

Abstract
Selective oxidation of higher alcohols using heterogeneous catalysts is an important reaction in the synthesis of fine chemicals with added value. Though the process for primary alcohol oxidation is industrially established, there is still a lack of fundamental understanding considering the complexity of the catalysts and their dynamics under reaction conditions, especially when higher alcohols and liquid‐phase reaction media are involved. Additionally, new materials should be developed offering higher activity, selectivity, and stability. This can be achieved by unraveling the structure–performance correlations of these catalysts under reaction conditions. In this regard, researchers are encouraged to develop more advanced characterization techniques to address the complex interplay between the solid surface, the dissolved reactants, and the solvent. In this mini‐review, we report some of the most important approaches taken in the field and give a perspective on how to tackle the complex challenges for different approaches in alcohol oxidation while providing insight into the remaining challenges.
Keywords: alcohol oxidation, experimental challenges, mechanisms, metal and oxide catalysts, operando characterization
Metals and oxides are two important types of heterogeneous catalysts applied in selective alcohol oxidation under thermal, electrocatalytic and photocatalytic conditions. Considering the intrinsic differences in these methods, different approaches exist to prepare the catalysts and study them under such reaction conditions. In this minireview, important advances and the remaining challenges for more in‐depth characterization of the above‐mentioned catalysts are described to provide a perspective for future works in the field of selective alcohol oxidation.

1. Introduction and Scope
Selective oxidation of primary alcohols is an environmentally friendly and important reaction to synthesize organic oxygenated compounds. [1] From an industrial point of view, the gas‐phase synthesis of formaldehyde from methanol has been conducted commercially using mixed oxide catalysts for many years. [2] Unlike this process, aerobic oxidation of other mono‐alcohols, diols, or generally higher alcohols to other added value fine chemicals and intermediates such as acetals are typically challenging and require multi‐step processes. [3] The use of glycerol as a starting material for obtaining commodity chemicals has gained much attention. [4] Especially, the oxidative dehydrogenation of glycerol to acrylic acid is of major interest, because the latter is a widely used monomer for the synthesis of resins and superabsorbents. [5]
To perform this reaction efficiently and economically, one deals with several choices such as reaction medium phases, batch vs. flow operation, the type of oxidants, and the catalysts. Using O2 as an abundant source is highly desirable but imposes a technical hindrance since its activation is typically a major challenge. [6] In addition, it is possible to apply thermo‐, electro‐ or photocatalysis to induce this catalytic reaction each of which introduces new challenges and important questions. Considering the complexity of the reaction mechanism in each of the mentioned approaches and the dynamics of the heterogeneous catalysts under reaction conditions, especially when a liquid phase is involved, we still lack a fundamental understanding of the phenomena occurring at the solid‐fluid interface. This essentially encourages us to develop systematic approaches to study different catalysts, particularly with in‐operando techniques. On the other hand, exploring new materials with desired properties in terms of activity, selectivity and stability is needed. In this minireview, we highlight some of the recent and important developments on supported metals and metal‐oxides, the fundamental challenges related to studying them under different reaction modes, and the perspectives into establishing advanced techniques to address the complexity issues in alcohol oxidation.
2. Catalysts for Alcohol Oxidation
2.1. Supported nanoparticulate and metal‐based catalysts
Typically, supported metal catalysts are highly active for alcohol oxidation and consequently, reactions are commonly studied under relatively mild conditions. Whilst alcohol oxidation over metal oxide catalysts has been studied for over half a century, the employment of supported nanoparticulate catalysts can be considered modern by comparison. Seminal studies from Mallat, [7] Kaneda, [8] and Corma [9] were the first to demonstrate the potential of using such catalysts for alcohol oxidation. Since then, countless more contributions have been made and alcohols are now considered by many to be ideal model substrates for the study of selective oxidation, aiding understanding of property‐performance relationships in supported metal catalysts. Despite the relative modernity, the abundance of work published on the use of supported nanoparticulate catalysts for alcohol oxidation has led to the publication of several key reviews. [10]
It has been demonstrated that supported noble metals, particularly Au, Pd and Pt, are highly effective in alcohol oxidation and continue to be widely studied since the early work described above. [11] These often display high activity and selectivity and increased resistance to leaching or poisoning by substrates and by‐products compared to their non‐noble metal counterparts. However, there are also many notable examples of non‐noble metal catalysts for alcohol oxidation. [12] Given that there is such an abundance of literature available in this area, we have dedicated this section of the review to highlight the most promising catalysts and the novel approaches used to enhance our understanding of the properties which influence the alcohol oxidation performance of such catalysts.
Given that the active sites in supported metal catalysts are often well defined, the use of higher reaction temperatures can dramatically reduce catalyst lifetime and facilitate analogous competitive reactions, reducing reaction selectivity. [13] For these reasons, most of the works encompassing the use of such catalysts are conducted in the liquid phase. That said, studies into the gas‐phase oxidation of alcohols are plentiful, particularly over the last decade. Recently, Somorjai and co‐workers [14] reviewed the performance of Pt supported catalysts for alcohol oxidation in the gas‐ and liquid‐phase and demonstrated that, in general, higher turnover frequencies (TOFs) were observed in reactions conducted in the gas phase (Figure 1‐A). Interestingly, however, it was also determined that apparent activation energies, of both primary and secondary alcohols, were considerably lower in the liquid phase (Figure 1‐B), which the authors attributed to the promotional effect of water. A recent publication by Wei et al. [15] elegantly demonstrated this (Figure 1‐C) whereby the authors spiked aerobic benzyl alcohol oxidation experiments with water and observed significant rate enhancements over a Pd/MgAl‐layered double hydroxide catalyst. The kinetic relevance of water was further evidenced through its substitution with D2O and the observation of a significant kinetic isotope effect (KIE). This effect was attributed to water coverage on the supported Pd nanoparticles, promoting substrate adsorption and O2 activation. A similar promotional effect has also been observed in 1‐ and 2‐octanol oxidation over Pt/C catalysts and in the oxidation of a range of primary alcohols over bimetallic AuPt and AuPd catalysts. [16] Indeed, solvent effects are known to influence reactivity over supported nanoparticulate catalysts (Figure 1‐A); they can reduce performance through competitive adsorption [17] or indeed promote performance through facilitating kinetically relevant transformations. [18]
Figure 1.
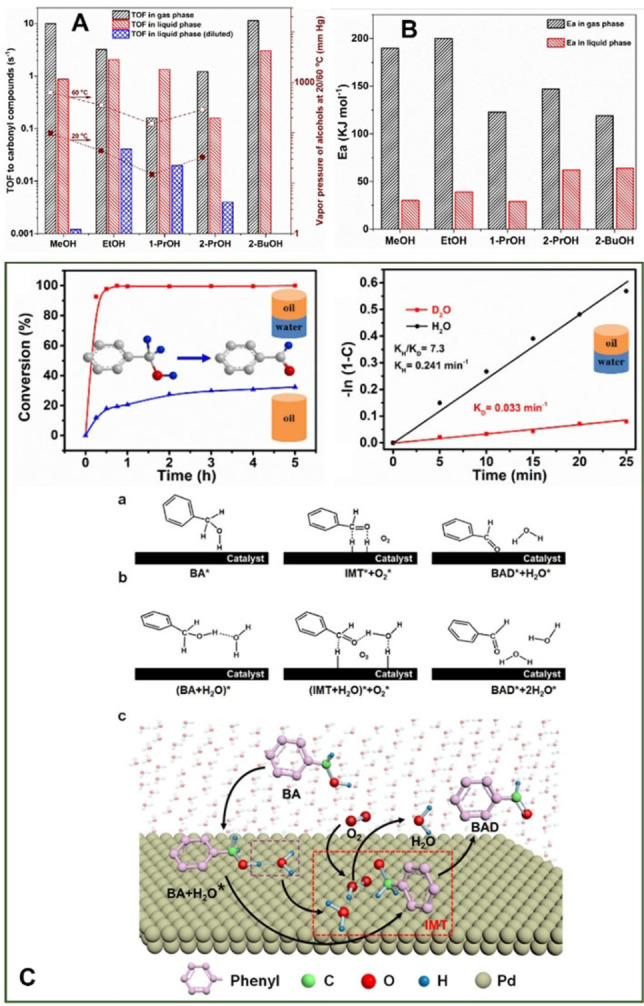
TOFs (A) and apparent activation energy (B) of supported Pt catalysts in the gas and liquid phases are compared; Figures republished from Ref. [14] with permission from the publisher (MDPI) under CC BY 4.0. Additional data from other publications were required to produce (A) and (B). [22] The influence of using a water co‐solvent on the reaction rate of aerobic benzyl alcohol oxidation is demonstrated (C); The conversion of benzyl alcohol is observed and a significant KIE is observed when water is substituted for D2O. A proposed mechanism illustrating how water promotes alcohol oxidation is also provided. The figures presented in (C) were republished from Ref. [23] Copyright 2014, American Chemical Society.
Liquid phase alcohol oxidation can be conducted using aqueous or organic solvents or under solvent‐free conditions. Regardless of the liquid environment employed, the rate‐limiting step (RDS) is widely acknowledged to be the activation of the C−H bond on the associated α‐carbon, although some studies have hypothesized that the initial dissociation of the alcohol also influences the rate. Aerobically, alcohol oxidation proceeds in three primary steps (Scheme 1): (1) dissociative adsorption of the alcohol moiety, forming a surface alkoxy intermediate; (2) activation of the C−H bond and (3) desorption of the formyl product and regeneration of the catalyst site. How the C−H bond is activated, a key activity descriptor, is highly dependent on the reaction conditions and oxidant used. Isotopic labeling experiments using R‐CD2OH confirmed that KIEs were consistent with the RDS being β‐hydride elimination (Figure 1‐B). [19] However, it should be noted that some researchers have speculated that in the presence of a base, C−H bond activation proceeds via proton abstraction (Figure 1‐A), [20] which likely explains why a substantially higher activity is consistently observed under alkaline conditions and is in agreement with the kinetic influence of base in other chemical transformations. [21]
Scheme 1.
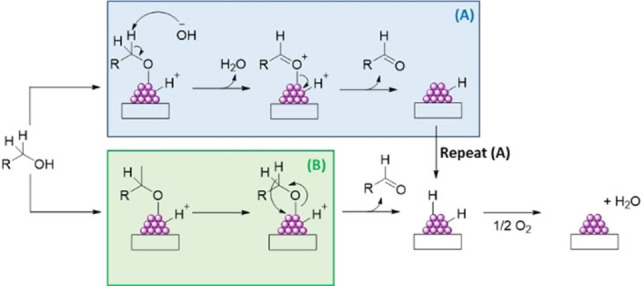
Aerobic, liquid phase alcohol oxidation on supported metal nanoparticles; the reaction mechanism proceeds differently under basic (A) and neutral (B) conditions.
The solvent employed can have further implications on the reaction mechanism, and notably the role of oxygen in the reaction. An important study by Davis and co‐workers, [24] investigating aerobic alcohol oxidation, confirmed using 18O2, that only marginal oxygen from the atmosphere was incorporated into corresponding acid products. Given that no activity was observed when O2 was replaced by N2, they speculated that the role of O2 was simply to scavenge electrons from the surface of the metal catalyst; an important but indirect involvement. This concept is further evidenced by examples where researchers have replaced O2 by sacrificial molecules. In such examples, electrons and protons formed from the dehydrogenation of alcohols are used to hydrogenate other reagents, [25] or can be used in subsequent reduction processes in cascade reactions. [26] Recently, this concept has been developed further with ‘acceptorless’ alcohol dehydrogenation becoming a rapidly developing field. Through optimization of the catalyst and conditions, formyl species and H2 can be produced quite efficiently (and selectively), [27] and could prove to be advantageous over the coming years, in the collective drive to replace conventional energy sources with clean processes. Under aerobic conditions, the formation of H2O2, as a by‐product, has been confirmed and is therefore probable that it and the intermediates formed during its synthesis, [28] must have some role on the reactivity, albeit marginal. This is pertinent as H2O2 has been shown to be an effective oxidant for alcohol oxidation when produced in situ. [28] Whilst there is evidence to suggest that under aerobic conditions aldehydic species, such as benzaldehyde [29] and ethanal, [30] can undergo radical auto‐oxidation, Davis and co‐workers showed using radical scavengers (tert‐butanol and 1,4‐benzoquinone) that there was limited evidence to suggest that the oxidative dehydrogenation of alcohols (over Fe−N−C and Pt/C catalysts) was promoted by hydroxyl or superoxide radicals. [31]
The physicochemical properties of the support material can also impact performance. Under neutral conditions, acid/base properties have been demonstrated to influence both activity and selectivity in alcohol oxidation. [32] Recent work has also suggested that exposed support facets, at the metal‐support interface, can influence performance. Feng and co‐workers demonstrated that differences in the rates of aqueous glycerol conversion and aldehyde/acid selectivity were exhibited when nanoparticles of alloyed AuPt were supported on TiO2 materials terminated at (101) and (001) facets. [33] The authors hypothesized (with evidence) that this was attributed to both an electronic effect and perhaps, more pertinently, a direct interaction between the substrate and support surface (Figure 2‐A), thus, directly influencing kinetically relevant surface transformations. The same group subsequently demonstrated that such effects extended to solvent‐free oxidation of long‐chain aliphatic alcohols and furthermore, highlighted how sensitive support surface structure is to the different atmospheres used in thermal treatments. [34] Researchers have also demonstrated that interfacial sites are critically important for O2 dissociation. [35] Niu, Li and co‐workers [36] were the first to demonstrate the significance of this for alcohol oxidation. Using model FeO/Pt(111) and Cu2O/Ag(111) catalysts, the authors demonstrated with STM and DFT measurements that activated oxygen, originating from facile O2 dissociation at interfacial sites, promoted alcohol adsorption and O−H bond activation. Furthermore, the authors elegantly demonstrated that these interfacial effects were independent of particle size (Figure 2‐B).
Figure 2.

Exposed support surface facets influence alcohol dehydrogenation performance over supported metal catalysts. (A) TiO2 terminating at (101) and (001) facets interact chemically with the substrate, assisting with adsorption and reactivity; Figure has been republished from Ref. [33]. Copyright 2019, American Chemical Society. (B) Reaction data demonstrates that interfacial sites are independent of particle/component sizes. Benzyl alcohol conversion (solid black square) and benzaldehyde selectivity (hollow blue circles) over metal particles, TMO nanoparticles, nano‐Ag/micro‐TMO, nano‐TMO/micro‐Ag and Ag‐TMO nanocomposites. This Figure has been republished under CC BY 4.0 from Ref. [36].
Particle size has been known to dramatically influence the reactivity of supported nanoparticulate catalysts for alcohol oxidation, which cannot simply be explained as a function of dispersion. However, a detailed understanding of why particle size impacts TOF so significantly has only recently emerged. Using liquid phase, aerobic benzyl alcohol oxidation as a model, Li, Lu and co‐workers studied the influence of Pd particle size on activity, over a series of supported Pd catalysts. [37] The TOF and ODH selectivity were both affected by particle size and the optimum particle size was established to be ca. 4 nm. Deviation from this diameter led to significant drop‐offs, particularly with respect to TOF (Figure 3‐A). The authors proposed that decreasing TOF, as particle size decreased, was attributed to electronic effects and, specifically, an increased metal work function which increased the binding strength of the substrate and reaction product(s). On the contrary, the authors speculated that the decreasing TOF, observed as the particle size was increased beyond the optimum value, was attributed solely to geometric effects. This would explain why the preparation method employed can have such a significant influence in the liquid phase oxidation of alcohols, with sol‐immobilized nanoparticulate catalysts often being particularly active and typically have mean particle diameters of between 2‐5 nm when stabilized by PVA. [38] This may suggest that the use of highly dispersed supported noble metal catalysts, and single‐site catalysts could be limited in alcohol oxidation compared to other fields. However, recent reports have suggested that such catalysts do indeed show promise in alcohol oxidation. [39] In particular, Wang and co‐workers demonstrated that single atom Au catalysts outperformed analogous nanocluster and nanoparticulate forms in benzyl alcohol oxidation (Figure 3‐B). [40] This was suggested to be due to a higher abundance of sites comprising oxygen vacancies (Ov) in the ceria support and adjacent Au3+/Au+, denoted as [O−Ov−Ce−O−Au], facilitating alcohol adsorption, dissociation of O−H bonds and subsequent beta‐hydride elimination. The authors confirmed the varied Au speciation and environments using a combination of techniques, which included extended X‐ray absorption fine structure (EXAFS), X‐ray photoelectron spectroscopy (XPS), Raman and Fourier transform‐infrared (FTIR) CO chemisorption. In‐situ IR spectroscopy was used to elucidate the adsorption and activation of benzyl alcohol over the series. As expected, a strong absorption characteristic of benzaldehyde was observed over the single atom catalysts, which was not observed over the CeO2 support and was only weakly observed over the nano cluster and nanoparticle analogues. Adsorption of benzyl alcohol over the CeO2 support was however detected, through observation of a cerium alkoxide species, highlighting the role of the support when adjacent to Au species. The single atom catalysts also displayed high reusability with almost unchanged activity, selectivity and speciation over 5 cycles. Atomically dispersed Pd has also proved to be highly active in the oxidation of allylic alcohols. [39c] Using EXAFS and X‐ray absorption near‐edge structure (XANES) analysis, Hackett et al. confirmed that the mesoporous alumina support stabilizes atomically dispersed Pd2+. The coordination environment was described as a four‐coordinate, pseudo‐square planar geometry. These isolated Pd centers were also confirmed using high‐angle annular dark field (HAADF) scanning transmission electron microscopy (STEM). Mesoporous alumina was found to be a far more effective support compared to conventional gamma‐alumina, which was attributed to a combination of its higher surface area and higher defect density providing ample Pd nucleation centers. These Pd species were also found to be stable over days of reaction, as confirmed by operando EXAFS measurements. Atomically dispersed non‐precious metal M−N−C catalysts have also been shown to be active with Cu demonstrating the highest activity in a range where M=Fe, Cu, Ni, Cr, Co. [31] The authors previously characterized the Fe−N−C catalyst by XPS, Mössbauer spectroscopy and in situ X‐ray absorption spectroscopy (XAS) to study the coordination environment, finding that ∼90 % of the Fe existed as atomically dispersed Fe−Nx species, which were previously linked with a high activity for the oxygen reduction reaction (ORR). [41] These species were found to be stable throughout the benzyl alcohol reaction, with only a minor drop in activity being assigned to strongly adsorbed species. Activity could be almost fully restored after a reductive treatment in H2. The higher activity of the Cu−N−C catalyst was attributed to faster β‐hydride elimination. Based on the aforementioned discussion, it is clear that a number of the areas discussed allude to the fact that electronic effects influence alcohol oxidation in supported nanoparticulate catalysts. This is a significant insight and requires further exploration. In 2012, Koper and co‐workers observed that CO chemisorbed to Au(111) could dramatically increase its catalytic activity towards alcohol oxidation. [42] The authors hypothesized that this enhancement was attributed to an electronic effect which, logically, they suggested promoted β‐hydride elimination. Later, Chen and co‐workers demonstrated that nitrogen doping of carbon support could dramatically promote alcohol oxidation over Co nanoparticles. [43] The authors attributed this enhancement to N in the carbon support, enriching positive charge build‐up in the Co nanoparticles which, in turn, promoted their ability to attract and activate O2, as well as aiding the removal of protons from Co−H intermediates. An in‐depth analysis of active site electronics on Au nanoparticles was recently conducted by Chandler and co‐workers. [44] Using the Hammett methodology, the authors were able to demonstrate a clear effect on turnover frequency arising from various extents of electron donation to Au from a range of metal oxide supports. Supports such as Al2O3 and SiO2 demonstrated a higher sensitivity to changes in substrate electronics compared with TiO2 and ZnO, indicating a more positive Au active site in the former, thus an increased ability to stabilize the hydride transfer transition state of the RDS. Crucially, this investigation has shown the Hammett method to be a powerful tool for assessing changes in active site electronics, and metal‐support interface (MSI) interactions, [45] without interference from other factors such as the number of active sites or particle size.
Figure 3.

