

Abstract
The recent development of ecological studies has been fueled by the introduction of massive information based on chromosome‐scale genome sequences, even for species for which genetic linkage is not accessible. This was enabled mainly by the application of Hi‐C, a method for genome‐wide chromosome conformation capture that was originally developed for investigating the long‐range interaction of chromatins. Performing genomic scaffolding using Hi‐C data is highly resource‐demanding and employs elaborate laboratory steps for sample preparation. It starts with building a primary genome sequence assembly as an input, which is followed by computation for genome scaffolding using Hi‐C data, requiring careful validation. This article presents technical considerations for obtaining optimal Hi‐C scaffolding results and provides a test case of its application to a reptile species, the Madagascar ground gecko (Paroedura picta). Among the metrics that are frequently used for evaluating scaffolding results, we investigate the validity of the completeness assessment of chromosome‐scale genome assemblies using single‐copy reference orthologues.
Keywords: BUSCO, chromosome‐scale genome assembly, completeness assessment, gene space, Hi‐C scaffolding, iconHi‐C
1. INTRODUCTION
Molecular ecology research often targets intra‐ or interspecific variations of information in DNA sequences. In eukaryotes, DNA molecules are found in cell nuclei as part of “chromatin”, a complex of proteins that modulates the conformation of chromosomal DNAs in the nuclear environment. Hi‐C is a method for the genome‐wide capture of such chromosome conformations and was originally developed for detecting the long‐range interaction of chromatins (Lieberman‐Aiden et al., 2009) (Figure 1). This method has more recently been applied to the scaffolding of genome sequences from diverse species (Burton et al., 2013; Kaplan & Dekker, 2013; Marie‐Nelly et al., 2014). In general, the more closely two genomic regions are located on DNA sequences, the more frequently they contact in 3D genomes in chromatin. In genome scaffolding using Hi‐C data, fragmentary sequences of genomic DNA are grouped, ordered, and oriented on the basis of chromatin contact frequency between different genomic regions. Collectively, the genome scaffolding based on this type of chromatin contacts captured in situ in nuclei by digestion‐ligation (“proximity ligation”) is called proximity‐guided assembly (PGA).
FIGURE 1.
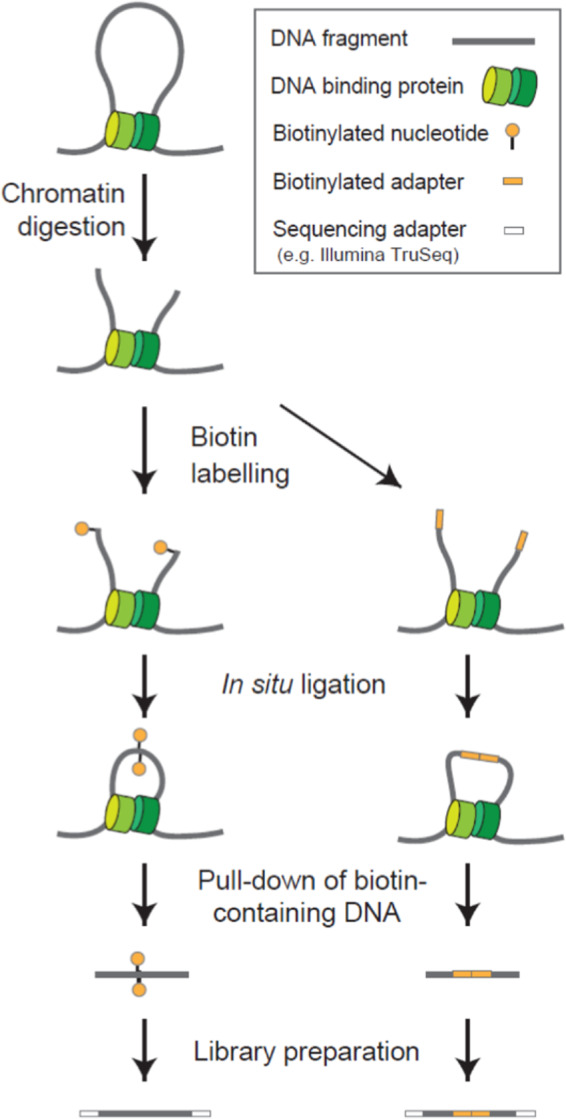
Overview of the workflow used for Hi‐C library preparation. Digestion of chromatin DNA is performed with restriction enzymes or DNA nuclease. DNA ends are labelled by a biotinylated nucleotide (left) or a biotinylated bridge adapter (right). Ligation is performed in situ in the nucleus, and biotin‐containing DNA is captured and used for the generation of sequencing libraries
Molecular ecology studies have been fueled by genome‐wide approaches for monitoring genetic diversity, which is most reliably achieved by the assembly of whole‐genome sequences using the output of modern DNA sequencers. Previously, sequences resulting from whole‐genome assembly were often flanked by long interspersed repeats and remained unassembled with any other sequence (Peona et al., 2018). Under this circumstance, chromosome‐scale sequences were obtained only through genetic linkage mapping, which requires a cross of identified mates and a sufficient number of offspring (Tang et al., 2015; Yoshitake et al., 2018), or optical mapping, which requires a large quantity of high‐molecular‐weight genomic DNA. After the introduction of PGA, Hi‐C scaffolding has become a major solution and has been adopted in mass genome sequencing projects to realize the reconstruction of chromosome‐scale sequences of genomic DNA (e.g., Rhie et al., 2021).
The utility of Hi‐C scaffolding is characterized by its handiness (compared with the resource‐demanding alternatives mentioned above), requiring only chromatin preparation from a single individual and short‐read sequencing on an ordinary sequencing platform. Nonetheless, performing successful Hi‐C scaffolding is not trivial. Most frequently, researchers outsource the whole process to a commercial company or an experienced collaborator, which may not allow them to optimize parameters pertaining to sample preparation and computation with repeated attempts. Alternatively, especially when cost‐saving is desired, researchers may perform the whole preparation by themselves; however, different parts of the process (tissue sampling, library preparation, sequencing, scaffolding, and output validation) may be performed by different individuals, rarely resulting in a self‐contained experience. For these reasons, technical tips regarding the whole process are not explicitly written or shared with academic researcher communities, although they may accumulate at facilities that take on mass genome sequencing projects. It should also be noted that Hi‐C requires the chromatin contained in cell nuclei, rather than extracted genomic DNA. This is often misunderstood, even by those who have a long experience with DNA sequencing, resulting in the unfavourable sampling and storage of materials.
In this review, we address the existing technical information about sample preparation protocols/kits and computational programs, and present technical factors for more successful Hi‐C scaffolding (Figure 2) based on our experience with diverse multicellular organisms (Kadota et al., 2020).
FIGURE 2.
