of 0.02 Da. Carbamidomethylation of cysteine was specified in Mascot as a fixed modification. N‐terminal acetylation of protein and oxidation of methionine were specified as variable modifications. Scaffold software (version 4.8, Proteome Software Inc., Portland, OR, USA) was used to validate MS/MS based peptide and protein identifications, and to perform dataset alignment. Peptide identifications established at lower than 90.0% probability by the Scaffold Local FDR algorithm were filtered out. Protein identifications were accepted if they could be established at > 95.0% probability and contained at least 2 identified peptides. Protein probabilities were assigned by the Protein Prophet algorithm [72]. Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony. Proteins sharing significant peptide evidence were grouped into clusters. Approximately 1500 proteins were identified, and the MS/MS spectra detected as unique for each protein (exclusive spectral counts) were used for statistical and GO analyses.
Statistical and GO analysis of mass spectrometry results
The exclusive spectral counts for each protein detected were used for statistical analysis. Individual t tests were performed between the control pull‐down and the A3C‐V5 pull‐down in untreated conditions (UP vs UA), and in etoposide‐treated cells (EP vs EA). The false discovery rate (FDR) was calculated using the Benjamini‐Hochberg method corrected for multiple comparisons. Only proteins with a FDR < 0.05 (n = 190) were included in the GO analysis. This analysis was performed with the PANTHER software [73] using the overrepresentation test with Bonferroni correction for multiple testing.
siRNA transfection
U2OS cells were placed in 6‐well plates at a density of 8∙104 cells·well−1, in 1 mL of media and immediately transfected using Lipofectamine RNAimax (Invitrogen, 13778‐075), according to the manufacturer's instructions. Briefly, for one well, 200 μL Opti‐MEM (Thermo Fisher Scientific, 11058021) was added in a sterile tube, and then, the siRNA oligos were added in the same tube and mixed. Next, 2 μL of Lipofectamine RNAi MAX was added and the contents were gently mixed. The tubes were incubated 15 min at room temperature. The mixture was added dropwise in the wells containing the cells. The next day, the media was changed and the transfection was repeated one more time. The following day, the media was changed and drugs were added for 48 h before harvesting the samples. Final siRNA concentration in the media was 20 nm. The following pools of siRNAs were purchased from Dharmacon: p53 (L‐003329‐00‐0005), nontargeting control pool (D‐001810‐10, used in Fig. 1D). The siRNA pool targeting A3C contained 30 siRNAs and was purchased from siTOOLS biotech (27350), and was used in parallel with a pool of 30 scrambled siRNA from the same company (si‐C005‐NEG001).
cDNA transfection
Cells were plated in 6‐well plates, at a density of 8∙104 cells·well−1 in 2 mL of media and left to adhere overnight. The next day, the media was aspirated, and 1 mL of fresh media was added. The cells were transfected using Lipofectamine 2000 according to the manufacturer's instructions. Briefly, for one well, 100 μL Opti‐MEM were mixed with 2 μg plasmid DNA in a sterile tube. In a different tube, 100‐μL Opti‐MEM were mixed with 2 μL of Lipofectamine 2000. The contents of the two tubes were gently mixed and incubated for 30 min at room temperature. The mixture was added dropwise in the well, and cells were analyzed 24–48 h later unless specified otherwise.
5‐bromo‐2′‐deoxyuridine (BrdU) incorporation assay
U2OS or HCT116 cells were plated and transfected as indicated. Twenty five minutes before the cells were harvested, 10 μm BrdU was added directly into culture media and the plates were placed back in the incubator. Afterward, the cells were harvested by trypsinization, washed once with cold PBS, and resuspended in 70% ethanol and stored in the dark at 4 °C. After 3 h, the cells were washed once with PBS and then resuspended in 500 μL 0.1 M HCl, 0.5% Triton X‐100 in PBS. After 10 min, the cells were pelleted by centrifugation (270 g for 5 min) and resuspended in cold water. Samples were then boiled for 10 min and then placed on ice for 10 min. One ml of 0.5% Triton X‐100 (in PBS) was added, and samples were spun down 270 g for 5 min. The supernatant was discarded and the pellets were resuspended in 50 μL antibody staining solution (PBS containing 1% BSA and 0.5% Tween) in which the primary anti‐BrdU antibody was added. Samples were incubated for 1 h in the dark. Afterward, 1.45 mL of PBS was added to the tubes, which were inverted 5 times. Samples were centrifuged and resuspended in 50 μL antibody staining solution containing the secondary antibody, and incubated for 30 min in the dark. Please note that in order to get a better resolution of BrdU‐positive cells, both the primary and secondary antibodies were FITC‐labeled. Afterward, 1.45 mL PBS was added and the tubes were inverted 5 times, and then centrifuged (270 g, 5 min). The pellets were resuspended in PBS containing 0.4 mg·mL−1 PI, and 0.3 mg·mL−1 RNase A. Tubes were incubated at 37 °C for 30 min and then analyzed by flow cytometry (FC‐500 Beckman Coulter machine; Beckman Coulter Eurocenter SA, Nyon, VD, Switzerland).
