

Abstract
South Africa is endemic for lumpy skin disease and is therefore reliant on various live attenuated vaccines for the control and prevention of the disease. In recent years, widespread outbreaks of vaccine‐like strains of lumpy skin disease virus (LSDV) were reported internationally, leading to an increase in the generation of full genome sequences from field isolates. In this study, the complete genomes of six LSDVs submitted during active outbreaks in the 1990s in South Africa were generated. Based on phylogenetic analysis, the six viruses clustered with vaccine strains in LSDV Subgroup 1.1 and are subsequently referred to as vaccine‐associated. The genetic differences between the phenotypically distinct vaccine and vaccine‐associated strains were 67 single nucleotide polymorphisms (SNPs). This study characterized the location and possible importance of each of these SNPs in their role during virulence and host specificity.
Keywords: emerging diseases, pathogenesis, sequences, vaccine, virus
1. INTRODUCTION
Although classical techniques for diagnosis of disease‐causing pathogens affecting both humans and animals most certainly still have their place, molecular tools, such as next generation sequencing (NGS), are proving invaluable for both disease diagnosis and control. Such is the case for the poxviral pathogen of cattle, lumpy skin disease virus (LSDV). Since its first isolation in South Africa in 1944, an extensive collection of epidemiological, diagnostic, control and genetic data relating to the virus and disease has been assimilated globally. This is largely attributed to the recent and rapid spread of the disease, previously thought to be confined to Africa, the Middle East, Eastern European countries and Asia (Sprygin, Artyuchova, et al., 2018; Vidanović et al., 2016). New outbreaks in these regions prompted the full genome sequencing of LSDV isolates collected in Greece (Agianniotaki, Mathijs, et al., 2017), Serbia (Toplak et al., 2017), Dagestan in Russia (Sprygin, Babin, et al., 2019), and an lumpy skin disease (LSD) vaccine virus during an active vaccination campaign in Croatia (Lojkic, Simic, Kresic, & Bedekovic, 2018). This significantly increased the existing full genome sequence data set of field isolates, viz. isolate NI‐2490 (Kenya; 1958) (Tulman et al., 2001) and isolate Warmbaths (South Africa; 1999; Kara et al., 2003).
A live attenuated virus (LAV) was generated in 1963 in South Africa at the Onderstepoort Veterinary Research Institute by serial passage of a Neethling‐type field LSDV in cell culture and on the chorioallantoic membranes of embryonated hen's eggs, with in vivo trials in cattle proving its safety and efficacy for use as a vaccine (Weiss, 1968). Regular and widespread use of the commercial formulation of this vaccine was effective in reducing the number of LSD outbreaks in subsequent years, especially in controlling the disease in endemic areas (Hunter & Wallace, 2001). However, more recently, linked to this vaccine and related LSD vaccines, certain shortcomings have been reported (e.g. lack of DIVA capabilities, severe post‐vaccination reactions and recombination with virulent field viruses) (Sprygin, Babin, et al., 2018). Protection against LSD is achieved by use of one of the commercially available Neethling‐type LAV strains, or by use of attenuated sheeppox or goatpox vaccine viruses (Coetzer, Tuppurainen, Babiuk, & Wallace, 2018; Gari et al., 2015; Kitching, 2003). Complete genome sequences of the commercially available Neethling‐type LAV strains of LSDV have been generated and submitted to the publicly accessible GenBank database (Douglass, van Der Walt, Omar, Munyanduki, & Williamson, 2019; Kara et al., 2003; Mathijs et al., 2016). Based on restriction fragment length polymorphism comparisons and subsequently, full genome sequencing of the Kenyan sheep‐ and goat‐pox O‐240 vaccine strain, it was elucidated that this strain was originally derived from a Neethling‐type LSDV (Black, Hammond, & Kitching, 1986; Tulman et al., 2001; Vandenbussche et al., 2016).
Various studies have been conducted to investigate the genome differences between virulent and vaccine LSDV strains in order to elucidate the genetic changes that result in the attenuation phenotype of the latter (Biswas et al., 2019; Douglass et al., 2019; Kara et al., 2003). With the exception of the single change in the superoxide dismutase (SOD) gene homolog encoded by LSDV ORF LW134 described by Douglass et al. (2019), all other studies have identified multiple open reading frames (ORFs) with both alternative reading frames and numerous single nucleotide differences, which may contribute to the attenuated phenotypes observed. Based on all the genetic differences between attenuated and virulent viruses, it is evident that multiple genes are involved and work in synergism to achieve host‐specific attenuation. Currently, the number of known genetic differences that could be targeted during genomics‐guided genetic engineering in development of a DIVA‐compliant, safe and effective recombinant LSD vaccine is vast and it would be costly and time‐consuming to evaluate them all using current technologies (Boshra et al., 2015).
Despite the tremendous success and effectiveness of using LAVs in preventing and controlling LSD, they have several inherent limitations (Hunter & Wallace, 2001; Klement et al., 2018). It is currently impossible to serologically differentiate between vaccinated animals and those showing seroconversion due to natural infection, leading to the development of various molecular tests capable of distinguishing between LAVs and naturally occurring field viruses (Agianniotaki, Chaintoutis, et al., 2017; Erster et al., 2019; Menasherow et al., 2014; Möller et al., 2019; Sprygin, Byadovskaya, et al., 2019). These tests are particularly important for use in countries non‐endemic for LSD, where use of vaccination may be prohibited and presence of any seropositive cases may lead to trade embargoes, etc. In addition, there is a growing body of evidence that LSD LAVs are undergoing reversion to virulence by either recombining with virulent field strains or through mutations in unstable genomic regions leading to full‐length gene restitution (Douglass et al., 2019). This shortcoming of LAVs is the reason why certain countries prohibit the use of commercial vaccines based on LAVs, but rather encourage control campaigns using inactivated, killed or heterologous vaccines. Outbreaks of severe LSD occurred in Russia in 2017, and the causative agents were identified to be genetically similar to the commercially available LAV strains (Kononov et al., 2019; Sprygin, Babin, et al., 2018). One virulent isolate from Saratov in Russia was identified as a multiple intragenic recombinant between commercially available Neethling‐attenuated vaccine virus and the Kenyan sheep‐ and goat‐pox O‐240 vaccine (Sprygin, Babin, et al., 2018). Since the exact cause of attenuation of the virus during cell culture adaptation is not known, the reversion of LAVs to virulent viruses is a major safety concern during vaccination campaigns (Tuppurainen et al., 2018). Even though South Africa is endemic for LSD, vaccination with either one of the commercial live attenuated Neethling‐type vaccines is advised, but not compulsory, while none of the heterologous vaccines are commercially available due to freedom from their respective parental viruses. Since vaccination is not compulsory in South Africa, the true extent of the number of animals vaccinated, the frequency of vaccination or which commercial vaccine was used on each farm or location is not known. Similarly, since not all suspected cases of LSD are laboratory confirmed, the true extent of wild‐type or vaccine‐related viruses circulating in South Africa is not known. Based on the observations in Europe and Russia of LAV vaccine strains involved in either reversion to virulence or recombination, it became essential to start investigating field isolates submitted in South Africa using full genome sequencing to identify the occurrence of similar events.
Phylogenetically, a clear distinction is observed between LSD vaccine viruses (Subgroup 1.1) and wild‐type viruses (Subgroup 1.2) as illustrated by Biswas et al. (2019). In this study, the complete genomes of six LSDVs were determined using NGS technologies. These viruses were obtained from clinical samples submitted during active LSD outbreaks in the Republic of South Africa between 1991 and 1997. They were submitted from three provinces and clustered phylogenetically with the vaccine LSDVs. One of the six vaccine‐associated viruses sequenced in this study, LSD‐248/93, has frequently been shown to elicit LSD clinical symptoms in animals and is therefore considered to be a virulent LSDV (Annandale, Holm, Ebersohn, & Venter, 2012; Tuppurainen et al., 2010, 2012, 2013). Since these virulent viruses clustered phylogenetically with the vaccine Subgroup 1.1, it was important to investigate nucleotide differences between them and the LAV strains. Various other studies have identified specific ORFs and their possible role in virulence, and while certain of these ORFs have been identified in this study, it is not true for others (Biswas et al., 2019; Douglass et al., 2019; Erster et al., 2019). The aim of this study was to investigate each of the observed nucleotide differences in order to predict their possible role in virulence.
2. MATERIALS AND METHODS
2.1. Virus strain culture, DNA isolation and sequencing
Information concerning the place of origin, year submitted and passage history of each of the viruses sequenced during this study are indicated in Table 1. Clinical samples were submitted to the University of Pretoria's Faculty of Veterinary Science for laboratory confirmation of suspected LSD outbreaks. Samples testing PCR‐positive for LSDV according to the method of Ireland and Binepal (1998) were subjected to virus isolation on cell culture.
Table 1.