Compilation of figures from previous studies highlighting that benzyl alcohol oxidation on supported metal catalysts is influenced by particle size, particle composition and electronic effects. (A) Particle size influences TOF in supported Pd catalysts republished with permission from Ref. [37], IAAS. (B) Supported Au single atoms (SA), nano clusters (NC) and nanoparticles (NP) influence conversion and TOF – republished from Ref. [40] Copyright 2019, American Chemical Society. (C) bimetallic AuPd catalysts are more efficient and selective to benzaldehyde than their monometallic counterparts (squares ‐Au/TiO2; circles – Pd/TiO2; AuPd/TiO2 – triangles; filled shapes – conversion; hollow shapes – benzaldehyde selectivity). Inlet – STEM‐EDS micrograph of bimetallic AuPd/TiO2 particle. Figures republished with permission from Ref. [46], Copyright 2006, AAAS. (D) confirmation that d charge at Pd sites is influenced by Au/Pd composition and that it correlates directly to TOF for 4‐methylbenzyl alcohol (black line), 4‐phenethyl alcohol (red line) and anisalcohol (blue line) oxidation. Figure republished from Ref. [49] under CC BY 4.0.
A major and ever‐growing area in alcohol oxidation is the use of bimetallic catalysts for increased performance. Hutchings and co‐workers first demonstrated the efficacy of Au−Pd/TiO2 catalysts for the oxidation of a range of alcohols including primary alcohols. [46] The increased activity compared to monometallic counterparts was suggested to be due to an electronic modification of an Au‐rich core to a Pd‐rich shell (Figure 3‐C). The authors also demonstrated that significant improvements in aldehyde selectivity were observed over the bimetallic AuPd catalysts, and later demonstrated that sequential aldehyde oxidation was inhibited by the presence of alcohol. [47] Since this, there have been many other reports of the enhanced activities of bimetallic catalysts comprising combinations of precious and non‐precious metals.[ 16b , 48 ] For Au‐ Pd catalysts, this enhancement is often ascribed to particle size, morphological and electronic effects, with the latter becoming an increasingly prominent area of study. Wan and co‐workers recently reported on the optimization of surface d‐charge in Au−Pd alloys and the subsequent effects on benzyl alcohol adsorption, oxidation and selectivity (Figure 3‐D). [49] This enhancement has been found to be present across a range of metal ratios and even with single‐atom doping of Pd into Au clusters. [50]
Given that the performance of supported metal catalysts in alcohol oxidation are evidently highly dependent on their defined structure and morphology, their stability under reaction conditions must be assessed. Numerous deactivation mechanisms have been proposed in the literature, which include support or supported metal leaching,[ 7 , 13 , 28a , 51 ] product inhibition, [52] active metal sintering and even over oxidation of the active metal component. [53] Many of these deactivation mechanisms are dependent on the reaction conditions used and the specific transformation under investigation.
Catalyst leaching, either from the supported metal component or the support itself, is commonly observed in liquid phase alcohol oxidation reactions and is often attributed to the solubility of the catalyst in the liquid medium, or the formation of soluble components under reactions conditions. [54] Hydroxylation of oxides, through interaction with water for example, can substantially increase solubility. Another common source of leaching is the chelation of supported metals with reaction products, particularly products possessing multiple acidic components.[ 28a , 55 ] Inductively coupled plasma mass spectrometry (ICP‐MS) and microwave plasma atomic emission spectrometry (MP‐AES) are incredibly effective at quantifying catalyst leaching, through examination of post reaction effluent. It is exceptionally difficult to suppress leaching, without making changes to the catalyst (through changing the support to induce a stronger metal‐support interaction) or by changing the reaction conditions (through addition of additives such as −OH). [13] This is a key problem facing liquid phase alcohol oxidations, and one which requires further study if industrialization of such processes is to be realized.
Another primary source of deactivation is product inhibition. Davis and co‐workers conducted a thorough investigation into this, by assessing how different functionalized additives influenced the rate of aqueous glycerol oxidation over supported Au and Pt catalysts. [52b] The authors determined that the addition of chelating species, such as sugar acids and polyols, led to significant deactivation. This was hypothesized to be attributed to the formation of ketones, enones and β‐carbonyl species in situ, adsorbing strongly to the metal active sites. More recent work by the same group, [52a] confirmed that over Pt catalysts, deactivation can be attributed to the formation of olefinic by‐products. The formation of these species, which were identified by surface enhanced Raman spectroscopy and solid state 13 C NMR, occurred through the decarbonylation of aldehyde. Despite the fact that kinetically, this side reaction was much slower, the olefinic by‐products were determined to be 2 orders of magnitude more effective at competing for the Pt active sites that the aldehydes and acids produced in the reaction. The authors also, rather interestingly, confirmed that these products could be easily removed from the catalyst by exposing the catalyst to a mild reductive heat treatment.
Evidently, the study of alcohol oxidation over supported nanoparticulate catalysts continues to be a hot topic in heterogeneous catalysts. It is clear that the initial works were predominantly focused towards understanding the mechanism and the physicochemical catalytic properties which promote alcohol oxidation. Armed with this increased understanding, research now appears more focused on using alcohol oxidation as a tool to study fundamental catalysis, particularly with relation to synergism and electronic effects, resulting in high‐performance catalysts. This is a continually evolving field of research, the findings of which could well have relevance and translate into other areas of heterogeneous catalysis. We hypothesize that DFT and theoretical calculation will play a big role in furthering understanding in this area, with supplemental experimental design and implementation. Nevertheless, there is still scope to enhance the use of such catalysts more abundantly industrially, if further methods of increasing catalyst stability are developed. The application of supported noble metal catalysts for gas‐phase alcohol oxidation is one area where this could make a significant influence.
2.2. Oxide‐based catalysts
Oxide‐based catalysts have been extensively used in partial oxidation reactions. One of the most commercially known industrial applications of oxide catalysts in alcohol oxidation is the methanol to formaldehyde process which uses iron molybdate as the catalyst. [2] Compared to metal catalysts, oxides have additional degrees of complexity when it comes to the identification of the active sites and the involved mechanisms. Their surfaces can be dynamically affected upon exposure to the reaction mixtures or various pretreatment environments much like the metal catalysts although the knowledge of such transformations and surface reconstructions is more limited with oxide catalysts. [56] As reported by Linnemann et al., [57] oxide catalysts used in electrocatalysis can also undergo nanoscale changes which are not trivial to understand yet play a huge role. Different combinatorial approaches and techniques should therefore be applied to elucidate these kinds of transformations and their impacts on the catalytic properties. On the other hand, the surface properties of oxide catalysts are defined and can be tuned by the bulk properties. Especially in the case of mixed oxides, the surface can be enriched by one or the other cation,[ 2a , 58 ] as schematically shown in Figure 4, which can eventually define the nature and the electronic state of the adsorption sites on the surface. Mixed oxides are particularly interesting as they provide a means to adjust the band structure [59] and the oxidation degree of the cationic species on the surface and therefore tuning of the active sites. [60] Also depending on composition, one oxide phase can be atomically dispersed in the other one, essentially creating an efficient catalyst as reported for V and Fe antimonates for oxidation of ethanol to acetaldehyde. [2a] Emerging attention has been also given to the preparation and application of high‐entropy noble‐metal‐free oxides involving several cations with great potential applications, generally in catalysis [61] and particularly in alcohol oxidation. [62]
Figure 4.

