

Abstract
Desminopathy is the most common intermediate filament disease in humans. The most frequent mutation causing desminopathy in patients is a R350P DES missense mutation. We have developed a rat model with an analogous mutation in R349P Des. To investigate the role of R349P Des in mechanical loading, we stimulated the sciatic nerve of wild‐type littermates (WT) (n = 6) and animals carrying the mutation (MUT) (n = 6) causing a lengthening contraction of the dorsi flexor muscles. MUT animals showed signs of ongoing regeneration at baseline as indicated by a higher number of central nuclei (genotype: P < .0001). While stimulation did not impact central nuclei, we found an increased number of IgG positive fibers (membrane damage indicator) after eccentric contractions with both genotypes (stimulation: P < .01). Interestingly, WT animals displayed a more pronounced increase in IgG positive fibers with stimulation compared to MUT (interaction: P < .05). In addition to altered histology, molecular signaling on the protein level differed between WT and MUT. The membrane repair protein dysferlin decreased with eccentric loading in WT but increased in MUT (interaction: P < .05). The autophagic substrate p62 was increased in both genotypes with loading (stimulation: P < .05) but tended to be more elevated in WT (interaction: P = .05). Caspase 3 levels, a central regulator of apoptotic cell death, was increased with stimulation in both genotypes (stimulation: P < .01) but more so in WT animals (interaction: P < .0001). Overall, our data indicate that R349P Des rats have a lower susceptibility to structural muscle damage of the cytoskeleton and sarcolemma with acute eccentric loading.
Keywords: exercise, filament, injury, intermediate, muscle, signaling
Abbreviations
- DC
detergent compatible
- DPX
dibutylphthalate polystyrene xylene
- EDL
extensor digitorum longus muscle
- EDTA
ethylenediaminetetraacetic acid
- EGTA
ethylene glycol tetraacetic acid
- H&E
hematoxylin and eosin
- HRP
horseradish peroxidase
- IP
intraperitoneal
- IgG
immunoglobulin G
- IPGTT
intraperitoneal glucose tolerance test
- LSB
Laemmli sample buffer
- MUT
R349P desmin mutant rats
- NGS
natural goat serum
- PVDF
polyvinylidene difluoride
- SLB
sucrose lysis buffer
- SUnSET
SUrface SEnsing of Translation
- TBST
Tris‐buffered saline
- TGX
Tris‐Glycine eXtended
- WT
wild‐type rats
1. INTRODUCTION
Desmin is the primary intermediate filament in skeletal and cardiac muscle. In concert with the three main filaments of the sarcomere, actin, myosin, and titin, it is involved in the organization of myofibrils and ensures structural integrity by forming three dimensional scaffolds that span the diameter of the muscle fiber. 1 Desmin is thought to surround z‐lines and connect them with the sarcolemma at the costamere. Through its binding partner synemin, desmin is also known to interact with the dystrophin associated glycoprotein complex, linking the z‐line to the extracellular matrix. 2 Beyond its structural role in skeletal muscle, there is also emerging evidence for several additional functions of desmin. Among its many interaction partners are chaperones and proteins involved in proteolysis, posttranslational modification, and mitochondrial function. 3 The complete lack of desmin in mice results in a progressive myopathy, characterized by z‐line streaming, reduced ability to produce force and misaligned myofibrils. 4 Myofibers of knockout mice have an increased susceptibility to break in the face of mechanical stress and show signs of degeneration and regeneration. 5 In the heart, the absence of desmin causes a concentric cardiomyocyte hypertrophy in addition to ventricular dilation and altered systolic function. 6 The heart of desmin knockout animals also shows decreased ejection fraction from both ventricles, reduced cardiac output and increased susceptibility to ventricular arrythmias. 5
Even though desmin knockout models allow for important insights into fundamental functions of the protein, clinically a complete lack of desmin is exceptionally rare. Most patients with a desmin associated myopathy suffer from a mutation to, rather than the absence of, desmin. 7 This is significant since one of the hallmarks of human desminopathy is the buildup of desmin positive protein aggregates in myofibers and these aggregates are thought to directly contribute to muscle degeneration. 8 The most prevalent mutation in humans is caused by the exchange of a single amino acid: arginine to proline at position 350 of DES (R350P). 9 , 10 In vitro, cells transfected with R350P DES were unable to form intermediate filament networks and started to buildup pathological protein aggregates in the cytoplasm, not unlike the aggregates that have been found in histological sections of patients. 9 R350P mutation causes a progressive muscle disease in patients, affecting skeletal and cardiac muscles. Distal and proximal limb and shoulder weakness, disturbed electrical signal conduction across the myocardium, dyspnea, and other cardio‐respiratory problems are among the clinical symptoms reported in families with the mutation. 9 Mice carrying an orthologous mutation in R349P showed protein aggregates and disorganized intermediate filament structures, resulting in muscle weakness and dilated cardiomyopathy. 11 More recently, we created a CRISPR‐Cas9 engineered knock‐in rat with the same mutation in order to have a model organism that is genetically and physiologically closer to humans. 12 At baseline, we found increased central nuclei in the skeletal muscle of mutants, a higher number of small muscle fibers and an altered fiber type distribution. 12 Various force transfer proteins were increased in mutant rats, potentially compensating for the lack of functional desmin. When challenged with two weeks of synergist ablation, the mutant animals demonstrated impaired adaptation, and less fiber hypertrophy, even though muscle mass and force did not differ significantly from wild‐type littermates. 12 This pointed to an inability of R349P rat muscle to maintain structural integrity in the face of mechanical stress and impaired adaptation relative to wild‐type muscle. We hypothesized that this could indicate that mechanical stress through physical activity could exacerbate the disease course in desminopathy. However, since synergist ablation is an extreme and chronic stimulus, the question remained whether the same would happen in an acute setting with a more physiological type of loading. This is of high clinical relevance, as currently no cure for desminopathy exists and physical therapy is the only treatment available.
To determine whether a mutation in desmin causes increased susceptibility to injury after acute exercise, we loaded the lower limbs of rats using eccentric contractions. We hypothesized that eccentric contractions would cause a greater injury response and increased muscle remodeling in desmin mutants compared to control animals.
2. METHODS
2.1. Animal model
All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of California, Davis, which is an AAALAC‐accredited institution. Animal housing was in accordance with recommendations of the Guide for the Care and Use of Laboratory Animals. Animals were housed in 12:12 hours light‐dark cycles, fed ad libitum in a conventional vivarium and were specific pathogen free. Male and female rats aged between 400 and 500 days were selected to study functional, histological, and biochemical differences between CRISPR‐Cas9 created knock‐in animals carrying a mutation in desmin (MUT; n = 6) or healthy, wild‐type littermates (WT; n = 6) after eccentric contractions of the tibialis anterior and extensor digitorum longus (EDL) muscle.
2.2. Stimulation protocol and muscle collections
Animals were anesthetized (2.5% isoflurane) and the sciatic nerve stimulated as described previously. 13 Briefly, the sciatic nerve was exposed by surgically opening the fascia between the vastus lateralis and the biceps femoris of the rats. The nerve was hooked onto a platinum electrode, connected to a Grass S5 stimulator (Grass Instruments, USA) and stimulated at a frequency of 100 Hz and 4‐6 V, causing eccentric contractions of the tibialis anterior and EDL muscles. 14 Each contraction lasted 2 seconds, with 10 seconds delay before the next contraction. After six repetitions, the animals rested for 1 minute, before the next set was performed. Every animal was stimulated for six sets of six repetitions, a total of 36 contractions. Following the end of the stimulation, the incision site was sutured, and the animals received 0.1 mg/kg buprenorphine (0.03 mg/mL solution; JHP Pharmaceuticals, USA) as analgesic. We collected the tibialis anterior and the EDL muscles 24 hours after conclusion of the stimulation.
2.3. Muscle force measurements
Four weeks before collection of the animals, a series of six isometric contractions was used to determine the maximal force of the ankle dorsi flexor muscles (tibialis anterior and EDL). The rats were anesthetized (2.5% isoflurane), placed in a supine position, and the right foot of the lower limb secured to a footplate attached to an Aurora Scientific 300B (Aurora, Canada) servomotor. The right leg was clamped in place so that both the knee and ankle were at an angle of 90°. The dorsi flexors were stimulated by needle electrodes inserted into the tibialis anterior muscle.
Torque was measured at stimulation frequencies of 20, 40, 60, 80, 100, and 125 Hz, with the force at 125 Hz being equivalent to the maximal force. Contractions lasted 200 ms, with 45 seconds rest between contractions. Data acquisition and analysis were performed using Dynamic Muscle Control and Dynamic Muscle Analysis software (Aurora, Canada).
2.4. Intraperitoneal glucose tolerance test
The intraperitoneal glucose tolerance test (IPGTT) was also done four weeks before the stimulation and collection of the animals. Animals were placed in cages with water and without food access from 6 PM on the previous day to ensure a postabsorptive state. The IPGTT was begun at 8 AM on the following morning by taking the fasted blood sugar values of every animal via a One Touch Ultra 2 device (Life Scan, USA). This measurement was immediately followed by injecting the rats with a solution of 2 g glucose × kg–1 body weight (0.3 g glucose/mL saline (0.9% NaCl)) intraperitoneally (i.p.). Following injection, blood glucose levels were determined at 15, 30, 60, 90, and 120 minutes, before the animals were returned to their original cages and returned to ad libitum feeding.