Lumpy skin disease viruses sequenced during this study
| Isolate name | Reference in article | Place of origin | Year submitted | Passage history | GenBank accession number |
|---|---|---|---|---|---|
| LSD‐103‐GP‐RSA‐1991 | LSD‐103/91 | Onderstepoort, Gauteng, RSA | 1991 | #2 | MN636839 |
| LSD‐58‐LP‐RSA‐1993 | LSD‐58/93 | Polokwane, Limpopo, RSA | 1993 | #3 | MN636838 |
| LSD‐220‐1‐NW‐RSA‐1993 | LSD‐220‐1/93 | Potchefstroom, North West, RSA | 1993 | #1 | MN636841 |
| LSD‐220‐2‐NW‐RSA‐1993 | LSD‐220‐2/93 | Potchefstroom, North West, RSA | 1993 | #1 | MN636842 |
| LSD‐248‐NW‐RSA‐1993 | LSD‐248/93 | Potchefstroom, North West, RSA | 1993 | #4 | MN636840 |
| LSD‐148‐GP‐RSA‐1997 | LSD‐148/97 | Onderstepoort, Gauteng, RSA | 1997 | #2 | MN636843 |
2.1.1. Virus isolation
The isolates, as listed in Table 1 (LSD‐103/91, LSD‐58/93, LSD‐220‐1/93, LSD‐220‐2/93, LSD‐248/93 and LSD‐148/97), include their place of origin, year submitted and passage history. The viruses, LSD‐103/91, LSD‐58/93, LSD‐220‐1/93, LSD‐220‐2/93 and LSD‐148/97, were isolated from tissue samples as described below (Tuppurainen, Venter, & Coetzer, 2005). The tissue samples were homogenized in PBS, followed by centrifugation at 2,000 rpm for 10 min (min). The supernatant was collected, and 500 µl of this supernatant was used to inoculate monolayers of primary bovine dermis (BD) cells. For virus isolation from heparin blood samples, isolate LSD‐248/93 was obtained from a heparin blood sample which was inoculated directly onto the monolayer of BD cells. The cells were prepared in 25 cm2 flasks, using Eagles MEM containing 5% foetal bovine serum (FBS) and 1 mg/ml gentamicin. After 24 hr, the cells were washed with PBS and fresh medium containing 2% FBS added. The flasks were incubated at 37°C for 14 days and observed daily for CPE. If no CPE was detected after this time, a blind passage was done onto fresh BD cells. The BD cells were prepared in 75 cm2 flasks, using Eagles MEM (Lonza) containing 10% FBS (OBP), 1 × GlutaMAX™ Supplement (Gibco) and 1 × antibiotics (100 µg/ml penicillin, 100 µg/ml streptomycin and 250 µg/ml amphotericin [Lonza]). Once the cells were 80%–90% confluent, an aliquot of an LSDV isolate was then added to them for infection. The flasks were rotated every 20 min for 1 hr. Thereafter, the medium was removed and replaced with fresh medium containing 2% FBS, 1 × GlutaMAX™ Supplement and 1 × antibiotics. The flasks were incubated at 37°C with 5% CO2 until 90% CPE was observed.
2.1.2. Viral DNA purification
The isolates were obtained from the University of Pretoria and used for full‐length genome sequencing. All were passaged once more on BD cells as described above, prior to three freeze–thaw cycles at −20°C. The medium containing the lysed cells and virus was then collected and subjected to sonication at 35 kHz for 10 min in a cooled water bath sonicator (Sonorex TK52, Bandelin, Germany). The tubes were then spun at 2,800 g for 5 min at 4°C. The supernatant was removed and kept on ice. The remaining pellet was re‐suspended in 3 ml of 1 × PBS (without Ca+ and Mg+) (Sigma) by vortexing the mixture, followed by sonication at 35 kHz for 5 min and then spun at 2,800 g for 5 min at 4°C. The pellet was discarded and the resulting supernatant pooled with the earlier supernatant containing virus, and then transferred to an Oakridge tube. The tubes were spun at 19 000 g for 60 min at 4°C. The supernatant was discarded, and the remaining pellet was re‐suspended in 500 µl of 1 × PBS. The 500 µl was then used for the extraction of viral DNA using the Invitrogen PureLink™ Viral RNA/DNA Mini Kit (Life Technologies) as per the manufacturer's instructions. The samples were eluted in 50 µl of nuclease‐free water and the viral DNA stored at –80°C prior to sequencing.
2.1.3. Next Generation Sequence analysis and genome annotation
Extracted DNA was submitted for NGS Illumina sequencing to the Agricultural Research Council‐Biotechnology Platform (ARC‐BTP) using the Nextera DNA library kit from Illumina on the HiSeq 2000 Illumina Sequencer. Between 0.5 and 2 Gb of HiSeq data was generated per sample. The HiSeq‐generated paired‐end reads were imported to CLC Genomics Workbench version 9.5., trimmed for quality by removing low‐quality reads (quality limit = 0.05) and Illumina adaptor sequences. The remaining sequences were de novo assembled into contigs, as well as mapped against the published reference sequence of KX764645_Neethling‐LSD‐Vaccine‐OBP and AF409137_Neethling‐LSD‐Warmbaths‐RSA‐2000. The consensus sequence of each assembly and mapping was used to generate a single full genome consensus sequence. The newly constructed consensus sequences were aligned to 14 published LSDV complete genome sequences, and a maximum likelihood phylogenetic tree was constructed under GTR (G + I, G = 4) with 1,000 bootstrap iterations using mega 6 (Tamura et al., 2011). The aligned nucleotide sequences were used to calculate the pairwise distances between each sequence and to identify single nucleotide polymorphisms (SNPs) between the vaccine and vaccine‐associated virus genomes. The new consensus sequences were annotated according to either the reference AF409137_Neethling‐LSD‐Warmbaths‐RSA‐2000 or KX764645_Neethling‐LSD‐Vaccine‐OBP sequences. The annotated sequences generated during this study were deposited in GenBank with accession numbers indicated in Table 1.
2.1.4. Genome comparison and protein domain predictions
A second alignment was generated containing the six vaccine‐associated, four vaccine sequences (KX764645_Neethling‐LSD‐Vaccine‐OBP, KX764643_Neethling‐LSD‐Vaccine‐ SIS‐Lumpyvax, MK441838_Neethling‐LSD‐Vaccine‐Herbivac‐ batch008 and KX764644_Neethling‐LSD‐Vaccine‐Herbivac) and an isolate from a recently vaccinated animal MG972412_ Neethling‐LSD‐Cro2016‐Croatia‐2016 (Lojkic et al., 2018). The 5′ and 3′ UTR regions were removed in order for the alignment to start at 91bps downstream of LW001 and end 7bps upstream of LW156. The sizes of each sequence in the alignment differ between 150,379 and 150,394. The number and position of each SNP between the vaccine and vaccine‐associated viruses were determined. The influence of each SNP on the reading frame or predicted amino acid (aa) exchange was determined and described in this study as either altering the reading frame, synonymous, non‐synonymous SNP or within the intragenic regions. Predicted ORFs with changes in reading frames or synonymous SNPs were translated using CLC Genomics Workbench version 9.5, and the putative aa sequences were analysed using the InterPro domain and protein family classification program from the European Bioinformatics Institute, Cambridge, GB (https://www.ebi.ac.uk/interpro).
2.1.5. PCR and Sanger sequence confirmation
Six of the mutations identified which might alter the reading frame of a specific ORF were confirmed with PCR and Sanger sequencing. A 1:10 dilution of the gDNA samples submitted for NGS was used as template in six individual PCRs along with the primers listed in Table 2. Dreamtaq DNA polymerase master mix (Life Technology) was added to a 20 µl reaction with 0.25 µmol/L of each primer at an annealing temperature of 53°C for 45 cycles. An aliquot of the resulting amplicons was analysed using 1% agarose gel electrophoresis, and the remainder of the reactions were submitted to Inqaba Biotechnical Industries (South Africa) for Sanger sequencing, using the primers incorporated during the generation of the amplicons. Sequence data were analysed and compared using CLC Genomics Workbench 9.5 (QIAGEN Aarhus, www.clcbio.com).
Table 2.
List of primers used during PCR and Sanger sequencing confirmation of mutations within six ORFs
| ORF investigated | Primer name | Primer sequence (5′–3′) |
|---|---|---|
| LW019 | LW019‐F | CAGTTCCCCACTTAATTAT |
| LW019‐R | CACTGGAAGGTAAAACTAAC | |
| LW086 | LW086‐F | CAACGTCGAAATTAGAGAGC |
| LW086‐R | GTTTTTTGATCTTATGATTTCTGT | |
| LW087 | LW087‐F | CTAACGAGTAATGAGATAATACA |
| LW087‐R | AGTTAAAGTTGTAGAAGGTGA | |
| LW114 | LW114‐F | GGTTTGTTTTAGATAAATGGG |
| LW114‐R | GGATATCACAATAAACCAACTT | |
| LW131 | LW131‐F | ATGGGTTTTTGAGTATGGG |
| LW131‐R | AAGTTATATCCTTTGCCTAA | |
| LW134 | LW134‐F | GACATTTTTAGGAGATCTTC |
| LW134‐R | TTACCATCACCTCTATAATTTG |
3. RESULTS
3.1. New LSDV genomes
The complete genome sequences of six additional LSDV isolates were determined and used in subsequent analysis. These viruses were submitted to the University of Pretoria, Faculty of Veterinary Science, for diagnostic confirmation of suspected outbreaks in South Africa. The submission dates varied from 1991 to 1997, and additional information concerning the origin, isolation and passage history of the viruses are described in Table 1. Between 3 × 106 and 1.8 × 107 HiSeq‐generated reads were used to de novo assemble the genomes as well as mapping back to the newly generated consensus sequences. Since the HiSeq NGS technology generates short reads (125 × 125 bp), the inverted terminal repeat regions were excluded from downstream analysis. Genomes between 150,391 and 150,430 bps were subsequently used to generate an alignment and analyse the phylogenetic relatedness of the isolates. Based on a maximum likelihood phylogenetic tree, the six isolates (LSD‐103/91, LSD‐58/93, LSD‐220‐1/93, LSD‐220‐2/93, LSD‐248/93 and LSD‐148/97) clustered with the commercial vaccine strains, KX764645_Neethling‐LSD‐Vaccine‐OBP, KX764643_Neethling‐LSD‐Vaccine‐SIS‐Lumpyvax, and KX764644_Neethling‐LSD‐Vaccine‐Herbivac (Figure 1). A high percentage sequence identity was observed within each of the two subgroup clusters (Subgroup 1.1 and 1.2). For example, the isolates within the vaccine cluster shared between 99.94% and 100% sequence identity between them, while the sequences in the wild‐type cluster shared between 99.86% and 100% sequence identity among themselves. In contrast, between 98.48% and 98.54% sequence identity were identified between isolates from the vaccine with the wild‐type cluster. The differences in percentage sequence identity between the two subgroup clusters were due to between 2,200 and 2,255 nucleotide differences, resulting in aa or reading frame changes affecting 124 of the 156 predicted ORFs. Based on the large number of SNPs and aa exchanges observed between the vaccine and wild‐type clusters, the differences between the vaccine and the six newly sequenced vaccine‐associated viruses were investigated for their possible role in virulence.