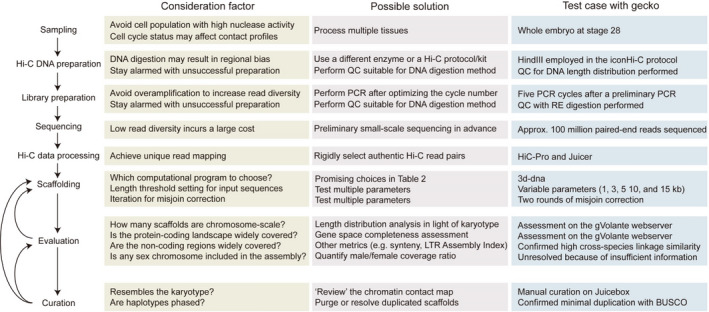
Technical considerations in Hi‐C scaffolding. The major points regarding technical considerations (left) are shown as hands‐on steps. Individual rows show possible solutions (middle) and our demonstration using the Madagascar ground gecko (right). See Naumova et al. (2013) for the detail of the potential effect of cell cycle status to chromatin contacts. See Kadota et al. (2020) for the method to estimate the optimal number of PCR cycles for library amplification
2. WHAT MAKES A DIFFERENCE IN CHROMOSOME‐SCALE GENOME SCAFFOLDING?
The analysis of chromatin dynamics, for which Hi‐C was originally developed, requires appropriate tissues/cells as materials for addressing specific biological questions; however, in Hi‐C scaffolding, the choice of materials is less important because it targets the reconstruction of the whole genome as the uniform goal, even when using different cell populations in an organism. One may expect that the use of numerous types of tissues will yield an optimal performance covering maximally diverse chromatin contacts. However, our previous attempt with this intention did not lead to improvement (Kadota et al., 2020). In general, the use of multiple tissues (in separate preparations) should increase the chance of obtaining a more successful library, and it is preferable to choose tissues with low endogenous nuclease activity (e.g., pancreas) (Takeshita et al., 2000) and those from which single cells can be prepared relatively easily for chromatin fixation (e.g., blood). For animals, tissues including muscle, blood, and liver are listed as promising choices in typical Hi‐C manuals and have been frequently used in published studies (e.g., Rhie et al., 2021). Basically, frozen tissues can serve as materials for Hi‐C library preparation, which should certainly lower the hurdle with species with low accessibility, a typical challenge in ecology. Table 1 summarizes the key laboratory steps in the preparation of chromatin, Hi‐C DNA, and libraries for sequencing, in that order. As a noncommercial choice, this table includes the traditional protocol by Rao et al. (2014), as well as a derivative of this protocol, iconHi‐C (Kadota et al., 2020), which resembles many others (e.g., Belaghzal et al., 2017). As of April 2021, four biochemical companies (Arima Genomics, Dovetail Genomics, Phase Genomics, and Qiagen) manufacture Hi‐C kits, which are formulated with different components and protocols. In general, conventional Hi‐C kits employ a restriction enzyme or a cocktail of multiple restriction enzymes, whereas Omni‐C employs a sequence‐independent endonuclease (Table 1). In Omni‐C, to capture more proximal contacts, disuccinimidyl glutarate (DSG) and formaldehyde are used for sample fixation (Nowak et al., 2005), which is now provided as a kit by Dovetail Genomics. Restriction enzyme digestion and ligation are performed in situ or on chromatin‐binding beads. Library preparation is performed by sonication followed by adapter ligation. The differences in specification between these kits/protocols include (1) choice of the DNA digestion method, (2) method of biotin incorporation, (3) adaptability of the sample quality control (QC) to the laboratory workflow, and (4) degree of amplification in library preparation (Table 1). Sufficient attention to these factors will issue an alert for unsuccessful sample preparation, such as insufficient chromatin fixation and insufficient DNA digestion, and will allow the retrieval of chromatin contacts with maximal diversity. Signs of unsuccessful samples will be alerted in QCs before sequencing (Kadota et al., 2020). When a species of interest has unusual biochemical properties in the selected tissues, genome size, and base composition, which affect the efficiency and uniformity of DNA fragmentation, the choice of the kit/protocol may be crucial (Figure 2). In sequencing Hi‐C libraries, one is usually recommended to obtain 100 million read pairs per Gb genome (Dudchenko et al., 2018; https://www.dnazoo.org/methods) as also suggested by typical Hi‐C kit manuals. Ultimately, the diversity of library molecules, which can be inferred with preliminary small‐scale sequencing (Kadota et al., 2020), determines the ideal number of read pairs to obtain.
TABLE 1.
Comparison of sample preparation for proximity‐based genome scaffolding
| Different specifications | In situ Hi‐C by Rao et al.a |
iconHi‐C (ver. 1.0)b |
Arima‐HiC Kit (Arima Genomics; ver. A160134 v01) |
Proximo Hi‐C (Animal) Prep Kit (Phase Genomics; ver. 4.0) |
Dovetail Hi‐C Kit (Dovetail Genomics; ver. 1.4) |
Omni‐C Proximity Ligation Assay Kit (Dovetail Genomics; ver. 1.3) |
EpiTect Hi‐C Kit (Qiagen; ver. 04/2019) |
|---|---|---|---|---|---|---|---|
| Crosslinking agent | Formaldehyde (final 1%) | Formaldehyde (final 1%) | Formaldehyde (final 2%) |
Crosslinking solution (included in the kit) |
Formaldehyde (final 1.5%) | DSG (final 30 mM)c and formaldehyde (final 1%) | Formaldehyde (final 1%) |
| Enzyme for chromatin DNA digestion | MboI (cuts at "GATC") | HindIII (cuts at "AAGCTT") or DpnII (cuts at "GATC") |
Cocktail of A1 and A2 enzymes (cut at "GATC" and "GANTC")c |
Sau3AI (cuts at "GATC") | DpnII (cuts at "GATC") | Nuclease enzyme mixc | Hi‐C digestion enzyme (cuts at "GATC") |
| Duration of restriction enzyme digestion | 2 h to overnight at 37℃ | Overnight at 37℃ | 30–60 min at 37℃ | 1 h at 37℃ | 1 h at 37℃ | 30 min at 30℃ | 2 h at 37℃ |
| Biotin‐labeling method | Incorporation of biotinylated nucleotide | Incorporation of biotinylated nucleotide | Incorporation of biotinylated nucleotide | Incorporation of biotinylated nucleotide | Incorporation of biotinylated nucleotide | Ligation of biotin‐containing bridge adapterc |
Incorporation of biotinylated nucleotide |
| Chromatin capture | N/A | N/A | N/A | By Recovery Beads (included in the kit)c | By Chromatin Capture Beads (included in the kit)c |
By Chromatin Capture Beads (included in the kit)c |
N/A |
| Ligation condition | 4 h at room temperature | 4–6 h at 16℃ | 15 min at room temperature | 4 h at 25℃ | 1–16 h at 16℃ |
30 min at 22℃ and 1 h at 22°Cc |
2 h at 16℃ |
| Reverse crosslinking |
Overnight or at least 1.5 h at 68℃ |
Overnight at 65℃ | 1.5–16 h at 68℃ | 1–18 h at 65℃ | 45 min at 68℃ | 45 min at 68℃ | 90 min at 80°Cc |
| Quality control (QC) of ligated DNA | No | Yes (by size distribution analysis) | Yes (yield of biotin incorporated DNA) | Yes (yield of biotin incorporated DNA) | Yes (yield of ligated DNA) | Yes (yield of ligated DNA) | No |
| Fragmentation of the ligated DNA | Yes (by sonication) | Yes (by sonication) | Yes (by sonication) | Yes (enzymatic; included in the kit)c | Yes (by sonication) | Noc | Yes (by sonication) |
| Removal of biotin from unligated ends |
No |
Yes | No | No | No | N/A | No |
| PCR cycles for sequencing library preparation | 4–12 cycles | Optimized for each libraryc |
Optimized for each libraryc |
12 cycles | 11 cycles | 12 cycles | 7 cycles |
| Library QC target | Not specified |
Yield and size distribution; digestion with NheI or ClaIc |
Yield and size distribution | Yield and size distribution | Yield and size distribution | Yield and size distribution | Yield and size distribution |
Table 2 summarizes the specification of the existing computational programs for Hi‐C scaffolding. Most of these were developed and maintained by academic parties, with the exception of HiRise, which is used exclusively in paid services by Dovetail Genomics (Putnam et al., 2016), and LACHESIS, which is no longer maintained (Burton et al., 2013). These programs implement different algorithms for using Hi‐C read alignment in scaffolding sequences (Ghurye et al., 2019). Apart from those core algorithmic differences, more superficial parameters with default settings that vary among programs can also largely affect the output, which includes a minimum input sequence length (see Kadota et al., 2020 for an example of a remarkable improvement using an altered length parameter setting) and the number of iterative cycles for misjoin correction (Figure 2). Some of the programs listed in Table 2 are used with certain specifications. FALCON‐Phase (Kronenberg et al., 2018) requires the output of the long read‐based assembly by FALCON‐Unzip (Chin et al., 2016), whereas ALLHiC, which was developed to overcome the difficulty in resolving polyploidy, requires a chromosome‐scale genome assembly or an associated gene annotation of a closely related species for phasing and scaffolding polyploid genomes (Zhang et al., 2019). More crucial key factors that are independent of program choice include the quality and continuity of the input genome assembly (reviewed in Whibley et al., 2021) and the amount of Hi‐C reads obtained after excluding improper fragments resulting from unintended ligation products (self‐ligation, religation, and unligation (“dangling end”); see the details in Kadota et al. (2020).