2.5. Histochemistry
Following collection, the frozen TA muscles were blocked and serial cross‐sections were cut at 10 μm. For hematoxylin and eosin (H&E) staining, sections were consecutively submerged in EtOH 90% and then tap water. 400 µL Mayer's hemalum solution (Merck, Darmstadt, Germany) was applied per slide and left to incubate for 6 minutes. Slides were then blued under running lukewarm tap water before being dipped in tap water for another 8 minutes. 300 µL 0.5% aqueous eosin γ‐solution (Merck, Darmstadt, Germany) were applied to each slide for 1 minute. Sections were dehydrated in sequential steps using 70%, 90%, and 100% EtOH. Sections were submerged in Xylene, air dried, and mounted with DPX Mountant for histology (Sigma, Darmstadt, Germany).
For immunohistochemistry, TA sections were fixed in acetone, washed, and blocked with 5% natural goat serum (NGS) for 30 minutes. For determination of fiber types, we used SC‐71 (myosin heavy chain 2A, mouse, IgG1, 1:250 in NGS), BF‐F3 (myosin heavy chain 2B, mouse, IgM, 1:250 in NGS), and a polyclonal laminin antibody (rabbit, IgG (H + L), 1:500 in NGS). For detection of desmin aggregates, a monoclonal desmin antibody (mouse, IgG2a, 1:200 in NGS) was used. Secondary antibodies (goat anti‐mouse Alexa Fluor 488 and 555, Life Technologies and goat‐anti‐rabbit AlexaFluor 647 for fiber typing, goat anti‐mouse Alexa Fluor 488 for desmin) were incubated for 30 minutes at room temperature. Anti‐myosin heavy chain antibodies were purchased from the Developmental Studies Hybridoma Bank (Iowa City, Iowa), anti‐laminin antibody was purchased from Sigma Aldrich (St Louis, Missouri), anti‐desmin antibody was purchased from Santa Cruz Biotechnology (Dallas, Texas).
Slides were imaged using a Leica DMi8 inverted microscope using the HC PL FLUOTAR 10x/0.32 PH1 objective (Leica Microsystems, Wetzlar, Germany). For comparative analysis, exposure length remained fixed for all samples. For all stains, overlapping images were stitched together such that the entire muscle could be analyzed.
Muscle fiber properties, fiber types, and central nuclei were analyzed using FIJI and SMASH (MATLAB) as described previously. 12
2.6. Western blots
Frozen powdered tibialis anterior muscles were homogenized in 200 µL sucrose lysis buffer (SLB; 50 mM Tris pH 7.5, 250 mM sucrose, 1 mM EDTA, 1 mM EGTA, 1% Triton X‐100, 1% protease inhibitor complex) on a vortexer for 60 minutes at 4℃. Following centrifugation at 10 000 g for 10 minutes, the supernatant was collected. To ensure that desmin levels in the supernatant are representative of the whole muscle, we conducted a control experiment on the pellet and solubilized it using a high salt buffer similar to what has been described by Solaro and colleagues (Figure S1). 15 Protein concentrations were determined in triplicates using the DC protein assay (Bio‐Rad, Hercules, CA, USA). Sample concentrations were adjusted using SLB. Following dilution in Laemmli sample buffer (LSB), 1 µg protein/µL were denatured at 100℃ for 5 minutes. Protein (10‐15 µg protein per lane) was loaded on 4%‐20% Criterion TGX Stain‐free gels (Bio‐Rad), run for 45 minutes at 200V and visualized after a UV‐induced 1‐minute reaction to produce fluorescence. Following quantification, proteins were transferred to nitrocellulose or polyvinylidene difluoride (PVDF) membrane at 100V for 30‐60 minutes, depending on the size of the protein of interest. Efficient transfer was confirmed using Ponceau staining of the membrane. Membranes were then air dried and directly incubated with the primary antibody, or washed and blocked in 1% fish skin gelatin dissolved in Tris‐buffered saline with 0.1% Tween‐20 (TBST) for 1 hour, rinsed and then incubated with the primary antibody overnight at 4⁰C. The next day, membranes were washed and incubated with HRP‐conjugated secondary antibodies at 1:5000 (goat) to 1:10 000 (mouse, rabbit) in 1% skim milk‐TBST for 1 hour at room temperature. Immobilon Western Chemiluminescent HRP substrate (Millipore, Hayward, CA, USA) was then applied to the membranes for protein visualization by chemiluminescence. Image acquisition and band quantification was performed using the ChemiDoc MP System and Image Lab 5.0 software (Bio‐Rad). Protein levels of each sample were calculated as band intensities relative to total protein as described previously (see Figure S2). 16 , 17 Biological duplicates of each group were loaded onto the gel in random order to avoid edge effects and lane bias. All samples of all groups (ie WT control, WT stim, MUT control, MUT stim) were run on the same gel for each protein probed; images of the bands in the figures display representative, biological duplicates for each condition. The following antibodies were used in this study at a concentration of 1 to 1000. Cell Signaling (Cell Signaling Technology, Danvers, MA): p70‐S6 Kinase 1 (p70S6K) (Thr389) (#9205; lot 16), ribosomal protein S6 (rpS6) (Ser240/244) (#5364), glycogen synthase (#3893; lot 2), microtubule‐associated proteins 1A/1B light chain 3B (LC3B) (#2775; lot 10), beclin 1 (#3738), autophagy related 7 (Atg 7) (#8558), heat shock protein β‐1 (HSP27) (#2402; lot 8), heat shock protein α (HSP90) (#4877; lot 5), Unc‐51 like autophagy activating kinase 1 (ULK1) (Ser757) (#14202; lot 1), sequestosome 1 (p62) (#5114; lot 4), nuclear factor κ B (NF‐kB) (Ser536) (#3033; lot14), annexin A2 (#8235; lot 2), C/EBP‐homologous protein (CHOP) (#5554), caspase 3 (#9662); Santa Cruz (Santa Cruz Biotechnology Inc, Dallas, TX): dystrophin (#365954; lot E2711), dysferlin (#16635; lot H162), desmin (#271677; lot F1913), glucose transporter type 1 (GLUT1) (#7903; lot 209), glucose transporter type 4 (GLUT4) (#53566; lot 2415), muscle LIM protein/cysteine and glycine‐rich protein 3 (mLIM) (#166930; lot E2814), septin 1 (#373925); Millipore Sigma (Merck Group): insulin receptor substrate 1 (IRS1) (#06‐248; lot 2465193), puromycin (MABE343); Enzo (Enzo Life Sciences Inc, Farmingdale, NY): αB crystallin (ADI‐SPA‐223; lot 05011812); Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IA): myosin heavy chain—Slow (#BA‐D5); Abcam (Abcam Inc, Eugene, OR): total oxidative phosphorylation (OXPHOS) (MS604‐300).
2.7. Protein synthesis
Global muscle protein synthesis was assessed using the SUrface SEnsing of Translation (SUnSET) method as described previously. 13 Puromycin was dissolved in sterile saline (0.9% NaCl) and delivered via i.p. injection (0.02 μmol puromycin × g–1 body weight) 30 minutes prior to muscle collection. Puromycin‐truncated peptides, reflecting the rate of global muscle protein synthesis, were analyzed by western blot as described above.
2.8. Mechanical testing on rat muscle
Prior to mechanical testing, rat EDL muscles were stored in −20℃ storage solution. 18 Within two weeks of being stored and blinded to the genotype, EDL muscles were placed in a petri dish filled with dissecting solution and pinned at the proximal and distal ends to maintain passive tension. 19 An average of three equal length bundles of fibers were cut from each EDL to be used for mechanical testing. Bundles were cut parallel to the myofiber angle and separately pinned from the rest of the EDL. Bundles were tied at the ends with 7‐0 sutures and loops were made to allow for attachment to the mechanical testing apparatus consisting of a 300C‐LR Dual‐Mode motor arm and force transducer (Aurora Scientific) in a well of dissecting solution at 22℃. Once bundles were secured inside the mechanical testing apparatus, they were stretched to an approximate slack length, a point at which there was minimal tension detected by the force transducer. The slack length (Lo) was measured using calipers at 4.19 ± 0.93 mm and was used in conjunction with bundle mass (m) and density (ρ) to calculate cross‐sectional area (CSA): CSA = m/(Lo × ρ). Passive mechanical testing consisted of a sequence of pre‐conditioning periods of cyclic strain followed by stress relaxation measurements at steps from 2.5% to 12.5% at 2.5% intervals. Strain was determined based on motor arm movement and does not account for end compliance which may overestimate strain of intact muscle. 20 , 21 The cyclic strain included five cycles of 2.5% strain cyclic at a frequency of 1 Hz and stress relaxation included a rapid 2.5% strain at 1 fiber length/s to the given strain and maintaining length for 120 seconds. A custom script in MATLAB was used to calculate the dynamic and elastic stiffness of the muscle bundles using a stress‐strain plot from the mechanical testing sequence. Elastic stiffness is the slope of the quadratic regression of elastic stress at 10% strain. Dynamic stiffness is the slope of the quadratic regression of the dynamic (maximum) stress at 10% strain. 22 The elastic index is the ratio between the elastic and dynamic stiffness. This analysis is similar to previous studies 22 , 23 , 24 ).