Schematic representation of cationic distribution in the bulk vs. the surface. Reprinted from Ref. [58], Copyright 2010, American Chemical Society
Another important parameter is the crystalline structure of the (mixed)‐oxides which has been shown to significantly affect the catalytic properties. For instance, Murayama et al. investigated the role of four different crystalline phases for the Mo3VOx catalysts (namely orthorhombic, trigonal, tetragonal, amorphous) in the allyl alcohol and found interesting trends for the selectivity towards aldehyde or acrylic acid products depending on the crystalline phase. [5] The reason can be attributed to the different surface terminations which are shown to greatly impact the catalytic activity and selectivity by tuning the exposed active sites. [63] The effect of crystalline structure can also be investigated by employing different types such as spinels [64] and perovskites. [65] In the former, one can manipulate the distribution of different cations on octa‐ or tetrahedral coordination with the aim to tune the catalytic properties. Jiang et al. have attributed the enhanced catalytic activity of CuCo2O4 catalyst in benzyl alcohol oxidation to the formation and presence of Cu3+ ions in some of the octahedral sites in addition to Cu2+. [59] Gurrala et al. studied the Cu, Co, and Mn containing spinel‐based mixed oxides catalysts for partial oxidation of benzyl alcohol with high selectivity towards benzaldehyde and found out that the activity strongly depends on the chemical composition. [66] Falk et al. studied the application of Co1+xFe2‐xO4 spinel oxides and identified the active sites in the catalytic oxidation of 2‐propanol solid/liquid and solid/gas interfaces. [67] The presence of oxygen vacancies and synergetic effects between the different cations within spinel structures have been reported to be important in tuning the catalytic activity.[ 64b , 68 ] Perovskites with the general formula of ABO3 (in which A represents a lanthanide, an alkali metal, or an alkaline earth metal and B represents a transition metal) form another class of oxides which have also attracted much attention due to their numerous interesting properties such as an abundance of oxygen vacancies, lattice oxygen mobility, thermal and mechanical stabilities.[ 65 , 69 ]
Other approaches have been also considered to tune the catalytic properties of oxide for alcohol oxidation through modifying the morphology or structural properties and nanosizing.[ 56 , 59 , 70 ] For instance, Hellier et al. reported the use of VOx/Fe2O3 core/shell catalysts with varying coverage of VOx for methanol to formaldehyde oxidation and reported that minimal exposure of Fe sites at the surface inhibits the full oxidation of methanol. [71] Highly ordered mesoporous Co3O4 have been prepared by Li et al. through silica‐templating methods to obtain a catalyst with a high surface area compared to a commercial Co3O4 sample and active in the oxidation of benzyl alcohol. [72] Gu et al. reported a one‐step template‐free solvothermal method to synthesize recyclable hollow carbon‐modified Fe3O4 catalyst active in the oxidation of benzyl alcohol. [73] Mesoporous NiO/Bi2WO6 nanocomposite has been synthesized by Pordel et al. and found to be an efficient catalyst for the oxidation of primary and secondary benzylic alcohols under mild conditions. [74] Hybrid Co3O4/MnO2 nanotube‐based catalysts were prepared by a simple hydrothermal synthesis method and showed superior catalytic properties in the oxidation of benzylic alcohols compared to the individual oxides. [75] In addition to the bulk oxides, supported oxides are also reported in alcohol oxidation. For instance, manganese oxide supported on MCM‐41 zeolite has been used in the oxidation of benzyl alcohol with peroxide. [76] RuIVCoIII mixed oxide supported on alumina is reported to be effective in the liquid‐phase oxidation of primary and secondary alcohols to their corresponding aldehyde and ketones using O2 or N2O as oxidants. [77] Vandadomolybdate positioned in MOF support has been used in the oxidation of primary aromatic alcohols with significant activity and selectivity due to the specific confinement effect and micropore structure, particularly enhancing O2 activation. [78]
The oxidation of alcohols with oxides can follow complex reaction paths especially considering the employed oxidants and the phase composition of the reaction medium. When O2 is used as the oxidant, its activation is typically the rate‐limiting step. In this regard, the reducibility of the oxide plays an important role in defining the catalyst performance when the Mars–van‐Krevelen mechanism is involved. [79] Two possible mechanisms with or without involving the breaking of the metal‐oxygen bond are schematically shown in Figure 5. Overall, several reactions are involved in the oxidation of alcohols including dehydrogenation, oxidative dehydrogenation, dehydration, and total oxidation, among which the first two are giving rise to the desired oxygenated products. [64] In order to promote the desired paths, one should consider not only redox properties of oxides but also their acid/base properties[ 60b , 80 ] as in some cases, they might even simultaneously contribute to the mechanism. [81] Therefore, it is highly desired to have tools to identify the nature of the active sites in oxide‐based catalysts.
Figure 5.
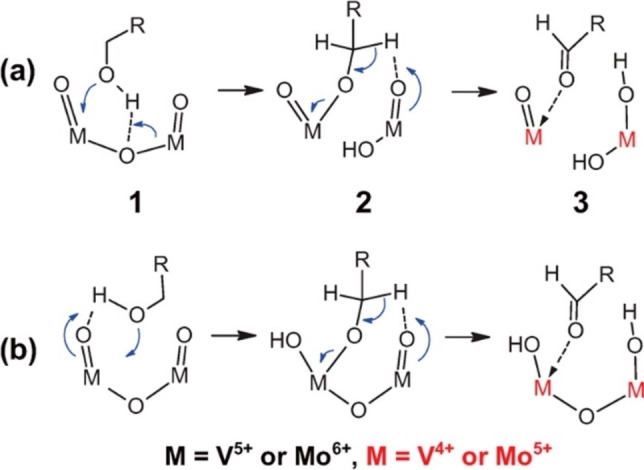
Suggested mechanisms for the oxidation of benzyl alcohol to benzaldehyde: with (a) and without (b) involving the opening of the M−O−M bond. Reprinted from Ref. [81] Copyright 2013, American Chemical Society.
In a pioneering work by Kulkarni and Wachs, adsorption of isopropanol and its oxidation has been used as a probe reaction to determine the number of active surface sites and their nature, redox or acidic, for a variety of bulk metal oxides. [82] Ten years later, Wachs and Routray reported that the absence of proper surface characterization techniques to fully identify the nature and number of the active sites still hampers the development of fundamental understating in the application of oxide catalysts. [83] All in all, considering the variety of possible compositions for these catalysts and the complexity of their surfaces, it is still required to develop a combination of advanced in situ or operando characterization tools aided by computational models along with the application of well‐defined model catalysts to achieve a meaningful spatial and temporal understanding of the nature of the active sites and their catalytic properties. [56] In the next sections, we will discuss more details about the different experimental approaches regarding alcohol oxidation using oxide catalysts and address some of the most important questions and challenges in this field.
3. Thermal Catalysis
3.1. Gas‐phase oxidation
Metal oxides with one or more transition metals are active compounds in the catalytic oxidation of alcohols. Iron molybdates are industrially employed as heterogeneous catalysts in the production of formaldehyde through the selective aerobic oxidation of methanol. [84] In this process, over 90 % of the product yield is ascribed to formaldehyde.[ 84a , 84b ] While carbon monoxide (CO) is the major by‐product, dimethyl ether and methylformiate as highly valuable chemicals are only obtained in small quantities. [84a] Although industrial Fe−Mo−O mixed oxides are prepared using Mo : Fe atomic ratios of around 3, the active phase shows similarities to that of the stoichiometric ferric molybdate Fe2(MoO4)3. [84a] X‐ray absorption spectroscopy (XAS) investigations have shown that the surface of a selective Fe2O3‐based catalyst is characterized by the presence of an oxide overlayer with octahedrally coordinated Mo sites. [85] Based on a randomized distribution model, it has been proposed that the active and selective site on Fe−Mo oxide catalysts is an ensemble that contains two adjacent Mo atoms. [86] In contrast, the presence of isolated Mo centers leads to the predominant formation of CO, while total oxidation is favored over a large ensemble of Fe sites.
Aliphatic C2‐C3 alcohols have been widely used as probe molecules in the context of selective oxidation to investigate the redox properties of solid oxide catalysts.[ 64b , 67 , 82 , 87 ] Ethanol oxidation over ceria nanoparticles with varying morphology showed high structural sensitivity. Results indicated that the type/stability of ethoxide intermediates, the onset temperature of the reaction, and the product selectivity pattern strongly depend on the type of exposed surface facets. [87e] Surface basicity, mobility of oxygen anions on different surface terminations, and variation in acid strength of the Ce cation sites related to changes in their oxygen coordination are among the factors that determine the catalytic activity and selectivity. Kinetic studies over mesoporous Co3O4 with varying pore structures revealed that large pores are beneficial for acetaldehyde desorption, which in turn limits the subsequent formation of total oxidation products. [87a]
Perovskites with ABO3 structure have been employed as catalysts in the context of ethanol oxidation.[ 87c , 87d , 87g , 87h ] Partial substitution of Mn with Ni into the solid solution of LaMnO3 perovskites generates Ni−O−Mn bridging lattice oxygen sites. Based on the catalytic performance in ethanol oxidation, these sites promote ethanol conversion, however, at the expense of acetaldehyde towards total oxidation products. [87c] According to the proposed mechanism shown in Figure 6, the catalytic cycle initiates with dissociative chemisorption of ethanol on the surface, thereby yielding an ethoxy at the transition metal site (Mn or Ni) and a surface hydroxyl at the neighboring lattice oxygen site. Further dehydration yields an acetaldehyde‐derived intermediate and an oxygen vacancy. At low temperatures, acetaldehyde is desorbed, while the reduced surface is re‐oxidized. In contrast, at high temperatures, the acetaldehyde intermediate may undergo association with the surface bridging oxygen (Mn−O−Mn, Ni−O−Mn, or Ni−O−Ni) to form an acetyl, which is then converted to carbon oxides, water, and methane.
Figure 6.
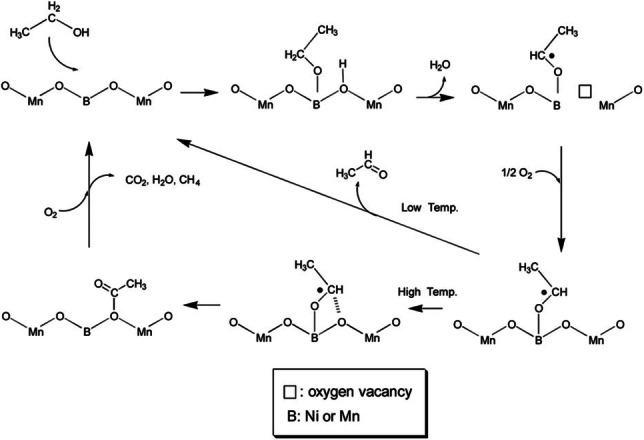
Proposed reaction pathway of gas‐phase ethanol oxidation over LaMn1‐xNixO3 perovskites. Reprinted with permission from Ref. [87c] Copyright 2014, Royal Society of Chemistry.
Reactivity studies based on LaMnO3 (100) thin films, which are enriched in Mn at the surface according to photon energy‐dependent XPS analysis, indicate that the formation of ethylene as a dehydration product is favored in an oxygen‐free atmosphere compared to acetaldehyde as the dehydrogenation product. [87g] In the presence of oxygen, the reactivity increases without affecting the selectivity pattern. Upon partial substitution of La with Sr, the overall activity of the surface‐enriched Sr‐based thin film decreases. This is most likely related to a decreased interaction of adsorbates at the binding sites. However, the aldehyde‐to‐alkene ratio increases, which clearly suggests that vacancies have a strong impact on product selectivity. A kinetic investigation over powdered LaMnO3‐based perovskites showed that the surface reaction between ethoxy species and dissociatively adsorbed oxygen is the rate‐determining step. [87d] The employed set of rate equations based on the Langmuir‐Hinshelwood‐Hougen‐Watson formalism were derived by lumping eight elementary reactions obtained in agreement with the Mars–van‐Krevelen redox cycle.
Operando studies are highly valuable since they give insight into the chemical state of a catalyst under working conditions. In the context of ethanol oxidation, Zhang et al. showed that the alcohol dissociates into ethoxy species and surface hydroxyls based on ambient pressure XPS (AP‐XPS) studies over SrTiO3 (100). [87h] In the absence of gas‐phase oxygen, the former are directly oxidized to acetates, which undergo further oxidation towards CO2 and H2O. Upon the addition of oxygen, the formation of total oxidation products is strongly enhanced. These observations indicate that lattice oxygen at the surface determines the oxidation rate of ethoxy species to acetates. A similar investigation was performed by Diulus et al. for isopropanol oxidation over stoichiometric, unreconstructed SnO2(110)‐(1×1) surfaces. [87b] AP‐XPS experiments applying isopropanol pressures below 3 mbar and various alcohol‐to‐oxygen ratios at different reaction temperatures allowed a systematic investigation of the chemical states of adsorbed species at the single‐crystal surface. AP‐XPS valence‐band spectra indicate that the surface was partially reduced from Sn4+ to Sn2+ upon exposure to pure isopropanol, which is directly linked to the formation of oxygen vacancies at the surface. Analysis of the gas phase indicates that acetone is the main product under these conditions. Addition of molecular oxygen to the reaction mixture results in a substantial increase of the reaction kinetics. However, the catalytic process proceeds unselectively as an enhanced formation of total oxidation products was observed. Since vacancies are replenished by gas‐phase oxygen, the high oxidation state of tin cations at the surface is maintained. According to the authors, SnO2 bridging and in‐plane oxygen are regarded as the reactive oxygen species. The results obtained from the operando investigation suggest that the oxidation of isopropanol over stoichiometric SnO2(110)‐(1×1) proceeds according to the Mars‐van‐Krevelen mechanism.
An extensive study dealing with isopropanol oxidation over a wide variety of metal oxides was performed by Kulkarni and Wachs. [82] Product selectivity during selective oxidation of isopropanol under differential conditions reflected the redox/acidic nature of the active surface sites. Redox surface sites yield acetone, whereas acidic surface sites mainly catalyze the formation of propene. Isopropanol chemisorption on dehydroxylated oxide surfaces was employed to determine the number of active sites. On average, experimental values lied in the range between 2 to 4 μmol m−2. TOFs obtained for redox‐active catalysts varied by six orders of magnitude (102 to 10−4 s−1). A stronger effect was observed for those showing acidic activity since the TOFs varied by over eight orders of magnitude (101 to 10−7 s−1). A weak inverse correlation was encountered between the TOF for the redox pathway and low values for bulk heats of formation of the metal oxides per oxygen atom, which are characteristic of noble metal oxides. Similarly, a moderate inverse relation was found between the redox‐related TOFs and the surface isopropoxide intermediate decomposition temperature, especially at low decomposition temperatures. This indicates that highly reactive surface isopropoxide species are easily formed over surfaces of noble metal oxides. Since the selectivity of the metal oxide catalysts is independent of the TOFs, it becomes evident that the catalytic activity has no significant impact on the type of product generated during the catalytic cycle.
Cobalt‐based spinel‐type oxides are known for their outstanding catalytic performance in the aerobic oxidation of isopropanol. Phase‐pure crystalline spinel cobalt oxide Co3O4 nanoparticles obtained by the decomposition of cobalt acetylacetonate in oleylamine catalyze the oxidative dehydrogenation of 2‐propanol to acetone as the main product at temperatures as low as 100 °C. [64a] In the range up to 300 °C, a slight decrease in selectivity is observed due to the undesired formation of CO2. This type of catalyst achieves almost 100 % yield to acetone at around 160 °C, which is attributed to the high amount of exposed active Co3+ sites and to the reactive oxygen species that populate the catalyst surface. A pronounced deactivation affecting the low‐temperature channel, which can be restored by an oxidative post‐treatment, is associated with the formation of strongly bound carbonaceous species. This poisoning effect, however, has no impact on the high‐temperature reaction path. Density functional theory calculations not only identified 5‐fold‐coordinated octahedral surface Co5c 3+ as the active site but also determined that the oxidative dehydrogenation involving adsorbed atomic oxygen is the energetically most favored pathway. Similar results were encountered for CoFe2O4 nanoparticles obtained with the same synthetic approach. [64b] However, the overall activity for cobalt ferrite is lower compared to that of pure cobalt oxide spinel. This can be explained based on XPS before and after reaction, which indicates a dominant contribution of significantly less reactive Co2+ species at the surface. Another interesting difference between both catalysts is that propene is formed besides CO2 at higher temperatures. This observation suggests that cationic iron species, which are more acidic in nature compared to the cobalt ones, are also available at the catalytic active surface. In situ DRIFTS identified acetates as the responsible adsorbates for the deactivation of the low‐temperature channel, whereas carbonates are only spectator species at the surface. The deactivation phenomenon observed for the isotropic cobalt ferrite nanoparticles was also encountered for anisotropic CoFe2O4. [87f] The latter can be obtained by topotactic transformation of a layered double hydroxide precursor (LDH) containing equimolar amounts of Fe2+, Co2+ and Fe3+ into the spinel‐type oxide through calcination in air. During this process, in which Fe2+ cations are oxidized into Fe3+, highly porous monocrystalline platelets are formed. A commercial catalyst with the same structure and composition showed a higher low‐temperature activity towards acetone in comparison to the anisotropic cobalt ferrite. However, a major difference was encountered in terms of product selectivity at higher temperatures, since the ex‐LDH CoFe2O4 leads to the preferential formation of propene, while the high selectivity to acetone over the commercial sample essentially remains unaffected. This is related to the limited oxygen conversion over the anisotropic material that triggers alcohol dehydration at the expense of the dehydrogenation reaction. These findings underline that acid‐base/redox properties at different surface facets can be tuned by modification of the catalyst morphology.
The effect of co‐feeding water on the catalytic activity in the selective oxidation of 2‐propanol over Co3O4 was investigated by Falk et al. using a mesostructured spinel oxide prepared by nanocasting with the aid of SBA‐15 silica as a hard template. [67] The apparent activation energy of 2‐propanol oxidation determined in the gas phase while co‐feeding water is comparable to the value obtained for the same catalyst in the aqueous phase. In contrast, lower apparent activation energy without water co‐feeding indicates that a different chemical state of the active sites governs the catalytic turnover, which in turn leads to a different mechanism in the gas phase. In addition, the presence of water affects the catalytic activity by increasing the conversion of the low‐temperature channel and the selectivity as well as the stability at high temperatures. This enhancing effect of water for the oxide catalyst is contrary to results obtained over noble‐metal nanoparticles. [88]
Direct oxydehydration of glycerol to acrylic acid as a one‐step process has attracted particular attention in the past years. A suitable bifunctional catalyst should possess surface acidity to trigger glycerol dehydration to acrolein as well as adequate redox properties for the subsequent oxidation of acrolein to acrylic acid. [89] Multinary metal oxides containing vanadium, molybdenum, and tungsten have proved to fulfill these requirements. A binary MoV2O8 phase obtained in situ upon thermal treatment of a mixture of (NH4)6Mo7O24 and NH4VO3 in an oxygen‐containing atmosphere showed a 3.5 times higher activity compared to the separate metal oxides. [90] This is associated with the dynamic changes in the oxidation state of vanadium ions in the binary oxide and the formation of oxygen vacancies. The presence of SDS and CTAB ionic surfactants during hydrothermal synthesis of the MoV2O8 phase promoted a change in morphology, which results in the formation of rod‐shaped crystals. [91] With this synthetic approach, an increased macroporosity of the materials related to intercrystallite spaces was achieved. Rod‐like morphology stabilizes the active MoV2O8 crystalline phase, which in turn favors the redox process through the equilibrium between MoV2O8 and MoVO5 during the reaction. This avoids the migration of vanadium atoms to an amorphous phase, which results in a significant improvement of the catalytic performance. The SDS‐assisted synthesis leads to a catalysts that achieves 100 % glycerol conversion, a maximum selectivity of 57 % towards acrylic acid and a considerable decrease in COx production from 66 to 36 %. Chieregato et al. studied the oxydehydration reaction over complex W−Mo−V oxides to elucidate the role of each element in the oxide framework. [92] Based on the physicochemical properties and the catalytic performance, it became clear that tungsten dehydrated glycerol to form acrolein, vanadium oxidized acrolein to form acrylic acid, and molybdenum moderated the strongly oxidizing properties of vanadium. Similar findings were encountered by Omata et al. with respect to tungsten and vanadium while investigating the catalytic properties of W−V−Nb−O metal oxides. [93] High selectivity towards acrolein was obtained over orthorhombic‐like W−Nb−O in the transformation of glycerol, whereas acrylic acid is directly obtained over V‐modified W−Nb−O catalysts in an oxygen‐containing gas atmosphere. In the presence of phosphoric acid, not only does the Brønsted acidity increase but also the sequential oxidation of acrylic acid towards total oxidation products is suppressed due to the interaction of phosphoric acid with the V sites.
Future work in the gas‐phase oxidation of alcohols should aim at understanding the interplay between the complex real structure of a catalyst and the chemical potential applied during catalytic turnover. This will allow identifying crucial building blocks that are necessary to achieve full conversion while maintaining high selectivity towards valuable intermediates. For this purpose, it is imperative to perform operando studies that give access to the physicochemical properties of the working catalyst under relevant conditions. In addition, extensive reactivity studies are required in order to obtain information about the kinetic parameters that help to understand the underlying reaction mechanism. Finally, theoretical calculations are necessary in order to shed light on the high complexity often encountered in interpreting experimental data.
3.2. Liquid‐phase oxidation
Compared with gas‐phase oxidation, liquid‐phase oxidation is usually performed under milder conditions resulting in more selective transformations. [94] Liquid‐phase oxidation is also the method of choice to functionalize organic molecules with high boiling points, for example, aromatic alcohols like benzyl alcohol, 1‐phenylethanol, and their derivatives. [95] Extending the aromatic alcohols by one conjugated C=C bond, like in cinnamyl alcohol or related compounds, allows studying the chemoselective oxidation of the OH group or C=C bond.[ 69b , 95a , 95b , 95g , 95k , 95l , 95n ] Allylic alcohols usually have higher reactivity due to mesomeric stabilization of intermediates or transition states of the allylic OH group, while aliphatic alcohols such as isopropanol, 1‐butanol, or cyclohexanol are more difficult to oxidize.[ 67 , 95a , 95g , 95k , 95l , 95m ] In recent years, the liquid‐phase oxidation of alcohols over transition metal oxides have been studied extensively, using for example cobalt oxides,[ 95a , 95b , 95c ] iron oxides,[ 95d , 95e , 95f , 95g ] and mixed‐metal oxides based on Cr, Mn, Co, Fe, or Ni, often with spinel or perovskite structures.[ 95h , 95i , 95j , 95k , 95l , 95m ] The oxidants are either gaseous O2 or liquid peroxides such as H2O2 or tert‐butyl hydroperoxide (TBHP). When using molecular O2 as the oxidant, an additional gas phase has to be brought into contact with the liquid phase and the dispersed solid catalyst, resulting in two‐phase boundaries and an increased possibility of mass transfer limitations. The major difference between O2 and peroxides lies in the electronic state of the molecules. Since the electronic ground state of O2 is a triplet state, its reaction with singlet‐state substrates is forbidden by quantum mechanical selection rules. [96] Combined with the highly stable O=O bond and the positive free energy of the one‐electron transfer from O2 to superoxide, a high activation energy barrier results in oxidation reactions with molecular O2. [96b] By comparison, the partially reduced molecular oxygen species like superoxide and peroxide are considerably more reactive. The activation of O2 requires a spin‐transfer by partial reduction, while peroxide oxidants are typically activated by partial decomposition to reactive radicals.[ 69b , 95g , 95l , 96b , 97 ]
Compared with aerobic oxidation, the required reaction temperatures for alcohol oxidation with peroxides are lower due to the higher reactivity of peroxides, typically between ambient temperature and 90 °C.[ 69b , 95g , 95i , 95j , 95l , 95n ] Higher temperatures usually result in the unproductive decomposition of peroxides, known to start for TBHP at about 70 °C for example. [98] To compensate for the partial unproductive loss of oxidants, peroxides are used in excess to the reactants up to 10 times, usually 2–5 times.[ 69b , 95g , 95n , 99 ] The oxidation reactions with peroxides are usually performed at ambient pressure using acetonitrile,[ 69b , 95n , 97b ] water,[ 95e , 95i , 99 ] or tert‐butanol [95j] as solvents. The solvent‐free oxidation with TBHP has also been reported.[ 95g , 95l ] The oxidation reactions with O2 are performed at elevated temperatures, ranging from 60 to 200 °C.[ 67 , 95a , 95h ] O2 is added to the reaction mixture either by bubbling through the slurry at ambient pressure or by applying high pressures in an autoclave. If the oxidation with O2 cannot be performed solventless, typical solvents like water, acetonitrile, toluene, or N,N‐dimethylformamide (DMF) are used.[ 67 , 95d , 95h , 95k , 95n ]
Leaching is a critical issue, especially for large‐scale applications, and heterogeneous catalysts have been termed as “trojan horses” by Sheldon et al. [100] Thus, leaching of the active species into solution needs particularly to be considered in liquid‐phase oxidation of alcohols, especially when strongly coordinating solvents are used. Despite the importance of leaching, Sheldon pointed out that the resistance properties against leaching are not investigated thoroughly in all studies. Three scenarios in terms of leaching are described: 1) the metal component leaches from the solid catalyst but is not active as a homogeneous catalyst, 2) the metal component leaches and acts as an effective homogeneous catalyst, or 3) the reaction proceeds truly heterogeneously without metal leaching. [100] Because the leached amount is typically small, recycling experiments without significant loss of activity cannot completely prove the heterogeneity of the reaction. [100] In hot‐filtration experiments, the solid particles are filtrated from the reaction solution and the filtrate is exposed to the same reaction conditions as with catalyst to check whether alcohol conversion still progresses. It is vital that hot filtration is performed at the reaction temperature because re‐adsorption of the leached active species on the catalyst surface is possible upon cooling. [100] Nevertheless, this experiment still does not rule out scenario 1. Rigorous proof of heterogeneity is provided by analyzing the filtrate for leached species by ICP‐MS, ICP‐OES, or similar methods for trace analysis. Closely related to leaching of the active component is also the overall stability of the materials. Reusability studies are well suited to evaluate the stability of the catalysts and usually no change or a slight decrease in catalytic efficiency is found.[ 67 , 69b , 95c , 95e , 95n , 101 ] These changes can affect both the activity and the selectivity of the oxidation catalysts.[ 69b , 95e , 95n , 101 ] Typically used methods to characterize the spent catalysts are XRD to investigate phase transformations, electron microscopy methods paired with EDX spectroscopy to reveal morphological changes or changes in the local chemical composition, and XPS to monitor changes in surface oxidation state.[ 67 , 69b , 95n , 101 ]
Compared with gas‐phase reactions, using a solvent in liquid‐phase oxidation of alcohols makes the interaction forces considerably stronger and more variable. [102] Because the solvent is used in large amounts, its environmental impact and safety issues need to be evaluated critically. In addition, the stability and reactivity of the solvent under reaction conditions are also rather important. Solvents in liquid‐phase oxidations are often non‐innocent, being oxidized or reduced during the reaction, and may even serve as the actual oxidants.[ 102 , 103 ] Mallat and Baiker [102] identified an unusual high solvent specificity and unexpected byproducts as warning signs for non‐inert solvents. If the solvent is not considered in these cases, erroneous mechanistic conclusions are inevitable. [102] Dimethyl sulfoxide (DMSO) is a “sacrificial” solvent often used in oxidation reactions and can serve as the oxidant by deoxygenation, yet it is also easily oxidized by hydroperoxides. [102] Acetonitrile is often described as the best solvent for alcohol oxidations with hydroperoxides.[ 69b , 97b , 104 ] It is known that nitriles are reactive towards the decomposition of H2O2 and organic hydroperoxides such as TBHP,[ 102 , 105 ] forming an amide as the final, inactive product. Another commonly used solvent is DMF, which is a versatile solvent owing to its polar and aprotic nature. It can be the reactant in various organic transformations. [106] DMF and other N,N‐dialkyl amides are found in many studies as the best solvent for aerobic oxidation reactions with CoOx or FeOx catalysts and are sometimes a prerequisite for the oxidation to occur.[ 95n , 102 , 107 ] The reason for this high solvent specificity is a matter of intense debate. According to some authors, specificity is ascribed to the high dielectric constant of DMF or the high O2 solubility.[ 102 , 105 ] Other studies attribute the solvent effect to the coordination of Co ions by DMF, changing the electronic structure and thus benefiting the activation of O2.[ 105 , 107e , 107f , 107g ] It has been found that homogeneous CoII complexes in square‐planar coordination by chelating ligands require an additional axial base for the efficient binding of O2. [108] The possible role of DMF to reduce the catalyst and restart the catalytic cycle has also been postulated. [109] Furthermore, the formation of hydroperoxides from dialkyl amides upon exposure to O2, their decomposition and activity as oxidants have been studied in detail and are known to be catalyzed by transition metal cations.[ 102 , 103 , 110 ] The C−H bonds in N,N‐dialkyl amides are labile and easily broken by oxygen‐centered radicals, [111] making the H abstraction by radical intermediates possible.
An important aim in the liquid‐phase oxidation of alcohols over metal oxides is to correlate structure, composition, and resulting catalytic properties. Synergistic effects between different metals have been frequently observed and present a straightforward approach for catalytic improvement. In a study of mixed Ni−Co oxide catalysts supported on the nitrogen‐doped ordered mesoporous carbon FDU‐15, the strong interaction between NiO and Co3O4 nanoparticles made the mixed metal materials more active in aerobic oxidation of benzyl alcohol compared with the monometallic samples. [95h] Using a SrMnO3 perovskite catalyst in the aerobic oxidation of 1‐phenylethanol revealed synergy between Sr and Mn, resulting in better catalytic properties than oxides containing only Mn. [95k] As LaMnO3 was nearly inactive, the importance of the MnIV oxidation state was postulated. A series of A‐site doped La1‐xCexCoO3 perovskites were studied in the aerobic oxidation of benzyl alcohol and showed enhanced activity of La0.9Ce0.1CoO3 compared with LaCoO3. [95m] The substitution of Co ions in Co3O4 by Fe ions was found to be detrimental in the aerobic oxidation of isopropanol, benzyl alcohol, and cinnamyl alcohol.[ 67 , 95n ] For the selective oxidation of isopropanol over Co1+xFe2‐xO4 samples, ensembles of Co3+ cus (coordinatively unsaturated) sites were identified as the active sites, which are assumed to consist of more than six Co ions (Figure 7). [67] Contrary to aerobic oxidation reactions, synergistic effects were observed when small amounts of Fe were present in CoxFe3‐xO3 or LaCoxFe1‐xO3 catalysts used for the oxidation of benzyl alcohol and cinnamyl alcohol with TBHP.[ 69b , 95n ] A different approach is the utilization of the Mott‐Schottky effect to modify the electron density of cobalt nanoparticles supported on nitrogen‐rich carbon materials. [95c] The heterojunction between the Co nanoparticles and the semiconductive support alters the electronic properties of the system, and the resulting catalyst showed significantly enhanced catalytic properties in the oxidation of benzylic alcohols. [95c]
Figure 7.
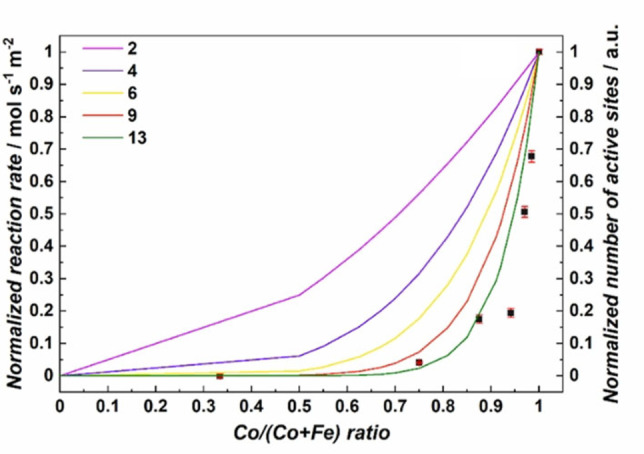
The normalized reaction rate of 2‐propanol oxidation plotted as a function of the Co/(Co+Fe) ratio of the Co1+xFe2‐xO4 samples and normalized number of active sites depending on the number of atoms forming an ensemble. Figure adapted under CC BY 4.0 from Ref. [67].
Extensive efforts have been dedicated to analyzing the reactants, products, catalysts, and intermediates in liquid‐phase oxidation reactions by in situ and operando measurements. By IR spectroscopy, it is possible to study different dioxygen species due to the characteristic stretching vibrations of adsorbed molecular oxygen, superoxide, and peroxide.[ 96a , 113 ] To identify a superoxide intermediate, in situ IR spectroscopy has been used in combination with isotope labeling using 18O2 over a SrMnO3 catalyst. [95k] By applying in situ ATR‐IR spectroscopy, the reaction progress in cyclohexene oxidation was monitored. [114] Vibrational spectroscopy furthermore enabled the study of the interplay of a Co‐based catalyst with DMF as the solvent. [107g] Interactions between catalyst and solvent were also studied by UV/Vis spectroscopy.[ 107e , 107g ] EPR spectroscopy allows identifying the paramagnetic oxygen species in the reaction mixtures derived from the oxidants.[ 96a , 109 , 113a , 115 ] By this method, it was possible to identify a Co‐superoxo complex as the key intermediate in aerobic oxidation reactions with a Co‐containing MOF catalyst. [109] EPR spectroscopy was also used to study the radical intermediates in the oxidation of benzyl alcohol over a cobalt catalyst in the presence of methanol. [112] EPR experiments using α‐(4‐pyridyl‐1‐oxide)‐N‐tert‐butylnitrone (POBN) as the spin‐trapping agent revealed the presence of ⋅CH2OH radicals in excess methanol, also proven by DFT simulations of its adduct with POBN and simulation of the EPR spectrum (Figure 8a&c). [112] If methanol was present as the limiting reactant, a more complex EPR spectrum was obtained (Figure 8b). By similar calculations, the additional presence of hydrogen radicals was confirmed and explained by the additional spectral features (Figure 8d). DFT and quantum chemical calculations are commonly implemented to support EPR results, achieving good agreement between experimental and simulated spectra.[ 112 , 116 ] DFT methods have also been used to calculate the adsorption energies of reactive intermediates like radicals and peroxides on CNT surfaces in liquid‐phase ethylbenzene oxidation. [117] In situ XAS methods were also employed to study oxidation reactions in the liquid phase, but have been mostly limited to noble metal catalysts up to now.[ 109 , 118 ]
Figure 8.