DNA repair reporter assays
The U2OS cells carrying the homologous recombination (DRGFP) and NHEJ (EJ5‐GFP) reporters were a generous gift from Professor J. Stark (City of Hope, Beckman Research Institute of the City of Hope, CA, 91010, USA). These cells were plated in 12‐well plates at a density of 4∙104 cells·well−1, and transfected immediately with the indicated siRNAs. The next day, the media was changed and the siRNA transfection was repeated. Twenty‐four hours later the media was changed and the cells were transduced with a lentivirus carrying the ISce‐I endonuclease (multiplicity of infection = 1). The next day, the media was aspirated and fresh media was added. After another 24 h, the percentage of GFP positive cells was investigated by flow cytometry using a Beckman Coulter CytoFLEX S machine.
Generation and validation of CRISPR‐Cas9‐mediated knockout clones
The generation of CRISPR/Cas9 APOBEC3C knockout cells was performed as previously described [74]. Briefly, U2OS cells were plated in 6‐well plates at an 8∙104 cells·well−1 density and cultured overnight for them to adhere. The next day, the cells were transfected with 2 μg of lenticrispr‐V2 plasmid carrying the Cas9 protein and the sgRNA targeting A3C (5′‐CTTCCAGTATCCATGTTACC‐3′). Although this is a lentiviral vector, it can also be used for transient transfection. The next day, the media was aspirated and fresh media was added that was supplemented with 2 μg·mL−1 of puromycin. The puromycin selection was performed to remove non transfected cells. After 3 days of selection, the cells were re‐plated in 96‐well plates at a 0.3 cell·well−1 density. Three weeks later, the clones were amplified and screened by western blotting for the absence of APOBEC3C. The screening was performed after treating the clones with 5 μm etoposide for 48 h to increase the expression of A3C if present. Two clones that presented no detectable levels of A3C were chosen for validation. Genomic DNA was extracted from wild‐type and A3C‐KO clones using the Blood & Cell Culture DNA Midi Kit (Qiagen, 13343). Genomic DNA was amplified with the following primers: FWD 5′ tcgtcggcagcgtcagatgtgtataagagacagCTGGTACACATCTTGGAGCC 3′ (#1506); REV 5′ gtctcgtgggctcggagatgtgtataagagacagCATTGGAGAACCATCTGCC 3′ (#1507). The lowercase letters indicate Illumina‐specific adapters for sequencing. The uppercase letters indicate APOBEC3C specific sequences. The amplicons were sent for next‐generation sequencing at the University of Lausanne Genomic Technologies (GTF) facility. The resulting fastq files were analyzed using the Cas‐Analyzer software [75].
Presto‐blue assay
U2OS cells were plated at a 4∙104 cells·well−1 density in 12‐well plates (1 mL media·well−1) and treated as indicated. At the time of analysis, 100 μL of Presto‐Blue viability reagent (Thermo Fisher Scientific, A13262) was added to each well and the plates were placed back in a 37 °C, 5% CO2 incubator for 30 min. The fluorescence was then read using a Cytation‐3 plate reader (excitation/emission of 560/590 nm). The value of the blank samples (media with no cells but with Presto‐Blue) was subtracted from all values, and the remaining absorbance values were plotted in the graphs.
Quantitative real‐time PCR
U2OS cells were plated and transfected with siRNAs as mentioned above. Total RNA was extracted using the High Pure RNA isolation kit (Roche, 11828665001) according to the manufacturer's instructions. One μg of RNA was reverse‐transcribed into cDNA using the Transcriptor High Fidelity cDNA Synthesis Kit (Roche, 5081955001). Fifty ng cDNA were used for qPCRs in combination with LightCycler 480 SYBR Green I Master mix (Roche 04707516001), using a ViiA 7 qPCR instrument. The relative expression of each gene was calculated as follows: 2−CT. Normalization was performed by dividing the obtained values by the value of GAPDH expression, and the resulting data were used to make the graph in Fig. 6A. Three independent experiments were performed. Primer sequences used for amplification are provided in Table S2.