2.9. Hydroxyproline determination of collagen content for muscle tissue
Collagen content was determined using a hydroxyproline assay. 25 Muscle tissue was placed in 1.7 mL tubes and weighed for wet mass. Then samples were placed on heating block with lids open for 30 minutes at 120℃ to sufficiently dry the tissue. Each sample was then weighed for dry mass and hydrolyzed in 200 µL of 6N HCl at 120℃ for 2 hours before drying for 1.5 hours at 120℃. The dried pellet was resuspended in 200 µL of hydroxyproline buffer (containing 173 mM citric acid, 140 mM acetic acid, 588 mM sodium acetate, 570 mM sodium hydroxide). The sample was further diluted 1:67 in hydroxyproline buffer. 150 µL of 14.1 mg/mL Chloramine T solution was added to each sample, then vortexed and incubated at room temperature for 20 minutes. 150 µL aldehyde‐perchloric acid containing 60% 1‐propanol, 5.8% perchloric acid, and 1 M 4‐(dimethylamino)benzaldehyde was then added to each sample, then vortexed and incubated at 60℃ for 15 minutes. The tubes were then cooled for 5‐10 minutes at room temperature. Samples were read at 550 nm on an Epoch Microplate Spectrophotometer (BioTek Instruments Limited, Winooski, VT). Hydroxyproline was converted to collagen mass assuming hydroxyproline contributes to 13.7% of the dry mass of collagen. 26
2.10. Statistics
Depending on the number of the groups compared, an unpaired t‐test or a two‐way ANOVA with a post hoc Tukey's multiple comparisons test was used to test the null hypothesis. For the two‐way ANOVA, the two main variables analyzed were “stimulation” (as in electrostimulation of the sciatic nerve) and “genotype” (WT littermate or desmin MUT). For the two‐way ANOVA, the p‐values for “stimulation”, “genotype”, and the interaction effect between the two variables will be reported; the results of any post hoc analyses may serve as an additional tool to directly compare groups when applicable and will be denoted as such. An alpha of P < .05 was deemed statistically significant and a P‐value between .05 and .1 was called a trend. Data in the text are reported as mean ± standard deviation, data in the figures are visually represented as scatter dot plot with error bars indicating standard error of the mean. All analyses were performed with Prism 8 (GraphPad, USA).
3. RESULTS
3.1. The animal model and the effect of electrostimulation
Immunohistochemical analysis of desmin revealed ubiquitous expression across myofibers in the WT animals, with particularly pronounced signals at the sarcolemma, while MUT animals rarely showed desmin outside of aggregates (Figure 1B). Body weight was not different between groups (WT = 495 ± 156 g; MUT = 526 ± 182 g; Figure 1C). When assessed via a 2‐way ANOVA, muscle mass of the tibialis anterior 24 hours after eccentric contractions was significantly higher in the stimulated leg compared to the contralateral control leg (stimulation: P < .05), with no genotype difference (genotype: P = .38) or interaction (interaction of stimulation × genotype: P =.74). Maximal dorsi flexion force relative to the body weight did not differ significantly between MUT (0.31 ± 0.2 mN/g body weight) and WT (0.39 ± 0.1 mN/g body weight; Figure 1E). Blood glucose levels after an i.p. injection of glucose differed for the variable time (time: P < .0001) and tended to be higher at each timepoint in MUT compared to WT group (genotype: P = .08) with no interaction effect between the two variables (time × genotype: P = .13; Figure 1F).
FIGURE 1.
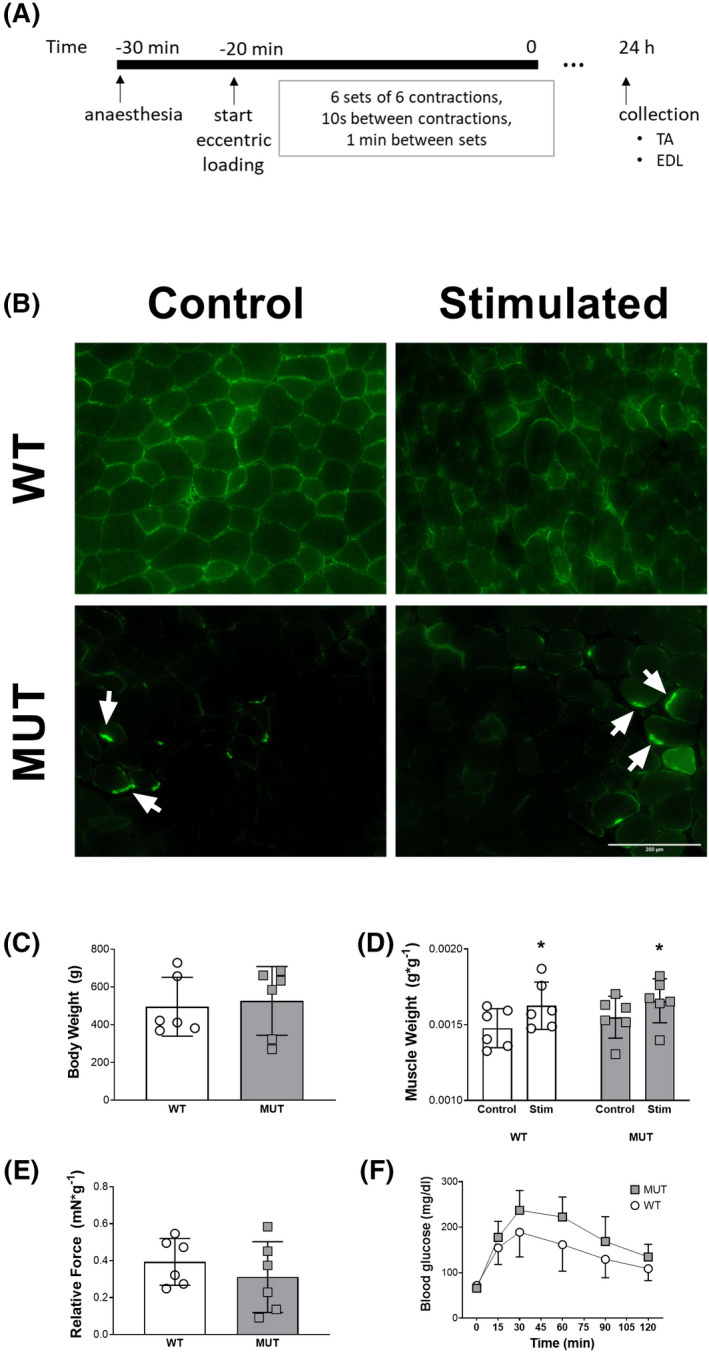
The animal model and the effect of electrostimulation. A, Eccentric stimulation and collection protocol. B, Immunohistological desmin staining of tibialis anterior sections. C, Body weight of the animals. D, Tibialis anterior mass in the contralateral control‐ and the stimulated leg of WT and MUT animals. E, Force during plantar flexion relative to the body weight of the animals. F, Intraperitoneal glucose tolerance test in WT and MUT animals. *denotes a statistically significant difference (Figure 1D, stimulation: P < .05)
3.2. Histological analysis of acute and chronic injury
Assessment of histological damage via H&E staining revealed increased chronic remodeling in the MUT compared to the WT animals (Figure 2A), as signified by a higher number of fibers with central nuclei (genotype: P < .0001; Figure 2B). Eccentric contractions did not significantly affect the number of central nuclei (stimulation: P = .22) and there was no interaction effect between stimulation and genotype (stimulation × genotype: P = .58; Figure 2B). Acute membrane injury following eccentric contractions, as assessed via IgG staining, showed a higher number of IgG positive fibers after stimulation (stimulation: P < .01), with the effect being more pronounced in WT animals compared to MUT (genotype: P < .05), resulting in an interaction effect (stimulation × genotype: P < .05; Figure 2D). No IgG positive fibers were found in the WT group at baseline, whereas one MUT animal had two IgG positive fibers. WT animals showed an average of 35 ± 30 IgG positive fibers per tibialis anterior cross‐section after electrostimulation, while MUT had 7 ± 9 IgG positive fibers (Figure 2D). To ensure that IgG was specific to injured muscle fibers and did not stain for interstitial space, vessels or fibrotic tissue, we performed H&E staining on serial sections (Figure S3).
FIGURE 2.
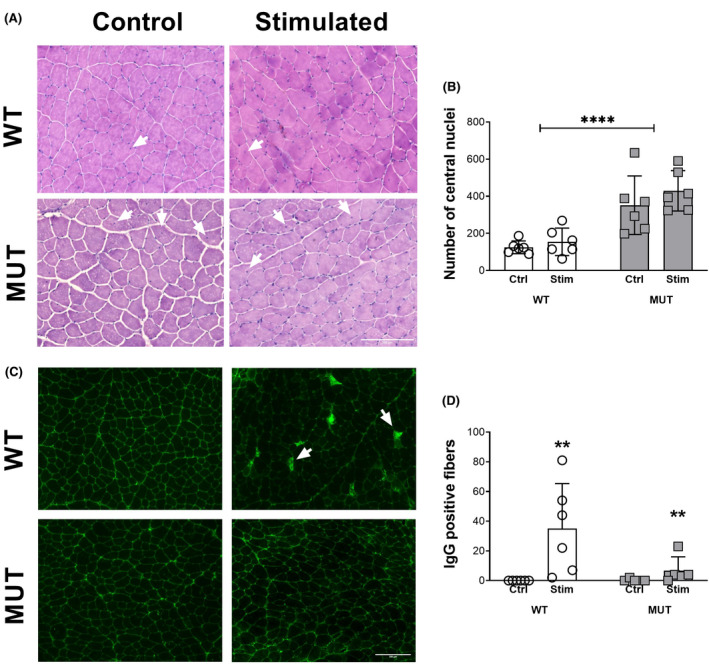
Histological analysis of acute and chronic injury. A, Hematoxylin and eosin staining in tibialis anterior sections of WT and MUT rats. White arrows denote examples of central nuclei. B, Quantification of the number of central nuclei across the entire section. C, IgG staining in tibialis anterior sections of WT and MUT rats. White arrows denote examples of IgG positive fibers. D, Quantification of IgG positive fibers across the entire section. ** and **** denote statistically significant differences (Figure 2B, genotype: P < .0001), (Figure 2D, stimulation: P < .01)
3.3. Muscle mechanics and collagen content
Complete deletion of desmin increases elastic stiffness of muscle bundles due to extracellular matrix aberrations. 27 However, there were no significant differences between WT and MUT EDL bundle passive mechanical properties. Unpaired t‐tests showed no significant differences between WT and MUT EDL bundle dynamic stiffness, elastic stiffness, and elastic index (dynamic: P = .22, elastic: P = .22; elastic index: P = .14; Figure 3A‐C). Collagen content in the tibialis anterior muscle was unchanged with stimulation (P = .26) and genotype (P = .56), and no interaction effect was present (P = .27; Figure 3D).