TABLE 2.
Comparison of computational programs for proximity‐based genome scaffolding. The programs are sorted in the descending order of the number of citations in the literature introducing the individual programs, with the exception of the programs that are not openly maintained (LACHESIS and HiRise at the bottom)
| Program | Description | Input data requirement | Other information |
|---|---|---|---|
| 3d‐dnaa,b |
Misjoin correction algorithm is applied to detect errors in the input assembly; compatible with multiple enzymes |
Accepts only Juicer mapper format |
The results can be reviewed and modified directly by JuiceBox |
| SALSA2c |
Uses the physical coverage of Hi‐C pairs to identify misassembled regions of the input assembly; compatible with multiple enzymes |
Generic bam (bed) file, assembly graph, unitig, 10x link files |
The results can be visualized by JuiceBox via the included script |
| ALLHiCd |
Scaffolding and phasing of a polyploid genome |
Hi‐C read pairs; (option) associated gene annotation or chromosome‐scale genome assembly for a closely related species | Generate the chromatin contact matrix to evaluate genome scaffolding |
| FALCON‐Phasee |
Scaffolding and phasing of a diploid genome |
Hi‐C read pairs; FALCON‐Unzip assembly | Output two phased full‐length pseudo‐haplotypes |
| HiCAssemblerf |
Misassemblies are corrected by iterative joining of high‐confidence scaffold paths |
Hi‐C matrix of h5 format created by HiCExplorer | Misassembled regions in the input assembly can be corrected by specifying the location in the program |
| instaGRAALg |
Overhauling the GRAAL program to allow efficient assembly of large genomes |
Hi‐C matrix of instaGRAAL format created by hicstuff or HiC‐Box | Requires NVIDIA CUDA and can be executed in a limited environment |
| LACHESISh |
No function to correct scaffold misjoins |
Generic bam format | Developer's support discontinued; intricate installation |
| HiRisei |
Employed in Dovetail Chicago/Hi‐C service |
Generic bam format | Open‐source version at GitHub not updated since 2015 |
Overall, there is no single gold‐standard method for library preparation and post‐sequencing scaffolding. When a need for troubleshooting is encountered, one can consider the technical points included in Figure 2, which may provide alternatives for possible improvement.
3. VALIDATION OF CHROMOSOME‐SCALE SCAFFOLDING OUTPUT
The goal of chromosome‐scale genome assembly is the reconstruction of actual nucleotide base lineups in DNA sequences. Assembly products can be rigidly evaluated by referring to any independent information on genome size, chromosomal organization, and location of individual genes, if available. It may not be widely known that a Hi‐C scaffolding output needs to be carefully evaluated and can often be manually modified by referring to the matrix of chromatin contact frequencies (Howe et al., 2020; also see below for an example of a reptile species), that is, the process called “review” in the manual of the program 3d‐dna (https://www.dnazoo.org/methods). In Hi‐C scaffolding, inversions and misjoins occur more frequently than in other scaffolding methods (Dudchenko et al., 2018; Ghurye et al., 2019). This is mainly because Hi‐C reads in pair do not instruct regarding the original fragment orientation in the genome, and the orientation of the sequences that are to be joined is reliably determined only when they are sufficiently long to harbour sufficient data points for chromatin contacts among them and other sequences. Therefore, it is also important to choose a scaffolding program that assumes and facilitates “review” in a dedicated editor, such as JuiceBox (Dudchenko et al., 2018). The visualized chromatin contact map indicates the parts to modify with outstanding signals distant from the diagonal line that do not fit in the intensified signals (intrachromosomal contacts) demarcated in squares (Figure 3a). Such outstanding signals caused by sequence misjoins or disjoins can be resolved by relocating the relevant scaffolds in the contact map (e.g., Figure 3a,b). After the “review”, the program HiC‐Hiker can reduce the error rate further by considering not only the junctions between two adjacent contigs, but also multiple neighbouring contigs (Nakabayashi & Morishita, 2020).
FIGURE 3.

Genome assembly of the Madagascar ground gecko. (a) Hi‐C contact map. The intensities of chromatin contacts quantified in Hi‐C data (red) are indicated in the matrix of different genomic regions. The blue frames indicate the putative chromosomal units. (b) An example of manual curation. The white frames indicate the scaffold units before Hi‐C scaffolding. In a part of the magnified view of the contact map shown in (a), the two input scaffolds indicated by the dashed lines on the left were judged to be derived from a single scaffold on the right. (c, d) Snail plots of the genome assembly before (c) and after (d) Hi‐C scaffolding. These plots were produced using BlobTools2 (Challis et al., 2020). The light‐grey spiral at the center shows the cumulative record count on a log scale, with the white lines indicating successive orders of digits. The distribution of scaffold lengths is shown in dark grey with the plot radius scaled to the longest scaffold of the assembly, and the ranges in orange and light orange indicate the N50 and N90 lengths, respectively. The blue area in the outer layer shows the distribution of GC, AT, and N percentages in the base composition of each scaffold unit
In reality, no comprehensive answer is available for checking the output of de novo genome sequencing. However, karyotypes, namely the number and size of chromosomes prepared from single cells, serve as valuable references for these aspects, and should ideally be made available prior to the assessment of Hi‐C scaffolding results (see Uno et al., 2020 for an example of this sort for sharks with scarce karyotyping reports). If chromosomal gene mapping records or optical mapping results also exist, they can be used as a reference for validation—namely, validating the order of the chromosome segments, for example, using protein‐coding genes with such prior records as markers (see Kadota et al., 2020 for an example). Several early studies employed an existing genome assembly of a closely related species for validation (Dong et al., 2013; Worley et al., 2014); however, this incurred uncontrollable risks because one cannot discern the artifacts to be corrected from natural cross‐species differences. It should be noted that sex chromosome pairs (X/Y or Z/W) may not be assembled with high precision, especially when they have regions that are similar to each other, which are known as pseudoautosomal regions (PAR) (Liu et al., 2019). Sex chromosomes or their segments can be identified by an outstanding ratio of read coverage between a male and a female, if additional whole genome sequencing reads covering both sexes are available (Palmer et al., 2019). Another typical concern is allelic redundancy. Unless one aims to separate different alleles (“haplotype phasing”), it is advisable to discard highly similar sequences with allelic differences (“haplotigs”) before performing Hi‐C, because they can confuse Hi‐C read mapping and result in insufficient scaffolding in those regions.