Nucleoli isolation
For purification of nucleoli, we followed a protocol developed by from Li et al. [76]. Briefly, HCT116 cells were grown in a 15 cm culture plate until 95% confluent. The media was discarded and the cells were washed 3 times with ice‐cold solution I (0.5 M sucrose, 3 mm MgCl2, supplemented with EDTA‐free protease inhibitor). After the last wash, the cells were scraped and the total volume was adjusted to 3 mL by adding solution I. The cell suspension was kept on ice and sonicated (Vibra‐Cell 75186, 50% amplitude) for 10 s pulses, with 10 s of break in between. For this cell type, 9 cycles of sonication were needed until all cells were disrupted and nucleoli were released. The cell suspension was overlaid on top of 3 mL of solution II (1 M sucrose, 3 mm MgCl2, supplemented with EDTA‐free protease inhibitor) and the tubes were centrifuged for 10 min at 1800 g, at 4 °C. The supernatant was discarded and the pellet was resuspended in 3 mL solution I, which was again overlaid on 3 mL solution II. The centrifugation was repeated as before. The supernatant was discarded as much as possible and the pellet containing the nucleoli was resuspended in 100 μL of 1X Laemmli buffer [70] and boiled for 10 min. This sample was used for western blotting as nucleolar fraction.
Chemicals
The following chemicals were purchased from Sigma‐Aldrich (Thermo Fisher): etoposide (E1383), and cisplatin (P4394), bleomycin (B8416), ATM inhibitor KU‐60019 (SML1416‐5MG), and Hydroxyurea (H8627), PIPES (P6757). Puromycin was acquired from Thermo Fisher Scientific (A1113802). Interferon α (ab48750) and interferon β (ab71475) were purchased form Abcam (Lucerna‐chem, Luzern, Switzerland); interferon γ from Roche (11040596001). Hoechst 33342 was purchased from Invitrogen (H3570). The c0mplete protease inhibitor (11836145001) and PhosStop phosphatase inhibitor (04906837001) were purchased from Roche. Esp‐3I restriction enzyme was acquired from Thermo Fisher Scientific (ER0451). Rapid ligation and dephosphorylation kit were purchased from Roche (ER0451).
Antibodies
The APOBEC3C antibody was purchased from GeneTex (Lucerna‐chem) (GTX102164 lot 40968). The one recognizing phospho‐H2A.X at serine 139 was from Millipore (Merck, Darmstadt, Germany) (5636, lot 2554898). The anti‐phospho‐ATM Ser1981 (81292, lotGR217573‐6) and anti‐nucleolin (22758, lotGR3269917‐1) antibodies were acquired from Abcam. The antibodies against phospho‐p53(ser15) (9284, lot 6),total p53 (9282, lot2), and beta‐actin (4970, lot 14) were purchased from Cell Signaling Technology (Danvers, MA, USA). The anti‐ASH2L antibody was from Bethyl (LuBioScience, Zürich, Switzerland; A300‐489 lot2). The anti‐V5 antibody used for immunoprecipitation was acquired from Thermo Fisher Scientific (R960, lot 1875853). The phospho‐STAT1 antibody was purchased from Santa Cruz (Santa Cruz, CA, USA; sc7988C, lot H0612). Anti‐GFP antibody was purchased from Roche (1014460001, lot 11063100).
Plasmids
The pcDNA3 plasmid (#1) is a mammalian expression vector.
The hAPOBEC3C‐V5.dn3 plasmid (#981) encodes a C‐terminally V5‐tagged human A3C version in the pcDNA3 backbone. It was made as follows: the human APOBEC3C‐V5 cDNA (NM_014508.3) with a C‐terminal V5‐tag was synthetized by Genscript with EcoRI (5′) and XhoI (3′) restriction enzyme sites flanking it. This fragment was ligated into pcDNA3 that was digested with the same enzymes.
The LentiCRISPR V2 (#868) lentiviral plasmid encodes a FLAG‐tagged nuclear location sequence‐bearing Cas9 protein. It also contains a site for the insertion of a single guide RNA (sgRNA) of interest (see next plasmid). It was a gift from Dr. Feng Zhang (Addgene plasmid #52961; http://n2t.net/addgene:52961; RRID:Addgene_52961) [77].
The sg(3)‐hAPOBEC3C.lti plasmid (#1058) is a lentiviral vector coding for Cas9 and for a single guide RNA targeting the A3C gene. It was produced following established procedures [74, 77]. Briefly, the following two oligonucleotides were annealed and subcloned into LentiCRISPR V2 (#868) opened with the Esp3I restriction enzyme and dephosphorylated. Oligonucleotides (forward and reverse, respectively): 5′‐ caccgCTTCCAGTATCCATGTTACC‐3′ and 5′‐aaacGGTAACATGGATACTGGAAGc‐3′. The upper case letters correspond to the sgRNA targeting exon 3 of A3C. The lower case letters correspond to the remaining overhangs on which forward and reverse oligonucleotides anneal.