Figure 1.
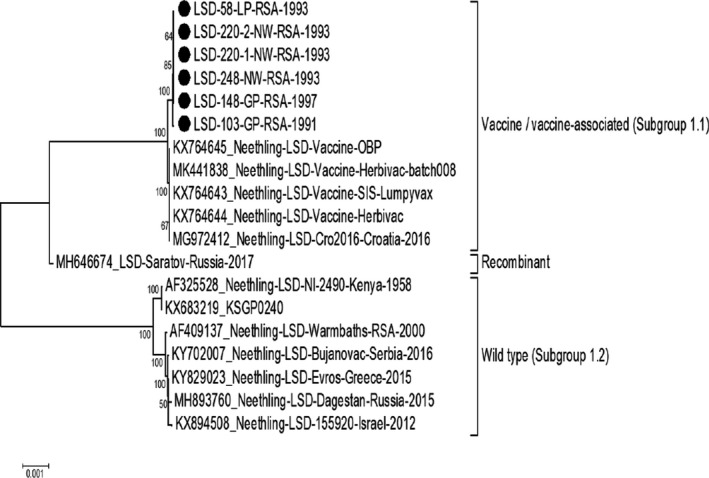
Maximum likelihood phylogenetic analysis of new LSDV genomes (marked with black dots) in relation to sequences obtained from GenBank
3.2. Virulent vaccine‐associated viruses
The six vaccine‐associated viruses were isolated from clinical samples submitted as part of diagnostic testing for confirmation of LSD outbreaks. Isolate LSD‐248/93 has been extensively used as a virulent virus in order to induce LSD‐associated pathogenicity in animals towards a better understanding of the disease and control (Annandale et al., 2012; Tuppurainen et al., 2010, 2012, 2013). There were 67 SNPs identified between the vaccine and vaccine‐associated viruses, and these were classified as follows: homopolymers in intragenic regions (n = 12); SNPs in intragenic regions (n = 6); SNPs altering the reading frame (n = 8); synonymous SNPs (n = 21); and non‐synonymous SNPs (n = 20) (Tables 1, 2, 3, 4, 5). Individual mutations will be discussed based on the aforementioned classifications.
Table 3.
Mutations identified between vaccine strains and vaccine‐associated viruses involved in altering the reading frame. The position of each SNP is provided for KX764645_Neethling‐LSD‐Vaccine‐OBP and AF409137_Neethling‐LSD‐Warmbaths‐RSA‐2000
| # | Nucleotide in vaccine strains (KX764645_Neethling‐LSD‐Vaccine‐OBP) | Nucleotide in vaccine‐associated viruses | Nucleotide in wild type (AF409137_Neethling‐LSD‐Warmbaths‐RSA‐2000) | ORF affected | Amino acid position in protein |
|---|---|---|---|---|---|
| 1 | 13,403 (–) | A | 13,412 (A) | LW019 | 124 |
| 2 | 68,066 (A) | – | 68,095 (–) | LW086 | 206 |
| 3 | 81,108 (T) | C | 81,131 (C) | LW087 | 198 |
| 4 | 81,110 (T) | A | 81,133 (A) | LW087 | 198 |
| 5 | 81,111 (T) | – | 81,134 (–) | LW087 | 198 |
| 6 | 105,629 (A) | – | 105,695 (–) | LW114 | 168 |
| 7 | 119,429 (–) | T | 119,523 (T) | LW131 | 83 |
| 8 | 124,248 (A) | – | 124,343 (–) | LW134 | 721 |
Table 4.
Non‐synonymous SNPs identified between vaccine strains and vaccine‐associated viruses. Amino acid exchanges are listed as follows: vaccine virus (>) vaccine‐associated virus. The position of each SNP is provided for KX764645_Neethling‐LSD‐Vaccine‐OBP and AF409137_ Neethling‐LSD‐Warmbaths‐RSA‐2000
| # | Nucleotide in vaccine strains (KX764645_Neethling‐LSD‐Vaccine‐OBP) | Nucleotide in vaccine‐associated viruses | Nucleotide in wild type (AF409137_Neethling‐LSD‐Warmbaths‐RSA‐2000) | ORF affected | Amino acid position in protein | Resulting change (>amino acid exchanges) |
|---|---|---|---|---|---|---|
| 1 | 8,074 (G) | T | 8,080 (G) | LW011 | 11 | A > D |
| 2 | 13,408 (C) | T | 13,418 (C) | LW019 | 123 | D > N |
| 3 | 18,562 (C) | T | 18,582 (C) | LW026 | 121 | A > T |
| 4 | 19,534 (T) | A | 19,554 (T) | LW027 | 438 | K > N |
| 5 | 21,567 (T) | C | 21,584 (C) | LW028 | 135 | T > A |
| 6 | 47,010 (A) | T | 47,033 (T) | LW052 | 33 | N > K |
| 7 | 50,168 (T) | C | 50,191 (C) | LW057 | 272 | I > V |
| 8 | 71,040 (G) | A | 71,064 (G) | LW079 | 128 | R > K |
| 9 | 77,038 (A) | T | 77,062 (T) | LW083 | 663 | K > N |
| 10 | 80,547 (G) | A | 80,570 (A) | LW087 | 12 | G > D |
| 11 | 81,255 (–) | A | 81,277 (A) | LW087 | C‐Terminal: ‐>S: Same as wild type | |
| 12 | 81,258 (–) | A | 81,272 (A) | LW087 | C‐Terminal: ‐> L: Same as wild type | |
| 13 | 91,639 (A) | G | 91,712 (G) | LW098 | 652 | I > T |
| 14 | 91,925 (C) | T | 92,009 (T) | LW098 | 553 | G > D |
| 15 | 104,904 (T) | G | 104,971 (G) | LW112 | 347 | S > R |
| 16 | 135,777 (T) | C | 135,894 (T) | LW144a | 119 | F > L |
| 17a | 136,177 (‐) | T | 136,295 (T) | LW144 | L > FI | |
| 18a | 136,177 (‐) | A | 136,295 (–) | LW144 | L > FI | |
| 19a | 136,177 (‐) | T | 136,295 (–) | LW144 | L > FI | |
| 20 | 140,948 (T) | C | 140,999 (T) | LW147 | 143 | I > T |
ªMutation confirmed with PCR and Sanger sequencing.
Table 5.