Methods for evaluating large genome assemblies have been long debated, and no single metric allows an overall assessment (Bradnam et al., 2013; Rhie et al., 2021; Thrash et al.,2020; Veeckman et al., 2016). Scaffolding programs insert tracts of undetermined bases (“N”) between the sequences joined by Hi‐C data, and it should be noted that “N” is implicitly set to a uniform length throughout a genome by individual programs (for example, inserting 500 Ns is the default setting in 3d‐dna and SALSA2).
In the evaluation of the output of de novo genome assembly, the metrics N50 length and NG50 length are frequently used (Bradnam et al., 2013). These metrics apply to scaffold sequences and contig sequences, with the latter indicating sequences without any intervening ambiguous bases (“N”). The N50 and NG50 length denotes the length of the shortest sequence at 50% of the total sequence length in the genome assembly and the genome size, respectively. Basically, a larger N50 or NG50 length entails a more continuous genome assembly. However, the optimal N50 or NG50 length is inherently defined by the karyotype of the species of interest. For the human genome, the N50 of the optimal genome assembly is approximately 154 Mbp, while it is limited to approximately 15 Mbp for the sea lamprey, with more than 100 small, dot‐like chromosomes (2n = 168; Potter & Rothwell, 1970). For this unique karyotype, N50 length cannot be substantially larger than 15 Mbp. Even larger N50 lengths for this species or its close relatives would indicate over‐assembly, which can be the result of the limited number of in silico chromosome fusions. Very importantly, the overall sequence length statistics, such as N50 and NG50, do not reflect the sequence content and its precision. To fulfill this task, one of the metrics proposed most recently was the quantification of reconstructed long terminal repeat (LTR) retrotransposons (LTR Assembly Index, LAI) (Ou et al., 2018). This metric, however, is accessible only when prior information of LTR motif sequences is available, and can be useful if the genome of question inherently harbours the type of LTRs assumed by this program with a certain abundance.
The demand for a more accurate assessment method is increasing as genome sequences of unprecedented quality and continuity emerge. When evaluating genome assemblies, one needs to perform a multifaceted assessment using different metrics (for details, see Rhie et al., 2021), including the coverage of the protein‐coding gene space, which is widely used as a central metric (Figure 2). The following section will focus on how the use of the metric for scoring the completeness of protein‐coding genes should be adapted to the prevailing chromosome‐scale genome assembly production.
4. LIMITATION OF GENE SPACE COMPLETENESS ASSESSMENT
The measurement of gene space completeness was used as a metric of genome assembly quality even before 2010, when most of the available genome assemblies did not reach a chromosomal scale. The only maintained program for this purpose in that period, CEGMA (Parra et al., 2009), was originally developed for identifying a set of protein‐coding genes in a given de novo genome assembly, to be used as a gene set for training gene prediction programs (Parra et al., 2007). Later, the support for CEGMA was discontinued, which was subsequently almost completely replaced by BUSCO (Simão et al., 2015). Generally, when no other option is available as a benchmark solution, users need to be warned about potential misleading reports from the single solution. As previously reported for the benchmarking of multiple sequence alignments (Iantorno et al., 2014), developers and users of genome assembly assessment tools should be fully informed about the perils of misleading assessments.
Since its first release in 2015, BUSCO has been rapidly upgraded to version 2 in 2016, version 3 in 2017, version 4 in 2019, and version 5 in January 2021. BUSCO assumes the use of its accompanying reference gene set derived from OrthoDB (Kriventseva et al., 2019), and both the reference gene set and the pipeline for searching reference genes have been upgraded. This sort of benchmark program is expected to serve as a reliable standard on which genome assemblies can be uniformly compared. Most recently, the BUSCO pipeline was upgraded to version 5 and adopted a new component program for gene search, MetaEuk (Levy Karin et al., 2020), which sometimes yields largely different values compared with the earlier versions 2 and 3 (these two versions superficially perform in the same way because version 3 was a refactored version of version 2).
Another persistent concern with BUSCO is the criterion for choosing reference single‐copy genes (see Korlach et al., 2017)—genes that are absent from genome‐wide sequences of some species (no more than 10% of all of the species considered) are included in the reference orthologue set. Some genes that were secondarily lost during evolution can also be implicitly queried and judged as missing from the genome assembly because of incomplete sequencing or assembly, which results in underestimation of genome assembly completeness. Such an inaccurate assessment of elaborately produced genome assemblies severely hampers the establishment of reasonable decisions in research. To circumvent this systematic inaccuracy, we previously developed the gene set core vertebrate genes (CVG) that contained only the genes retained as single copies in all 29 rigorously selected vertebrate species (Hara et al., 2015). This gene set is included as an option at our original web application, gVolante (Nishimura et al., 2017, 2019), in which different BUSCO versions (including its latest version 5), as well as CEGMA, are available.
Apart from the concerns mentioned above, scoring orthologue detection beyond cross‐species differences is not trivial. As a baseline that is independent of this factor, we assessed the nearly complete human genome assembly CHM13 v1.0 (https://github.com/nanopore‐wgs‐consortium/chm13) released by the Telomere‐to‐Telomere consortium (https://sites.google.com/ucsc.edu/t2tworkinggroup/home; Nurk et al., 2021)—the completeness assessment of this assembly is expected to be nearly 100% if no technical limitations arise. This assessment of the human CHM13 v1.0 assembly resulted in 79 genes judged as missing out of 5,310 BUSCO reference orthologues for Tetrapoda (1.49%) by BUSCO version 5, and one out of 233 CVGs (0.43%) by CEGMA. We tentatively analysed the properties of these 79 reference genes that were judged as missing in OrthoDB v9 and v10 and checked manually the nucleotide sequences of the human CHM13 v1.0 genome assembly for the existence of their orthologues. Importantly, this search revealed that all 79 genes existed in the CHM13 v1.0 assembly (Table S1) and proved BUSCO’s false‐negative detections. This suggests a systematic underestimation of completeness assessment scores by BUSCO, which may be worth exploring further on a larger scale.
Importantly, in this human CHM13 genome assembly (version 1.0), the five remaining gaps are known to be localized in nonprotein‐coding regions—more precisely, ribosomal DNA arrays in the acrocentric arm regions of five chromosomes. The orthologues that were judged as missing in the assessment above are thought to have escaped the gene detection process of the BUSCO pipeline. It is possible that such false negatives occur when a queried orthologue is too divergent to fit within a range recognized as an orthologue by BUSCO or has sequences that are too long or repetitive (even in introns or flanking non‐coding regions) to be scanned properly by the programs implemented inside BUSCO, namely, TBLASTN and Augustus.