EPR investigation of radical intermediates formed in the oxidation of benzyl alcohol over a cobalt catalyst in the presence of methanol, (a) experimental and simulated EPR spectrum with methanol in excess, (b) experimental and simulated EPR spectrum with methanol as limiting reactant, (c) and (d) calculated structures and EPR parameters of the adducts of POBN with a⋅CH2OH or hydrogen radical, respectively. Figure reproduced with permission from Ref. [112] Copyright 2018, the Royal Society of Chemistry.
To understand the complex mechanistic interplay of oxidants, reactants, solvents, and catalysts, careful analyses of all reaction products are crucial to avoid overlooking unexpected reaction paths. The abovementioned spectroscopic methods, mostly in situ IR and EPR spectroscopy, offer the opportunity to detect highly reactive intermediates. A straightforward approach to identify free radical reactions and specific radicals is the addition of radical scavengers to the reaction solution.[ 69b , 95c , 95n , 107a , 107b , 107e , 109 ] Organic molecules to quench radicals are substituted phenols like hydroquinone or butylated hydroxytoluene. [119] Inspired by photochemistry, scavengers for specific radicals are also used, such as benzoquinone for superoxide or alcohols like tert‐butanol or isopropanol for hydroxyl radicals. [120]
In the future, the selective alcohol oxidation catalyzed by metal oxides in the liquid phase needs to focus on unraveling the real structure of the catalysts beyond the composition, which already requires thorough ex situ investigations of the metal oxides. To achieve an enhanced understanding of reaction pathways and mechanistic details, control experiments and in situ analysis methods such as IR and EPR spectroscopy need to be combined with theoretical calculations. Since catalysts are often metastable and the structure depends critically on the chemical environment, in situ and operando techniques combined with computational modeling are required to identify the active sites exposed by the metal oxide catalysts. [121] Further improvements of in situ and operando characterization techniques including XRD, XPS, electron microscopy, and XAS for liquid‐phase applications are needed to gain more insight in the real structure of the working oxide catalysts. [121] Molecularly defined models representing structural motifs of the solid metal oxides can provide further insight and build a bridge to molecular catalysis. [122]
4. Electrocatalysis
The oxidation of alcohol receives great interest among electrochemists. Works investigating the electrochemical oxidation of short‐chain alcohols such as methanol or ethanol have attracted particular attention, [123] as it can serve as the anode reaction in direct alcohol fuel cells. [124] Here, complete oxidation of the alcohol is desired, in order to achieve maximum utilization of the chemical energy stored in the alcohol. Hence, much of the works inspired by fuel cell applications has focused on electrodes featuring platinum containing catalysts, [124] which oxidize alcohols at least partially to CO2. [125] In contrast to platinum electrods, transition metal oxides (TMO), [126] as well as gold electrodes, [127] do not promote the cleavage of the C−C bond readily and oxidation of primary alcohols usually stops at the corresponding carboxylic acid, while secondary alcohols are oxidized to the corresponding ketones. [128] The tendency towards partial oxidation has rendered gold and transition metal oxides uninteresting anode materials for fuel cell applications. However, the electrochemical oxidation of alcohols at transition metal oxides is highly interesting for synthetic purposes and the reaction can be employed for a large number of intricate and valuable chemicals (e. g., ascorbic acid). [128] Note also that the oxidation reaction can be specific towards the alcohol functional group, which allows the oxidation of allylic alcohols to α,β‐unsaturated ketones. [128] The electrochemical oxidation of alcohols at nickel anodes for synthetic purposes has been reviewed by Schäfer. [128]
The key advantage of electrochemical alcohol oxidation compared to its thermal catalytic counterpart lies in the fact that it can serve as the counter reaction to the electrochemical hydrogen evolution, that is, alcohol oxidation can substitute the oxygen evolution reaction (OER) at the anode [129] which yields only a low value by‐product. Instead, the oxidation of chemicals like HMF [126a] and glycerol [129] that are available in bulk quantities could yield value added products. In addition, alcohol oxidation at the anode would reduce along with the cell voltage the energy costs of water electrolysis. [129] For this reason, the electrochemical oxidation of alcohols, in particular, the oxidation of HMF to FDCA has attracted much interest. The latter reaction proceeds with high current density and efficiency at a number of electrode materials: (hydr)oxides such as NiFe layered double hydroxides [126a] , NiOOH,[ 126c , 126d ] CoOOH,[ 126d , 130 ] FeOOH, [126d] NixCo3‐xO4, [126e] Cu(OH)2. [126f] Since alkaline water electrolyzers employ nickel anodes, [131] stability issues due to catalyst corrosion are not expected to pose a major challenge when shifting the anode reaction to electrochemical alcohol oxidation. Indeed, the reduced potential at the anode might even allow the use of cheaper anode materials such as iron, which is prone to undergo corrosion during electrochemical oxygen evolution. [132] In view of the potential technological importance of electrochemical alcohol oxidation at transition metal oxides, it is worthwhile to briefly review what is known about the mechanism of this reaction as this might spark improved catalyst design.
When studying the oxidation of various alcohols at Ni, Ag, Cu and Co anodes, Fleischman et al. noted that the oxidation of organic compounds coincides with the oxidation of the electrode surface.[ 126g , 133 ] That is, the oxidation of the alcohol proceeds at potential that are associated with the transition Ni(OH)2→NiO(OH), Co(OH)2→CoO(OH), AgI 2O→(AgIAgIII)O2 and CuO→Cu2O3, respectively. [133] This is illustrated for nickel anodes by the measurements presented in Figure 9, [134] where the curves represent the linear sweep voltammograms (LSVs) taken at the Ni anode in the blank electrolyte (black) and in in the presence of the organic compounds (red). Acetaldehyde (A) and benzaldehyde (B) are considered here alcohols because the electroactive species for aldehyde oxidation is the diol.[ 128 , 135 ] The peak in the blank electrolyte at 0.45 V is due to the oxidation of Ni(OH)2 to NiO(OH), [134] whereas OER dominates at potentials beyond 0.6 V. [134] The additional current in the red curves of Figure 9A and B indicates the oxidation of acetaldehyde and benzaldehyde, which parallels the oxidation of the electrode surface. This observation was interpreted in terms of an indirect mechanism in which the higher surface oxide acts as the oxidizing agent of the alcohol and is reduced in the process.[ 126g , 133 , 134 ] This view is supported by cyclic voltammetry (CV) studies: the peak associated with the reduction of the higher surface oxide is diminished in the presence of alcohols compared to a CV taken in the blank electrolyte.[ 126g , 133 , 136 ] The reduction of the higher surface oxide in the presence of the alcohol is also evidenced by a rapid decrease of the open circuit potential to a value corresponding to that of the lower surface oxide. [134]
Figure 9.
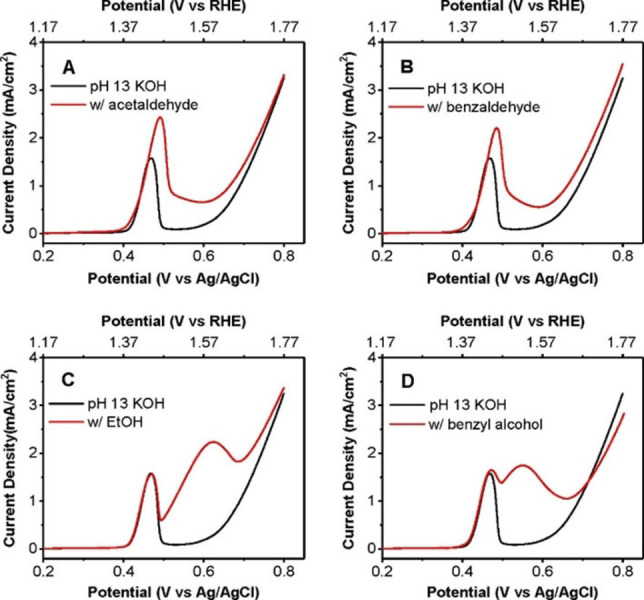
LSVs using a Ni(OH)2 working electrode in a pH 13 solution without (black) and with (red) 10 mM acetaldehyde (A), 10 mM benzaldehyde (B), 10 mM ethanol (C), and 10 mM benzyl alcohol (scan rate 10 mV/s; D). Reprinted with permission from Ref. [134] Copyright 2020, American Chemical Society.
However, the indirect mechanism was also criticized, as alcohol oxidation does not always coincide with the formation of the higher surface oxide.[ 126c , 134 , 137 ] This is shown in Figure 9C and D for the oxidation of ethanol and benzyl alcohol at Ni anodes, which occurs at potentials more positive than the formation of the surface oxide. Hence, a so‐called potential dependent pathway of alcohol oxidation has been proposed.[ 126c , 134 ] However, Bender et al. have shown that ethanol oxidation occurs at a potential where the average valency of Ni in the surface oxide is about +3.3, [134] suggesting a reaction between ethanol and Ni4+ species (instead of Ni3+). Since Ni4+ forms at more positive potentials, [138] the oxidation of ethanol may still follow an indirect pathway. [134]
Still, a mechanism that considers the higher surface oxide merely a charge transfer shuttle, in which surface atoms can freely alternate between different valence states, cannot account for all experimental observations. First, the average oxidation state of the surface oxide should drop upon exposure to alcohol. [136] However, XANES measurements could not confirm this for Co3O4, [136] which maintained its oxidation state upon the addition of glycerol. [136] Second, it would be expected that the activity of the TMO correlates with the potential at which the electrode surface is oxidized. Studying mixed Co/Ni oxides of various compositions, Sun et al. were able to enhance the performance of the catalyst for methanol [139] and ethanol [140] oxidation by adjusting the Ni content to 46 %. Yet, for a range of materials featuring different Ni contents, no obvious correlation between the potential of surface oxidation and catalytic performance exists.[ 139 , 140 ] Hence, other aspects of alcohol oxidation such as the adsorption of the alcohol[ 133 , 134 , 139 , 140 ] and the cleavage of the C−H bond[ 133 , 134 ] must be promoted by the higher surface oxide, too, and might be more important than the charge transfer.
The need for alcohol adsorption is evident from the dependence of the current due to alcohol oxidation on the alcohol concentration: Following the behavior of an adsorption isotherm the current first increases with the alcohol concentration and then enters a plateau upon further addition of the alcohol. [133] Also, electrochemical impedance spectra taken at various TMOs highlight the importance of alcohol adsorption and more importantly, of the desorption of the reaction product.[ 139 , 140 ] Note in this context that it is plausible that the formation of carboxylic acids leads to the blockage of active surface sites, as they tend to adsorb strongly on metal oxides. [141]
Figure 10 shows the limiting current of alcohol oxidation plotted against the OH− concentration in the electrolyte. In the context of Figure 10, limiting current refers to the current plateau that is reached at high potentials when alcohol oxidation is studied by cyclic voltammetry in agitated electrolytes. It is shown for 6 different alcohols that the limiting current of alcohol oxidation increases first linearly with the OH− concentration and, then, levels off to reach a plateau value at sufficiently high OH− concentrations. [137b] Comparison of curve e (ethanol) with curve g (propargyl alcohol) shows that the effect of the OH− concentration depends on the identity of the alcohol. That is, the OH− concentration at which the limiting current of alcohol oxidation levels off depends on the nature of the alcohol, whereas the linear increase of the limiting current at low OH− concentrations is independent of the identity of the alcohol. This is also true for the concentration of the alcohol (compare curve e with curve f and curve g with curve h). The pH at which the limiting current of alcohol oxidation becomes independent of the OH− concentration depends on the concentration of the alcohol in the electrolyte. However, the initial linear increase of the limiting current at low OH− concentration is not affected by the alcohol concentration. Robertson suggested that the initial linear increase of the limiting current is due to mass transport limitation of OH− to the electrode surface. [137b] That is, at a certain current density consumption of OH− during alcohol oxidation outpaces diffusion of OH− to the electrode surface. Hence, the limiting current can only increase further when mass transport of OH− to the surface is enhanced by increasing the OH− concentration in the electrolyte. Once the reaction rate is no longer limited by the availability of OH− at the electrode surface the limiting current will not continue to increase with electrolyte pH. However, the mechanistic role of OH− in alcohol oxidation at transition metal oxides s might be more complicated than that of a mere reactant: It is widely accepted that alcohol oxidation at Au electrodes, where the current due to alcohol oxidation increases with the electrolyte pH as well, proceeds via the oxidation of the alcoholate. [127] That is, a sufficient OH− concentration is required to form the electroactive species in the first place. [127] This was derived from a general trend (with exceptions such as methanol or CF3CH2OH) [127b] that the current due to alcohol formation increases as the pKa‐value of the alcohol increases. Similar studies are missing for alcohol oxidation at transition metal oxides, but considering the effect of the OH− on the reaction kinetics, the involvement of the alcoholate as the electroactive species is also plausible here. This is particularly true when it is considered that the negatively charged alcoholate should be better suited to compete with carboxylates for adsorption sites than the neutral alcohol. Indeed, based on DFT calculations Bender et al. suggested that the oxidation of ethanol at NiO(OH) proceeds via the adsorption of ethanolate. [134]
Figure 10.
Peak current for the oxidation of alcohols at a polished nickel anode as a function of [OH−]. Compounds are as follows: (a) 0.055 M pentan‐1‐ol; (b) 0.219 M butan‐1‐ol (c) 0.266 M propanol, (d) 0.782 M isopropanol, (e) 0.343 M ethanol, (f) 0.857 M ethanol; (g) 0.338 M propargyl alcohol, (h) 0.676 M propargyl alcohol. Reprinted with permission from Ref. [137b] Copyright 1980, Elsevier.
The oxidation of alcohols to carbonyl compounds or carboxylic acids requires the abstraction of one or two H‐atoms from the OH‐group bearing C‐atom. The oxidation of the alcohol therefore involves the cleavage of the C−H bond. In fact, based on the primary kinetic isotope effect observed for the oxidation of methanol it can be derived that this is the rate determining step of alcohol oxidation at transition metal oxides. [133] The rate constant for the reaction decreases by a factor of 7 when the C−H bond is replaced by a C−D bond. [133] Also, DFT calculations suggest that the adsorption of the ethanolate to the NiO(OH) surface is followed by the transfer of a hydride ion to the oxide surface. [134] This falls in line with the oxidation of alcohols at Au electrode, where a primary kinetic isotope effect was observed in the case of 2‐propanol. [142] Furthermore, at gold electrodes where the readiness of alcohols to undergo oxidation depends on the C−H‐bond strength: secondary alcohols featuring a lower C−H‐bond strength than primary alcohols undergo oxidation more readily. [143] In account of the strong C−H‐bond in CF3CH2OH no oxidation of this compound can be achieved at gold electrodes in spite of its high pKa‐value. [127b]
Although it is known that the higher surface oxide must promote alcohol adsorption and facilitate the cleavage of the C−H bond, no structure‐activity relationship has yet been established. Comparative studies have shown that the activity of TMOs decreases in the order Cu2O3, NiO(OH), (AgIAgIII)O2, CoO(OH), [133] that the activity of FeO(OH) is lower than that of NiO(OH) or CoO(OH), [42] and that the activity of NiCo2O4 surpasses that of NiO or CoO.[ 139 , 140 , 144 ] However, to achieve the rational design of improved catalysts, a better understanding of how the properties of the transition metal oxide affect different aspects of the reaction is needed.
5. Photocatalysis
Besides wastewater treatment, [145] H2 production by overall water splitting, [146] and photo reforming of oxygenates, [147] also selective alcohol oxidation[ 147 , 148 ] represents an environmentally friendly and efficient application of heterogeneous semiconducting photocatalysts. Alcohols from bio‐sources are interesting reactant molecules for hydrogen production in the quest to set up a carbon‐neutral and thus environmentally benign process. [149] With a report on qualitative approaches towards useful photocatalytic materials, Parkin and coworkers provided a critical perspective on some of the main factors affecting the assessment of photocatalytic materials with application potential. [150] They conclude that although the area of photocatalysis may have reached maturity from a fundamental perspective, the research efforts in photocatalyst design and engineering are still in their infancy. Thus, the search for new materials, especially alternatives for TiO2, is a field of explorative nature.
Interesting candidates include perovskites with the general formula ABO3. Here, SrTiO3 can be regarded as an ideal model compound due to its non‐stoichiometry, polarizability, and similar bandgap as TiO2 (3.25 eV). [151] Its wide range of applications comprising photocatalysis, thermoelectric materials, capacitors and anode materials in solid oxide fuel cells aroused interest and led to the development of various synthesis routes. SrTiO3 production and modification via polycondensation of metal chelate complexes and alkoxysilanes, [151] radical polymerization with acrylic precursors, [152] or electric field experiments [153] proved to provide good control of the oxide properties. To achieve a high activity under solar light irradiation, doping and/or the deposition of cocatalysts are inevitable. As an example, Rh‐ or Fe‐doped SrTiO3 loaded with Pt nanoparticles achieved high apparent quantum efficiency (49.5 % and 13.2 %) in selective benzyl alcohol oxidation under visible light.[ 148a , 148b ]
Spinel ferrites with the general formula MFe2O4 (M=Zn2+, Fe2+, Co2+, Ni2+, Mn2+, Cu2+) are also of great interest in photocatalysis due to their band structure, low toxicity, and interesting magnetic properties. Its capability to alter its physical properties and morphologies renders CoFe2O4 a significant ferrite. [154] It crystallizes in a face‐centered cubic (fcc) structure with a unit cell of eight formula units. In its normal spinel structure, Co2+ is tetragonally coordinated and Fe3+ occupies the octahedral sites. The inverse spinel structure has Fe3+ on the tetragonal sites whereas the octahedral sites are equally occupied by Co2+ and Fe3+. [145] The structure of real CoFe2O4 lies in between these two structures and it is described by the inversion parameter d, which refers to the fraction of tetrahedral sites occupied by Fe3+ ions. Thus, the formula can be written as (Co2+ 1‐dFe3+ d)A[Co2+ d Fe3+ 2‐d]BO4. [155] In order to overcome basic problems regarding the application of pure CoFe2O4 in photocatalysis like leaching, agglomeration of nanoparticles and low activity in high and low pH media, researchers focus on novel synthesis methods to enhance the photocatalytic performance of CoFe2O4.[ 145 , 154 , 156 ] Heterojunction formation with a second semiconductor is known as the generation of an interfacial band arrangement between both semiconductors. A proper connection may result in decreased charge carrier recombination and, correspondingly, in higher photocatalytic efficiency. Radovanovic et al. [157] demonstrated the superior efficiency of CoFe2O4/Ag2MoO4 (82 %) compared with CoFe2O4 (12 %) or Ag2MoO4 (48 %) separately in selective benzyl alcohol oxidation. Its magnetic properties also simplify the separation of CoFe2O4‐containing photocatalysts from the reaction mixture thus increasing reusability.
In contrast to thermal catalysis, the investigation of heterogeneous photocatalytic reactions needs a light source to illuminate the photocatalyst requiring a sufficient understanding of photon transport phenomena including radiation absorption and scattering by the catalyst particles. [158] Several publications on proper photocatalytic reactor and experimental design [159] supply useful information and guidelines to achieve results from which reliable conclusions can be drawn.
Various concepts regarding set‐ups and reactors have been applied so far. A few examples of set‐ups and photoreactors are displayed in Scheme 2. Busser et al. [160] used a homemade double‐walled glass reactor where the light source (Hg immersion lamp) was placed inside the reactor, surrounded by the catalyst suspension. In this way, the complete light intensity was forced to pass through the liquid containing the dispersed photocatalyst and the dissolved reactant. A semi‐batch operation mode allowed continuous flushing of the suspension and quantitative online monitoring of the evolving gaseous products, for example allowing the in situ photo‐deposition of cocatalysts for the hydrogen evolution reaction in the presence of methanol as reductant.
Scheme 2.