The GFP‐hAPOBEC3C‐V5 plasmid (#1060) encodes a fusion protein between GFP and the human A3C tagged at the C‐terminus with the V5 sequence. It was created by subcloning the EcoRI/ApaI insert of hAPOBEC3C‐V5.dn3 (#981) into the pEGFP‐C2 vector (#111) opened with the same enzymes.
The hAPOBEC3C[1‐166]‐V5.dn3 plasmid (#1059) encodes the human A3C protein lacking the last 24 amino acids and tagged at the C‐terminus with a V5 sequence. It was generated using the Q5® site‐directed mutagenesis kit with hAPOBEC3C‐V5.dn3 plasmid (#981) as template and the following two oligonucleotides: 5′‐GGCAAGCCCATCCCTAAC‐3′ and 5′‐CTCATTATCATTGTACACAAAGTTTTCC‐3′.
The GFP‐hAPOBEC3C[1‐166]‐V5 plasmid (#1061) was created by subcloning the EcoRI/ApaI insert of plasmid hAPOBEC3C[1‐166].dn3 (#1059) into pEGFP‐C2 (#111) opened with the same enzymes.
The hNPM1‐DsRed.cmv plasmid (#1062) codes for a fusion protein between human nucleophosmin 1 and DsRed. It is a gift from Mary Dasso [78] (Addgene plasmid # 34553; pCMV‐NPM1‐DsRed).
The mApple‐h53BP1[1220‐1709].lti plasmid (#1063) encodes a fusion protein between the red fluorescent mApple protein from Discosoma sp. and amino acids 1220‐1709 of the human 53BP1 protein. This construct accumulates at double‐strand DNA breaks. This plasmid is a gift from Ralph Weissleder [42] (Addgene plasmid # 69531; pLVX‐Apple‐53BP1trunc).
The hTP53.lti (#384) and hTP53(S15A).lti (#511) lentiviral vectors code for wild‐type human p53 and the corresponding S15A mutant, respectively [79].
The HA‐NLS‐scSceI‐IRES‐BFP.pCVL (#1053) is used for the expression of the ISce‐I endonuclease into cells. It was a gift from Andrew Scharenberg (Addgene plasmid #45574; pCVL.SFFV. Kozak.HA.NLS. SceOpt.IRES.BFP; http://n2t.net/addgene:45574; RRID:Addgene_45574) [80].
Conflict of interest
The authors declare no conflict of interest.
Author contributions
DC, MCCR, and CW involved in conception and design of study. DC and GD performed acquisition of data. DC and CW involved in analysis and/or interpretation of data, and drafted the manuscript. DC, GD, MCCR, and CW revised the manuscript and approved the submitted version.
Peer Review
The peer review history for this article is available at https://publons.com/publon/10.1111/febs.16202.
Supporting information
Table S1. A3C‐interacting proteins The table lists the proteins, identified by mass spectrometry, that were pulled down from cells transfected with either pcDNA3 or hAPOBEC3C‐V5.dn3 that were treated or not with etoposide for 48 hours.
Table S2. Primers used for quantitative real‐time PCR.
Acknowledgements
We are thankful to the Cellular Imaging Facility and Genomic Technologies Facility at the University of Lausanne for the resources provided and their technical help. This work was supported by institutional funding (Faculty of Biology and Medicine of the University of Lausanne) to CW. Open Access Funding provided by Universite de Lausanne.
Data accessibility
The data described in this work are contained within the manuscript except for the raw mass spectrometry data that are available at the online proteomic repository consortium ProteomeXchange [81] (http://www.proteomexchange.org) with identifier PXD020105 and https://doi.org/10.6019/PXD020105.