Basically, genome assemblies with higher continuity are expected to yield higher completeness scores (see Jauhal & Newcomb, 2021); however, the scores tend to be rather saturated as long as the assessment targets the genomic space marked by a limited number of protein‐coding genes. In resorting to protein‐coding gene completeness, one needs to pay closer attention to the mitigation of false negatives and false positives, by choosing a more appropriate orthologue set and parameters for orthologue search. It is also instrumental to perform an independent assessment of gene coverage in genome assemblies by mapping raw RNA‐seq reads or the transcript contig sequences derived from them to the genome assembly sequences (for details, see Rhie et al., 2021).
5. ARE THEY CHROMOSOMES? CONSIDERATIONS IN ASSEMBLY FINALIZATION
The typical practice of genome assembly finalization includes the process of removing unnecessary sequences, such as unambiguous contaminants and organelle genomes. Herein, a possible discrepancy between the number of resultant chromosome‐scale sequences and the haploid/diploid chromosome number needs to be addressed. This should be followed by the renumbering of the sequences and other amendments required at sequence submission to public databases (https://www.ncbi.nlm.nih.gov/genbank/genomesubmit/). It remains controversial whether the products of chromosome‐scale genome assemblies can be called “chromosomes”. A semantic criticism in this context is that chromosomes consist of not only DNA, but also other components, mainly proteins. It should also be cautioned that “chromosome‐scale” sequences built by Hi‐C scaffolding alone are prone to errors and should be continuously improved by other approaches—it may be risky to regard “Hi‐C karyotyping” as replacing conventional cytogenetic analyses of karyotypes. To evoke a careful distinction, a set of terms including “C‐scaffold” (for chromosome‐scale genome scaffold, instead of “chromosome”) and “scaffotype” (a set of chromosome‐scale scaffolds, instead of “karyotype”) was introduced to avoid confusion (Lewin et al., 2019). Apart from these concerns about semantics and QC, the utility of chromosome‐scale genome sequences opens up new frontiers of molecular‐level biology affecting a wide variety of fields involving diverse species (reviewed in Deakin et al., 2019).
6. TEST CASE OF THE MADAGASCAR GROUND GECKO
As a test case, we dissected the chromosome‐scale genome assembly of the Madagascar ground gecko (Paroedura picta) by referring to the technical consideration factors raised above (Figure 2). The karyotype of this species is 2n = 36 (Main et al., 2012), and the genome size based on the nuclear DNA content is 1.80 Gbp (Hara et al., 2018). Molecular sequence data provision for this animal was initiated with transcriptome analysis (Hara et al., 2015), which was followed by short‐read genome assembly (Hara et al., 2018). For loss‐of‐function experiments, genome editing with CRISPR/Cas9 was recently demonstrated in a reptilian species (Rasys et al., 2019). To promote the potential of this species in question‐driven biological studies, the genome assembly of this species has been incorporated as one of the target species into the guide RNA designing tool CRISPRdirect (https://crispr.dbcls.jp/) (Naito et al., 2015). This resource is expected to facilitate the use of this animal in diverse life science studies that demand loss‐of‐function experiments.
The chromosome‐scale genome scaffolding of the Madagascar ground gecko benefited from the supply of embryos (see Supporting Information Methods for the detailed procedure). Chromatin preparation from the small embryonic sample allowed the improvement of sequence continuity without sacrificing adult animals—the N50 scaffold length increased from 4.1 to 109.0 Mbp (Table 3). This scaffolding performance was achieved with only about 100 million read pairs, which is half of the data size usually recommended in the specification of commercial kits (100 million read pairs per Gb of genome). This could be because the diversity of the read obtained from our Hi‐C library was sufficiently high. Precise control of library quality before sequencing was a prerequisite for this efficient data production (Figure S1).
TABLE 3.
Improvement of the Madagascar ground gecko genome assembly. BUSCO's Tetrapoda gene set consisting of 5310 orthologues was used to assess gene space completeness with BUSCO v5
| Metric | Draft v1.0 (Hara et al., 2018) | Hi‐C scaffolds v2.0 (this study) |
|---|---|---|
| Total length (Mbp) | 1,694 | 1,562 |
| N50 scaffold length (Mbp) | 4.1 | 109.0 |
| Largest scaffold length (Mbp) | 33.7 | 184.3 |
| Number of scaffolds >1 Mbp | 297 | 18 |
| % of sum length of sequences >10 Mbp | 26.6 | 96.5 |
| % of sum length of sequences >1 Mbp | 73.3 | 96.5 |
| Number (%) of reference orthologues detected as “complete” | 4,575 (86.16) | 4,577 (86.20) |
| Number (%) of reference orthologues detected as ‘fragmented’ or “complete” | 4,960 (93.41) | 4,969 (93.58) |
| Number (%) of reference orthologues detected as “duplicated” | 45 (0.8%) | 38 (0.7%) |
| Number (%) of reference orthologues recognized as “missing” | 350 (6.59%) | 341 (6.42%) |
As the input for this Hi‐C scaffolding demonstration aimed at obtaining the first chromosome‐scale genome assembly for the taxon Gekkota (as of May 2021), we employed three draft genome assemblies: (1) the traditional short‐read shotgun assembly, (2) the Chromium supernova assembly using linked reads, and (3) the combination of the two former data types, as well as scaffolding with paired‐end RNA‐seq reads (Figure S2). Each of these three starting assemblies was scaffolded using Hi‐C reads by varying the input sequence length threshold, as included in Figure 2. We derived 15 chromosome‐level assemblies, and a total of 18 assemblies, including the three starting nonchromosome‐scale assemblies, were subjected to the comparison of sequence length statistics (Figure S3) and completeness assessment with BUSCO, which did not produce remarkable differences between the assemblies as often observed in the assessment of chromosome‐scale sequences (see above). Remarkably, varying input sequence length thresholds largely affected the scaffolding output (Figure S3). Applying the small length of 1,000 bp always produced suboptimal output, while the large length of 15,000 bp, the default of some scaffolding programs, did not produce the best output, either (Figures S1 and S3). In the variable output, we evaluated multiple aspects including component sequence length distribution and identified an assembly with optimal or nearly optimal results in all of N50 scaffold length, largest scaffold length, and the proportion of the sum scaffold length for the total assembly size (Assembly 6 in Figures S1 and S3). This assembly was subjected to manual curation (“review”; see above), to derive a sequence assembly for a public release. The manual interventions performed therein included a recovery of the linkage between two small scaffolds, to form a putative single middle‐sized chromosome sequence (Figure 3a,b). Importantly, in assessing the genome assembly of this species, a cross‐species comparison referring to a chromosome‐scale genome assembly was not helpful, because species outside the taxon Gekkota (e.g., anole lizard) diverged more than 150 million years ago (Hara et al., 2018). Conversely, our review was performed by referring to the previously published records of gene mapping using fluorescence in situ hybridization (FISH) on a different species of Gekkota (Supporting Information Methods), which assisted the retrieval of the correct order of chromosome segments based on the location of protein‐coding genes.