Different set‐ups (I, II, III, and IV) and reactors (V, VI, VII, and VIII) in photocatalytic research. (I) Visible‐light photocatalytic aza‐Henry reaction in a glass microreactor (a), photocatalytic membrane reactor for oxidation of citronellol (b), and O2 generation with immobilized TDCPP in a scCO2 continuous‐flow system (c), reproduced with permission from Ref. [174] Copyright 2018, Wiley; (II) Set‐up for photocatalytic overall liquid phase water splitting, reprinted with permission from Ref. [160] Copyright 2012, Wiley; (III) Set‐up for photocatalytic Deacon reaction in the gas phase reprinted with permission from Ref. [161] Copyright 2020, American Chemical Society; (IV) Typical reaction chamber for photocatalytic organic conversion, reproduced with permission from Ref. [162] Copyright 2019, Royal Society of Chemistry; (V) Three distributor/collector designs for flow equipartition: manifold‐type design with a lateral inlet and outlet (a), bifurcation/bifurcation design (b) and bifurcation/chamber design (c) adapted with permission from Ref. [176] (further permissions related this material should be directed to the ACS), along with schematic representation of gas–liquid segmented flow in a microchannel (d), reproduced with permission from Ref. [174] Copyright 2018, Wiley; (VI) Semi‐batch photoreactor; (VII) Flat‐plate photoreactor equipped with flow channel, reprinted with permission from Ref. [161] Copyright 2020, American Chemical Society; (VIII) Possible designs for ALD coated continuous flow reactors: regular continuous flow reactor (a), semi‐baffled continuous flow reactor (b), fully baffled flow reactor (c), and micro‐channel reactor (d), reprinted with permission from Ref. [162] Copyright 2019, Royal Society of Chemistry.
Noël describes the use of continuous flow microreactors which have large interfacial areas due to a high surface to volume ratio, less mass transfer limitation issues and optimized illumination conditions. [158] In the photocatalytic aerobic oxidation of thiols to disulfides over TiO2, the group found a tremendous increase of the overall reaction rate compared with a classical batch reactor.
Rath et al. [161] designed a novel corrosion‐resistant heatable flat‐plate reactor for the aerobic HCl oxidation over TiO2 in the gas phase. The photocatalyst deposited as a thin layer on a quartz glass plate was placed in the reactor and illuminated using a UV‐LED array. This arrangement allowed a precise measurement of the catalyst layer thickness and of UV transmission and irradiance at the catalyst surface. Therefore, an accurate calculation of the apparent quantum yield for each catalyst was possible. The use of ALD for innovative preparation of photocatalysts and photocatalytic reactor designs was reviewed recently by Eswar et al. [162]
Whereas the photocatalytic mechanism of water splitting has up to now been thoroughly investigated by Domen and coworkers, [163] investigations on the mechanism of photocatalytic alcohol oxidation are also a recent topic of interest. In this respect, Mul et al. [164] provided an interesting contribution on the selective photocatalytic oxidation of cyclohexanol to cyclohexanone in which they propose a detailed mechanism. Quantum efficiency is a specific topic involving charge carrier generation and recombination. An example of a thorough and detailed investigation regarding this aspect concerning photocatalytic methanol reforming was published by Kubacka and coworkers. [165]
In literature, different approaches to elucidate photocatalytic mechanisms are described in order to achieve a fundamental understanding of the involved processes, providing the basis for a knowledge‐based improvement. Some authors concentrate on the organic chemistry occurring on the catalyst surface or in the reaction medium and include the catalyst just as a source for providing reactive charge carriers. In this respect, Schnee et al. [166] focused on the role of surface formate species on a TiO2 surface as a crucial intermediate in the photocatalytic oxidation of methanol. Using ultra‐fast time‐resolved quantum cascade laser diagnostics, the group monitored the kinetic behavior of formate species with the formation of CO2 and methyl formate and revealed that the surface formate reaction is the rate‐determining step of the process. Understanding the role of different solvent molecules for practical solid‐liquid heterogeneous photocatalytic reactions is critical for determining the pathway of the reactions in the presence of a liquid phase. As an example, the influence of the solvating water molecules was demonstrated by Xu et al. [167]
A different approach is to include the photocatalyst and all processes regarding charge carrier generation and recombination into a combined experimental and theoretical approach. Xiong et al. [148d] describe the different reactive oxygen species and challenges regarding the selectivity to desired products in the photocatalytic oxidation of organic substrates. In their review, six strategies, namely band gap engineering, metal loading, hybrid materials, defect engineering, morphology control and crystallization control, are described to control the production of reactive oxygen species, suppress undesired side‐reactions and, therefore, improve selectivity.
Since the beginning of the century, operando spectroscopy has played an increasing role in the analysis of catalysts. [168] This development made it possible to see “catalysts in action”. Whereas in situ methods work under catalytic conditions, the operando mode couples in situ spectroscopy with determining catalytic activity and selectivity. Thus, analytical tools and catalytic testing are combined.
Wachs and coworkers performed important pioneering work and demonstrated the operando technique as a powerful tool for catalyst characterization. [169] A recent interesting example concerns the characterization of metal catalysts supported on reducible metal oxides such as CeO2 and TiO2 [170] , allowing the determination of the nature of unique structural properties of the support. In this way, oxygen vacancies, reversible valence changes or surface hydroxyl groups are analyzed as a function of the operating conditions thus enabling the establishment of structure‐activity relationships. The use of operando X‐Ray spectroscopic techniques to investigate electrochemical hydrogen and oxygen evolution reactions including methanol and ethanol oxidation has been recently summarized. [171]
Important milestones were defined by the work of Weckhuysen et al., who recently pointed out the necessity to combine operando spectroscopy with nanosensors for spatial resolution on the catalyst particles and in the reactor. [172] In this way, it is possible to monitor the state of the catalyst and to optimize temperature and regeneration protocols. They highly recommend the technique for applications in areas such as liquid‐phase catalysis, electrocatalysis, and photocatalysis. The application of operando techniques in photocatalysis is still at an early stage, but several profound studies have been published in the last couple of years by Fernández‐García and coworkers. [173] They clearly point out the necessity to match the sample volume illuminated by the excitation radiation with the volume probed by spectroscopy. Thus, dark areas have to be clearly separated from illuminated areas. Moreover, their analysis of the state of the art for the use of operando tools in photocatalysis involves XPS as a surface‐sensitive technique as well as spectroscopies devoted to the study of the reactant, catalyst interface and particularly to adsorbed surface species.
Xiong et al. [148d] identify several major challenges comprising the assessment of the specific role of reactive oxygen species and role of defects (potential recombination sites), the appropriate use of in situ and operando techniques, the improvement of theory, which is still far away from experiments to close this gap, and the application of better methods for efficient catalyst design including the use of single‐atom catalysts. Fernández‐García and coworkers urge that the effect of light should be implemented in kinetic modeling schemes. [173c]
Additionally, future research should focus particularly on differences between illuminated and dark conditions, the identification of (adsorbed) intermediates and their interaction with the catalyst and molecules in the liquid phase, and the separation of processes occurring in the liquid phase and on the surface. In this respect, in particular stability is an important issue that should be addressed more closely. The photocatalyst is a dynamic system and several processes can occur during irradiation and in the dark. [175] Reasons for deactivation can be either photocorrosion (unwanted reduction or oxidation of the (co‐)catalyst), leaching of the cocatalyst, and/or deposition of reaction intermediates/byproducts on the surface. Due to an imbalance of charges generated during irradiation oxidation states of the photoabsorber and cocatalysts might change and this can reduce their stability leading to leaching. Special strategies have to be developed in order to counteract photocorrosion, for example through doping. [174] Blocking of active sites by unwanted intermediates on the surface might be an issue, for example in photocatalytic methanol reforming. A thorough and detailed investigation of reaction mechanisms is recommended to allow control over and avoid unwanted side reactions. [176]
6. Conclusions and Outlook
In this minireview, we reported some of the advances in the field of alcohol oxidation and addressed the most important challenges in designing efficient heterogeneous catalysts and understanding their fundamental properties. Several examples were shown to highlight the complexities in different approaches taken for alcohol oxidations. Considering the introduced methods, one should take note of the fundamental differences in the approaches taken in designing of electro/photocatalysts versus thermocatalysts, as well as studying their reaction mechanisms. However, and regardless of the type and the mode of the reaction, it is necessary to design reliable model catalysts and study them using probe molecules that reflect the complexity of the higher alcohol reactants. The fundamental understanding of metals, in general, is more advanced compared to the oxides, where several challenges still exist in identifying the mechanisms and the nature of the active sites. Nevertheless, it is required in both cases to develop reliable operando characterization techniques to build up the structure−performance correlations. In addition, identifying the reaction mechanisms and the involved intermediate species at the surface are of high importance, especially when a liquid reaction medium is used. In doing so, one should note the possible structural transformations of the catalysts, whether in the bulk or on the surface, to fully address the complexities involved in this reaction. Identifying the structural defects, their formation or transformation during the course of the reaction and their contribution to the overall activity is still a challenging task yet of significant importance. In this regard, modeling of the catalysts and their evolution under reaction conditions with advanced calculation tools is also required in order to elucidate possible involved mechanisms and the nature of the active sites. The authors of this minireview are collaborating on several projects to advance the existing knowledge in this field.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Sharif Najafishirtari received his M.Sc. in chemical engineering from the University of Tehran (Iran) in 2010. After a few years of working in the chemical industry, he reinitiated his research career and obtained his dual degree Ph.D., as a Marie Skłodowska‐Curie early‐stage researcher, from the Italian Institute of Technology in collaboration with the University of Genova (Italy) and University of Regensburg (Germany) in 2017. His Ph.D. work specifically focused on synthesizing tailored metal/oxide nanocrystals and their applications as model catalysts to build up structure‐activity correlations. Currently, he is a postdoctoral researcher at the University of Duisburg‐Essen where he is developing projects for spectroscopy‐based operando characterization of oxide catalysts in partial oxidation reactions.

Biographical Information
Klaus Friedel Ortega holds a ‘Diploma’ in industrial engineering with a major in technical chemistry from the Technische Universität Berlin. In 2014, he completed his PhD in the group of Prof. Schlögl at the Fritz Haber Institute of the Max Planck Society in Berlin. He then joined the group of Prof. Behrens at the University of Duisburg‐Essen as a postdoctoral researcher. In 2019, he was appointed tenured academic lecturer. Since 2020 he is working as a group leader for heterogeneous catalysis in the group of Prof. Behrens at the University of Kiel. His research focuses on the development of novel nanostructured catalysts for oxidation and hydrogenation/dehydrogenation reactions.

Biographical Information
Mark Douthwaite is a postdoctoral research associate at the Cardiff Catalysis Institute under the supervision of Professors Graham Hutchings and Stuart Taylor. His primary interests are focused on the design, synthesis and characterization of heterogeneous catalysts for selective oxidation.

Biographical Information
Samuel Pattisson is a postdoctoral research associate at the Cardiff Catalysis Institute working with Prof. Graham Hutchings. His research is focused on the design, evaluation and characterization of novel heterogeneous catalysts for the selective and total oxidation of molecules for academically and industrially relevant applications.

Biographical Information
Graham J. Hutchings is Regius Professor of Chemistry at Cardiff University. His early career was with ICI and AECI Ltd where he became interested in gold catalysis. In 1984 he moved to academia and has held chairs at the Universities of Witwatersrand, Liverpool, and Cardiff. His main interests are in oxidation catalysis with a special interest in catalysis by gold. He was elected a Fellow of the Royal Society in 2009, a Member of Academia Europaea in 2010, and a Founding Fellow of the Learned Society of Wales in 2010. He was awarded the Davy Medal of the Royal Society in 2013, the ENI Award for Advanced Environmental Solutions in 2017, the RSC Faraday Lectureship and Prize in 2018, a CBE in the Queen's Birthday Honours List in 2018 and the Michel Boudart Award in 2021.

Biographical Information
Christoph Bondue obtained his Diploma in chemistry at Rheinische Friedrich‐Wilhelms‐Universität Bonn in 2011. Afterward, he joined the group for Prof. H. Baltruschat (Bonn), where he obtained his PhD. In 2016 he started a Postdoc in the Group of Prof. M.T.M. Koper at Universiteit Leiden and joined in 2020 the group of Prof. K. Tschulik at Ruhr‐Universität Bochum, where he is currently a Fellow of the Marie Skłodowska‐Curie COFUND Program. His research is focused on the interaction between electrode surface and substrate molecule and its effect on electrocatalytic reactions.

Biographical Information
Kristina Tschulik holds a ‘Diplom’ in Chemistry from TU Dresden (Germany) and performed her doctoral studies on magnetic field‐assisted structured electrodeposition at IFW Dresden (Germany) until 2012. Afterward, she joined the University of Oxford (UK) as a Marie Skłodowska‐Curie IEF postdoctoral researcher, working on single nanoparticle electrochemistry and quantitative physicochemical analysis of electrochemical reactions. In 2015, Kristina moved to Ruhr University Bochum (Germany) as a Junior Professor, where she has held the Chair of Analytical Chemistry II since 2018. Her research focuses on characterizing the physical properties and intrinsic chemical (re‐)activity of functional nanomaterials for renewable energy technologies, based on advanced electrochemical and spectro‐electrochemical methods.

Biographical Information
Daniel Waffel studied chemistry at the Ruhr‐University Bochum and received his B.Sc. and M.Sc. in 2016 and 2018, respectively. Since 2018 he is a Ph.D. student at the Laboratory of Industrial Chemistry, Ruhr‐University Bochum under the guidance of Prof. Martin Muhler. His research is focused on liquid‐phase oxidation reactions of alcohols and alkenes with mixed metal oxides as heterogeneous catalysts.