References
- 1. Dagenais GR, Leong DP, Rangarajan S, Lanas F, Lopez‐Jaramillo P, Gupta R, Diaz R, Avezum A, Oliveira GBF, Wielgosz A et al. Variations in common diseases, hospital admissions, and deaths in middle‐aged adults in 21 countries from five continents (PURE): a prospective cohort study. Lancet 395, 785–794. [DOI] [PubMed] [Google Scholar]
- 2. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA & Jemal A (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68, 394–424. [DOI] [PubMed] [Google Scholar]
- 3. Cheung‐Ong K, Giaever G & Nislow C (2013) DNA‐damaging agents in cancer chemotherapy: serendipity and chemical biology. Cell Chem Biol 20, 648–659. [DOI] [PubMed] [Google Scholar]
- 4. Woods D & Turchi JJ (2013) Chemotherapy induced DNA damage response: convergence of drugs and pathways. Cancer Biol Ther 14, 379–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Khanna A (2015) DNA damage in cancer therapeutics: a boon or a curse? Can Res 75, 2133. [DOI] [PubMed] [Google Scholar]
- 6. Jarmuz A, Chester A, Bayliss J, Gisbourne J, Dunham I, Scott J & Navaratnam N (2002) An anthropoid‐specific locus of orphan C to U RNA‐editing enzymes on chromosome 22. Genomics 79, 285–296. [DOI] [PubMed] [Google Scholar]
- 7. Salter JD, Bennett RP & Smith HC (2016) The APOBEC protein family: united by structure, divergent in function. Trends Biochem Sci 41, 578–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chiu Y‐L & Greene WC (2008) The APOBEC3 cytidine deaminases: an innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu Rev Immunol 26, 317–353. [DOI] [PubMed] [Google Scholar]
- 9. Refsland EW & Harris RS (2013) The APOBEC3 family of retroelement restriction factors. Curr Top Microbiol Immunol 371, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen J & MacCarthy T (2017) The preferred nucleotide contexts of the AID/APOBEC cytidine deaminases have differential effects when mutating retrotransposon and virus sequences compared to host genes. PLoS Comput Biol 13, e1005471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nik‐Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, Jones D, Hinton J, Marshall J, Stebbings LA et al. (2012) Mutational processes molding the genomes of 21 breast cancers. Cell 149, 979–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Burns MB, Lackey L, Carpenter MA, Rathore A, Land AM, Leonard B, Refsland EW, Kotandeniya D, Tretyakova N, Nikas JB et al. (2013) APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 494, 366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, Kiezun A, Kryukov GV, Carter SL & Saksena G (2013) An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet 45, 970–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Buisson R, Langenbucher A, Bowen D, Kwan EE, Benes CH, Zou L & Lawrence MS (2019) Passenger hotspot mutations in cancer driven by APOBEC3A and mesoscale genomic features. Science 364, eaaw2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Taylor BJM, Nik‐Zainal S, Wu YL, Stebbings LA, Raine K, Campbell PJ, Rada C, Stratton MR & Neuberger MS (2013) DNA deaminases induce break‐associated mutation showers with implication of APOBEC3B and 3A in breast cancer kataegis. eLife 2, e00534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Landry S, Narvaiza I, Linfesty DC & Weitzman MD (2011) APOBEC3A can activate the DNA damage response and cause cell‐cycle arrest. EMBO Rep 12, 444–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Seplyarskiy VB, Soldatov RA, Popadin KY, Antonarakis SE, Bazykin GA & Nikolaev SI (2016) APOBEC‐induced mutations in human cancers are strongly enriched on the lagging DNA strand during replication. Genome Res 26, 174–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nowarski R, Wilner OI, Cheshin O, Shahar OD, Kenig E, Baraz L, Britan‐Rosich E, Nagler A, Harris RS, Goldberg M et al. (2012) APOBEC3G enhances lymphoma cell radioresistance by promoting cytidine deaminase‐dependent DNA repair. Blood 120, 366–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Prabhu P, Shandilya SMD, Britan‐Rosich E, Nagler A, Schiffer CA & Kotler M (2016) Inhibition of APOBEC3G activity impedes double‐stranded DNA repair. FEBS J 283, 112–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Anderson BD, Ikeda T, Moghadasi SA, Martin AS, Brown WL & Harris RS (2018) Natural APOBEC3C variants can elicit differential HIV‐1 restriction activity. Retrovirology 15, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wittkopp CJ, Adolph MB, Wu LI, Chelico L & Emerman M (2016) A single nucleotide polymorphism in human APOBEC3C enhances restriction of lentiviruses. PLoS Pathog 12, e1005865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nguyen