In the resulting genome assembly, the number of chromosome‐scale scaffolds with a length >1 Mbp was 18, which is almost the same as the haploid number of chromosomes (n = 18 for XX/ZZ or 19 for XY/ZW; note that the sex chromosome organization in this species is unknown) (Figure 3a). The percentage of sequences longer than 1 Mbp in the entire assembly was 96.5%, indicating that most of the sequence information is incorporated into the resulting chromosome‐sized scaffolds (Table 3). The resulting Madagascar ground gecko genome assembly was assessed to cover 93.58% of the BUSCO’s reference orthologues for the taxon Tetrapoda (4,969 out of 5,310 genes) that were judged as being complete or fragmented by BUSCO version 5 (Table 3). The number of reference orthologues detected as complete increased by two genes after Hi‐C scaffolding (Table 3). The low percentage of the orthologues detected as duplicated (<1%) shows that the assembly harbours almost no redundancy caused by duplicated haplotypes. Alleged contaminated sequences from organelles or other organisms were removed from the assembly prior to the public release.
The resulting chromosome‐scale genome assembly of the Madagascar ground gecko, which was introduced as an example of Hi‐C scaffolding, will serve as a basis for various studies focusing on the ecology and evolution of this species, as well as other molecular‐level biological studies performed in comparison with other amniote species, including mammals and birds.
AUTHOR CONTRIBUTIONS
Shigehiro Kuraku conceived the study and drafted the manuscript. Yuta Ohishi, Shigehiro Kuraku, Mitsutaka Kadota, Osamu Nishimur, and Kazuaki Yamaguchi analysed the data reviewed in this article. Yuki Naito and Kazuaki Yamaguchi set up public data use. All authors contributed to the final writing of the manuscript.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest statement.
Supporting information
Supplementary Material
ACKNOWLEDGEMENTS
We thank the other members of the DNA Analysis Facility managed by the Laboratory for Phyloinformatics, RIKEN BDR for daily collaboration, which provided the insights outlined in this article, and Hiroshi Kiyonari for overall coordination of biological studies on gecko. This work was supported by intramural grants from RIKEN, including the All‐RIKEN “Epigenome Manipulation Project”, and a Grant‐in‐Aid for Scientific Research (B) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) (20H03269) to SK.
Yamaguchi, K. , Kadota, M. , Nishimura, O. , Ohishi, Y. , Naito, Y. , & Kuraku, S. (2021). Technical considerations in Hi‐C scaffolding and evaluation of chromosome‐scale genome assemblies. Molecular Ecology, 30, 5923–5934. 10.1111/mec.16146
Kazuaki Yamaguchi, Mitsutaka Kadota and Osamu Nishimura contributed equally to this study.
Contributor Information
Yuki Naito, Email: y-naito@dbcls.rois.ac.jp.
Shigehiro Kuraku, Email: skuraku@nig.ac.jp.
Data Availability Statement
The resultant Madagascar ground gecko genome assembly is available at NCBI Genome under the BioProject PRJDB5392 and DDBJ under the assembly session ID BDOT02000000. Raw genome, Hi‐C, and transcriptome sequence reads are deposited in the DDBJ under the accession IDs in Tables S1 and S2.
REFERENCES
- Baudry, L. , Guiglielmoni, N. , Marie‐Nelly, H. , Cormier, A. , Marbouty, M. , Avia, K. , Mie, Y. L. , Godfroy, O. , Sterck, L. , Cock, J. M. , Zimmer, C. , Coelho, S. M. , & Koszul, R. (2020). instaGRAAL: chromosome‐level quality scaffolding of genomes using a proximity ligation‐based scaffolder. Genome Biology, 21(1), 148. 10.1186/s13059-020-02041-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belaghzal, H. , Dekker, J. , & Gibcus, J. H. (2017). Hi‐C 2.0: An optimized Hi‐C procedure for high‐resolution genome‐wide mapping of chromosome conformation. Methods, 123, 56–65. 10.1016/j.ymeth.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradnam, K. R. , Fass, J. N. , Alexandrov, A. , Baranay, P. , Bechner, M. , Birol, I. , & Korf, I. F. (2013). Assemblathon 2: evaluating de novo methods of genome assembly in three vertebrate species. Gigascience, 2(1), 10. 10.1186/2047-217x-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton, J. N. , Adey, A. , Patwardhan, R. P. , Qiu, R. , Kitzman, J. O. , & Shendure, J. (2013). Chromosome‐scale scaffolding of de novo genome assemblies based on chromatin interactions. Nature Biotechnology, 31(12), 1119–1125. 10.1038/nbt.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challis, R. , Richards, E. , Rajan, J. , Cochrane, G. , & Blaxter, M. (2020). BlobToolKit – interactive quality assessment of genome assemblies. G3 (Bethesda), 10(4), 1361–1374. 10.1534/g3.119.400908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin, C. S. , Peluso, P. , Sedlazeck, F. J. , Nattestad, M. , Concepcion, G. T. , Clum, A. , Dunn, C. , O'Malley, R. , Figueroa‐Balderas, R. , Morales‐Cruz, A. , Cramer, G. R. , Delledonne, M. , Luo, C. , Ecker, J. R. , Cantu, D. , Rank, D. R. , & Schatz, M. C. (2016). Phased diploid genome assembly with single‐molecule real‐time sequencing. Nature Methods, 13(12), 1050–1054. 10.1038/nmeth.4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deakin, J. E. , Potter, S. , O'Neill, R. , Ruiz‐Herrera, A. , Cioffi, M. B. , Eldridge, M. D. B. , Fukui, K. , Marshall Graves, J. A. , Griffin, D. , Grutzner, F. , Kratochvíl, L. , Miura, I. , Rovatsos, M. , Srikulnath, K. , Wapstra, E. , & Ezaz, T. (2019). Chromosomics: bridging the gap between genomes and chromosomes. Genes (Basel), 10(8), 10.3390/genes10080627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, Y. , Xie, M. , Jiang, Y. , Xiao, N. , Du, X. , Zhang, W. , Tosser‐Klopp, G. , Wang, J. , Yang, S. , Liang, J. , Chen, W. , Chen, J. , Zeng, P. , Hou, Y. , Bian, C. , Pan, S. , Li, Y. , Liu, X. , Wang, W. … Wang, W. (2013). Sequencing and automated whole‐genome optical mapping of the genome of a domestic goat (Capra hircus). Nature Biotechnology, 31(2), 135–141. 10.1038/nbt.2478. [DOI] [PubMed] [Google Scholar]
- Dudchenko O., Batra S. S., Omer A. D., Nyquist S. K., Hoeger M., Durand N. C., Shamim M. S., Machol I., Lander E. S., Aiden A. P., & Aiden E. L. (2017). De novo assembly of the Aedes aegypti genome using Hi‐C yields chromosome‐length scaffolds. Science, 356(6333), 92–95. 10.1126/science.aal3327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudchenko, O. , Shamim, M. S. , Batra, S. S. , Durand, N. C. , Musial, N. T. , Mostofa, R. , Pham, M. , Glenn St Hilaire, B. , Yao, W. , Stamenova, E. , Hoeger, M. , Nyquist, S. K. , Korchina, V. , Pletch, K. , Flanagan, J. P. , Tomaszewicz, A. , McAloose, D. , Pérez Estrada, C. , Novak, B. J. … Aiden, E. L. (2018). The Juicebox Assembly Tools module facilitates de novo assembly of mammalian genomes with chromosome‐length scaffolds for under $1000. bioRxiv, 254797. 10.1101/254797 [DOI] [Google Scholar]