Biographical Information
Baoxiang Peng received his B.Sc. and M.Sc. in 2005 and 2008 at Tsinghua University and Ph.D. in 2012 at the Technical University of Munich. After three years of working experience as a chemical engineer in the chemical industry, he joined the Max Planck Institute for Chemical Energy Conversion and Ruhr‐University Bochum as a group leader in 2015. His current research interest is three‐phase redox catalysis under mild conditions.

Biographical Information
Michel Deitermann studied chemistry at Ruhr‐University Bochum (Germany) and received his B.Sc degree in 2018 and M.Sc in 2020. Since 2020, he is working as a doctoral student in the laboratory of industrial chemistry at the Ruhr‐University Bochum under the supervision of Prof. Martin Muhler. His current research topic is heterogeneous semiconductor‐based photocatalysis in the liquid and gas phase.

Biographical Information
G. Wilma Busser is a postdoctoral researcher at the Laboratory of Industrial Chemistry of the Ruhr University Bochum under the supervision of Prof. Dr. Martin Muhler. Her research focuses on heterogeneous redox catalysis in the liquid and gas phase including photocatalysis.

Biographical Information
Martin Muhler received his Ph.D. in 1989 from the Freie Universität Berlin. After a postdoc period at Haldor Topsoe A/S from 1989 to 1991, he returned to Prof. Ertl's group and finished his habilitation in Industrial Chemistry in 1996 (Technische Universität Berlin). In 1996 he was appointed full professor in Industrial Chemistry at the Ruhr‐University Bochum. His research is focused on heterogeneous redox catalysis comprising selective reduction and oxidation, electrocatalysis, and photocatalysis.

Biographical Information
Malte Behrens obtained his doctoral degree at Kiel University in the group of Prof. Wolfgang Bensch in 2006 in the field of solid‐state chemistry and joined the Fritz‐Haber‐Institut in Berlin to work as a postdoc with Robert Schlögl afterward. He stayed in that institute as a group leader and finished his habilitation in 2013 at TU Berlin. In 2014, he was appointed professor for inorganic chemistry at the University of Duisburg‐Essen. In 2020, he joined Kiel University as a full professor of solid‐state chemistry and catalysis. His research focuses on structure‐activity relationships and new materials for heterogeneous hydrogenation and oxidation catalysis.