Synonymous SNPs identified between vaccine strains and vaccine‐associated viruses. The position of each SNP is provided for KX764645_Neethling‐LSD‐Vaccine‐OBP and AF409137_ Neethling‐LSD‐Warmbaths‐RSA‐2000
| # | Nucleotide in vaccine strains (KX764645_Neethling‐LSD‐Vaccine‐OBP) | Nucleotide in vaccine‐associated viruses | Nucleotide in wild type (AF409137_Neethling‐LSD‐Warmbaths‐RSA‐2000) | ORF affected | Amino acid position in protein | Resulting amino acid change |
|---|---|---|---|---|---|---|
| 1 | 11,507 (C) | T | 11,512 (C) | LW017 | 12 | L = L |
| 2 | 39,546 (T) | C | 39,567 (T) | LW045 | 212 | T = T |
| 3 | 43,846 (T) | C | 43,866 (C) | LW049 | 249 | G = G |
| 4 | 56,951 (C) | G | 56,977 (C) | LW066 | 60 | A = A |
| 5 | 61,240 (T) | C | 61,269 (T) | LW071 | 416 | G = G |
| 6 | 64,167 (C) | T | 64,196 (T) | LW072 | 68 | S = S |
| 7 | 71,998 (G) | A | 72,022 (G) | LW083 | 572 | A = A |
| 8 | 81,723 (A) | G | 81,747 (A) | LW088 | 488 | P = P |
| 9 | 85,571 (C) | T | 85,595 (T) | LW090 | 65 | A = A |
| 10 | 85,990 (A) | T | 86,014 (A) | LW091 | 85 | S = S |
| 11 | 87,868 (G) | A | 87,892 (A) | LW094 | 441 | G = G |
| 12 | 103,332 (A) | G | 103,405 (A) | LW111 | 60 | S = S |
| 13 | 104,442 (C) | T | 104,509 (T) | LW112 | 193 | P = P |
| 14 | 109,433 (G) | A | 109,499 (G) | LW116 | 863 | L = L |
| 15 | 109,664 (T) | C | 109,730 (T) | LW116 | 940 | G = G |
| 16 | 118,583 (T) | C | 118,713 (T) | LW129 | 61 | D = D |
| 17 | 125,940 (C) | T | 126,035 (T) | LW135 | 231 | S = S |
| 18 | 131,803 (C) | T | 131,901 (T) | LW144a | 25 | G = G |
| 19 | 133,229 (T) | C | 13,332 (T) | LW145 | 574 | C = C |
| 20 | 136,014 (A) | G | 136,132 (G) | LW151 | 540 | E = E |
| 21 | 147,891 (A) | T | 147,946 (A) | LW152 | 389 | I = I |
3.3. Mutations altering the reading frame
Eight mutations were identified to alter the predicted reading frames of six ORFs (Table 3). Each of the 8 mutations was confirmed using PCR and Sanger sequencing of the six vaccine‐associated isolates, wild‐type Neethling‐LSD‐Warmbaths‐RSA‐2000 and Neethling‐LSD‐Vaccine‐OBP vaccine strain. With the exception of ORF LW019, each of the changes resulted in predicted proteins similar to the wild‐type viruses. In LW019, both the vaccine and wild‐type viruses were predicted to translate two individual proteins (LW019a and LW019b) of different sizes. The wild‐type LW019b (264 amino acids) contained both the BTB/POZ and BTB/Kelch domains, while LW019a (290 aa) had the Kelch‐type beta‐propeller. In contrast, the vaccine virus produced a shorter LW019b (150 aa) containing only the BTB/POZ domain and a larger LW019a (440 aa) with both BTB/Kelch and Kelch‐type beta‐propeller domains (Figure 2a). The vaccine‐associated viruses had an (A) nucleotide insertion compared to the vaccine genome, but similar to the wild‐type viruses at aa position 124. This abolishes the truncation of LW019b at position 150 and instead resulted in a reading frame that predictably encodes a single 569 aa protein (Figure 2a). This ORF LW019, producing a single protein, is unique to the vaccine‐associated viruses described here. Another unique feature of the LW019 ORF from the vaccine‐associated viruses was a non‐synonymous SNP located at position 123, exchanging the aspartate (D) aa found in both vaccine and virulent viruses with asparagine (N) (Figure 2a). The D123N exchange occurs at the start of the BTB‐Kelch domain, yet the influence of this exchange on the function of the protein is not known.
Figure 2.

Graphical representation of six LSDV ORFs and their predicted domains using InterProScan predictions based on VACV protein names and functionality, where known. The influence of each SNP on the reading frames of either vaccine, vaccine‐associated or wild‐type LSDVs is indicated. (a) ORF LW019. A complete 569 aa ORF is observed in the vaccine‐associated strains, compared to the two individual ORFs (LW019a and LW019b) predicted for either the wild‐type (AF409137_Neethling‐LDS‐Warmbaths‐RSA‐2000) or vaccine (KX764645_Neethling‐LSD‐Vaccine‐OBP) strains. (b) ORF LW086. This ORF within the vaccine‐associated and virulent wild‐type viruses is predicted to code for a 213 aa protein, compared to a 210 aa truncated protein in the vaccine strain. (c) ORF LW087. Similar to LW086, the vaccine‐associated and virulent wild‐type ORFs are predicted to produce a 253 aa protein and the vaccine virus strain a truncated 200 aa protein. In addition, both the vaccine‐associated and wild‐type viruses have a D at position 12 in the N‐terminal domain, compared to the G of the vaccine virus. (d) ORF LW114. The vaccine virus predictably encodes a longer 179 aa protein, while both the vaccine‐associated and wild‐type viruses encode truncated 168 aa proteins. (e) ORF LW131. The vaccine‐associated and wild‐type viruses are predicted to code for a complete 161 aa superoxide dismutase‐like protein, whereas for the vaccine viruses a termination signal leads to a shorter 108 amino acid protein. (f) ORF LW134. The wild‐type virus is predicted to produce a complete 2025 aa protein, while the vaccine‐associated viruses produced a 2024 aa protein, due to a deletion of the asparagine (N) at position 2020 (N2020del). By contrast, the vaccine virus should produce two proteins of 721 and 1,243 amino acids
3.4. Single nucleotide polymorphisms
The 21 synonymous SNPs between the vaccine and vaccine‐associated viruses were detected in 20 ORFs (Table 5). Of the 21 SNPs, 13 were unique to the vaccine‐associated viruses and eight similar to the virulent wild‐type viruses. The majority of these SNPs were located in ORFs central to the LSDV genome. In contrast, 20 non‐synonymous SNPs in 18 ORFs were identified with nine of them unique to the vaccine‐associated and 11 identical to the virulent wild‐type viruses (Table 4). The non‐synonymous mutations were equally distributed across the LSDV genome.
3.5. Mutations within the intragenic regions
Twelve mutations associated with homopolymer repeat sequences and six additional SNPs were identified within the intragenic regions of the vaccine‐associated viruses. Of the 18 SNPs, 13 were identical to the wild‐type viruses and the others unique to the vaccine‐associated viruses (Table 6). The majority of the mutations were insertions of a nucleotide within the vaccine‐associated viruses.
Table 6.
Mutations within the intragenic regions identified between vaccine strains and vaccine‐associated viruses. The position of each SNP is provided for KX764645_Neethling‐LSD‐Vaccine‐OBP and AF409137_Neethling‐LSD‐Warmbaths‐RSA‐2000
| # | Nucleotide in vaccine strains (KX764645_Neethling‐LSD‐Vaccine‐OBP) | Nucleotide in vaccine‐associated viruses | Nucleotide in wild type (AF409137_Neethling‐LSD‐Warmbaths‐RSA‐2000) | Resulting change | Intragenic between ORFs | Positions before ORF |
|---|---|---|---|---|---|---|
| 1 | 1,329 (–) | A | 1,349 (A) | Homopolymer (Poly A = 9) | LW002 and LW003 | Before LW002 |
| 2 | 8,160 (–) | T | 8,168 (T) | Homopolymer (Poly T = 11) | LW011 and LW012 | −56 before LW011 |
| 3 | 9,956 (–) | A | 9,960 (–) | Homopolymer (Poly A = 9) | LW013 and LW014 | −43 before LW013 |
| 4 | 10,753 (T) | — | 10,758 (T) | Homopolymer (Poly T = 9) | LW015 and LW016 | −28 before LW015 |
| 5 | 15,627 (–) | T | 15,644 (T) | Indel | LW022 and LW023 | −144 before LW022 |
| 6 | 16,006 (–) | A | 16,025 (A) | Homopolymer (Poly A = 8 – 10) | LW023 and LW024 | −67 before LW023 |
| 7 | 16,006 (–) | A | 16,026 (A) | Homopolymer (Poly A = 8–10) | LW023 and LW024 | −67 before LW023 |
| 8 | 22,614 (A) | G | 22,632 (G) | SNP | LW029 and LW030 | −6 before LW029 |
| 9 | 27,947 (–) | T | 27,970 (T) | Homopolymer (Poly T = 11) | LW034 and LW035/LW036 | −31 before LW034 |
| 10 | 57,977 (–) | T | 58,006 (T) | Homopolymer (Poly T = 7) | LW067 and LW068 | −43 before LW068 |
| 11 | 110,317 (A) | ‐ | 110,383 (A) | Indel | LW116 and LW117 | |
| 12 | 113,978 (–) | A | 114,044 (A) | Homopolymer (Poly A = 11) | LW122 and LW123 | −21 before LW123 |
| 13 | 113,978 (–) | A | 114,045 (A) | Homopolymer (Poly A = 11) | LW122 and LW123 | −22 before LW123 |
| 14 | 122,088 (–) | T | 122,182 (T) | Indel | LW133 and LW134 | −10 before LW134 |
| 15 | 122,088 (–) | C | 122,183 (C) | Indel | LW133 and LW134 | −9 before LW134 |
| 16 | 122,088 (–) | T | 122,184 (T) | Indel | LW133 and LW134 | −8 before LW134 |
| 17 | 139,041 (–) | T | Homopolymer (Poly T = 7) | LW145 and LW146 | −180 before LW146 | |
| 18 | 149,398 (T) (not OBP, but all other vaccine strains) | — | 149,453 (T) | Homopolymer (Poly T = 9) | LW154 and LW155 |
4. DISCUSSION
4.1. Mutations altering the reading frame
Nine frameshift mutations were previously identified in vaccine and three in virulent LSDV genomes (Kara et al., 2003). In this study, 8 mutations were identified that alter the reading frames of six ORFs (Table 3). Six of these ORFs (LW019, LW086, LW087, LW131 and LW134) were previously described by Kara et al. (2003), and every mutation observed here resulted in the alteration to a reading frame identical to the virulent wild‐type virus, with the exception of the one identified within ORF LW019. In contrast to both wild‐type and vaccine viruses, the newly described vaccine‐associated strains have a single LW019 ORF, predicted to produce a single Kelch‐like protein. Only isolate Neethling LSDV NI‐2490‐Kenya‐1958 (AF325528) and its laboratory‐derived strain KSGPO240 (KX683219) have a similar single predicted 569 amino acid protein.