- Durand, N. C. , Shamim, M. S. , Machol, I. , Rao, S. S. , Huntley, M. H. , Lander, E. S. , & Aiden, E. L. (2016). Juicer provides a one‐click system for analyzing loop‐resolution Hi‐C experiments. Cell Syst, 3(1), 95–98. 10.1016/j.cels.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghurye, J. , Rhie, A. , Walenz, B. P. , Schmitt, A. , Selvaraj, S. , Pop, M. , Phillippy, A. M. , & Koren, S. (2019). Integrating Hi‐C links with assembly graphs for chromosome‐scale assembly. PLoS Computational Biology, 15(8), e1007273. 10.1371/journal.pcbi.1007273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara, Y. , Takeuchi, M. , Kageyama, Y. , Tatsumi, K. , Hibi, M. , Kiyonari, H. , & Kuraku, S. (2018). Madagascar ground gecko genome analysis characterizes asymmetric fates of duplicated genes. BMC Biology, 16(1), 40. 10.1186/s12915-018-0509-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara, Y. , Tatsumi, K. , Yoshida, M. , Kajikawa, E. , Kiyonari, H. , & Kuraku, S. (2015). Optimizing and benchmarking de novo transcriptome sequencing: from library preparation to assembly evaluation. BMC Genomics, 16, 977. 10.1186/s12864-015-2007-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe K., Chow W., Collins J., Pelan S., Pointon D.‐L., Sims Y., Torrance J., Tracey A., & Wood J. (2021). Significantly improving the quality of genome assemblies through curation. GigaScience, 10(1). 10.1093/gigascience/giaa153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iantorno, S. , Gori, K. , Goldman, N. , Gil, M. , & Dessimoz, C. (2014). Who watches the watchmen? An appraisal of benchmarks for multiple sequence alignment. Methods in Molecular Biology, 1079, 59–73. 10.1007/978-1-62703-646-7_4. [DOI] [PubMed] [Google Scholar]
- Jauhal, A. A. , & Newcomb, R. D. (2021). Assessing genome assembly quality prior to downstream analysis: N50 versus BUSCO. Molecular Ecology Resources, 21(5), 1416–1421. 10.1111/1755-0998.13364. [DOI] [PubMed] [Google Scholar]
- Kadota, M. , Nishimura, O. , Miura, H. , Tanaka, K. , Hiratani, I. , & Kuraku, S. (2020). Multifaceted Hi‐C benchmarking: what makes a difference in chromosome‐scale genome scaffolding? Gigascience, 9(1), 10.1093/gigascience/giz158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan, N. , & Dekker, J. (2013). High‐throughput genome scaffolding from in vivo DNA interaction frequency. Nature Biotechnology, 31(12), 1143–1147. 10.1038/nbt.2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korlach, J. , Gedman, G. , Kingan, S. B. , Chin, C. S. , Howard, J. T. , Audet, J. N. , Cantin, L. , & Jarvis, E. D. (2017). De novo PacBio long‐read and phased avian genome assemblies correct and add to reference genes generated with intermediate and short reads. Gigascience, 6(10), 1–16. 10.1093/gigascience/gix085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriventseva, E. V. , Kuznetsov, D. , Tegenfeldt, F. , Manni, M. , Dias, R. , Simão, F. A. , & Zdobnov, E. M. (2019). OrthoDB v10: sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Research, 47(D1), D807–d811. 10.1093/nar/gky1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronenberg, Z. N. , Hall, R. J. , Hiendleder, S. , Smith, T. P. L. , Sullivan, S. T. , Williams, J. L. , & Kingan, S. B. (2018). FALCON‐Phase: Integrating PacBio and Hi‐C data for phased diploid genomes. bioRxiv, 327064. 10.1101/327064. [DOI] [Google Scholar]
- Levy Karin, E. , Mirdita, M. , & Söding, J. (2020). MetaEuk‐sensitive, high‐throughput gene discovery, and annotation for large‐scale eukaryotic metagenomics. Microbiome, 8(1), 48. 10.1186/s40168-020-00808-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewin, H. A. , Graves, J. A. M. , Ryder, O. A. , Graphodatsky, A. S. , & O'Brien, S. J. (2019). Precision nomenclature for the new genomics. Gigascience, 8(8), 10.1093/gigascience/giz086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman‐Aiden, E. , van Berkum, N. L. , Williams, L. , Imakaev, M. , Ragoczy, T. , Telling, A. , Amit, I. , Lajoie, B. R. , Sabo, P. J. , Dorschner, M. O. , Sandstrom, R. , Bernstein, B. , Bender, M. A. , Groudine, M. , Gnirke, A. , Stamatoyannopoulos, J. , Mirny, L. A. , Lander, E. S. , & Dekker, J. (2009). Comprehensive mapping of long‐range interactions reveals folding principles of the human genome. Science, 326(5950), 289–293. 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, R. , Low, W. Y. , Tearle, R. , Koren, S. , Ghurye, J. , Rhie, A. , Phillippy, A. M. , Rosen, B. D. , Bickhart, D. M. , Smith, T. P. L. , Hiendleder, S. , & Williams, J. L. (2019). New insights into mammalian sex chromosome structure and evolution using high‐quality sequences from bovine X and Y chromosomes. BMC Genomics, 20(1), 1000. 10.1186/s12864-019-6364-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Main, H. , Scantlebury, D. P. , Zarkower, D. , & Gamble, T. (2012). Karyotypes of two species of Malagasy ground gecko (Paroedura: Gekkonidae). African Journal of Herpetology, 61(1), 81–90. 10.1080/21564574.2012.667837. [DOI] [Google Scholar]
- Marie‐Nelly, H. , Marbouty, M. , Cournac, A. , Flot, J. F. , Liti, G. , Parodi, D. P. , Syan, S. , Guillén, N. , Margeot, A. , Zimmer, C. , & Koszul, R. (2014). High‐quality genome (re)assembly using chromosomal contact data. Nature Communications, 5, 5695. 10.1038/ncomms6695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito, Y. , Hino, K. , Bono, H. , & Ui‐Tei, K. (2015). CRISPRdirect: software for designing CRISPR/Cas guide RNA with reduced off‐target sites. Bioinformatics, 31(7), 1120–1123. 10.1093/bioinformatics/btu743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakabayashi, R. , & Morishita, S. (2020). HiC‐Hiker: a probabilistic model to determine contig orientation in chromosome‐length scaffolds with Hi‐C. Bioinformatics, 36(13), 3966–3974. 10.1093/bioinformatics/btaa288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumova, N. , Imakaev, M. , Fudenberg, G. , Zhan, Y. , Lajoie, B. R. , Mirny, L. A. , & Dekker, J. (2013). Organization of the mitotic chromosome. Science, 342(6161), 948–953. 10.1126/science.1236083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura, O. , Hara, Y. , & Kuraku, S. (2017). gVolante for standardizing completeness assessment of genome and transcriptome assemblies. Bioinformatics, 33(22), 3635–3637. 10.1093/bioinformatics/btx445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura, O. , Hara, Y. , & Kuraku, S. (2019). Evaluating genome assemblies and gene models using gVolante. Methods in Molecular Biology, 1962, 247–256. 10.1007/978-1-4939-9173-0_15. [DOI] [PubMed] [Google Scholar]