DH, Gummuluru S & Hu J (2007) Deamination‐independent inhibition of hepatitis B virus reverse transcription by APOBEC3G. J Virol 81, 4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Horn AV, Klawitter S, Held U, Berger A, Vasudevan AAJ, Bock A, Hofmann H, Hanschmann K‐MO, Trösemeier J‐H, Flory E et al. (2014) Human LINE‐1 restriction by APOBEC3C is deaminase independent and mediated by an ORF1p interaction that affects LINE reverse transcriptase activity. Nucleic Acids Res 42, 396–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Periyasamy M, Singh AK, Gemma C, Kranjec C, Farzan R, Leach DA, Navaratnam N, Pálinkás HL, Vértessy BG, Fenton TR et al. (2017) p53 controls expression of the DNA deaminase APOBEC3B to limit its potential mutagenic activity in cancer cells. Nucleic Acids Res 45, 11056–11069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Menendez D, Nguyen T‐A, Snipe J & Resnick MA (2017) The cytidine deaminase APOBEC3 family is subject to transcriptional regulation by p53. Mol Cancer Res 15, 735–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Blackford AN & Jackson SP (2017) ATM, ATR, and DNA‐PK: the trinity at the Heart of the DNA damage response. Mol Cell 66, 801–817. [DOI] [PubMed] [Google Scholar]
- 27. Golding SE, Rosenberg E, Valerie N, Hussaini I, Frigerio M, Cockcroft XF, Chong WY, Hummersone M, Rigoreau L, Menear KA et al. (2009) Improved ATM kinase inhibitor KU‐60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol Cancer Ther 8, 2894–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rodova M, Jayini R, Singasani R, Chipps E & Islam MR (2013) CMV promoter is repressed by p53 and activated by JNK pathway. Plasmid 69, 223–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ngo TD, Partin AC & Nam Y (2019) RNA specificity and autoregulation of DDX17, a modulator of microRNA biogenesis. Cell Rep 29, 4024–4035.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ray Chaudhuri A & Nussenzweig A (2017) The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat Rev Mol Cell Biol 18, 610–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pierce AJ, Johnson RD, Thompson LH & Jasin M (1999) XRCC3 promotes homology‐directed repair of DNA damage in mammalian cells. Genes Dev 13, 2633–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gunn A & Stark JM (2012) I‐SceI‐based assays to examine distinct repair outcomes of mammalian chromosomal double strand breaks in DNA repair protocols (Bjergbæk L, ed), pp. 379–391. Humana Press, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- 33. Cerikan B, Shaheen R, Colo GP, Gläßer C, Hata S, Knobeloch K‐P, Alkuraya FS, Fässler R & Schiebel E (2016) Cell‐intrinsic adaptation arising from chronic ablation of a key rho GTPase regulator. Dev Cell 39, 28–43. [DOI] [PubMed] [Google Scholar]
- 34. Andersen JS, Lyon CE, Fox AH, Leung AKL, Lam YW, Steen H, Mann M & Lamond AI (2002) Directed proteomic analysis of the human nucleolus. Curr Biol 12, 1–11. [DOI] [PubMed] [Google Scholar]
- 35. Nicol SM, Causevic M, Prescott AR & Fuller‐Pace FV (2000) The nuclear DEAD box RNA helicase p68 interacts with the nucleolar protein fibrillarin and colocalizes specifically in nascent nucleoli during telophase. Exp Cell Res 257, 272–280. [DOI] [PubMed] [Google Scholar]
- 36. Zhou Y, Ma J, Bhusan Roy B, Wu JY‐Y, Pan Q, Rong L & Liang C (2008) The packaging of human immunodeficiency virus type 1 RNA is restricted by overexpression of an RNA helicase DHX30. Virology 372, 97–106. [DOI] [PubMed] [Google Scholar]
- 37. Lackey L, Law EK, Brown WL & Harris RS (2013) Subcellular localization of the APOBEC3 proteins during mitosis and implications for genomic DNA deamination. Cell Cycle 12, 762–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schnell U, Dijk F, Sjollema KA & Giepmans BNG (2012) Immunolabeling artifacts and the need for live‐cell imaging. Nat Methods 9, 152–158. [DOI] [PubMed] [Google Scholar]
- 39. Li M‐W, Zhou L & Lam H‐M (2015) Paraformaldehyde fixation may lead to misinterpretation of the subcellular localization of plant high mobility group box proteins. PLoS ONE 10, e0135033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kosugi S, Hasebe M, Tomita M & Yanagawa H (2009) Systematic identification of cell cycle‐dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc Natl Acad Sci USA 106, 10171–10176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Scott MS, Troshin PV & Barton GJ (2011) NoD: a nucleolar localization sequence detector for eukaryotic and viral proteins. BMC Bioinformatics 12, 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yang KS, Kohler RH, Landon M, Giedt R & Weissleder R (2015) Single cell resolution in vivo imaging of DNA damage following PARP inhibition. Sci Rep 5, 10129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mirza‐Aghazadeh‐Attari M, Mohammadzadeh A, Yousefi B, Mihanfar A, Karimian A & Majidinia M (2019) 53BP1: a key player of DNA damage response with critical functions in cancer. DNA Repair (Amst) 73, 110–119. [DOI] [PubMed] [Google Scholar]