The ORFs LW086 and LW087 both contain a Nudix hydrolase signature domain, found in the MutT proteins, similar to the vaccinia virus (VACV) D9 and D10 mRNA‐decapping enzymes (Bessman, Frick, & O’Handley, 1996). Both these enzymes have been shown to play a role in the virulence of VACV by altering or inhibiting the expression of host proteins, through the degradation of mRNA by means of decapping the methylated cap attached to mRNA (Parrish & Moss, 2007). The two neighbouring proteins function in synergism in order to down‐regulate the host mRNA production, even though D9 is expressed early and D10 late during virus infection (Liu, Wyatt, Orandle, Minai, & Moss, 2014; Parrish, Resch, & Moss, 2007). Vaccine‐associated and virulent wild‐type viruses encode LW086 ORFs of 213 and ORF LW087 of 253 aa, in contrast to the truncated versions in vaccine viruses of 210 and 200 amino acids, respectively (Figure 2b,c). Even though none of these truncations disrupted the predicted Nudix boxes, they would be predicted to alter the tertiary five beta‐stranded structure of LW086 and LW087 (Bessman et al., 1996) (Figure 2b,c). A non‐synonymous substitution changed aa position 12 of LW087 from glycine (G) to an acidic aspartate (D) in both the virulent wild‐type and vaccine‐associated viruses (Figure 2c). The influence of this exchange on the biological function and cellular location of the protein is not known.
In contrast to the previous ORFs, LW114 was truncated at 168 amino acids in the vaccine‐associated and virulent wild‐type viruses compared to the 179 aa protein of the vaccine strains (Figure 2d). The latter has an additional C‐terminal sequence of IVKKPFKNNNG. LW114 encodes a ribonuclease H‐like domain found in Holliday junction resolvases such as A22 of VACV, with which it has a 56% sequence identity. The vaccinia virus A22 has been shown to function as dimers during association with Holiday Junction DNA during the cleavage of such structures (Garcia, Otero, Lebowitz, Schuck, & Moss, 2006).
A significant change at aa position 83 of ORF LW131 in the vaccine‐associated viruses abolished the truncation observed in vaccine viruses (generating a 108 aa SOD), restoring the protein to its full length, as found for the virulent wild‐type viruses (a 161 aa protein) (Figure 2e). Even though SODs are highly conserved among pox viruses, it was the first VACV core protein described not to play a role in virulence or virus replication (Almazan, Tscharke, & Smith, 2001). It is not yet clear if the large deletion within LW131 in the LSD vaccine viruses produces a functional SOD protein.
The last of the six predicted ORFs with an altered reading frame difference was LW134 (Table 3). The predicted protein is similar to the VACV serpin protein B22R, coding for a complete 2025 aa protein in the virulent wild‐type viruses, and due to the reading frame shift a 2024, aa protein in the vaccine‐associated viruses (Figure 2f). In contrast, the vaccine viruses are predicted to produce two proteins, one of 721 amino acids containing the N‐terminal domain and the larger, a protein of 1,243 amino acids inclusive of the C‐terminal transmembrane and cytoplasmic regions (Figure 2f). Complete or partial deletion of the B22R homologues in other poxviruses has indicated that it is nonessential for modulating the immune responses, yet it has anti‐inflammatory and anti‐apoptotic properties (Blake et al., 1995; Kettle, Blake, Law, & Smith, 1995; Legrand et al., 2005).
Two ORFs with alternative reading frames in both the vaccine and wild‐type viruses (LW013 and LW026) remained unchanged in the vaccine‐associated viruses. ORF LW144, with a truncation in the vaccine virus, also codes for two proteins in the vaccine‐associated viruses, similar to the vaccine strains (Kara et al., 2003).
4.1.1. Single nucleotide polymorphisms
Of the 41 SNPs within ORFs identified between the vaccine and vaccine‐associated viruses, 21 were synonymous and 20 non‐synonymous. Although the synonymous SNPs are interesting for virus evolution, the focus of this study was to investigate the possible role non‐synonymous SNPs and their resulting amino acid exchanges might have on virulence, and thus, only these will be discussed in more detail.
The first non‐synonymous SNP unique to the vaccine‐associated viruses was in LW011, the predicted G‐protein‐coupled chemokine receptor. This SNP resulted in an exchange of alanine (A) at position 11 with aspartate (D). This substitution (A11D) occurred in the signal peptide region of the non‐cytoplasmic N‐terminal domain, essential for translocating the protein and its seven transmembrane regions to the cell membrane (Figure 3a). The impact of this mutation on the translocation and subsequent functioning of the LSDV G‐protein chemokine needs to be investigated. This unique mutation did not occur in the virulent vaccine‐like strains identified during the 2017 outbreaks in Russia (Kononov et al., 2019). Since this mutation does not occur in any of the other virulent LSDVs, the importance of it during virulent phenotype is questioned and should be elucidated.
Figure 3.
Graphical representation of the predicted domains within five ORFs, based on InterProScan using VACV protein names and functions. The location of each observed non‐synonymous SNP within these reading frames is indicated with an arrow at its position with the aa predicted in the genomes of the LSD vaccine viruses first and the aa exchange present in the vaccine‐associated viruses second. (a) ORF LW011. The G‐coupled chemokine receptor protein with N‐terminal signal, transmembrane and non‐cytoplasmic domains, followed by the G‐coupled chemokine receptor domain with six intermitted transmembrane regions. (b) ORF LW028. This phosphatidylcholine‐hydrolysing phospholipase D (PLD) has the characteristic multiple PLD domains, with two PLD4 and one PLDc3 domain. (c) ORF LW083. The vaccinia D5 homologue has a C‐terminal with AAA + ATPase, Walker A and B domains and a helicase motif C belonging to the superfamily III. (d) ORF LW144. In the wild‐type virulent LSDV strain, a single 550 aa protein is encoded for. In contrast, the vaccine and vaccine‐associated viruses the reading frame is divided to encode two proteins, LW144a with either 269 or 270 amino acids and LW144b consisting of 281 amino acids. The Kelch‐like proteins produced by the vaccine and vaccine‐associated viruses are divided so that LW144a contains the BTB/POZ and BTB‐kelch domains and LW144b the Kelch‐like beta‐propeller with four Kelch 1 repeat regions. (e) ORF LW147. The ankyrin repeat domain has seven ankyrin repeat regions followed by a PRANC C‐terminal domain
Several hydrophobic–polar mutations were observed in LW026, LW028 and LW147 (Table 4). The A121T exchange in the Pox‐F11 Rho signalling inhibitor encoded for by ORF LW026 is unique to the vaccine‐associated viruses. No additional information or domain predictions are available for this protein, and therefore, the impact of this mutation on virulent phenotype is not known. In contrast, the T135A exchange in the palmitylated extracellular enveloped virus membrane protein predicted for ORF LW028 is identical between the virulent wild‐type and vaccine‐associated viruses (Table 5). This protein has the characteristic multiple phosphatidylcholine‐hydrolysing phospholipase D (PLD) repeat domains (Koonin, 1996). The T135A mutation occurs within the first PLD4 catalytic motif, yet the influence of this single mutation on the virulent phenotype is not known (Figure 3b).
The neighbouring LW027 encodes for a 639 aa homologue of VACV F12 (Tulman et al., 2001). The latter is important in virion transport to the cell surface due to its kinesin light chain, and specifically, the cargo‐binding tetratricopeptide repeats (TPRs) contained therein (Morgan et al., 2010). A comparison between VACV‐F12 and LW027 of LSDV shows conservation at the six TPRs and WD motif. The vaccine‐associated viruses possess a unique aa exchange at position 438 of a positive lysine (K) to asparagine (N). This K438N is within the cargo‐binding region, between TPR‐5 and TPR‐6, where LSDV has multiple consecutive positive amino acids compared to the VACV polar uncharged ones. The implication of this substitution on virion export function of the vaccine‐associated viruses needs to be elucidated.
The ORF LW052 encodes a 110 aa protein, predicted to belong to the G3 family of pox virus proteins (Tulman et al., 2001). The exact function of this protein is unknown, but it is predicted to play a role in virus entry as a component of the entry‐fusion complex through its association with L5 (Wolfe & Moss, 2011). The protein has an N‐terminal non‐cytoplasmic domain followed by a transmembrane region, and from position 25 a cytoplasmic domain (Senkevich, Ojeda, Townsley, Nelson, & Moss, 2005). In the vaccine‐associated and virulent wild‐type viruses, a positive lysine (K) is located at position 33, compared to the uncharged asparagine (N) in the vaccine viruses (Table 4). This N33K aa exchange results in a significant increase in the predicted antigenicity profile of the region, using the algorithm described by Welling, Weijer, van der Zee, and Welling‐Wester (1985; Figure 4). Of the 12 known proteins involved in the virus entry‐fusion complex, only LW052 was identified to differ between the vaccine‐associated, virulent and vaccine viruses. The impact of this aa exchange in LW052 on virus entry or the functionality of the virus entry‐fusion complex is not known.
Figure 4.
Graphical representation of the predicted domains present in LW052 and their relationship to the calculated antigenicity profiles based on the method of Welling. The antigenicity profile of AF409137_Neethling‐LSD‐Warmbaths‐RSA‐2000 is in blue, vaccine strain KX764645_Neethling‐LSD‐Vaccine‐OBP in red and that of the vaccine‐associated LSD‐248‐NW‐RSA‐1993 in black
Two conservative aa exchanges were observed in LW057 and LW079. The first was an isoleucine (I) exchange at the C‐terminal aa position 372 of vaccine viruses to valine (V) in the vaccine‐associated viruses. This V372I non‐synonymous SNP was also observed for the virulent wild‐type viruses (Table 4). The second exchange is of an arginine (R) at position 128 of LW079 within the vaccine viruses to a lysine (K) in the vaccine‐associated viruses. This substitution and the resulting R128K aa exchange are unique to the vaccine‐associated viruses (Table 4).