FIGURE 3.
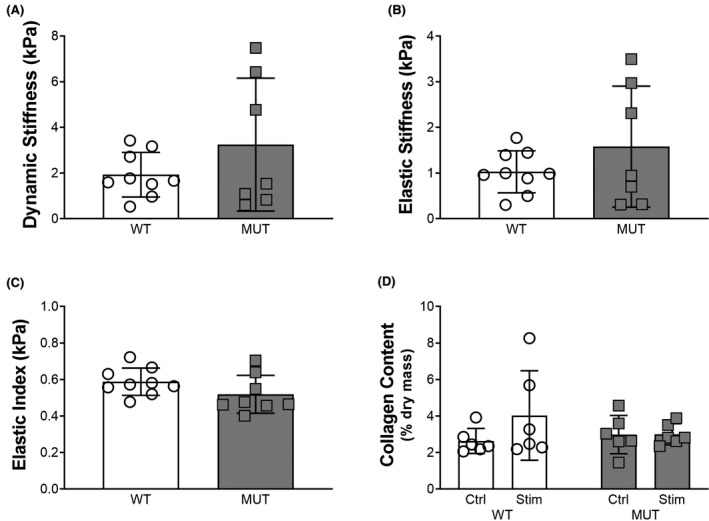
Muscle mechanics and collagen content. A, Dynamic stiffness of EDL muscle bundles in WT and MUT rats. B, Elastic stiffness of EDL muscle bundles in WT and MUT rats. C, The elastic index (ratio of elastic to dynamic stiffness) of EDL muscle bundles in WT and MUT rats. D, Collagen content in tibialis anterior muscles after stimulation and in the contralateral control leg of WT and MUT rats
3.4. Force transfer and membrane injury
Next, we investigated the effect of eccentric contractions on protein levels associated with structural integrity, force transfer and membrane injury in skeletal muscle of WT and MUT animals. Desmin protein levels at baseline were about fourfold as high in the WT compared to the MUT group. Stimulation had a significant effect on desmin levels in both groups (P < .05; Figure 4A). However, WT desmin increased to an average of 6.3‐fold, while MUT, despite lower baseline levels, increased to only 2.7‐fold compared to baseline. This resulted in desmin levels being about ninefold as high in WT compared to MUT after stimulation (post hoc: P < .01; Figure 4A). Consequently, there was a significant effect for genotype (P < .01) and an interaction between genotype and exercise (P < .05; Figure 4A). Interestingly, this pronounced increase in protein levels via western blotting was in contrast with the immunohistochemical data, where desmin appeared to decrease with stimulation (Figure 1B). The membrane injury associated protein dysferlin showed no effect for stimulation (P = .46) or genotype (P = .34; Figure 4B). Despite this, there was a significant interaction effect between stimulation and genotype (P < .05), as dysferlin levels tended to decrease with stimulation in the WT group, while they increased with stimulation in the MUT group (Figure 4B). Annexin a2 levels did not reach statistical significance for stimulation (P = .19; Figure 4C), genotype (P = .12) or an interaction effect (P = .8; Figure 4C). Dystrophin levels increased following stimulation in both groups (P < .05) with no effect of genotype (P = .33) and no interaction effect (P = .44; Figure 4D). Muscle LIM protein levels at baseline were on average 4.3‐fold as high in the MUT compared to the WT group (post hoc: P < .05; Figure 4E). Stimulation increased mLIM levels in both groups (P < .0001; Figure 4E). The WT group increased to about 5.3‐fold with stimulation while the MUT group increased to twofold compared to baseline (Figure 4E). Consequently, there was a significant effect of the genotype of the animals (P < .001) but no interaction effect between stimulation and the genotype (P = .97; Figure 4E). Levels of slow myosin heavy chain protein (MHCs) increased with stimulation in both groups (P < .01), with no effect of the genotype (P = .17) and no interaction effect (P = .44; Figure 4F). All raw data and statistical results of our western blots (Figures 4, 5, 6) are available in Table S1.
FIGURE 4.
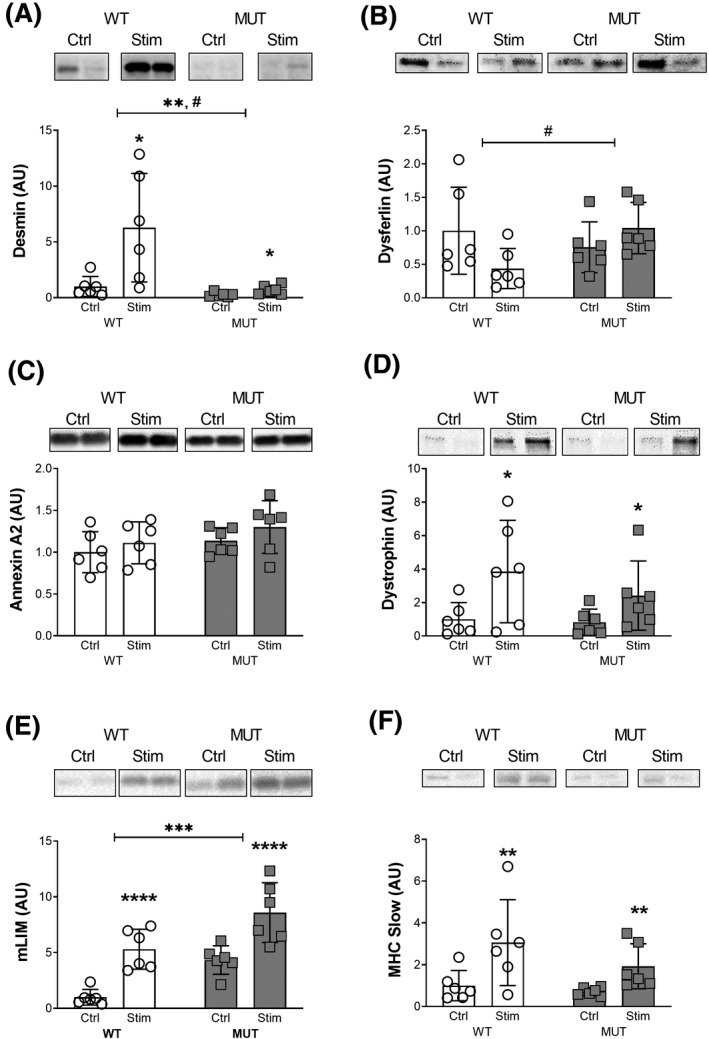
Force transfer and membrane injury. Western Blot analysis of protein levels: A, Desmin. B, Dysferlin. C, Annexin A2. D, Dystrophin. E, Muscle LIM Protein/Cysteine and Glycine‐rich Protein 3 (mLIM). F, Myosin Heavy Chain (MHC) Slow. #, *, **, *** and **** denote statistically significant differences (Figure 4A, stimulation: P < .05, genotype: P < .01, interaction: P < .05), (Figure 4B, interaction: P < .05), (Figure 4D, stimulation: P < .05), (Figure 4E, stimulation: P < .0001, genotype: P < .001), (Figure 4F, stimulation: P < .01). Protein levels were normalized to total protein content, bands display representative biological duplicates of each condition. For each protein, all samples were run together on the same gel
FIGURE 5.
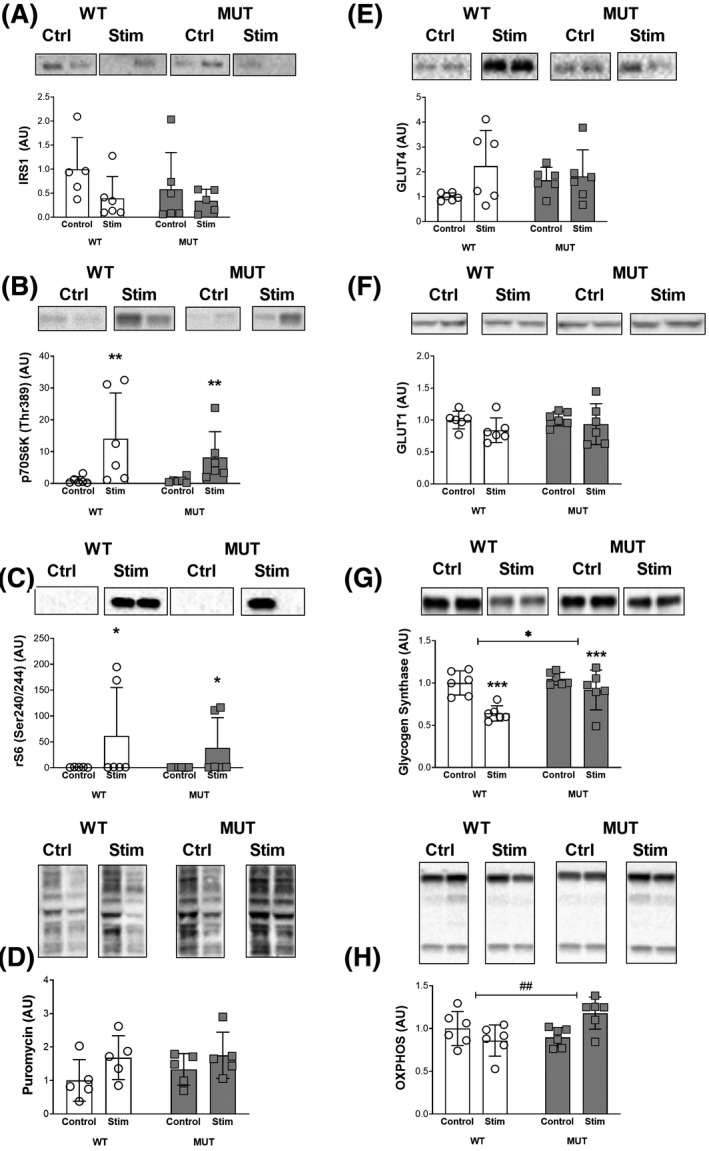
Skeletal muscle metabolism and remodeling. Western blot analysis of protein levels: A, Total insulin receptor substrate 1 (IRS1). B, Phospho‐p70 ribosomal S6 kinase 1 (p70S6K) (Thr389). C, Phospho‐ribosomal protein S6 (rp S6) (Ser240/244). D, Puromycin. E, Glucose transporter type 4 (GLUT4). F, Glucose transporter type 1 (GLUT1). G, Glycogen synthase. H, Total oxidative phosphorylation (OXPHOS). ##, *, ** and *** denote statistically significant differences (Figure 5B, stimulation: P < .01), (Figure 5C, stimulation: P < .05), (Figure 5G, stimulation: P < .001, genotype: P < .05), (Figure 4H, interaction: P < .01). Protein levels were normalized to total protein content, bands display representative biological duplicates of each condition. For each protein, all samples were run together on the same gel
FIGURE 6.