- 44. Loughery J, Cox M, Smith LM & Meek DW (2014) Critical role for p53‐serine 15 phosphorylation in stimulating transactivation at p53‐responsive promoters. Nucleic Acids Res 42, 7666–7680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Milewska A, Kindler E, Vkovski P, Zeglen S, Ochman M, Thiel V, Rajfur Z & Pyrc K (2018) APOBEC3‐mediated restriction of RNA virus replication. Sci Rep 8, 5960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. He WJ, Chen R, Yang Z & Zhou Q (2006) Regulation of two key nuclear enzymatic activities by the 7SK small nuclear RNA. Cold Spring Harb Symp Quant Biol 71, 301–311. [DOI] [PubMed] [Google Scholar]
- 47. Salamango DJ, Becker JT, McCann JL, Cheng AZ, Demir Ö, Amaro RE, Brown WL, Shaban NM & Harris RS (2018) APOBEC3H subcellular localization determinants define zipcode for targeting HIV‐1 for restriction. Mol Cell Biol 38, e00356–e418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Larsen DH, Hari F, Clapperton JA, Gwerder M, Gutsche K, Altmeyer M, Jungmichel S, Toledo LI, Fink D, Rask M‐B et al. (2014) The NBS1–Treacle complex controls ribosomal RNA transcription in response to DNA damage. Nat Cell Biol 16, 792–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Harding SM, Boiarsky JA & Greenberg RA (2015) ATM dependent silencing links nucleolar chromatin reorganization to DNA damage recognition. Cell Rep 13, 251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yang Y, Chen Y, Zhang C, Huang H & Weissman SM (2002) Nucleolar localization of hTERT protein is associated with telomerase function. Exp Cell Res 277, 201–209. [DOI] [PubMed] [Google Scholar]
- 51. Lee JH, Lee YS, Jeong SA, Khadka P, Roth J & Chung IK (2014) Catalytically active telomerase holoenzyme is assembled in the dense fibrillar component of the nucleolus during S phase. Histochem Cell Biol 141, 137–152. [DOI] [PubMed] [Google Scholar]
- 52. Iarovaia OV, Minina EP, Sheval EV, Onichtchouk D, Dokudovskaya S, Razin SV & Vassetzky YS (2019) Nucleolus: a central hub for nuclear functions. Trends Cell Biol 29, 647–659. [DOI] [PubMed] [Google Scholar]
- 53. Yang C, Liu X, Gao Q, Cheng T, Xiao R, Ming F, Zhang S, Jin M, Chen H, Ma W et al. (2018) The nucleolar protein LYAR facilitates ribonucleoprotein assembly of influenza A virus. J Virol 92, e01042–e1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hiscox JA, Wurm T, Wilson L, Britton P, Cavanagh D & Brooks G (2001) The coronavirus infectious bronchitis virus nucleoprotein localizes to the nucleolus. J Virol 75, 506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pyper JM, Clements JE & Zink MC (1998) The nucleolus is the site of Borna disease virus RNA transcription and replication. J Virol 72, 7697–7702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Horn AV, Klawitter S, Held U, Berger A, Jaguva Vasudevan AA, Bock A, Hofmann H, Hanschmann K‐MO, Trösemeier J‐H, Flory E et al. (2013) Human LINE‐1 restriction by APOBEC3C is deaminase independent and mediated by an ORF1p interaction that affects LINE reverse transcriptase activity. Nucleic Acids Res 42, 396–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Aynaud MM, Suspène R, Vidalain PO, Mussil B, Guétard D, Tangy F, Wain‐Hobson S & Vartanian JP (2012) Human tribbles 3 protects nuclear DNA from cytidine deamination by APOBEC3A. J Biol Chem 287, 39182–39192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chen Y, Shen B, Zheng X, Long Q, Xia J, Huang Y, Cai X, Wang D, Chen J, Tang N et al. (2020) DHX9 interacts with APOBEC3B and attenuates the anti‐HBV effect of APOBEC3B. Emerg Microb Infect 9, 366–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. McCann JL, Klein MM, Leland EM, Law EK, Brown WL, Salamango DJ & Harris RS (2019) The DNA deaminase APOBEC3B interacts with the cell‐cycle protein CDK4 and disrupts CDK4‐mediated nuclear import of Cyclin D1. J Biol Chem 294, 12099–12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Veith S, Schink A, Engbrecht M, Mack M, Rank L, Rossatti P, Hakobyan M, Goly D, Hefele T, Frensch M et al. (2019) PARP1 regulates DNA damage‐induced nucleolar‐nucleoplasmic shuttling of WRN and XRCC1 in a toxicant and protein‐specific manner. Sci Rep 9, 10075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Meder VS, Boeglin M, de Murcia G & Schreiber V (2005) PARP‐1 and PARP‐2 interact with nucleophosmin/B23 and accumulate in transcriptionally active nucleoli. J Cell Sci 118, 211. [DOI] [PubMed] [Google Scholar]