The VACV D5 protein is the product of one of three early viral genes constitutively expressed for its involvement in DNA synthesis (Traktman, Sridhar, Condit, & Roberts, 1984). It has an AAA + ATPase with both Walker A and B domains, as well as helicase motif C, belonging to the superfamily III (SFIII) helicase family (Evans, Klemperer, Ghosh, & Traktman, 1995). Various temperature‐sensitive D5 mutants have been generated in this regard, and their functions in replication and recombination were determined (Evans & Traktman, 1992). Additional site‐directed mutants involving D5 were produced to investigate the role these amino acids have on the structure and function of the protein (Boyle, Arps, & Traktman, 2007). The closely related homologue of D5 in LSDV is encoded by ORF LW083. At position 663, the vaccine‐associated, virulent wild type and VACV code for an asparagine (N), while the vaccine viruses code for a positive lysine (K) (Table 4). The majority of the previously mapped mutations in D5 resulting in the temperature‐sensitive mutants were located in the N‐terminal region, for which functionality has not yet been determined. The four conserved motifs (Walker A and Walker B boxes, motif C and the AAA + motif) and one of the previously mapped temperature‐sensitive mutations (Ets69) are all located in the C‐terminal region, from position 490 onwards (Figure 3c). The K663N exchange occurred in the C‐terminal region, after the four conserved motifs and before the temperature‐sensitive mutant Ets69 at position 682 (Figure 3c). The impact of K663N on the biological or structural function of LW083 in LSDV needs to be investigated using site‐directed mutagenesis. Previous studies showed that the VACV protein A20 interacts as the scaffold with D4, H5 and D5 to form a functional replication complex. The binding of A20 to D5 involves amino acids 201 to 251 of the A20 protein (Ishii & Moss, 2002). The LSDV homologue of A20 is encoded by the LW112 ORF, where both synonymous and non‐synonymous SNPs were identified (Tables 4 and 5). The latter results from exchange of a positive arginine (R), observed at position 347 in vaccine‐associated, virulent wild‐type, VACV and certain LSDV vaccine strains, with a serine (S) in the KX764645_Neethling‐LSD‐Vaccine‐OBP and MH646674_LSD‐Saratov‐Russia‐2017 viruses. Mutant VACV variants where the same positive region was substituted with alanine (A) resulted in viable viruses with no observable phenotypic changes (Punjabi et al., 2001). Therefore, the effect of this S347R mutation, as well as K663N in LW083, on the formation and functionality of the DNA replication complex needs to be investigated.
Transcription of early viral genes is dependent on the early transcription factor protein complex comprised of the 70 kDa small subunit of LW084 and the 82 kDa large subunit of LW098 (Li & Broyles, 1993; Tulman et al., 2001). The latter protein has two non‐synonymous mutations at positions 553 and 652, both of which are identical in the vaccine‐associated and virulent wild‐type viruses, in comparison with the vaccine viruses. The vaccine strains have a glycine (G) at position 553, while the vaccine‐associated viruses have negatively charged aspartate (D). The second mutation at position 652 exchanges a non‐polar isoleucine in the vaccine viruses with a polar threonine (T) (Table 4). The importance of the LW098 homologue in VACV, A8, during early transcription and virion morphogenesis has been described (Hu, Wolffe, Weisberg, Carroll, & Moss, 1998), yet the impact of these mutations in LSDV is currently unknown and warrants further investigation.
As previously mentioned, ORF LW144 encodes for two overlapping proteins, LW144a and LW144b (269/270 and 281 amino acids), in the vaccine and vaccine‐associated viruses and a single 550 aa protein in the wild‐type viruses (Figure 3d). The vaccine‐associated viruses have a conservative aa exchange at position 119 of leucine (L) from phenylalanine (F), that is unique to this group of viruses. This exchange is located within the BTB‐Kelch‐like domain of the LW144a (Figure 3d). At position 252 of LW144a, the vaccine viruses have a leucine (L), while both the vaccine‐associated and wild‐type viruses have a phenylalanine (F) and an insertion of isoleucine (I). This insertion resulted in the vaccine‐associated viruses encoding a 270 aa LW144a in contrast to the 269 aa encoded by the vaccine viruses (Figure 3d). The L252FI exchange occurs between the BTB‐Kelch‐like and Kelch‐type beta‐propeller domains, and therefore, the influence of this mutation is not known.
The last confirmed non‐synonymous mutation was observed in ORF LW147, encoding a protein with predicted ankyrin repeat regions. The aa exchange at position 143 is a polar threonine (T) from a non‐polar isoleucine (I) in both the vaccine and wild‐type viruses. This I143T exchange is unique to the vaccine‐associated viruses and occurs between ankyrin repeat regions 3 and 4 (Figure 3e). Since this is a newly described exchange, the influence of it on the structure and function should be investigated.
4.1.2. Mutations within the intragenic regions
The study identified 18 SNPs within intragenic regions, with 12 associated with homopolymer repeat sequences. The composition of homopolymers is difficult to determine accurately, irrespective of the sequencing technology or platform used. The combination of new NGS technologies and different error correction software tools aims at solving these problems. Since different methods were used to generate the consensus sequences for each of the viruses, each of the mutations was validated by visual inspecting the reads mapped to the position, rather than through Sanger sequencing. The mutations within the intragenic region were mostly indels and predominantly located in early promoter regions, thereby extending the promoter spacer length and possibly increasing the expression level of the ORFs under the control of these set promoters (Di Pilato, Sanchez‐Sampedro, Mejias‐Perez, Sorzano, & Esteban, 2015). Expression of ORFs that might be affected by increased spacer regions is as follows: LW002, LW011, LW013, LW022, LW023, LW034, LW068, LW123, LW134 and LW146. In contrast, there are deletions of a single nucleotide in the promoter regions of ORFs LW015 and LW155 of vaccine‐associated viruses compared to certain vaccine strains.
5. CONCLUSION
Since South Africa is endemic for LSD, it has utilized LAVs since the 1970s to control major disease outbreaks. Annual vaccination of animals is advised, but is not obligatory, resulting in indeterminate data concerning the overall usage, frequency and circulation of LAVs in the animal population. Reports of ‘vaccine failures’ are received occasionally, and thus, it is important to determine which factors may be at play, including genetic stability of field and vaccine strains. In order to fill this caveat, retrospective sequencing of the complete genomes of historic isolates of LSDV submitted during active outbreaks is underway. This study introduced the complete genomes of six field‐derived LSDVs, which clustered phylogenetically with the LAV viruses. The nucleotide differences between these virulent vaccine‐associated and attenuated vaccine viruses were investigated and their possible roles in virulence discussed. Of the 67 SNPs observed between the vaccine‐associated and vaccine viruses, 40 were identical to the virulent wild‐type viruses. This 60% similarity to virulent phenotypes is indicative of selection‐driven genetic drift. Since 31% of the mutations resulted in no aa exchanges, the remaining SNPs were scrutinized for their possible role in virulence. Of the remaining 46 SNPs, 18 occurred in the intragenic regions and may play a role in the regulation of gene transcription and protein levels, leaving 28 non‐synonymous SNPs in 17 ORFs as possible causes for the reversion of the attenuated phenotypes to virulence. These 17 ORFs encode putative proteins involved in mRNA regulation, DNA synthesis, protein regulation, virus entry, cellular apoptosis, virion formation and transport. It is important that the role of each of these ORFs be investigated individually as well as in synergism for their potential role in virulence, and especially reversion to virulence from an attenuated phenotype.
DATA SHARING AND ACCESSIBILITY STATEMENT
The data that support the findings of this study were deposited in GenBank (https://www.ncbi.nlm.nih.gov/genbank/ ) with accession numbers indicated in Table 1.
ETHICS STATEMENT
The relevant ethics approval was obtained for the study (ARC‐OVI approval number AEC 20.17) and the required permit from the South African Department Agriculture, Land Reform and Rural Development (formerly known as ‘DAFF’) (Section 20 permit no. 12/11/1/1 [649]).
CONFLICT OF INTEREST
The authors declare there are no conflicting interests in relation to this paper.
ACKNOWLEDGEMENTS
The authors would like to thank the South African Gauteng Department of Agriculture and Rural Development for funding this work, with contributing funding from the European Union's Horizon 2020 research and innovation programme under grant agreement number 773701, and the South African Agricultural Research Council for their continued support.