Acknowledgements
The authors acknowledge funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 388390466 – TRR 247. We also gratefully acknowledge the financial support from the Mercator Research Center Ruhr (MERCUR, Pe‐2018‐0034). S.N. and M.B. acknowledge support by the Open Access Publication Fund of the University of Duisburg‐Essen. C.B. acknowledges that he received funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska‐Curie grant agreement No 801459 – FP‐RESOMUS and was funded by the Deutsche Forschungsgemeinschaft (DFG) under Germany's Excellence Strategy – EXC 2033 – 390677874 – RESOLV. Open Access funding enabled and organized by Projekt DEAL.
S. Najafishirtari, K. Friedel Ortega, M. Douthwaite, S. Pattisson, G. J. Hutchings, C. J. Bondue, K. Tschulik, D. Waffel, B. Peng, M. Deitermann, G. W. Busser, M. Muhler, M. Behrens, Chem. Eur. J. 2021, 27, 16809.
Contributor Information
Dr. Sharif Najafishirtari, Email: sharif.najafishirtari@uni-due.de.
Prof. Dr. Malte Behrens, Email: malte.behrens@uni-due.de, Email: mbehrens@ac.uni-kiel.de.
References
- 1. Wang Q., Chen L., Guan S., Zhang X., Wang B., Cao X., Yu Z., He Y., Evans D. G., Feng J., Li D., ACS Catal. 2018, 8, 3104–3115. [Google Scholar]
- 2.
- 2a. Mehlomakulu B., Nguyen T. T. N., Delichère P., van Steen E., Millet J. M. M., J. Catal. 2012, 289, 1–10; [Google Scholar]
- 2b. Yeo B. R., Pudge G. J. F., Bugler K. G., Rushby A. V., Kondrat S., Bartley J., Golunski S., Taylor S. H., Gibson E., Wells P. P., Brookes C., Bowker M., Hutchings G. J., Surf. Sci. 2016, 648, 163–169. [Google Scholar]
- 3.
- 3a. Zhang S., Li J. P. H., Zhao J., Wu D., Yuan B., Hernández W. Y., Zhou W.-J., He T., Yu Y., Yang Y., Ordomsky V., Li T., Nano Res. 2020, 14, 479–485; [Google Scholar]
- 3b. Zhang L.-L., Hao K., Wang R.-K., Ma X.-Q., Liu T., Song L., Yu Q., Wang Z.-W., Zeng J.-M., Zeng R.-C., Front. Mater. 2020, 14, 52–61. [Google Scholar]
- 4. Katryniok B., Paul S., Dumeignil F., ACS Catal. 2013, 3, 1819–1834. [Google Scholar]
- 5. Murayama T., Katryniok B., Heyte S., Araque M., Ishikawa S., Dumeignil F., Paul S., Ueda W., ChemCatChem 2016, 8, 2415–2420. [Google Scholar]
- 6. Slot T. K., Eisenberg D., van Noordenne D., Jungbacker P., Rothenberg G., Chem. Eur. J. 2016, 22, 12307–11. [DOI] [PubMed] [Google Scholar]
- 7. Mallat T., Baiker A., Catal. Today 1994, 19, 247–283. [Google Scholar]
- 8.
- 8a. Kaneda K., Fujii M., Morioka K., J. Org. Chem. 1996, 61, 4502–4503; [DOI] [PubMed] [Google Scholar]
- 8b. Yamaguchi K., Mori K., Mizugaki T., Ebitani K., Kaneda K., J. Am. Chem. Soc. 2000, 122, 7144–7145. [Google Scholar]
- 9.
- 9a. Abad A., Concepcion P., Corma A., Garcia H., Angew. Chem. Int. Ed. 2005, 44, 4066–9; [DOI] [PubMed] [Google Scholar]
- 9b. Corma A., Domine M. E., Chem. Commun. 2005, 4042–4. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Mallat T., Baiker A., Chem. Rev. 2004, 104, 3037–58; [DOI] [PubMed] [Google Scholar]
- 10b. Davis S. E., Ide M. S., Davis R. J., Green Chem. 2013, 15, 17–45; [Google Scholar]
- 10c. Villa A., Dimitratos N., Chan-Thaw C. E., Hammond C., Prati L., Hutchings G. J., Acc. Chem. Res. 2015, 48, 1403–12. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Sharma A. S., Kaur H., Shah D., RSC Adv. 2016, 6, 28688–28727; [Google Scholar]
- 11b. Carabineiro S. A. C., Front. Chem. 2019, 7, 702; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11c. Davis S. E., Ide M. S., Davis R. J., Green Chem. 2013, 15, 17–45; [Google Scholar]
- 11d. Chan-Thaw C., Savara A., Villa A., Catalysts 2018, 8. [Google Scholar]
- 12. Parmeggiani C., Matassini C., Cardona F., Green Chem. 2017, 19, 2030–2050. [Google Scholar]
- 13. Douthwaite M., Huang X., Iqbal S., Miedziak P. J., Brett G. L., Kondrat S. A., Edwards J. K., Sankar M., Knight D. W., Bethell D., Hutchings G. J., Catal. Sci. Technol. 2017, 7, 5284–5293. [Google Scholar]
- 14. Liu F., Wang H., Sapi A., Tatsumi H., Zherebetskyy D., Han H.-L., Carl L., Somorjai G., Catalysts 2018, 8. [Google Scholar]
- 15. Wei Q., Yu C., Song X., Zhong Y., Ni L., Ren Y., Guo W., Yu J., Qiu J., J. Am. Chem. Soc. 2021, 143, 6071–6078. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Frassoldati A., Pinel C., Besson M., Catal. Today 2011, 173, 81–88; [Google Scholar]
- 16b. Dimitratos N., Villa A., Wang D., Porta F., Su D., Prati L., J. Catal. 2006, 244, 113–121. [Google Scholar]
- 17.
- 17a. D′Agostino C., Feaviour M. R., Brett G. L., Mitchell J., York A. P. E., Hutchings G. J., Mantle M. D., Gladden L. F., Catal. Sci. Technol. 2016, 6, 7896–7901; [Google Scholar]
- 17b. Huang K., Fu H., Shi W., Wang H., Cao Y., Yang G., Peng F., Wang Q., Liu Z., Zhang B., Yu H., J. Catal. 2019, 377, 283–292. [Google Scholar]
- 18. Dodekatos G., Ternieden J., Schünemann S., Weidenthaler C., Tüysüz H., Catal. Sci. Technol. 2018, 8, 4891–4899. [Google Scholar]
- 19.
- 19a. Fristrup P., Johansen L. B., Christensen C. H., Catal. Lett. 2007, 120, 184–190; [Google Scholar]
- 19b. Ide M. S., Davis R. J., J. Catal. 2013, 308, 50–59. [Google Scholar]
- 20.
- 20a. Douthwaite M., Powell N., Taylor A., Ford G., López J. M., Solsona B., Yang N., Sanahuja-Parejo O., He Q., Morgan D. J., Garcia T., Taylor S. H., ChemCatChem 2020, 12, 3097–3107; [Google Scholar]
- 20b. Carrettin S., McMorn P., Johnston P., Griffin K., Hutchings G. J., Chem. Commun. 2002, 696–7. [DOI] [PubMed] [Google Scholar]
- 21. Ouellette R. J., Rawn J. D., 9 - Haloalkanes and Alcohols: Introduction to Nucleophilic Substitution and Elimination Reactions, in Organic Chemistry, R. J. Ouellette, J. D. Rawn, Editors., Elsevier, Boston. 2014, p. 287–331. [Google Scholar]
- 22.
- 22a. Wang H., An K., Sapi A., Liu F., Somorjai G. A., Catal. Lett. 2014. 144, 1930–1938; [Google Scholar]
- 22b. Tatsumi H., Liu F., Han H.-L., Carl L. M., Sapi A., Somorjai G. A., J. Phys. Chem. C 2017, 121, 7365–7371; [Google Scholar]
- 22c. Sapi A., Liu F., Cai X., Thompson C. M., Wang H., An K., Krier J. M., Somorjai G. A., Nano Lett. 2014, 14, 6727–30; [DOI] [PubMed] [Google Scholar]
- 22d. Liu F., Han H.-L., Carl L. M., Zherebetskyy D., An K., Wang L.-W., Somorjai G. A., J. Phys. Chem. C 2018, 123, 7577–7583; [Google Scholar]
- 22e. Wang H., Sapi A., Thompson C. M., Liu F., Zherebetskyy D., Krier J. M., Carl L. M., Cai X., Wang L. W., Somorjai G. A., J. Am. Chem. Soc. 2014, 136, 10515–20. [DOI] [PubMed] [Google Scholar]
- 23. Wei Q., Yu C., Song X., Zhong Y., Ni L., Ren Y., Guo W., Yu J., Qiu J., J. Am. Chem. Soc. 2021. 143, 6071–6078. [DOI] [PubMed] [Google Scholar]
- 24. Zope B. N., Hibbitts D. D., Neurock M., Davis R. J., Science 2010, 330, 74–8. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Tang Z., Cao H., Tao Y., Heeres H. J., Pescarmona P. P., Appl. Catal. B 2020, 263; [Google Scholar]
- 25b. Tang Z., Liu P., Cao H., Bals S., Heeres H. J., Pescarmona P. P., ACS Catal. 2019, 9, 9953–9963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Climent M. J., Corma A., Iborra S., Sabater M. J., ACS Catal. 2014, 4, 870–891. [Google Scholar]
- 27.
- 27a. Zhang J., Jiang Q., Yang D., Zhao X., Dong Y., Liu R., Chem. Sci. 2015, 6, 4674–4680; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27b. Jaiswal G., Landge V. G., Jagadeesan D., Balaraman E., Nat. Commun. 2017, 8, 2147; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27c. Cui X., Huang Z., van Muyden A. P., Fei Z., Wang T., Dyson P. J., Sci. Adv. 2020, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.
- 28a. Underhill R., Douthwaite M., Lewis R. J., Miedziak P. J., Armstrong R. D., Morgan D. J., Freakley S. J., Davies T., Folli A., Murphy D. M., He Q., Akdim O., Edwards J. K., Hutchings G. J., Res. Chem. Intermed. 2021, 47, 303–324; [Google Scholar]
- 28b. Lyu J., Niu L., Shen F., Wei J., Xiang Y., Yu Z., Zhang G., Ding C., Huang Y., Li X., ACS Omega 2020, 5, 16865–16874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sankar M., Nowicka E., Carter E., Murphy D. M., Knight D. W., Bethell D., Hutchings G. J., Nat. Commun. 2014, 5, 3332. [DOI] [PubMed] [Google Scholar]
- 30. Mostrou S., Nagl A., Ranocchiari M., Fottinger K., van Bokhoven J. A., Chem. Commun. 2019, 55, 11833–11836. [DOI] [PubMed] [Google Scholar]
- 31. Xie J., Yin K., Serov A., Artyushkova K., Pham H. N., Sang X., Unocic R. R., Atanassov P., Datye A. K., Davis R. J., ChemSusChem 2017, 10, 359–362. [DOI] [PubMed] [Google Scholar]
- 32. Villa A., Campisi S., Mohammed K. M. H., Dimitratos N., Vindigni F., Manzoli M., Jones W., Bowker M., Hutchings G. J., Prati L., Catal. Sci. Technol. 2015, 5, 1126–1132. [Google Scholar]
- 33. Yang P., Pan J., Liu Y., Zhang X., Feng J., Hong S., Li D., ACS Catal. 2018, 9, 188–199. [Google Scholar]
- 34. Yang P., Douthwaite M., Pan J., Zheng L., Hong S., Morgan D. J., Gao M., Li D., Feng J., Hutchings G. J., Catal. Sci. Technol. 2021. [Google Scholar]
- 35. Zhang X., Yang P., Liu Y., Pan J., Li D., Wang B., Feng J., J. Catal. 2020, 385, 146–159. [Google Scholar]
- 36. Zhao G., Yang F., Chen Z., Liu Q., Ji Y., Zhang Y., Niu Z., Mao J., Bao X., Hu P., Li Y., Nat. Commun. 2017, 8, 14039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang H., Gu X. K., Zheng X., Pan H., Zhu J., Chen S., Cao L., Li W. X., Lu J., Sci. Adv. 2019, 5, eaat6413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Villa A., Wang D., Veith G. M., Vindigni F., Prati L., Catal. Sci. Technol. 2013, 3. [Google Scholar]
- 39.
- 39a. Miyamura H., Matsubara R., Miyazaki Y., Kobayashi S., Angew. Chem. Int. Ed. 2007, 46, 4151–4; [DOI] [PubMed] [Google Scholar]
- 39b. Ishida T., Nagaoka M., Akita T., Haruta M., Chem. Eur. J. 2008, 14, 8456–60; [DOI] [PubMed] [Google Scholar]
- 39c. Hackett S. F., Brydson R. M., Gass M. H., Harvey I., Newman A. D., Wilson K., Lee A. F., Angew. Chem. Int. Ed. 2007, 46, 8593–6. [DOI] [PubMed] [Google Scholar]
- 40. Lei L., Liu H., Wu Z., Qin Z., Wang G., Ma J., Luo L., Fan W., Wang J., ACS Appl. Nano Mater. 2019, 2, 5214–5223. [Google Scholar]
- 41. Serov A., Artyushkova K., Niangar E., Wang C., Dale N., Jaouen F., Sougrati M.-T., Jia Q., Mukerjee S., Atanassov P., Nano Energy 2015, 16, 293–300. [Google Scholar]
- 42. Rodriguez P., Kwon Y., Koper M. T., Nat. Chem. 2011, 4, 177–82. [DOI] [PubMed] [Google Scholar]
- 43. Su H., Zhang K. X., Zhang B., Wang H. H., Yu Q. Y., Li X. H., Antonietti M., Chen J. S., J. Am. Chem. Soc. 2017, 139, 811–818. [DOI] [PubMed] [Google Scholar]
- 44. Kumar G., Tibbitts L., Newell J., Panthi B., Mukhopadhyay A., Rioux R. M., Pursell C. J., Janik M., Chandler B. D., Nat. Chem. 2018, 10, 268–274. [DOI] [PubMed] [Google Scholar]
- 45. Mahdavi-Shakib A., Sempel J., Babb L., Oza A., Hoffman M., Whittaker T. N., Chandler B. D., Austin R. N., ACS Catal. 2020, 10, 10207–10215. [Google Scholar]
- 46. Enache D. I., Edwards J. K., Landon P., Solsona-Espriu B., Carley A. F., Herzing A. A., Watanabe M., Kiely C. J., Knight D. W., Hutchings G. J., Science 2006, 311, 362–5. [DOI] [PubMed] [Google Scholar]
- 47. Sankar M., Nowicka E., Carter E., Murphy D. M., Knight D. W., Bethell D., Hutchings G. J., Nat. Commun. 2014, 5, 3332. [DOI] [PubMed] [Google Scholar]
- 48.
- 48a. Hou W., Dehm N., Scott R., J. Catal. 2008, 253, 22–27; [Google Scholar]
- 48b. Della Pina C., Falletta E., Rossi M., J. Catal. 2008, 260, 384–386; [Google Scholar]
- 48c. Meenakshisundaram S., Nowicka E., Miedziak P. J., Brett G. L., Jenkins R. L., Dimitratos N., Taylor S. H., Knight D. W., Bethell D., Hutchings G. J., Faraday Discuss. 2010, 145, 341–356. [Google Scholar]
- 49. Zhu X., Guo Q., Sun Y., Chen S., Wang J. Q., Wu M., Fu W., Tang Y., Duan X., Chen, Wan Y., Nat. Commun. 2019, 10, 1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.
- 50a. Xie S., Tsunoyama H., Kurashige W., Negishi Y., Tsukuda T., ACS Catal. 2012, 2, 1519–1523; [Google Scholar]
- 50b. Chivers B. A., Scott R. W. J., Catal. Sci. Technol. 2020, 10, 7706–7718. [Google Scholar]
- 51.
- 51a. Sádaba I., López Granados M., Riisager A., Taarning E., Green Chem. 2015, 17, 4133–4145; [Google Scholar]
- 51b. Yang W., Vogler B., Lei Y., Wu T., Environ. Sci.: Water Res. Technol. 2017, 3, 1143–1151. [Google Scholar]
- 52.
- 52a. Xie J., Duan P., Kaylor N., Yin K., Huang B., Schmidt-Rohr K., Davis R. J., ACS Catal. 2017, 7, 6745–6756; [Google Scholar]
- 52b. Zope B. N., Davis R. J., Green Chem. 2011, 13; [Google Scholar]
- 52c. Skupien E., Berger R., Santos V., Gascon J., Makkee M., Kreutzer M., Kooyman P., Moulijn J., Kapteijn F., Catalysts 2014, 4, 89–115; [Google Scholar]
- 52d. D'Agostino C., Armstrong R. D., Hutchings G. J., Gladden L. F., ACS Catal. 2018, 8, 7334–7339; [Google Scholar]
- 52e. Ide M. S., Falcone D. D., Davis R. J., J. Catal. 2014, 311, 295–305. [Google Scholar]
- 53.
- 53a. Dirkx J., J. Catal. 1981. 67(1), 14–20; [Google Scholar]
- 53b. Mallat T., Baiker A., Chem. Rev. 2004, 104, 3037–58. [DOI] [PubMed] [Google Scholar]
- 54. Fu J., He Q., Miedziak P. J., Brett G. L., Huang X., Pattisson S., Douthwaite M., Hutchings G. J., Chem. 2018, 24, 2396–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Armstrong R. D., Hirayama J., Knight D. W., Hutchings G. J., ACS Catal. 2018, 9, 325–335. [Google Scholar]
- 56. Polo-Garzon F., Bao Z., Zhang X., Huang W., Wu Z., ACS Catal. 2019, 9, 5692–5707. [Google Scholar]
- 57. Linnemann J., Kanokkanchana K., Tschulik K., ACS Catal. 2021, 11, 5318–5346. [Google Scholar]
- 58. Routray K., Zhou W., Kiely C. J., Wachs I. E., ACS Catal. 2010, 1, 54–66. [Google Scholar]
- 59. Jiang Y.-w., Chai K., Wang Y.-q., Zhang H.-d., Xu W., Li W., Shi Y., ACS Appl. Nano Mater. 2019, 2, 4435–4442. [Google Scholar]
- 60.
- 60a. Malmusi A., Velasquez Ochoa J., Tabanelli T., Basile F., Lucarelli C., Agnoli S., Carraro F., Granozzi G., Cavani F., Appl. Catal. A 2019, 570, 139–147; [Google Scholar]
- 60b. Bankar B. D., Advani J. H., Biradar A. V., ChemistrySelect 2021, 6, 3814–3821. [Google Scholar]
- 61. Sarkar A., Wang Q., Schiele A., Chellali M. R., Bhattacharya S. S., Wang D., Brezesinski T., Hahn H., Velasco L., Breitung B., Adv. Mater. 2019, 31, e1806236. [DOI] [PubMed] [Google Scholar]
- 62. Feng D., Dong Y., Zhang L., Ge X., Zhang W., Dai S., Qiao Z. A., Angew. Chem. Int. Ed. 2020, 59, 19503–19509. [DOI] [PubMed] [Google Scholar]
- 63. Polo-Garzon F., Yang S. Z., Fung V., Foo G. S., Bickel E. E., Chisholm M. F., Jiang D. E., Wu Z., Angew. Chem. Int. Ed. 2017, 56, 9820–9824; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 9952–9956. [Google Scholar]
- 64.
- 64a. Anke S., Bendt G., Sinev I., Hajiyani H., Antoni H., Zegkinoglou I., Jeon H., Pentcheva R., Roldan Cuenya B., Schulz S., Muhler M., ACS Catal. 2019, 9, 5974–5985; [Google Scholar]
- 64b. Anke S., Falk T., Bendt G., Sinev I., Hävecker M., Antoni H., Zegkinoglou I., Jeon H., Knop-Gericke A., Schlögl R., Roldan Cuenya B., Schulz S., Muhler M., J. Catal. 2020, 382, 57–68. [Google Scholar]
- 65.
- 65a. Alkan B., Cychy S., Varhade S., Muhler M., Schulz C., Schuhmann W., Wiggers H., Andronescu C., ChemElectroChem 2019, 6, 4266–4274; [Google Scholar]
- 65b. Zhu J., Zhao Y., Tang D., Zhao Z., Carabineiro S. A. C., J. Catal. 2016, 340, 41–48. [Google Scholar]
- 66. Gurrala L., Nagpure A. S., Gurav H. R., Chilukuri S., ChemistrySelect 2018, 3, 3751–3761. [Google Scholar]
- 67. Falk T., Budiyanto E., Dreyer M., Pflieger C., Waffel D., Büker J., Weidenthaler C., Ortega K. F., Behrens M., Tüysüz H., Muhler M., Peng B., ChemCatChem 2021, 13, 2942–2951. [Google Scholar]
- 68.
- 68a. Li D., Ruan F., Jin Y., Ke Q., Cao Y., Wang H., Wang T., Song Y., Cui P., Catal. Sci. Technol. 2019, 9, 418–424; [Google Scholar]
- 68b. Xu Y., Qu Z., Ren Y., Dong C., Appl. Surf. Sci. 2021, 560. [Google Scholar]
- 69.
- 69a. Zhu H., Zhang P., Dai S., ACS Catal. 2015, 5, 6370–6385; [Google Scholar]
- 69b. Waffel D., Alkan B., Fu Q., Chen Y. T., Schmidt S., Schulz C., Wiggers H., Muhler M., Peng B., ChemPlusChem 2019, 84, 1155–1163; [DOI] [PubMed] [Google Scholar]
- 69c. Kubo J., Ueda W., Mater. Res. Bull. 2009, 44, 906–912. [Google Scholar]
- 70.
- 70a. Ali R., Nour K., Al-warthan A., Siddiqui M. R. H., Arabian J. Chem. 2015, 8, 512–517; [Google Scholar]
- 70b. Thao N. T., Nhu N. T., J. Sci. Adv. Mater. Devices 2018, 3(3), 289–295; [Google Scholar]
- 70c. Akinnawo C. A., Bingwa N., Meijboom R., Catal. Commun. 2020, 145; [Google Scholar]
- 70d. Li X., Zheng J., Liu S., Zhu T., J. Colloid Interface Sci. 2019, 555, 667–675. [DOI] [PubMed] [Google Scholar]
- 71. Hellier P., Wells P. P., Gianolio D., Bowker M., Top. Catal. 2018, 61, 357–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Li M., Fu X., Peng L., Bai L., Wu S., Kan Q., Guan J., ChemistrySelect 2017, 2, 9486–9489. [Google Scholar]
- 73. Gu Y., Lu P., Zhan W., Zhang Y., Sun L., Chen G., Long Z., J. Porous Mater. 2020, 27, 701–705. [Google Scholar]
- 74. Pordel S., Rabbani M., Rahimi R., Heidari-Golafzani M., Azad A., Solid State Sci. 2020, 107. [Google Scholar]
- 75. Reddy V. G., Jampaiah D., Chalkidis A., Sabri Y. M., Mayes E. L. H., Bhargava S. K., Catal. Commun. 2019, 130. [Google Scholar]
- 76. Wu G., Gao Y., Ma F., Zheng B., Liu L., Sun H., Wu W., Chem. Eng. J. 2015, 271, 14–22. [Google Scholar]
- 77. Stuchinskaya T., Musawir M., Kozhevnikova E., Kozhevnikov I., J. Catal. 2005, 231, 41–47. [Google Scholar]
- 78. Zhu Q., Yin S., Zhou M., Wang J., Chen C., Hu P., Jiang X., Zhang Z., Li Y., Ueda W., ChemCatChem 2021, 13, 1763–1771. [Google Scholar]
- 79.
- 79a. Arena F., Gumina B., Lombardo A. F., Espro C., Patti A., Spadaro L., Spiccia L., Appl. Catal. B 2015, 162, 260–267; [Google Scholar]
- 79b. Nguyen V. D. N., Yun D., Sakulchaicharoen N., Herrera J. E., Int. J. Chem. React. Eng. 2018, 16. [Google Scholar]
- 80. Torres S., Palacio R., López D., Appl. Catal. A 2021, 621. [Google Scholar]
- 81. Amakawa K., Kolen'ko Y. V., Villa A., Schuster M. E., Csepei L.-I., Weinberg G., Wrabetz S., Naumann d'Alnoncourt R., Girgsdies F., Prati L., Schlögl R., Trunschke A., ACS Catal. 2013, 3, 1103–1113. [Google Scholar]
- 82. Kulkarni D., Wachs I. E., Appl. Catal. A 2002, 237, 121–137. [Google Scholar]
- 83. Wachs I. E., Routray K., ACS Catal. 2012, 2, 1235–1246. [Google Scholar]
- 84.
- 84a. Soares A. P. V., Portela M. F., Kiennemann A., Catal. Rev. 2005, 47, 125–174; [Google Scholar]
- 84b. Weissermel K. A., Hans-Jürgen, Industrial Organic Chemistry, 3rd completely rev. ed., VCH: Weinheim, New York, 1997, p. 37; [Google Scholar]
- 84c. House M., Carley A., Bowker M., J. Catal. 2007, 252, 88–96; [Google Scholar]
- 84d. Bowker M., Carley A. F., House M., Catal. Lett. 2007, 120, 34–39. [Google Scholar]
- 85. Brookes C., Wells P. P., Dimitratos N., Jones W., Gibson E. K., Morgan D. J., Cibin G., Nicklin C., Mora-Fonz D., Scanlon D. O., Catlow C. R. A., Bowker M., J. Phys. Chem. C 2014, 118, 26155–26161. [Google Scholar]
- 86. Bowker M., House M., Alshehri A., Brookes C., Gibson E. K., Wells P. P., Catal., Struct. React. 2015, 1, 95–100. [Google Scholar]
- 87.
- 87a. Chen S., Xie H., Zhou G., Ceram. Int. 2019, 45, 24609–24617; [Google Scholar]
- 87b. Diulus J. T., Elzein R., Addou R., Herman G. S., J. Chem. Phys. 2020, 152, 054713; [DOI] [PubMed] [Google Scholar]
- 87c. Hou Y.-C., Ding M.-W., Liu S.-K., Wu S.-K., Lin Y.-C., RSC Adv. 2014, 4; [Google Scholar]
- 87d. Jiang B.-S., Chang R., Lin Y.-C., Ind. Eng. Chem. Res. 2012; [Google Scholar]
- 87e. Li M., Wu Z., Overbury S. H., J. Catal. 2013, 306, 164–176; [Google Scholar]
- 87f. Ortega K. F., Anke S., Salamon S., Özcan F., Heese J., Andronescu C., Landers J., Wende H., Schuhmann W., Muhler M., Lunkenbein T., Behrens M., Chem. Eur. J. 2017, 23, 12443–12449; [DOI] [PubMed] [Google Scholar]
- 87g. Zhang Y., Mullins D. R., Savara A., J. Phys. Chem. C 2020, 124, 3650–3663; [Google Scholar]
- 87h. Zhang Y., Savara A., Mullins D. R., J. Phys. Chem. C 2017, 121, 23436–23445. [Google Scholar]