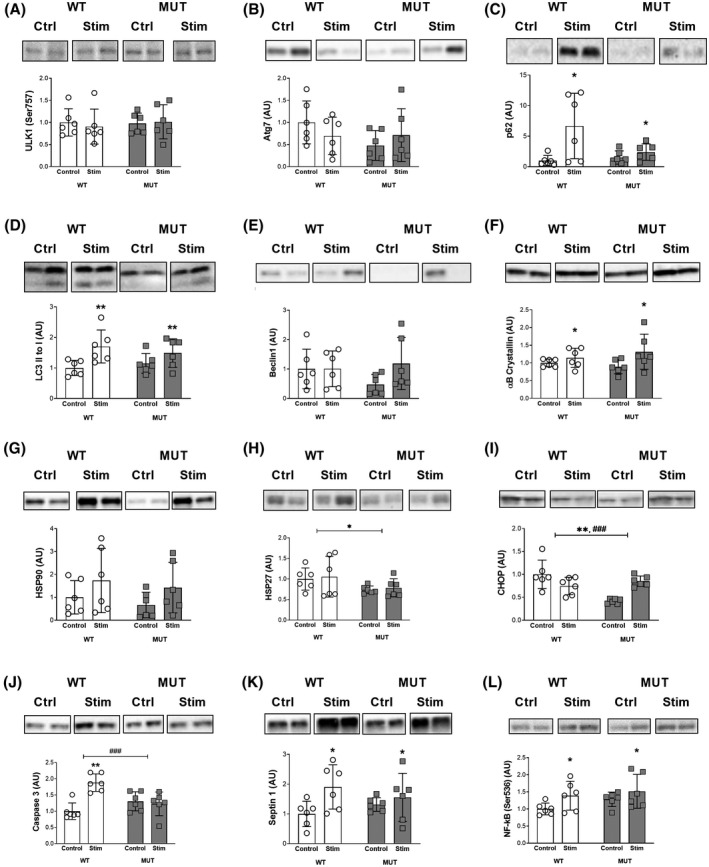
Cell damage, degradation, and apoptosis. Western blot analysis of protein levels: A, Phospho‐Unc‐51 like autophagy activating kinase 1 (ULK1) (Ser757). B, Autophagy related 7 (Atg 7). C, Sequestosome 1 (p62). D, Microtubule‐associated proteins 1A/1B light chain 3B (LC3B). E, Beclin1. F, αB crystallin. G, Heat shock protein‐alpha (HSP90). H, Heat shock protein β‐1 (HSP27). I, C/EBP‐homologous protein (CHOP). J, Caspase 3. K, Septin 1. L, Nuclear factor κB (NF‐kB) (Ser536). ###, *, ** and *** denote statistically significant differences (Figure 6C, stimulation: P < .05), (Figure 6D, stimulation: P < .01), (Figure 6F, stimulation: P < .05), (Figure 6H, genotype: P < .05), (Figure 6I, genotype: P < .01, interaction: 0.001), (Figure 6J, stimulation: P < .01, interaction: P < .001), (Figure 6K, stimulation: P < .05), (Figure 6L, stimulation: P < .05). Protein levels were normalized to total protein content, bands display representative biological duplicates of each condition. For each protein, all samples were run together on the same gel
3.5. Skeletal muscle metabolism and remodeling
Immunoblotting revealed differences in anabolic and metabolic signaling pathways between WT and MUT animals at baseline and after eccentric contractions. Total IRS levels tended to decrease after eccentric contractions (stimulation: P = .1) independent of the mutation (genotype: P = .35; stimulation × genotype: P = .46; Figure 5A). Phosphorylated p70S6K1 (Thr389) levels increased with stimulation (P < .01) but showed no effect of genotype (P = .39) or an interaction effect (P = .39; Figure 5B). Phosphorylated ribosomal protein S6 (rS6 Ser240/244) levels tended to increase with stimulation (P = .05) but similarly showed no effect of genotype (P = .63) or an interaction effect (P = .64; Figure 5C). Global muscle protein synthesis as assessed via puromycin trended to be elevated 24 hours after stimulation (P = .06). Despite 33% higher baseline values in MUT compared to WT, there was no effect for genotype (P = .48) or an interaction effect (P = .64; Figure 5D).
For metabolic proteins, GLUT4 levels increased to about 220% in the WT group and by only 9% in the MUT group with stimulation, causing a trend toward significance for stimulation (P = .09; Figure 5E). While baseline levels in the MUT group were 66% higher than in the WT group, variability in GLUT4 levels masked any effect for the genotype (P = .75) and the interaction effect did not reach statistical significance (P = .17; Figure 5E). GLUT1 levels decreased with stimulation but did not reach statistical significance (P = .16) and showed no effect for genotype (P =.5) or an interaction effect (P = .67; Figure 5F). Glycogen synthase decreased with stimulation in both groups (P < .001) but to a lesser extent in the MUT group, causing an effect for genotype (P < .05) and a trend for an interaction effect (P =.08), with GS levels after stimulation being significantly higher in MUT than WT animals (post hoc: P < .05, Figure 5G). Total oxidative phosphorylation protein levels showed no effect for stimulation (P = .33) or genotype (P = .14) but a significant interaction effect between stimulation and genotype (P < .01), as protein levels tended to decrease with stimulation in the WT group yet increased with stimulation in the MUT group (Figure 5H).
3.6. Cell damage, degradation, and apoptosis
Upstream autophagy signaling through phosphorylated ULK1 (Ser757) was not affected by stimulation (P = .84), genotype (P = .76) and there was no interaction effect between the two (P = .64; Figure 6A). Atg7 levels tended to decrease in the WT group with stimulation, while they increased in the MUT group (P = .85; Figure 6B). However, due to high variability this did not lead to a significant effect for the genotype (P = .2) or an interaction effect (P = .18; Figure 6B). Baseline levels of the autophagy substrate p62 were about 50% higher in the MUT compared to the WT group (Figure 6C). Stimulation increased p62 levels in both groups (P < .05), with the levels in the WT group being 6.7‐fold and in the MUT group being 1.6‐fold higher than baseline (Figure 6C). This resulted in no statistical effect of the genotype (P = .12) but a trend towards an interaction effect of stimulation x genotype (P = .05; Figure 6C). The ratio of LC3 II to LC3 I was increased with stimulation in both groups (P < .01) without any effect of the genotype (P = .85) or an interaction effect (P = .29; Figure 6D). Beclin1 protein levels at baseline averaged 53% lower in the MUT compared to the WT group (post hoc: P = .52; Figure 6E). With stimulation, Beclin1 levels increased to 2.5‐fold in the MUT group but stayed unchanged in the WT group (Figure 6E). However, due to high variability neither stimulation (P = .19), genotype (P = .52) nor the interaction effect (P = .2) reached statistical significance (Figure 6E). CCAAT‐enhancer‐binding protein homologous protein (CHOP) levels at baseline were 58% higher in the WT compared to the MUT group (post hoc: P < .001) and decreased with stimulation in the WT animals while they increased with stimulation in the MUT animals (Figure 6I). This led to significant genotype (P < .01) and interaction effects (P < .001), while stimulation was not different (P = .27; Figure 6I). We found a significant effect of stimulation on Caspase 3 levels (P < .01; Figure 6J). However, this was mostly due to the WT group which increased protein levels by 88% following stimulation, while the MUT group decreased Caspase 3 levels by 6% (Figure 6J). Consequently, there was no effect for the genotype (P = .17) but an interaction effect between stimulation and genotype (P < .001; Figure 6J). Similarly, we found a significant effect of stimulation on Septin1 levels (P < .05), but the increase trended to occur only in the WT group (post hoc: P = .07), while no increase in the MUT group was observed (post hoc: P = .75; Figure 6K). However, no effect of the genotype (P = .95) or an interaction effect (P = .19) was found (Figure 6K). Alpha‐B crystallin (αB crystallin) levels increased with stimulation in both groups (P < .05) without an effect of genotype (P = .8) or an interaction effect (P = .28; Figure 6F). Heat shock protein 90 (HSP90) levels trended to increase with stimulation (P = .08) but did not differ based on genotype (P = .44) and showed no interaction effect (P = .98; Figure 6G). Stimulation had no effect on heat shock protein 27 (HSP27) levels in either of the groups (P = .69); however, levels were about 25% higher in the WT compared to the MUT group resulting in a significant effect for genotype (P < .05) without an interaction effect (P = .96; Figure 6H). Phosphorylated NF‐κB (Ser536) levels showed a comparable pattern, as there was a significant effect of stimulation (P < .05) that appeared primarily driven by a 39% increase in the WT group, while the MUT group only increased by 18% with stimulation (Figure 6L). There was no significant effect for genotype (P = .17) and no interaction between genotype and stimulation (P = .6; Figure 6L).