- Nowak, D. E. , Tian, B. , & Brasier, A. R. (2005). Two‐step cross‐linking method for identification of NF‐kappaB gene network by chromatin immunoprecipitation. BioTechniques, 39(5), 715–725. 10.2144/000112014. [DOI] [PubMed] [Google Scholar]
- Nurk, S. , Koren, S. , Rhie, A. , Rautiainen, M. , Bzikadze, A. V. , Mikheenko, A. , Vollger, M. R. , Altemose, N. , Uralsky, L. , Gershman, A. , Aganezov, S. , Hoyt, S. , Diekhans, M. , Logsdon, G. , Alonge, M. , Antonarakis, S. , Borchers, M. , Bouffard, G. G. , Brooks, S. Y. … Phillippy, A. M. (2021). The complete sequence of a human genome. bioRxiv, 2021.2005.2026.445798. 10.1101/2021.05.26.445798 [DOI] [Google Scholar]
- Ou, S. , Chen, J. , & Jiang, N. (2018). Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Research, 46(21), e126. 10.1093/nar/gky730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer, D. H. , Rogers, T. F. , Dean, R. , & Wright, A. E. (2019). How to identify sex chromosomes and their turnover. Molecular Ecology, 28(21), 4709–4724. 10.1111/mec.15245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra, G. , Bradnam, K. , & Korf, I. (2007). CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics, 23(9), 1061–1067. 10.1093/bioinformatics/btm071. [DOI] [PubMed] [Google Scholar]
- Parra, G. , Bradnam, K. , Ning, Z. , Keane, T. , & Korf, I. (2009). Assessing the gene space in draft genomes. Nucleic Acids Research, 37(1), 289–297. 10.1093/nar/gkn916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peona, V. , Weissensteiner, M. H. , & Suh, A. (2018). How complete are "complete" genome assemblies?‐An avian perspective. Molecular Ecology Resources, 18(6), 1188–1195. 10.1111/1755-0998.12933. [DOI] [PubMed] [Google Scholar]
- Potter, I. C. , & Rothwell, B. (1970). The mitotic chromosomes of the lamprey, Petromyzon marinus L. Experientia, 26(4), 429–430. 10.1007/bf01896930. [DOI] [PubMed] [Google Scholar]
- Putnam, N. H. , O'Connell, B. L. , Stites, J. C. , Rice, B. J. , Blanchette, M. , Calef, R. , Troll, C. J. , Fields, A. , Hartley, P. D. , Sugnet, C. W. , Haussler, D. , Rokhsar, D. S. , & Green, R. E. (2016). Chromosome‐scale shotgun assembly using an in vitro method for long‐range linkage. Genome Research, 26(3), 342–350. 10.1101/gr.193474.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao, S. S. , Huntley, M. H. , Durand, N. C. , Stamenova, E. K. , Bochkov, I. D. , Robinson, J. T. , Sanborn, A. L. , Machol, I. , Omer, A. D. , Lander, E. S. , & Aiden, E. L. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell, 159(7), 1665–1680. 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasys, A. M. , Park, S. , Ball, R. E. , Alcala, A. J. , Lauderdale, J. D. , & Menke, D. B. (2019). CRISPR‐Cas9 gene editing in lizards through microinjection of unfertilized oocytes. Cell Reports, 28(9), 2288–2292 e2283. 10.1016/j.celrep.2019.07.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renschler, G. , Richard, G. , Valsecchi, C. I. K. , Toscano, S. , Arrigoni, L. , Ramírez, F. , & Akhtar, A. (2019). Hi‐C guided assemblies reveal conserved regulatory topologies on X and autosomes despite extensive genome shuffling. Genes & Development, 33(21–22), 1591–1612. 10.1101/gad.328971.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhie, A. , McCarthy, S. A. , Fedrigo, O. , Damas, J. , Formenti, G. , Koren, S. , Uliano‐Silva, M. , Chow, W. , Fungtammasan, A. , Kim, J. , Lee, C. , Ko, B. J. , Chaisson, M. , Gedman, G. L. , Cantin, L. J. , Thibaud‐Nissen, F. , … Jarvis, E. D. (2021). Towards complete and error‐free genome assemblies of all vertebrate species. Nature, 592(7856), 737–746. 10.1038/s41586-021-03451-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão, F. A. , Waterhouse, R. M. , Ioannidis, P. , Kriventseva, E. V. , & Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics, 31(19), 3210–3212. 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- Takeshita, H. , Mogi, K. , Yasuda, T. , Nakajima, T. , Nakashima, Y. , Mori, S. , Hoshino, T. , & Kishi, K. (2000). Mammalian deoxyribonucleases I are classified into three types: Pancreas, parotid, and pancreas‐parotid (mixed), based on differences in their tissue concentrations. Biochemical and Biophysical Research Communications, 269(2), 481–484. 10.1006/bbrc.2000.2300. [DOI] [PubMed] [Google Scholar]
- Tang, H. , Zhang, X. , Miao, C. , Zhang, J. , Ming, R. , Schnable, J. C. , Schnable, P. S. , Lyons, E. , & & Lu, J. (2015). ALLMAPS: robust scaffold ordering based on multiple maps. Genome Biology, 16(1), 3. 10.1186/s13059-014-0573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrash, A. , Hoffmann, F. , & Perkins, A. (2020). Toward a more holistic method of genome assembly assessment. BMC Bioinformatics, 21(Suppl 4), 249. 10.1186/s12859-020-3382-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uno, Y. , Nozu, R. , Kiyatake, I. , Higashiguchi, N. , Sodeyama, S. , Murakumo, K. , Sato, K. , & Kuraku, S. (2020). Cell culture‐based karyotyping of orectolobiform sharks for chromosome‐scale genome analysis. Commun Biol, 3(1), 652. 10.1038/s42003-020-01373-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeckman, E. , Ruttink, T. , & Vandepoele, K. (2016). Are we there yet? Reliably estimating the completeness of plant genome sequences. The Plant Cell, 28(8), 1759–1768. 10.1105/tpc.16.00349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whibley, A. , Kelley, J. L. , & Narum, S. R. (2021). The changing face of genome assemblies: Guidance on achieving high‐quality reference genomes. Molecular Ecology Resources, 21(3), 641–652. 10.1111/1755-0998.13312. [DOI] [PubMed] [Google Scholar]
- Worley, K. C. , Warren, W. C. , Rogers, J. , Locke, D. , Muzny, D. M. , Mardis, E. R. , & Analysis, C. (2014). The common marmoset genome provides insight into primate biology and evolution. Nature Genetics, 46(8), 850–857. 10.1038/ng.3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshitake, K. , Igarashi, Y. , Mizukoshi, M. , Kinoshita, S. , Mitsuyama, S. , Suzuki, Y. , Saito, K. , Watabe, S. , & Asakawa, S. (2018). Artificially designed hybrids facilitate efficient generation of high‐resolution linkage maps. Scientific Reports, 8(1), 16104. 10.1038/s41598-018-34431-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X. , Zhang, S. , Zhao, Q. , Ming, R. , & Tang, H. (2019). Assembly of allele‐aware, chromosomal‐scale autopolyploid genomes based on Hi‐C data. Nat Plants, 5(8), 833–845. 10.1038/s41477-019-0487-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
The resultant Madagascar ground gecko genome assembly is available at NCBI Genome under the BioProject PRJDB5392 and DDBJ under the assembly session ID BDOT02000000. Raw genome, Hi‐C, and transcriptome sequence reads are deposited in the DDBJ under the accession IDs in Tables S1 and S2.