van Schalkwyk A, Kara P, Ebersohn K, et al. Potential link of single nucleotide polymorphisms to virulence of vaccine‐associated field strains of lumpy skin disease virus in South Africa. Transbound Emerg Dis. 2020;67:2946–2960. 10.1111/tbed.13670
REFERENCES
- Agianniotaki, E. I. , Chaintoutis, S. C. , Haegeman, A. , Tasioudi, K. E. , De Leeuw, I. , Katsoulos, P. D. , … Dovas, C. I. (2017). Development and validation of a TaqMan probe‐based real‐time PCR method for the differentiation of wild type lumpy skin disease virus from vaccine virus strains. Journal of Virological Methods, 249, 48–57. 10.1016/j.jviromet.2017.08.011 [DOI] [PubMed] [Google Scholar]
- Agianniotaki, E. I. , Mathijs, E. , van den Bussche, F. , Tasioudi, K. E. , Haegeman, A. , Iliadou, P. , … de Clercq, K. (2017). Complete genome sequence of the lumpy skin disease virus isolated from the first reported case in Greece in 2015. Genome Announcement C, 5, e00550–e617. 10.1128/genomeA.00550-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almazan, F. , Tscharke, D. C. , & Smith, G. L. (2001). The vaccinia virus superoxide dismutase‐like protein (A45R) is a virion component that is nonessential for virus replication. Journal of Virology, 75, 7018–7029. 10.1128/JVI.75.15.7018-7029.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annandale, C. H. , Holm, D. E. , Ebersohn, K. , & Venter, E. H. (2012). Seminal transmission of lumpy skin disease virus in heifers. Transboundary and Emerging Diseases, 61, 443–448. 10.1111/tbed.12045 [DOI] [PubMed] [Google Scholar]
- Bessman, M. J. , Frick, D. N. , & O’Handley, S. F. (1996). The MutT proteins or “Nudix” hydrolases, a family of versatile, widely distributed “housecleaning” enzymes. Journal of Biological Chemistry, 271, 25059–25062. 10.1074/jbc.271.41.25059 [DOI] [PubMed] [Google Scholar]
- Biswas, S. , Noyce, R. S. , Babiuk, L. A. , Lung, O. , Bulach, D. M. , Bowden, T. R. , … Evans, D. H. (2019). Extended sequencing of vaccine and wild‐type capripoxvirus isolates provide insights into genes modulating virulence host range. Transboundary and Emerging Diseases, 67, 80–97. 10.1111/tbed.13322 [DOI] [PubMed] [Google Scholar]
- Black, D. N. , Hammond, J. M. , & Kitching, R. P. (1986). Genomic relationship between Capripoxviruses. Virus Research, 5, 277–292. 10.1016/0168-1702(86)90024-9 [DOI] [PubMed] [Google Scholar]
- Blake, N. W. , Kettle, S. , Law, K. M. , Gould, K. , Bastin, J. , Townsend, A. R. M. , & Smith, G. L. (1995). Vaccinia virus serpins B13R and B22R do not inhibit antigen presentation to class I‐restricted cytotoxic T lymphocytes. Journal of General Virology, 76, 2393–2398. 10.1099/0022-1317-76-9-2393 [DOI] [PubMed] [Google Scholar]
- Boshra, H. , Truong, T. , Nfon, C. , Bowden, T. R. , Gerdts, V. , Tikoo, S. , … Babiuk, S. (2015). A lumpy skin disease virus deficient of an IL‐10 gene homologue provides protective immunity against virulent capripoxvirus challenge in sheep and goats. Antiviral Research, 123, 39–49. 10.1016/j.antiviral.2015.08.016 [DOI] [PubMed] [Google Scholar]
- Boyle, K. A. , Arps, L. , & Traktman, P. (2007). Biochemical and genetic analysis of the vaccinia virus D5 protein: Multimerization‐dependent ATPase activity is required to support viral DNA replication. Journal of Virology, 81, 844–859. https://dx.doi.org/10.1128%2FJVI.02217‐06 . 10.1128/JVI.02217-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coetzer, J. A. W. , Tuppurainen, E. , Babiuk, S. , & Wallace, D. B. (2018). Lumpy Skin Disease. In “Infectious Diseases of Livestock. Part 2”, Anipedia. https://anipedia.org/resources/lumpy‐skin‐disease/1201.
- Di Pilato, M. , Sanchez‐Sampedro, L. , Mejias‐Perez, E. , Sorzano, C. O. S. , & Esteban, M. (2015). Modification of promoter spacer length in vaccinia virus as a strategy to control the antigen expression. Journal of General Virology, 96, 2360–2371. 10.1099/vir.0.000183 [DOI] [PubMed] [Google Scholar]
- Douglass, N. , van der Walt, A. , Omar, R. , Munyanduki, H. , & Williamson, A.‐L. (2019). The complete genome sequence of the lumpy skin disease virus vaccine Herbivac LS reveals a mutation in the superoxide dismutase gene homolog. Archives of Virology, 164, 3107–3109. 10.1007/s00705-019-04405-8 [DOI] [PubMed] [Google Scholar]
- Erster, O. , Guini Rubinstein, M. , Menasherow, S. , Ivanova, E. , Venter, E. , Šekler, M. , … Stram, Y. (2019). Importance of the lumpy skin disease virus (LSDV) LSDV126 gene in differential diagnosis and epidemiology and its possible involvement in attenuation. Archives of Virology, 164, 2285–2295. 10.1007/s00705-019-04327-5 [DOI] [PubMed] [Google Scholar]
- Evans, E. , Klemperer, N. , Ghosh, R. , & Traktman, P. (1995). The vaccinia virus D5 protein, which is required for DNA replication, is a nucleic acid‐independent Nucleoside Triphosphatase. Journal of Virology, 69, 5353–5361. 10.1128/JVI.69.9.5353-5361.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, E. , & Traktman, P. (1992). Characterization of vaccinia virus DNA replication mutants with lesions in D5 gene. Chromosoma, 102, S72–S82. 10.1007/BF02451789 [DOI] [PubMed] [Google Scholar]
- Garcia, A. D. , Otero, J. , Lebowitz, J. , Schuck, P. , & Moss, B. (2006). Quaternary structure and cleavage specificity of a poxvirus Holliday Junction resolvase. Journal of Biological Chemistry, 281, 11618–11626. 10.1074/jbc.M600182200 [DOI] [PubMed] [Google Scholar]
- Gari, G. , Abie, G. , Gizaw, D. , Wubete, A. , Kidane, H. , Asgedom, H. , … Tuppurainen, E. S. M. (2015). Evaluation of the safety, immunogenicity and efficacy of the three capripox virus vaccines strains against lumpy skin disease virus. Vaccine, 33, 3256–3261. 10.1016/j.vaccine.2015.01.035 [DOI] [PubMed] [Google Scholar]
- Hu, X. , Wolffe, E. J. , Weisberg, A. S. , Carroll, L. J. , & Moss, B. (1998). Repression of the A8L gene, encoding the early transcription factor 82‐kilodalton subunit, inhibits morphogenesis of vaccinia virions. Journal of Virology, 72, 104–112. PMC109354. 10.1128/JVI.72.1.104-112.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter, P. , & Wallace, D. B. (2001). Lumpy skin disease in southern Africa: A review of the disease and aspects of control. Journal of the South African Veterinary Association, 72, 68–71. 10.4102/jsava.v72i2.619 [DOI] [PubMed] [Google Scholar]
- Ireland, D. C. , & Binepal, Y. S. (1998). Improved detection of capripoxvirus in biopsy samples by PCR. Journal of Virological Methods, 74, 1–7. 10.1016/S0166-0934(98)00035-4 [DOI] [PubMed] [Google Scholar]
- Ishii, K. , & Moss, B. (2002). Mapping interaction sites of the A20R protein component of the vaccinia virus DNA replication complex. Virology, 303, 232–239. 10.1006/viro.2002.1721 [DOI] [PubMed] [Google Scholar]
- Kara, P. D. , Afonso, C. L. , Wallace, D. B. , Kutish, G. F. , Abolnik, C. , Lu, Z. , … Rock, D. L. (2003). Comparative sequence analysis of the South African vaccine strain and two virulent field isolates of lumpy skin disease virus. Archives of Virology, 148, 1335–1356. 10.1007/s00705-003-0102-0 [DOI] [PubMed] [Google Scholar]
- Kettle, S. , Blake, N. W. , Law, K. M. , & Smith, G. L. (1995). Vaccinia virus serpins B13R (SPI‐2) and B22R (SPI‐1) encodes Mr 38.5 and 40K, intracellular polypeptides that do not affect virus virulence in a murine intranasal model. Virology, 206, 13–147. 10.1016/s0042-6822(95)80028-x [DOI] [PubMed] [Google Scholar]
- Kitching, R. P. (2003). Vaccines for lumpy skin disease, sheep pox and goat pox. Developmental Biology, 144, 161–167. [PubMed] [Google Scholar]
- Klement, E. , Broglia, A. , Antoniou, S.‐E. , Tsiamadis, V. , Plevrakie, E. , Petrović, T. , … Cortiñas Abrahantes, J. (2018). Neethling vaccine proved highly effective in controlling lumpy skin disease epidemics in the Balkans. Preventive Veterinary Medicine, in press. 10.1016/j.prevetmed.2018.12.001 [DOI] [PubMed] [Google Scholar]
- Kononov, A. , Byadovskaya, O. , Kononova, S. , Yashin, R. , Zinyakov, N. , Mischenko, V. , … Sprygin, A. (2019). Detection of vaccine‐like strains of lumpy skin disease virus in outbreaks in Russia in 2017. Archives of Virology, 164, 1575–1585. 10.1007/s00705-019-04229-6 [DOI] [PubMed] [Google Scholar]
- Koonin, E. V. (1996). A duplicated catalytic motif in a new superfamily of phosphohydrolases and phospholipid synthases that includes poxvirus envelope proteins. Trends in Biochemical Sciences, 21, 242–243. 10.1016/s0968-0004(96)30024-8 [DOI] [PubMed] [Google Scholar]
- Legrand, F. A. , Verardi, P. H. , Chan, K. S. , Peng, Y. , Jones, L. A. , & Yilma, T. D. (2005). Vaccinia virus with a serpin gene deletion and expressing INF‐ɤ induce potent immune responses without detectable replication in vivo . Proceedings of the National Academy of Sciences of the United States of America, 102, 2940–2945. 10.1073/pnas.0409846102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. , & Broyles, S. S. (1993). The DNA‐dependent ATPase activity of vaccinia virus early gene transcription factor is essential for its transcription activation function. Journal of Biological Chemistry, 268, 20016. [PubMed] [Google Scholar]