- 62. Schärer OD (2013) Nucleotide excision repair in eukaryotes. Cold Spring Harb Perspect Biol 5, a012609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sung P & Klein H (2006) Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nat Rev Mol Cell Biol 7, 739–750. [DOI] [PubMed] [Google Scholar]
- 64. Zimmer AM, Pan YK, Chandrapalan T, Kwong RWM & Perry SF (2019) Loss‐of‐function approaches in comparative physiology: is there a future for knockdown experiments in the era of genome editing? J Exp Biol 222, jeb175737. [DOI] [PubMed] [Google Scholar]
- 65. El‐Brolosy MA & Stainier DYR (2017) Genetic compensation: a phenomenon in search of mechanisms. PLoS Genet 13, e1006780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jalal C, Uhlmann‐Schiffler H & Stahl H (2007) Redundant role of DEAD box proteins p68 (Ddx5) and p72/p82 (Ddx17) in ribosome biogenesis and cell proliferation. Nucleic Acids Res 35, 3590–3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Holmes RK, Malim MH & Bishop KN (2007) APOBEC‐mediated viral restriction: not simply editing? Trends Biochem Sci 32, 118–128. [DOI] [PubMed] [Google Scholar]
- 68. Sharma S, Patnaik SK, Thomas Taggart R, Kannisto ED, Enriquez SM, Gollnick P & Baysal BE (2015) APOBEC3A cytidine deaminase induces RNA editing in monocytes and macrophages. Nat Commun 6, 6881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sharma S, Patnaik SK, Taggart RT & Baysal BE (2016) The double‐domain cytidine deaminase APOBEC3G is a cellular site‐specific RNA editing enzyme. Sci Rep 6, 39100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685. [DOI] [PubMed] [Google Scholar]
- 71. Shevchenko A, Tomas H, Havlis J, Olsen JV & Mann M (2006) In‐gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc 1, 2856–2860. [DOI] [PubMed] [Google Scholar]
- 72. Nesvizhskii AI, Keller A, Kolker E & Aebersold R (2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem 75, 4646–4658. [DOI] [PubMed] [Google Scholar]
- 73. Mi H, Muruganujan A & Thomas PD (2013) PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res 41, D377–D386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Heulot M, Chevalier N, Puyal J, Margue C, Michel S, Kreis S, Kulms D, Barras D, Nahimana A & Widmann C (2016) The TAT‐RasGAP317‐326 anti‐cancer peptide can kill in a caspase‐, apoptosis‐, and necroptosis‐independent manner. Oncotarget 7, 64342–64359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Park J, Lim K, Kim J‐S & Bae S (2016) Cas‐analyzer: an online tool for assessing genome editing results using NGS data. Bioinformatics 33, 286–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Li ZF & Lam YW (2015) A new rapid method for isolating nucleoli. Methods Mol Biol 1228, 35–42. [DOI] [PubMed] [Google Scholar]
- 77. Sanjana NE, Shalem O & Zhang F (2014) Improved vectors and genome‐wide libraries for CRISPR screening. Nat Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Yun C, Wang Y, Mukhopadhyay D, Backlund P, Kolli N, Yergey A, Wilkinson KD & Dasso M (2008) Nucleolar protein B23/nucleophosmin regulates the vertebrate SUMO pathway through SENP3 and SENP5 proteases. J Cell Biol 183, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Annibaldi A, Heulot M, Martinou JC & Widmann C (2014) TAT‐RasGAP317‐326‐mediated tumor cell death sensitization can occur independently of Bax and Bak. Apoptosis 19, 719–733. [DOI] [PubMed] [Google Scholar]
- 80. Kuhar R, Gwiazda KS, Humbert O, Mandt T, Pangallo J, Brault M, Khan I, Maizels N, Rawlings DJ, Scharenberg AM et al. (2014) Novel fluorescent genome editing reporters for monitoring DNA repair pathway utilization at endonuclease‐induced breaks. Nucleic Acids Res 42, e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Perez‐Riverol Y, Csordas A, Bai J, Bernal‐Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M et al. (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47, D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. A3C‐interacting proteins The table lists the proteins, identified by mass spectrometry, that were pulled down from cells transfected with either pcDNA3 or hAPOBEC3C‐V5.dn3 that were treated or not with etoposide for 48 hours.
Table S2. Primers used for quantitative real‐time PCR.
Data Availability Statement
The data described in this work are contained within the manuscript except for the raw mass spectrometry data that are available at the online proteomic repository consortium ProteomeXchange [81] (http://www.proteomexchange.org) with identifier PXD020105 and https://doi.org/10.6019/PXD020105.