- 88. Wang H., Sapi A., Thompson C. M., Liu F., Zherebetskyy D., Krier J. M., Carl L. M., Cai X., Wang L. W., Somorjai G. A., J. Am. Chem. Soc. 2014, 136, 10515–20. [DOI] [PubMed] [Google Scholar]
- 89. Paula A. S., Possato L. G., Ratero D. R., Contro J., Keinan-Adamsky K., Soares R. R., Goobes G., Martins L., Nery J. G., Microporous Mesoporous Mater. 2016, 232, 151–160. [Google Scholar]
- 90. Possato L. G., Cassinelli W. H., Meyer C. I., Garetto T., Pulcinelli S. H., Santilli C. V., Martins L., Appl. Catal. A 2017, 532, 1–11. [Google Scholar]
- 91. Rasteiro L. F., Vieira L. H., Santilli C. V., Martins L., RSC Adv. 2018, 8, 11975–11982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Chieregato A., Soriano M. D., Garcia-Gonzalez E., Puglia G., Basile F., Concepcion P., Bandinelli C., Lopez Nieto J. M., Cavani F., ChemSusChem 2015, 8, 398–406. [DOI] [PubMed] [Google Scholar]
- 93. Omata K., Matsumoto K., Murayama T., Ueda W., Catal. Today 2016, 259, 205–212. [Google Scholar]
- 94. Gavriilidis A., Constantinou A., Hellgardt K., Hii K. K., Hutchings G. J., Brett G. L., Kuhn S., Marsden S. P., React. Chem. Eng. 2016, 1, 595–612. [Google Scholar]
- 95.
- 95a. Jagadeesh R. V., Junge H., Pohl M. M., Radnik J., Bruckner A., Beller M., J. Am. Chem. Soc. 2013, 135, 10776–82; [DOI] [PubMed] [Google Scholar]
- 95b. Jagadeesh R. V., Stemmler T., Surkus A. E., Bauer M., Pohl M. M., Radnik J., Junge K., Junge H., Bruckner A., Beller M., Nat. Protoc. 2015, 10, 916–26; [DOI] [PubMed] [Google Scholar]
- 95c. Su H., Zhang K. X., Zhang B., Wang H. H., Yu Q. Y., Li X. H., Antonietti M., Chen J. S., J. Am. Chem. Soc. 2017, 139, 811–818; [DOI] [PubMed] [Google Scholar]
- 95d. Geng L., Zhang X., Zhang W., Jia M., Liu G., Chem. Commun. 2014, 50, 2965–7; [DOI] [PubMed] [Google Scholar]
- 95e. Xiao S., Zhang C., Chen R., Chen F., New J. Chem. 2015, 39, 4924–4932; [Google Scholar]
- 95f. Obermayer D., Balu A. M., Romero A. A., Goessler W., Luque R., Kappe C. O., Green Chem. 2013, 15; [Google Scholar]
- 95g. Martins N., Martins L., Amorim C., Amaral V., Pombeiro A., Catalysts 2017, 7; [Google Scholar]
- 95h. Fu X., Wu S., Li Z., Yang X., Wang X., Peng L., Hu J., Huo Q., Guan J., Kan Q., RSC Adv. 2016, 6, 57507–57513; [Google Scholar]
- 95i. Tan J., Liu X. B., Chen W. F., Hu Y. L., ChemistrySelect 2019, 4, 8477–8481; [Google Scholar]
- 95j. Zhao C., Wu J., Yang L., Fan G., Li F., Ind. Eng. Chem. Res. 2017, 56, 4237–4244; [Google Scholar]
- 95k. Kawasaki S., Kamata K., Hara M., ChemCatChem 2016, 8, 3247–3253; [Google Scholar]
- 95l. Singh S. J., Jayaram R. V., Synth. Commun. 2011, 42, 299–308; [Google Scholar]
- 95m. Zhu J., Zhao Y., Tang D., Zhao Z., Carabineiro S. A. C., J. Catal. 2016, 340, 41–48; [Google Scholar]
- 95n. Waffel D., Budiyanto E., Porske T., Büker J., Falk T., Fu Q., Schmidt S., Tüysüz H., Muhler M., Peng B., J. Mol. Catal. 2020, 498. [Google Scholar]
- 96.
- 96a. Jones R. D., Summerville D. A., Basolo F., Chem. Rev. 2002, 79, 139–179; [Google Scholar]
- 96b. Neumann R., Inorg. Chem. 2010, 49, 3594–601. [DOI] [PubMed] [Google Scholar]
- 97.
- 97a. bin Saiman M. I., Brett G. L., Tiruvalam R., Forde M. M., Sharples K., Thetford A., Jenkins R. L., Dimitratos N., Lopez-Sanchez J. A., Murphy D. M., Bethell D., Willock D. J., Taylor S. H., Knight D. W., Kiely C. J., Hutchings G. J., Angew. Chem. Int. Ed. 2012, 51, 5981–5; [DOI] [PubMed] [Google Scholar]
- 97b. Räisänen M. T., Al-Hunaiti A., Atosuo E., Kemell M., Leskelä M., Repo T., Catal. Sci. Technol. 2014, 4, 2564–2573. [Google Scholar]
- 98. Liu H., Gu L., Zhu P., Liu Z., Zhou B., Procedia Eng. 2012, 45, 574–579. [Google Scholar]
- 99. Campbell M. L., Sulejmanovic D., Schiller J. B., Turner E. M., Hwu S.-J., Whitehead D. C., Catal. Sci. Technol. 2016, 6, 3208–3213. [Google Scholar]
- 100. Sheldon R. A., Wallau M., Arends I. W. C. E., Schuchardt U., Acc. Chem. Res. 1998, 31, 485–493. [Google Scholar]
- 101. Büker J., Alkan B., Fu Q., Xia W., Schulwitz J., Waffel D., Falk T., Schulz C., Wiggers H., Muhler M., Peng B., Catal. Sci. Technol. 2020, 10, 5196–5206. [Google Scholar]
- 102. Mallat T., Baiker A., Catal. Sci. Technol. 2011, 1. [Google Scholar]
- 103. Dhar S. K., Abdelaziz J., Cozzi P., Jasien P., Mason C., Zalenas R., J. Chem. Soc. Chem. Commun. 1984. [Google Scholar]
- 104.
- 104a. Chàvez J. E., Crotti C., Zangrando E., Farnetti E., J. Mol. Catal. A 2016, 421, 189–195; [Google Scholar]
- 104b. Kani I., Bolat S., Appl. Organomet. Chem. 2016, 30, 713–721. [Google Scholar]
- 105. Sebastian J., Jinka K., Jasra R., J. Catal. 2006, 244, 208–218. [Google Scholar]
- 106.
- 106a. Ding S., Jiao N., Angew. Chem. Int. Ed. 2012, 51, 9226–37; [DOI] [PubMed] [Google Scholar]
- 106b. Muzart J., Tetrahedron 2009, 65, 8313–8323. [Google Scholar]
- 107.
- 107a. Liang J., Zhang Q., Wu H., Meng G., Tang Q., Wang Y., Catal. Commun. 2004, 5, 665–669; [Google Scholar]
- 107b. Opre Z., Mallat T., Baiker A., J. Catal. 2007, 245, 482–486; [Google Scholar]
- 107c. Patil M. V., Yadav M. K., Jasra R. V., J. Mol. Catal. A 2007, 277, 72–80; [Google Scholar]
- 107d. Tang B., Lu X. H., Zhou D., Tian P., Niu Z. H., Zhang J. L., Chen X., Xia Q. H., Catal. Commun. 2013, 31, 42–47; [Google Scholar]
- 107e. Tang Q., Zhang Q., Wu H., Wang Y., J. Catal. 2005, 230, 384–397; [Google Scholar]
- 107f. Xu G., Xia Q. H., Lu X. H., Zhang Q., Zhan H. J., J. Mol. Catal. A 2007, 266, 180–187; [Google Scholar]
- 107g. Zhan H. J., Xia Q. H., Lu X. H., Zhang Q., Yuan H. X., Su K. X., Ma X. T., Catal. Commun. 2007, 8, 1472–1478. [Google Scholar]
- 108.
- 108a. Carter M. J., Rillema D. P., Basolo F., J. Am. Chem. Soc. 2002, 96, 392–400; [Google Scholar]
- 108b. Chen D., Martell A. E., Inorg. Chem. 2002, 26, 1026–1030; [Google Scholar]
- 108c. Eichhorn E., Rieker A., Speiser B., Stahl H., Inorg. Chem. 1997, 36, 3307–3317. [DOI] [PubMed] [Google Scholar]
- 109. Beier M. J., Kleist W., Wharmby M. T., Kissner R., Kimmerle B., Wright P. A., Grunwaldt J. D., Baiker A., Chem. Eur. J. 2012, 18, 887–98. [DOI] [PubMed] [Google Scholar]
- 110.
- 110a. Drago R. S., Riley R., J. Am. Chem. Soc. 2002, 112, 215–218; [Google Scholar]
- 110b. Lock M. V., Sagar B. F., J. Chem. Soc. B 1966; [Google Scholar]
- 110c. Sagar B. F., J. Chem. Soc. B 1967; [Google Scholar]
- 110d. Sagar B. F., J. Chem. Soc. B 1967. [Google Scholar]
- 111.
- 111a. Salamone M., Milan M., DiLabio G. A., Bietti M., J. Org. Chem. 2013, 78, 5909–17; [DOI] [PubMed] [Google Scholar]
- 111b. Salamone M., Milan M., DiLabio G. A., Bietti M., J. Org. Chem. 2014, 79, 7179–84. [DOI] [PubMed] [Google Scholar]
- 112. Nandan D., Zoppellaro G., Medřík I., Aparicio C., Kumar P., Petr M., Tomanec O., Gawande M. B., Varma R. S., Zbořil R., Green Chem. 2018, 20, 3542–3556. [Google Scholar]
- 113.
- 113a. Tovrog B. S., Kitko D. J., Drago R. S., J. Am. Chem. Soc. 2002, 98, 5144–5153; [Google Scholar]
- 113b. Vaska L., Acc. Chem. Res. 2002, 9, 175–183. [Google Scholar]
- 114. Büker J., Huang X., Bitzer J., Kleist W., Muhler M., Peng B., ACS Catal. 2021, 11, 7863–7875. [Google Scholar]
- 115. Wen G., Gu Q., Liu Y., Schlogl R., Wang C., Tian Z., Su D. S., Angew. Chem. Int. Ed. 2018, 57, 16898–16902; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 57, 16898–16902. [DOI] [PubMed] [Google Scholar]
- 116.
- 116a. Zhou W.-J., Wischert R., Xue K., Zheng Y.-T., Albela B., Bonneviot L., Clacens J.-M., De Campo F., Pera-Titus M., Wu P., ACS Catal. 2013, 4, 53–62; [Google Scholar]
- 116b. Neese F., eMagRes 2017, 6, 1–22. [Google Scholar]
- 117. Luo J., Peng F., Yu H., Wang H., Zheng W., ChemCatChem 2013, 5, 1578–1586. [Google Scholar]
- 118.
- 118a. Grunwaldt J., J. Catal. 2003, 213, 291–295; [Google Scholar]
- 118b. Parlett C. M. A., Gaskell C. V., Naughton J. N., Newton M. A., Wilson K., Lee A. F., Catal. Today 2013, 205, 76–85. [Google Scholar]
- 119.
- 119a. Kancheva V. D., Eur. J. Lipid Sci. Technol. 2009, 111, 1072–1089; [Google Scholar]
- 119b. Michalik M., Poliak P., Lukes V., Klein E., Phytochemistry 2019, 166, 112077; [DOI] [PubMed] [Google Scholar]
- 119c. Thavasi V., Bettens R. P., Leong L. P., J. Phys. Chem. A 2009, 113, 3068–77. [DOI] [PubMed] [Google Scholar]
- 120.
- 120a. Chen P., Wang F., Zhang Q., Su Y., Shen L., Yao K., Chen Z. F., Liu Y., Cai Z., Lv W., Liu G., Chemosphere 2017, 172, 193–200; [DOI] [PubMed] [Google Scholar]
- 120b. Lin W., Xie X., Wang X., Wang Y., Segets D., Sun J., Chem. Eng. J. 2018, 349, 708–718; [Google Scholar]
- 120c. Mureseanu M., Chivu V., Osiac M., Ciobanu M., Bucur C., Parvulescu V., Cioatera N., Catal. Today 2021, 366, 164–176; [Google Scholar]
- 120d. Zhao G., Bonke S. A., Schmidt S., Wang Z., Hu B., Falk T., Hu Y., Rath T., Xia W., Peng B., Schnegg A., Weng Y., Muhler M., ACS Sustainable Chem. 2021, 9, 5422–5429; [Google Scholar]
- 120e. Zhao G., Busser G. W., Froese C., Hu B., Bonke S. A., Schnegg A., Ai Y., Wei D., Wang X., Peng B., Muhler M., J. Phys. Chem. Lett. 2019, 10, 2075–2080. [DOI] [PubMed] [Google Scholar]
- 121. Topsøe H., J. Catal. 2003, 216, 155–164. [Google Scholar]
- 122. McAlpin J. G., Stich T. A., Ohlin C. A., Surendranath Y., Nocera D. G., Casey W. H., Britt R. D., J. Am. Chem. Soc. 2011, 133, 15444–52. [DOI] [PubMed] [Google Scholar]
- 123.
- 123a. Cuesta A., Curr. Opin. Electrochem. 2017, 4, 32–38; [Google Scholar]
- 123b. Rizo R., Pérez-Rodríguez S., García G., ChemElectroChem 2019, 6, 4725–4738; [Google Scholar]
- 123c. Lai S. C. S., Lebedeva N. P., Housmans T. H. M., Koper M. T. M., Top. Catal. 2007, 46, 320–333; [Google Scholar]
- 123d. Lai S. C., Koper M. T., Faraday Discuss. 2008, 140, 399–416; discussion 417–37; [DOI] [PubMed] [Google Scholar]
- 123e. Cuesta A., Escudero M., Lanova B., Baltruschat H., Langmuir 2009, 25, 6500–7; [DOI] [PubMed] [Google Scholar]
- 123f. Guillén-Villafuerte O., García G., Arévalo M. C., Rodríguez J. L., Pastor E., Electrochem. Commun. 2016, 63, 48–51. [Google Scholar]
- 124. Antolini E., J. Power Sources 2007, 170, 1–12. [Google Scholar]
- 125.
- 125a. Abd-El-Latif A. A., Mostafa E., Huxter S., Attard G., Baltruschat H., Electrochim. Acta 2010, 55, 7951–7960; [Google Scholar]
- 125b. Wang H., Löffler T., Baltruschat H., J. Appl. Electrochem. 2001, 31, 759–765. [Google Scholar]
- 126.
- 126a. Liu W.-J., Dang L., Xu Z., Yu H.-Q., Jin S., Huber G. W., ACS Catal. 2018, 8, 5533–5541; [Google Scholar]
- 126b. Zhang N., Zou Y., Tao L., Chen W., Zhou L., Liu Z., Zhou B., Huang G., Lin H., Wang S., Angew. Chem. 2019, 131, 16042–16050; [DOI] [PubMed] [Google Scholar]
- 126c. Taitt B. J., Nam D.-H., Choi K.-S., ACS Catal. 2018, 9, 660–670; [Google Scholar]
- 126d. Latsuzbaia R., Bisselink R., Anastasopol A., van der Meer H., van Heck R., Yagüe M. S., Zijlstra M., Roelands M., Crockatt M., Goetheer E., Giling E., J. Appl. Electrochem. 2018, 48, 611–626; [Google Scholar]
- 126e. Gao L., Bao Y., Gan S., Sun Z., Song Z., Han D., Li F., Niu L., ChemSusChem 2018, 11, 2547–2553; [DOI] [PubMed] [Google Scholar]
- 126f. Nam D.-H., Taitt B. J., Choi K.-S., ACS Catal. 2018, 8, 1197–1206; [Google Scholar]
- 126g. Fleischmann M., Korinek K., Pletcher D., J. Chem. Soc. Perkin Trans. 2 1972,1396–1403. [Google Scholar]
- 127.
- 127a. Lai S. C. S., Kleijn S. E. F., Öztürk F. T. Z., van Rees Vellinga V. C., Koning J., Rodriguez P., Koper M. T. M., Catal. Today 2010, 154, 92–104; [Google Scholar]
- 127b. Kwon Y., Lai S. C., Rodriguez P., Koper M. T., J. Am. Chem. Soc. 2011, 133, 6914–7; [DOI] [PubMed] [Google Scholar]
- 127c. Beyhan S., Uosaki K., Feliu J. M., Herrero E., J. Electroanal. Chem. 2013, 707, 89–94. [Google Scholar]
- 128. Schäfer H.-J., Oxidation of organic compounds at the nickel hydroxide electrode. in Electrochemistry I., Heidelberg: Springer Berlin Heidelberg, Berlin, 1987. [Google Scholar]
- 129. Simoes M., Baranton S., Coutanceau C., ChemSusChem 2012, 5, 2106–24. [DOI] [PubMed] [Google Scholar]
- 130. Zhang R., Jiang S., Rao Y., Chen S., Yue Q., Kang Y., Green Chem. 2021, 23, 2525–2530. [Google Scholar]
- 131.
- 131a. Santos A. L., Cebola M.-J., Santos D. M. F., Energies 2021, 14; [Google Scholar]
- 131b. Badruzzaman A., Yuda A., Ashok A., Kumar A., Inorg. Chim. Acta 2020, 511. [Google Scholar]
- 132. Zayat B., Mitra D., Irshad A., Rajan A. S., Narayanan S. R., Curr. Opin. Electrochem. 2021, 25. [Google Scholar]
- 133. Fleischmann M., Korinek K., Pletcher D., J. Electroanal. Chem. Interfacial Electrochem. 1971, 31, 39–49. [Google Scholar]
- 134. Bender M. T., Lam Y. C., Hammes-Schiffer S., Choi K. S., J. Am. Chem. Soc. 2020, 142, 21538–21547. [DOI] [PubMed] [Google Scholar]
- 135. Van Effen R. M., Evans D. H., J. Electroanal. Chem. Interfacial Electrochem. 1980, 107, 405–418. [Google Scholar]
- 136. Sun S., Sun L., Xi S., Du Y., Anu Prathap M. U., Wang Z., Zhang Q., Fisher A., Xu Z. J., Electrochim. Acta 2017, 228, 183–194. [Google Scholar]
- 137.
- 137a. Houache M. S. E., Hughes K., Ahmed A., Safari R., Liu H., Botton G. A., Baranova E. A., ACS Sustainable Chem. Eng. 2019, 7, 14425–14434; [Google Scholar]
- 137b. Robertson P. M., J. Electroanal. Chem. Interfacial Electrochem. 1980, 111, 97–104; [Google Scholar]
- 137c. Vértes G., Horányi G., J. Electroanal. Chem. Interfacial Electrochem. 1974, 52, 47–53. [Google Scholar]
- 138. Smith R. D., Berlinguette C. P., J. Am. Chem. Soc. 2016, 138, 1561–7. [DOI] [PubMed] [Google Scholar]
- 139. Sun S., Xu Z. J., Electrochim. Acta 2015, 165, 56–66. [Google Scholar]
- 140. Sun S., Zhou Y., Hu B., Zhang Q., Xu Z. J., J. Electrochem. Soc. 2015, 163, H99-H104. [Google Scholar]
- 141. Jadhav S., Open Chemistry 2011, 9, 369–378. [Google Scholar]
- 142. Beltowska-Brzezinska M., Vielstich W., Electrochim. Acta 1977, 22, 1313–1314. [Google Scholar]
- 143. Holze R., Beŧowska-Brzezinska M., Electrochim. Acta 1985, 30, 937–939. [Google Scholar]
- 144. Anu Prathap M. U., Srivastava R., Nano Energy 2013, 2, 1046–1053. [Google Scholar]
- 145. Sonu, Dutta V., Sharma S., Raizada P., Hosseini-Bandegharaei A., Kumar Gupta V., Singh P., J. Saudi Chem. Soc. 2019, 23, 1119–1136. [Google Scholar]
- 146.
- 146a. Busser G. W., Mei B., Pougin A., Strunk J., Gutkowski R., Schuhmann W., Willinger M. G., Schlogl R., Muhler M., ChemSusChem 2014, 7, 1030–4; [DOI] [PubMed] [Google Scholar]
- 146b. Lukic S., Menze J., Weide P., Busser G. W., Winterer M., Muhler M., ChemSusChem 2017, 10, 4190–4197. [DOI] [PubMed] [Google Scholar]
- 147. Yao Y., Gao X., Li Z., Meng X., Catalysts 2020, 10. [Google Scholar]
- 148.
- 148a. Zhao G., Busser G. W., Froese C., Hu B., Bonke S. A., Schnegg A., Ai Y., Wei D., Wang X., Peng B., Muhler M., J. Phys. Chem. Lett. 2019, 10, 2075–2080; [DOI] [PubMed] [Google Scholar]
- 148b. Hu Y., Zhao G., Pan Q., Wang H., Shen Z., Peng B., Busser G. W., Wang X., Muhler M., ChemCatChem 2019, 11, 5139–5144; [Google Scholar]
- 148c. Shen Z., Hu Y., Li B., Zou Y., Li S., Wilma Busser G., Wang X., Zhao G., Muhler M., J. Energy Chem. 2021, 62, 338–350; [Google Scholar]
- 148d. Xiong L., Tang J., Adv. Energy Mater. 2021, 11; [Google Scholar]
- 148e. Yang X., Zhang S., Li P., Gao S., Cao R., J. Mater. Chem. A 2020, 8, 20897–20924. [Google Scholar]
- 149. Barba-Nieto I., Caudillo-Flores U., Gómez-Cerezo M. N., Kubacka A., Fernández-García M., Chem. Eng. J. 2020, 398. [Google Scholar]
- 150. Quesada-Cabrera R., Parkin I. P., Front. Chem. 2020, 8, 817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Kayaalp B. E., Lee Y. J., Kornowski A., Gross S., D′Arienzo M., Mascotto S., RSC Adv. 2016, 6, 90401–90409. [Google Scholar]
- 152. Kayaalp B., Klauke K., Biesuz M., Iannaci A., Sglavo V. M., D'Arienzo M., Noei H., Lee S., Jung W., Mascotto S., J. Phys. Chem. C 2019, 123, 16883–16892. [Google Scholar]
- 153.
- 153a. Kayaalp B., Klauke K., Biesuz M., Iannaci A., Sglavo V. M., D'Arienzo M., Noei H., Lee S., Jung W., Mascotto S., J. Phys. Chem. C 2019, 123, 16883–16892; [Google Scholar]
- 153b. Klauke K., Kayaalp B., Biesuz M., Iannaci A., Sglavo V. M., D′Arienzo M., Lee S., Seo J., Jung W., Mascotto S., ChemNanoMat 2019, 5, 948–956. [Google Scholar]
- 154. Vinosha P. A., Manikandan A., Preetha A. C., Dinesh A., Slimani Y., Almessiere M. A., Baykal A., Xavier B., Nirmala G. F., J. Supercond. Novel Magn. 2021, 34, 995–1018. [Google Scholar]
- 155. Selima S. S., Bayoumy W. A., Khairy M., Mousa M. A., Research Square preprint 2021. [Google Scholar]
- 156. Hafeez H. Y., Lakhera S. K., Narayanan N., Harish S., Hayakawa Y., Lee B. K., Neppolian B., ACS Omega 2019, 4, 880–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Ghobadifard M., Mohebbi S., Radovanovic P. V., New J. Chem. 2020, 44, 2858–2867. [Google Scholar]
- 158. Noël T., Photochemical Processes in Continuous-Flow Reactors. World Scientific Publishing Europe Ltd, London, 2017. [Google Scholar]
- 159.
- 159a. Kudo A., Miseki Y., Chem. Soc. Rev. 2009, 38, 253–78; [DOI] [PubMed] [Google Scholar]
- 159b. Takanabe K., Domen K., Green 2011, 1; [Google Scholar]
- 159c. Hisatomi T., Takanabe K., Domen K., Catal. Lett. 2014, 145, 95–108; [Google Scholar]
- 159d. Qureshi M., Takanabe K., Chem. Mater. 2016, 29, 158–167. [Google Scholar]
- 160. Busser G. W., Mei B., Muhler M., ChemSusChem 2012, 5, 2200–6. [DOI] [PubMed] [Google Scholar]
- 161. Rath T., Bloh J. Z., Lüken A., Ollegott K., Muhler M., Ind. Eng. Chem. Res. 2020, 59, 4265–4272. [Google Scholar]
- 162. Eswar N. K. R., Singh S. A., Heo J., J. Mater. Chem. A 2019, 7, 17703–17734. [Google Scholar]
- 163.
- 163a. Wang Z., Li C., Domen K., Chem. Soc. Rev. 2019, 48, 2109–2125; [DOI] [PubMed] [Google Scholar]
- 163b. Chen S., Takata T., Domen K., Nat. Rev. Mater. 2017, 2. [Google Scholar]
- 164. Karimi Estahbanati M. R., Feilizadeh M., Babin A., Mei B., Mul G., Iliuta M. C., Chem. Eng. J. 2020, 382. [Google Scholar]
- 165. Fontelles-Carceller O., Muñoz-Batista M. J., Rodríguez-Castellón E., Conesa J. C., Fernández-García M., Kubacka A., J. Catal. 2017, 347, 157–169. [Google Scholar]
- 166. Schnee J., Daturi M., El-Roz M., Catal. Sci. Technol. 2020, 10, 5618–5627. [Google Scholar]
- 167. Xu B. B., Zhou M., Zhang R., Ye M., Yang L. Y., Huang R., Wang H. F., Wang X. L., Yao Y. F., J. Phys. Chem. Lett. 2020, 11, 3738–3744. [DOI] [PubMed] [Google Scholar]
- 168. Agostini G., Radnik J., Catalysts 2020, 10. [Google Scholar]
- 169.
- 169a. Wachs I. E., Roberts C. A., Chem. Soc. Rev. 2010, 39, 5002–17; [DOI] [PubMed] [Google Scholar]
- 169b. Zhao C., Wachs I. E., J. Phys. Chem. C 2008, 112, 11363–11372. [Google Scholar]
- 170. Wang F., Jiang J., Wang B., Catalysts 2019, 9. [Google Scholar]
- 171. Nageswaran V. M. V. G., Front. Chem. 2020, 8, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Hartman T., Geitenbeek R. G., Wondergem C. S., van der Stam W., Weckhuysen B. M., ACS Nano 2020, 14, 3725–3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.
- 173a. Munoz-Batista M. J., Motta Meira D., Colon G., Kubacka A., Fernandez-Garcia M., Angew. Chem. Int. Ed. 2018,. 57, 1199–1203; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 57, 1199–1203; [DOI] [PubMed] [Google Scholar]
- 173b. Caudillo-Flores U., Muñoz-Batista M. J., Kubacka A., Fernández-García M., ChemPhotoChem 2018, 2, 777–785; [Google Scholar]
- 173c. Caudillo-Flores U., Barba-Nieto I., Munoz-Batista M. J., Kubacka A., Fernandez-Garcia M., Top. Curr. Chem. 2019, 377, 24. [DOI] [PubMed] [Google Scholar]
- 174. Straathof N. J. W., Noël T., Accelerating Visible-Light Photoredox Catalysis in Continuous-Flow Reactors, in Visible Light Photocatalysis in Organic Chemistry. Wiley-VCH, Weinheim, 2018, 389–413. [Google Scholar]
- 175. Mei B., Han K., Mul G., ACS Catal. 2018, 8, 9154–9164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Zhao F., Cambie D., Janse J., Wieland E. W., Kuijpers K. P. L., Hessel V., Debije M. G., Noel T., ACS Sustainable Chem. Eng. 2018, 6, 422–429. [DOI] [PMC free article] [PubMed] [Google Scholar]