4. DISCUSSION
The primary goal of this study was to investigate the effect of an acute bout of lengthening contractions on skeletal muscle in rats with a mutation in desmin (R349P). We hypothesized that the desmin mutation would make animals more susceptible to acute load induced muscle damage. However, we found that even though rats with the desmin mutation showed more signs of chronic injury, these animals demonstrated fewer signs of acute injury and appeared somewhat protected from acute load‐induced muscle damage compared to their wild‐type littermates.
We have previously published an intervention with the same strain of animals, where we assessed the phenotype of young adult WT and MUT animals at baseline and after 14 days of functional overload through synergist ablation. 12 Like the current work, at baseline we found a higher number of central nuclei in MUT rats and increased force transfer proteins in skeletal muscle. Central nuclei are a histological hallmark of muscle regeneration that become visible 3‐7 days after traumatic injury or are chronically higher in the context of congenital muscular dystrophies. 28 , 29 , 30 Increase in number of central nuclei has been reported for various desmin mutations including the R349P knock‐in. 31 , 32 , 33 In line with this, we interpret the increased central nuclei in our model as a sign of chronic remodeling as a result of an inability to maintain structural integrity in the absence of functional desmin. Over time, this would result in a compensatory increase in other force transfer proteins. After synergist ablation, we found an impaired ability of MUT muscle to increase myofiber size. 12 Based on our data, we hypothesized that R349P mutant animals had a decreased ability to cope with a severe exercise stimulus compared to WT littermates possibly because they were more prone to membrane and myofibrillar injury.
To test this hypothesis, in the current study we exposed older WT and MUT rats to an acute bout of eccentric loading and collected the tissue at 24 hours later. We chose older animals, around 400 to 500 days, to better mimic the human condition, which normally presents in patients in their 40s. Body‐ and muscle weight were similar between WT and MUT animals, with the latter slightly increased in both genotypes after stimulation, most likely due to eccentric exercise‐induced edema. Maximal dorsi flexion force relative to body weight was not statistically different between the groups, albeit force from the MUT animals averaged 28% lower. In line with the findings of our previous study and the work of others, 11 , 12 we found desmin to be almost undetectable via western blot and histological sections of MUT animals using commercially available antibodies, except for occasional protein aggregates in the subsarcolemmal space. This can likely be explained through the relatively mild clinical presentation in the mutant animals, which have not reached a fully dystrophic phenotype yet, as well as through the lack of an antibody that is specifically raised against R349P desmin. 11 Interestingly, like other models of muscular dystrophies where glucose metabolism is impaired, 34 , 35 we found signs of glucose intolerance. This finding may be related to the relatively frequently observed occurrence of type 2 diabetes in human patients suffering from desminopathy and is in line with recent findings that have reported a direct association between the dystrophin glycoprotein complex, desmin and the insulin receptor. 36
With respect to histological indicators of muscle damage and remodeling, we found increased central nuclei in MUT compared to WT rats. This is in line with our previous findings in younger animals where we also observed a higher number of central nuclei in individuals with the R349P mutation of desmin. Lengthening contractions tended to mildly increase the number of central nuclei in WT and MUT, but the effect was small and did not reach statistical significance. This is not surprising, as central nuclei are thought to be a sign of regeneration that peaks ~7 days after injury in rodents rather than after acute muscle damage. 37 These data suggest that baseline levels of fiber regeneration are higher in muscle carrying the R349P mutation in desmin.
To assess acute membrane damage in response to eccentric loading, we counted fibers demonstrating intracellular IgG in histological sections. When membranes are damaged and become permeable, IgG infiltrates the sarcoplasm and can be visualized inside the myofiber. An unaccustomed bout of lengthening contractions is reported to cause substantial membrane damage. 38 Interestingly, while lengthening contractions caused an increase to fivefold the number of IgG positive fibers in WT rats, contrary with our original hypothesis, R349P rats showed a smaller increase in IgG positive fibers, suggesting that rats with the R349P mutation of desmin are less, not more, susceptible to acute load‐induced muscle damage. Lieber and colleagues have shown that following eccentric loading desmin protein is lost from histological sections of WT muscles 39 , 40 and desmin knockout muscle fibers are less susceptible to membrane damage. 41 The authors hypothesized that the lack of desmin makes muscle fibers more compliant, permitting less force transmission between sarcomeres and thus injury. 41 In contrast, most data from desminopathy models and patients indicate increased damage in myofibers with a mutation in, or complete absence of, desmin. Specifically, biomechanical testing of isolated primary myotubes from patients with a mutation in R350P as well as isolated murine myofibers with the analogous R349P mutation showed a higher stiffness and greater vulnerability to passive stretch induced rupture. 11 , 42 One possible explanation for this discrepancy is that the mutation results in changes to individual fibers that are not reflected in the behavior of the whole muscle. This hypothesis is consistent with the fact that desmin is a force transfer protein linking the contractile machinery of a single fiber to the extracellular matrix. Mutation of desmin could therefore result in altered linkage between sarcomeres, resulting in increased stiffness at the fiber level, but if force transfer between fibers is decreased, the result would be decreased stiffness at the whole muscle level.
To test this hypothesis, we determined the dynamic and elastic stiffness of EDL muscle bundles from WT and MUT rats. We did not see a significant difference in muscle stiffness resulting from the R349P mutation. Similarly, collagen content was not different in WT and MUT skeletal muscle. As such, whole muscle mechanical properties and connective tissue content are unlikely to explain why MUT muscle showed a decreased susceptibility to acute load‐induced injury in our model. Since these data overall show the greatest alignment with the early observations of Lieber et al which were also conducted in the context of eccentric contractions, 41 it appears possible that the observation that desmin knockout or mutant muscle is less susceptible to injury could be specific to the mode of contraction.
Indeed, in addition to desmin the giant protein titin has recently emerged as a key regulator of muscle contraction and elasticity. 43 For example, the increase in muscle force during eccentric contractions despite a deteriorating force‐length relationship could be attributed to titin and, potentially, actin‐titin binding. 43 , 44 As such, a compensatory change to titin in R349P desmin mutant rats could potentially explain why the animals were protected from membrane damage through eccentric contractions even though passive and active muscle stiffness were unaltered compared to wild‐type animals. Since titin is progressively shortened during development resulting in increased passive stiffness, 45 , 46 , 47 it is possible that the chronic injury in the R349P muscles results in a longer isoform of titin, lower passive tension, and therefore less injury following acute eccentric loading. A last possible explanation for the lack of mechanical differences present in our study could be the temperature at which our samples were processed. 48
To investigate whether structural components aside from the three main filaments are responsible for the histopathological differences observed in the mutant muscle, we measured levels of proteins associated with force transfer and membrane damage. Desmin levels in our previous study in 150 to 200 days old animals showed higher levels in WT compared to MUT at baseline, as would be expected based on that commercially available antibodies are less reactive with the mutated protein. 12 Interestingly, this discrepancy at baseline was not as pronounced in the current study with 400 to 500 days old animals. While on average still almost fourfold higher in the WT compared to MUT rats, some of the older WT did not have detectable desmin at baseline either. This suggests an age‐related decrease in desmin levels in the tibialis anterior of our rats that might mask some of the differences between WT and R349P rats. In this study, WT animals increased desmin levels to over sixfold and MUT animals to over twofold following eccentric contractions of the tibialis anterior. This biochemical increase in protein is in contrast with the decrease seen histologically. One explanation for this discrepancy could be that following eccentric loading desmin is released from its costameric location and is more soluble, resulting in less signal histologically and more free desmin that can be isolated biochemically during immunoblotting. In respect to membrane damage, dysferlin protein showed a distinct response to stimulation in WT and MUT (Figure 4B). Dysferlin tended to decrease in WT, whereas it increased in MUT animals. We saw greater membrane damage in WT rats after eccentric loading in this study, as indicated by a higher number of IgG positive fiber, raising the question whether the decrease in dysferlin is a natural function of injured muscle. In contrast, we have previously shown that in old rats (28 months), dysferlin mRNA levels increase between 24 and 72 hours in response to an unloading‐reloading protocol. 37 In a human trial of 12 weeks endurance exercise, on the other hand, we found dysferlin protein levels to be decreased in healthy individuals after training. This makes the results at hand difficult to interpret in respect to whether they are adaptive or pathological. Muscle LIM has been postulated as a master regulator of cardiac and skeletal muscle. 49 It has been suggested to have a mechano‐signaling role in association with the z line and costameres as well as stabilizing and crosslinking actin filaments into bundles. 50 , 51 We found mLIM to be over fourfold as high at baseline in MUT compared to WT animals. This is in line with our previous data in younger animals. 12 At the same time, the increase in mLIM with stimulation was greater in the WT (to about fivefold) compared to the MUT animals (to about twofold). Other force transfer‐ and membrane damage associated protein levels such as dystrophin or annexin a2 showed no difference that would explain the increased membrane permeability in WT animals with stimulation.