- Liu, S.‐W. , Wyatt, L. S. , Orandle, S. M. , Minai, M. , & Moss, B. (2014). The D10 decapping enzyme of vaccinia virus contributes to the decay of cellular and viral mRNAs and to virulence in mice. Journal of Virology, 88, 202–211. https://dx.doi.org/10.1128%2FJVI.02426‐13 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lojkic, I. , Simic, I. , Kresic, N. , & Bedekovic, T. (2018). Complete genome sequence of a lumpy skin disease virus strain isolated from the skin of a vaccinated animal. Genome Announce, 6, e00482‐18. https://doi. org/10.1128/genomeA.00482‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathijs, E. , Vandenbussche, F. , Haegeman, A. , King, A. , Nthangeni, B. , Potgieter, C. , … De Clercq, K. (2016). Complete genome sequences of the Neethling‐like lumpy skin disease virus strains obtained directly from three commercial live attenuated vaccines. Genome Announcements, 4, e01255–e1316. 10.1128/genomeA.01255-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menasherow, S. , Rubinstein‐Giuni, M. , Kovtunenko, A. , Eyngor, Y. , Fridgut, O. , Rotenberg, D. , … Stram, Y. (2014). Development of an assay to differentiate between virulent and vaccine strains of lumpy skin disease virus (LSDV). Journal of Virological Methods, 199, 95–101. 10.1016/j.jviromet.2013.12.013 [DOI] [PubMed] [Google Scholar]
- Möller, J. , Moritz, T. , Schlottau, K. , Krstevski, K. , Hoffmann, D. , Beer, M. , & Hoffmann, B. (2019). Experimental lumpy skin disease virus infection of cattle: Comparison of a field strain and a vaccine strain. Archives of Virology, 64, 2931–2941. 10.1007/s00705-019-04411-w [DOI] [PubMed] [Google Scholar]
- Morgan, G. W. , Hollinshead, M. , Ferguson, B. J. , Murphy, B. J. , Carpentier, D. C. J. , & Smith, G. L. (2010). Vaccinia protein F12 has structural similarity to Kinesin light chain and contains a motor binding motif required for virion export. PLOS Pathogen, 6, e1000785. 10.1371/journal.ppat.1000785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish, S. , & Moss, B. (2007). Characterization of a second Vaccinia virus mRNA‐decapping enzyme conserved in poxviruses. Journal of Virology, 81, 12973–12978. 10.1128/JVI.01668-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish, S. , Resch, W. , & Moss, B. (2007). Vaccinia virus D10 protein has mRNA decapping activity, providing a mechanism for control of host and viral gene expression. Proceedings of the National Academy of Sciences of the United States of America, 104, 2139–2144. 10.1073/pnas.0611685104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punjabi, A. , Boyle, K. , de Masi, J. , Grubisha, O. , Unger, B. , Khanna, M. , & Traktman, P. (2001). Clustered charge‐to‐alanine mutagenesis of the vaccinia virus A20 gene: Temperature‐sensitive mutants have a DNA‐minus phenotype and are defective in the production of possessive DNA polymerase activity. Journal of Virology, 75, 12308–12318. 10.1128/JVI.75.24.12308-12318.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senkevich, T. G. , Ojeda, S. , Townsley, A. , Nelson, G. E. , & Moss, B. (2005). Poxvirus multiprotein entry‐fusion complex. Proceedings of the National Academy of Sciences of the United States of America, 102, 18572–18577. 10.1073/pnas.0509239102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprygin, A. , Artyuchova, E. , Babin, Y. , Prutnikov, P. , Kostrova, E. , Byadovskaya, O. , & Kononov, A. (2018). Epidemiological characterization of lumpy skin disease outbreaks in Russia in 2016. Transboundary and Emerging Diseases, 65, 1514–1521. https://doi. org/10.1111/tbed.12889. [DOI] [PubMed] [Google Scholar]
- Sprygin, A. , Babin, Y. , Pestova, Y. , Kononova, S. , Byadovskaya, O. , & Kononov, A. (2019). Complete Genome sequence of the lumpy skin disease virus recovered from the first outbreak in the Northern Caucasus region of Russia in 2015. Microbiology Resource Announcements, 8, e01733–e1818. 10.1128/MRA.01733-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprygin, A. , Babin, Y. , Pestova, Y. , Kononova, S. , Wallace, D. B. , van Schalkwyk, A. , … Kononov, A. (2018). Analysis and insights into recombination signals in lumpy skin disease virus recovered in the field. PLoS One, 13, e0207480. 10.1371/journal.pone.0207480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprygin, A. , Byadovskaya, O. , Kononova, S. , Zakharov, V. , Pestova, Y. , Prutnikov, P. , & Kononov, A. (2019). A real‐time PCR screening assay for the universal detection of lumpy skin disease virus DNA. BMC Research Notes, 12, 371. 10.1186/s13104-019-4412-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura, K. , Peterson, D. , Peterson, N. , Stecher, G. , Nei, M. , & Kumar, S. (2011). MEGA5: Molecular evolutionary genetic analysis using maximum likelihood, evolutionary distance and maximum parsimony methods. Molecular Biology and Evolution, 28, 2731–2739. 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toplak, I. , Petrovic, T. , Vidanovic, D. , Lazic, S. , Sekler, M. , Manic, M. , … Kuhar, U. (2017). Complete genome sequence of lumpy skin disease virus isolate SERBIA/Bujanovac/2016, Detected during an Outbreak in the Balkan Area. Genome Announce, 5, e00882–e917. 10.1128/genomeA.00882-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traktman, P. , Sridhar, P. , Condit, C. , & Roberts, B. E. (1984). Transcriptional mapping of the DNA polymerase gene of vaccinia virus. Journal of Virology, 49, 125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulman, E. R. , Afonso, C. L. , Lu, Z. , Zsak, L. , Kutish, G. F. , & Rock, D. L. (2001). Genome of lumpy skin disease virus. Journal of Virology, 75, 7122–7130. https://dx.doi.org/10.1128%2FgenomeA.00550‐17 . 10.1128/JVI.75.15.7122-7130.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuppurainen, E. , Antoniou, S.‐E. , Tsiamadis, E. , Topkaridou, M. , Labus, T. , Debeljak, Z. , … Broglia, A. (2018). Field observations and experiences gained from the implementation of control measures against lumpy skin disease in South‐East Europe between 2015 and 2017. Preventive Veterinary Medicine, S0167–5877(18), 30548–30558. 10.1016/j.prevetmed.2018.12.006 [DOI] [PubMed] [Google Scholar]
- Tuppurainen, E. S. M. , Lubinga, J. C. , Stoltsz, W. H. , Troskie, M. , Carpenter, S. T. , Coetzer, J. A. W. , … Oura, C. A. L. (2012). Mechanical transmission of lumpy skin disease virus by Rhipicephalus appendiculatus male ticks. Epidemiology and Infection, 141, 425–430. 10.1017/S0950268812000805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuppurainen, E. S. M. , Lubinga, J. C. , Stoltsz, W. H. , Troskie, M. , Carpenter, S. T. , Coetzer, J. A. W. , … Oura, C. A. L. (2013). Evidence of vertical transmission of lumpy skin disease virus in Rhipicephalus decoloratus ticks. Ticks and Tick‐Borne Diseases, 4, 329–333. 10.1016/j.ttbdis.2013.01.006 [DOI] [PubMed] [Google Scholar]
- Tuppurainen, E. S. M. , Stoltsz, W. H. , Troskie, M. , Wallace, D. B. , Oura, C. A. L. , Mellor, P. S. , … Venter, E. H. (2010). A potential role for Ixodid (hard) tick vectors in the transmission of lumpy skin disease virus in cattle. Transboundary and Emerging Diseases, 58, 93–104. 10.1111/j.1865-1682.2010.01184.x [DOI] [PubMed] [Google Scholar]
- Tuppurainen, E. S. M. , Venter, E. H. , & Coetzer, J. A. W. (2005). The detection of lumpy skin disease virus in samples of experimentally infected cattle using different diagnostic techniques. Onderstepoort Journal of Veterinary Research, 72, 153–164. 10.4102/ojvr.v72i2.213 [DOI] [PubMed] [Google Scholar]
- Vandenbussche, F. , Mathijs, E. , Haegeman, A. , Al‐Majali, A. , Van Borm, S. , & De Clercq, K. (2016). Complete genome sequence of Capripoxvirus strain KSGP 0240 from a commercial live attenuated vaccine. Genome Announcement, 4, e01114–e1116. https://10.1128/genomeA.01114‐16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidanović, D. , Šekler, M. , Petrović, T. , Debeljak, Z. , Vasković, N. , Matović, K. , & Hofmann, B. (2016). Real‐time PCR assays for the specific detection of field Balkan strains of lumpy skin disease virus. Acta Veterinaria, 66, 444–454. 10.1515/acve-2016-0038 [DOI] [Google Scholar]
- Weiss, K. E. (1968). Lumpy skin disease virus. Virology monographs (vol. 3, pp. 111–131). Vienna/New York: Springer‐Verlag. [Google Scholar]
- Welling, G. W. , Weijer, W. J. , van der Zee, R. , & Welling‐Wester, S. (1985). Prediction of sequential antigenic regions in proteins. FEBS Letters, 188, 215–218. 10.1016/0014-5793(85)80374-4 [DOI] [PubMed] [Google Scholar]
- Wolfe, C. L. , & Moss, B. (2011). Interaction between the G3 and L5 proteins of the vaccinia virus entry‐fusion complex. Virology, 412, 278–283. 10.1016/j.virol.2011.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]