To investigate whether the differences in the injury response and chronic remodeling can be found at the protein level, we examined signaling proteins associated with muscle protein turnover. Global protein synthesis, as assessed by puromycin, and translation initiation as assessed through p70S6K and its downstream substrate rS6, all tended to increase with stimulation, without any effect of the genotype. However, these signaling cascades are known to be dynamic and their activity is transient with loading. 13 Similarly, differences in protein synthesis between groups can be harder to measure following unaccustomed exercise, where a strong injury response is seen, whereas with a repeated exercise regimen the data are far more consistent. 52 As such, it is possible that we would have to use a technique such as stable isotope labeling and a repeated exercise design to detect differences in the synthesis and remodeling of contractile proteins between WT and MUT. Since we and others showed that muscle glucose metabolism can be affected in different types of neuromuscular disorders and we found a trend toward decreased glucose tolerance in MUT animals, we also looked at proteins regulating glucose uptake and processing. 35 , 53 , 54 We found that IRS1 levels tend to be lower in both genotypes after stimulation. This is in line with previous studies that found a transient insulin resistance with eccentric exercise in healthy individuals. 55 Interestingly, though, we found GLUT4 levels to increase to 2.2‐fold after stimulation in WT animals, while GLUT4 protein levels in MUT were largely unchanged. Part of the GLUT4 response can again be attributed to the fact that GLUT4 levels at baseline were already about 70% greater in the MUT animals. The higher GLUT4 at baseline is surprising given the trend for impaired glucose tolerance, which is often dependent on GLUT4 levels. 56 However, we and others have previously found that in conditions of severe muscle loss and disturbed muscle homeostasis such as nerve damage, 53 critical illness myopathy, 57 duchenne muscular dystrophy, 34 , 35 or dysferlinopathy, 58 , 59 GLUT4 protein levels can be normal or even slightly elevated. This occurs despite impaired translocation of GLUT4 to the sarcolemma and decreased glucose tolerance, suggesting a potential compensatory effect of protein levels to account for hampered function. The increase in mitochondrial proteins with stimulation in the MUT and decrease in WT animals further suggests a distinct metabolic phenotype with R349P mutation of desmin.
Since protein aggregates are a main hallmark for desminopathy and the aggregation is thought to be caused by an inability to adequately remove proteins, we investigated molecular markers of autophagy. The strongest indicator for a reduced ability to initiate autophagy in MUT animals were p62 levels. Both genotypes increased p62 with stimulation; however, WT animals increased to levels of about sevenfold while MUT animals only showed a 60% increase. Proteins associated with autophagy are notoriously difficult to interpret, because markers of upregulated autophagy undergo degradation through autophagy themselves, potentially resulting in increased or decreased protein levels depending on the timing of the measurement. 60 Under normal circumstances, the induction of autophagy results in a transient increase in cytosolic p62, which is degraded in the autophagosome resulting in a decrease of p62 levels. Since we collected the muscle tissue 24 hours after the acute, eccentric loading stimulus, the increased p62 levels in the WT likely indicate increased autophagic activity. This is further supported by the fact that the LC3 II to I ratio was increased with stimulation. Like p62, LC3 II and the ratio of LC3 II to I are thought to increase during the early phase of an autophagic stimulus but decrease with chronic changes to autophagy. 61 Both p62 and the LC3 II to I ratio increased with eccentric loading in both genotypes, suggesting that this is an acute scenario reflecting the onset of autophagy. As such, the greater increase in p62 and LC3II:I in the WT animals could either be a function of greater acute muscle damage, as indicated by a higher number of IgG positive fibers, or a greater ability to initiate autophagy in response to a remodeling stimulus in the R349P animals.
To delineate whether it is greater acute damage or an inherent ability for improved autophagy, we investigated several markers of cellular stress. In line with the idea that acute eccentric loading resulted in more injury in the WT animals while the MUT animals suffer from greater chronic stress, we found that Caspase 3 increased by 80% following eccentric loading in WT animals but was not affected by loading in the MUT animals. However, baseline Caspase 3 levels were about 30% higher in MUT compared to WT animals. Similarly, we found a ~ 40% increase in the inflammatory marker p‐NF‐κB (Ser536) in WT muscle following eccentric loading, whereas this marker was largely unchanged in MUT animals. Once again, baseline p‐NF‐κB levels were about 30% higher in the MUT compared to the WT rats. Together, these data suggest that acute eccentric loading resulted in greater muscle fiber injury in WT animals, whereas chronic markers of injury were higher in the MUT muscles.
To summarize, this is the first study that investigated the effect of an acute bout of eccentric contractions on skeletal muscle in rats harboring a mutation in R349P of desmin. MUT animals showed greater signs of ongoing, chronic muscle remodeling; however, in contrast with our hypothesis, MUT animals appeared to be protected from eccentric injury compared to WT animals. Mechanical testing did not reveal a significant difference in stiffness at the muscle bundle level that could explain why desmin mutants showed less injury. Molecular signaling supported the observation of greater susceptibility to acute injury in WT animals but higher chronic levels of stress in MUT animals. Consistent with this, Caspase 3 and p‐NF‐κB were higher at baseline in the MUT but increased more following eccentric loading in the WT animals. As such, we conclude that R349P mutation of desmin results in more injury from daily activities but protects the muscle from greater injury following significant acute eccentric loading. Whether this means that chronic daily exercise is better or worse for individuals with desminopathy remains to be determined.
CONFLICT OF INTEREST
HTL, AAM, AMA, RPW, LRS, and HZF declare that they have no conflict of interest. KB is paid by professional sports and Olympic teams to consult on tendon/performance issues and has received grants, honoraria, and travel support from PepsiCo for nutrition research.
AUTHOR CONTRIBUTIONS
HTL designed the experiment, performed the eccentric loading, western blots and drafted the manuscript. AAM contributed to the design of the experiment, performed histochemistry, and drafted the manuscript. AMA performed the hydroxyproline assay. RPW and LRS performed the mechanical testing of muscle bundles. HZF contributed to drafting of the manuscript. KB helped to design the experiment and drafted the manuscript.
Supporting information
Fig S1‐S3
Table S1
ACKNOWLEDGMENTS
The research was made possible by a generous gift from the Bertin‐Barbe Family.
Langer HT, Mossakowski AA, Avey AM, et al. A mutation in desmin makes skeletal muscle less vulnerable to acute muscle damage after eccentric loading in rats. FASEB J. 2021;35:e21860. 10.1096/fj.202100711RR
Henning T. Langer and Agata A. Mossakowski contributed equally to this work.
Funding information
The current study was funded by a generous gift from the Bertin‐Barbe Family to HZF, and KB. The work was further supported by the National Institute of Aging through project grants (R01AG056999 and R01AG45375). AAM was supported by a postdoctoral fellowship from Deutsche Forschungsgemeinschaft
REFERENCES
- 1. Capetanaki Y, Bloch RJ, Kouloumenta A, Mavroidis M, Psarras S. Muscle intermediate filaments and their links to membranes and membranous organelles. Exp Cell Res. 2007;313:2063‐2076. [DOI] [PubMed] [Google Scholar]
- 2. Bhosle RC, Michele DE, Campbell KP, Li Z, Robson RMJB. Interactions of intermediate filament protein synemin with dystrophin and utrophin. Biochem Biophys Res Commun. 2006;346:768‐777. [DOI] [PubMed] [Google Scholar]
- 3. Capetanaki Y. Desmin cytoskeleton: a potential regulator of muscle mitochondrial behavior and function. Trends Cardiovasc Med. 2002;12:339‐348. [DOI] [PubMed] [Google Scholar]
- 4. Capetanaki Y, Milner DJ, Weitzer G. Desmin in muscle formation and maintenance: knockouts and consequences. Cell Struct Funct. 1997;22:103‐116. [DOI] [PubMed] [Google Scholar]
- 5. Milner DJ, Weitzer G, Tran D, Bradley A, Capetanaki Y. Disruption of muscle architecture and myocardial degeneration in mice lacking desmin. J Cell Biol. 1996;134:1255‐1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lorenzon A, Beffagna G, Bauce B, et al. Desmin mutations and arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 2013;111:400‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Clemen CS, Herrmann H, Strelkov SV, Schroder R. Desminopathies: pathology and mechanisms. Acta Neuropathol. 2013;125:47‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Goebel HH, Bornemann A. Desmin pathology in neuromuscular diseases. Virchows Archiv B. 1993;64:127‐135. [DOI] [PubMed] [Google Scholar]
- 9. Bar H, Fischer D, Goudeau B, et al. Pathogenic effects of a novel heterozygous R350P desmin mutation on the assembly of desmin intermediate filaments in vivo and in vitro. Hum Mol Genet. 2005;14:1251‐1260. [DOI] [PubMed] [Google Scholar]
- 10. Walter MC, Reilich P, Huebner A, et al. Scapuloperoneal syndrome type Kaeser and a wide phenotypic spectrum of adult‐onset, dominant myopathies are associated with the desmin mutation R350P. Brain. 2007;130:1485‐1496. [DOI] [PubMed] [Google Scholar]
- 11. Clemen CS, Stockigt F, Strucksberg KH, et al. The toxic effect of R350P mutant desmin in striated muscle of man and mouse. Acta Neuropathol. 2015;129:297‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Langer HT, Mossakowski AA, Willis BJ, et al. Generation of desminopathy in rats using CRISPR‐Cas9. J Cachexia Sarcopenia Muscle. 2020;11:1364‐1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. West DWD, Baehr LM, Marcotte GR, et al. Acute resistance exercise activates rapamycin‐sensitive and‐insensitive mechanisms that control translational activity and capacity in skeletal muscle. J Physiology. 2016;594:453‐468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wong TS, Booth FW. Protein metabolism in rat tibialis anterior muscle after stimulated chronic eccentric exercise. J Appl Physiol. 1990;69:1718‐1724. [DOI] [PubMed] [Google Scholar]
- 15. Solaro RJ, Pang DC, Briggs FN. The purification of cardiac myofibrils with Triton X‐100. Biochem Biophys Acta. 1971;245:259‐262. [DOI] [PubMed] [Google Scholar]
- 16. Gilda JE, Gomes AV. Stain‐Free total protein staining is a superior loading control to β‐actin for Western blots. Anal Biochem. 2013;440:186‐188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Aldridge GM, Podrebarac DM, Greenough WT, Weiler IJ. The use of total protein stains as loading controls: an alternative to high‐abundance single‐protein controls in semi‐quantitative immunoblotting. J Neurosci Methods. 2008;172:250‐254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Roche SM, Gumucio JP, Brooks SV, Mendias CL, Claflin DR. Measurement of maximum isometric force generated by permeabilized skeletal muscle fibers. J Vis Exp. 2015. 10.3791/52695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shah S, Lieber R. Real‐time imaging and mechanical measurement of muscle cytoskeletal proteins. J Histochem Cytochem. 2003;51:19‐29. [DOI] [PubMed] [Google Scholar]
- 20. Galler S, Hilber K. Tension/stiffness ratio of skinned rat skeletal muscle fibre types at various temperatures. Acta Physiol Scand. 1998;162:119‐126. [DOI] [PubMed] [Google Scholar]
- 21. Goldman YE, Simmons RM. Control of sarcomere length in skinned muscle fibres of Rana temporaria during mechanical transients. J Physiol. 1984;350:497‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Smith LR, Barton ER. Collagen content does not alter the passive mechanical properties of fibrotic skeletal muscle in mdx mice. Am J Physiol Cell Physiol. 2014;306:C889‐C898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Burkholder TJ, Lieber RL. Sarcomere length operating range of vertebrate muscles during movement. J Exp Biol. 2001;204:1529‐1536. [DOI] [PubMed] [Google Scholar]
- 24. Brashear SE, Wohlgemuth RP, Gonzalez G, Smith LR. Passive stiffness of fibrotic skeletal muscle in mdx mice relates to collagen architecture. J Physiol. 2021;599:943‐962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Woessner JF. The determination of hydroxyproline in tissue and protein samples containing small proportions of this imino acid. Arch Biochem Biophys. 1961;93:440‐447. [DOI] [PubMed] [Google Scholar]
- 26. Creemers L, Jansen D, van Veen‐Reurings A, Van den Bos T, Everts VJB. Microassay for the assessment of low levels of hydroxyproline. Biotechniques. 1997;22:656‐658. [DOI] [PubMed] [Google Scholar]
- 27. Meyer GA, Lieber RL. Skeletal muscle fibrosis develops in response to desmin deletion. Am J Physiol‐Cell Physiol. 2012;302:C1609‐C1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schmalbruch H. The morphology of regeneration of skeletal muscles in the rat. Tissue Cell. 1976;8:673‐692. [DOI] [PubMed] [Google Scholar]
- 29. Benoit PW, Belt WD. Destruction and regeneration of skeletal muscle after treatment with a local anaesthetic, bupivacaine (Marcaine). J Anat. 1970;107:547‐556. [PMC free article] [PubMed] [Google Scholar]
- 30. Bönnemann CG, Wang CH, Quijano‐Roy S, et al. Diagnostic approach to the congenital muscular dystrophies. J Neuromuscular disorders. 2014;24:289‐311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Henderson M, De Waele L, Hudson J, et al. Recessive desmin‐null muscular dystrophy with central nuclei and mitochondrial abnormalities. Acta Neuropathol. 2013;125:917‐919. [DOI] [PubMed] [Google Scholar]
- 32. Shinde A, Nakano S, Sugawara M, et al. Expression of caveolar components in primary desminopathy. Neuromuscul Disord. 2008;18:215‐219. [DOI] [PubMed] [Google Scholar]
- 33. Diermeier S, Buttgereit A, Schuermann S, et al. Preaged remodeling of myofibrillar cytoarchitecture in skeletal muscle expressing R349P mutant desmin. Neurobiol Aging. 2017;58:77‐87. [DOI] [PubMed] [Google Scholar]
- 34. Bostock EL, Edwards BT, Jacques MF, et al. Impaired glucose tolerance in adults with duchenne and becker muscular dystrophy. Nutrients. 2018;10:1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schneider SM, Sridhar V, Bettis AK, et al. Glucose metabolism as a pre‐clinical biomarker for the golden retriever model of Duchenne muscular dystrophy. Mol Imag Biol. 2018;20:780‐788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Eid Mutlak Y, Aweida D, Volodin A, et al. A signaling hub of insulin receptor, dystrophin glycoprotein complex and plakoglobin regulates muscle size. Nat Commun. 2020;11:1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hughes DC, Marcotte GR, Baehr LM, et al. Alterations in the muscle force transfer apparatus in aged rats during unloading and reloading: impact of microRNA‐31. J Physiol. 2018;596(14):2883‐2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lovering RM, De Deyne PG. Contractile function, sarcolemma integrity, and the loss of dystrophin after skeletal muscle eccentric contraction‐induced injury. Am J Physiol‐Cell Physiol. 2004;286:C230‐C238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lieber RL, Schmitz MC, Mishra DK, Fridén J. Contractile and cellular remodeling in rabbit skeletal muscle after cyclic eccentric contractions. J Appl Physiol (1985). 1994;77:1926‐1934. [DOI] [PubMed] [Google Scholar]
- 40. Barash IA, Peters D, Fridén J, Lutz GJ, Lieber RL. Desmin cytoskeletal modifications after a bout of eccentric exercise in the rat. Am J Physiol‐Regul Integr Comp Physiol. 2002;283:R958‐R963. [DOI] [PubMed] [Google Scholar]
- 41. Sam M, Shah S, Fridén J, Milner DJ, Capetanaki Y, Lieber RL. Desmin knockout muscles generate lower stress and are less vulnerable to injury compared with wild‐type muscles. Am J Physiol‐Cell Physiol. 2000;279:C1116‐C1122. [DOI] [PubMed] [Google Scholar]
- 42. Bonakdar N, Luczak J, Lautscham L, et al. Biomechanical characterization of a desminopathy in primary human myoblasts. Biochem Biophys Res Commun. 2012;419:703‐707. [DOI] [PubMed] [Google Scholar]
- 43. Linke WA. Titin gene and protein functions in passive and active muscle. Annu Rev Physiol. 2018;80:389‐411. [DOI] [PubMed] [Google Scholar]
- 44. Tomalka A, Rode C, Schumacher J, Siebert T. The active force‐length relationship is invisible during extensive eccentric contractions in skinned skeletal muscle fibres. Proc Biol Sci. 2017;284:20162497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lahmers S, Wu Y, Call DR, Labeit S, Granzier H. Developmental control of titin isoform expression and passive stiffness in fetal and neonatal myocardium. Circ Res. 2004;94:505‐513. [DOI] [PubMed] [Google Scholar]
- 46. Brynnel A, Hernandez Y, Kiss B, et al. Downsizing the molecular spring of the giant protein titin reveals that skeletal muscle titin determines passive stiffness and drives longitudinal hypertrophy. eLife. 2018;7. 10.7554/elife.40532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ottenheijm CAC, Knottnerus AM, Buck D, et al. Tuning passive mechanics through differential splicing of titin during skeletal muscle development. Biophys J. 2009;97:2277‐2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. KarisAllen JJ, Veres SP. Effect of testing temperature on the nanostructural response of tendon to tensile mechanical overload. J Biomech. 2020;104:109720. [DOI] [PubMed] [Google Scholar]
- 49. Vafiadaki E, Arvanitis DA, Sanoudou D. Muscle LIM protein: master regulator of cardiac and skeletal muscle functions. Gene. 2015;566:1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Knöll R, Kostin S, Klede S, et al. A common MLP (muscle LIM protein) variant is associated with cardiomyopathy. Circ Res. 2010;106:695. [DOI] [PubMed] [Google Scholar]
- 51. Hoffmann C, Moreau F, Moes M, et al. Human muscle LIM protein dimerizes along the actin cytoskeleton and cross‐links actin filaments. Mol Cell Biol. 2014;34:3053‐3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Damas F, Phillips SM, Libardi CA, et al. Resistance training‐induced changes in integrated myofibrillar protein synthesis are related to hypertrophy only after attenuation of muscle damage. J Physiol. 2016;594:5209‐5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Langer HT, Afzal S, Kempa S, Spuler S. Nerve damage induced skeletal muscle atrophy is associated with increased accumulation of intramuscular glucose and polyol pathway intermediates. Sci Rep. 2020;10:1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Collis WJ, Engel WK. Glucose metabolism in five neuromuscular disorders. Neurology. 1968;18:915. [DOI] [PubMed] [Google Scholar]
- 55. Kirwan J, Hickner R, Yarasheski K, Kohrt W, Wiethop B, and Holloszy JO. Eccentric exercise induces transient insulin resistance in healthy individuals. J Appl Physiol (1985). 1992;72:2197‐2202. [DOI] [PubMed] [Google Scholar]
- 56. Bryant NJ, Govers R, James DE. Regulated transport of the glucose transporter GLUT4. Nat Rev Mol Cell Biol. 2002;3:267. [DOI] [PubMed] [Google Scholar]
- 57. Weber‐Carstens S, Schneider J, et al. Critical illness myopathy and GLUT4. Am J Respir Crit Care Med. 2013;187(4):387‐396. [DOI] [PubMed] [Google Scholar]
- 58. Xiao Y, Zhu H, Li L, et al. Global analysis of protein expression in muscle tissues of dermatomyositis/polymyosisits patients demonstrated an association between dysferlin and human leucocyte antigen A. Rheumatology. 2019;58(8):1474‐1484. [DOI] [PubMed] [Google Scholar]
- 59. Keller S. GC/MS‐and LC/MS‐Based Metabolic and Proteomic Analysis of Dysferlin‐Deficient Muscle from Patients and Animal Models. Berlin, Germany: Freie Universität Berlin; 2014. [Google Scholar]
- 60. Gottlieb RA, Andres AM, Sin J, Taylor DP. Untangling autophagy measurements: all fluxed up. Circ Res. 2015;116:504‐514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pigna E, Berardi E, Aulino P, et al. Aerobic exercise and pharmacological treatments counteract cachexia by modulating autophagy in colon cancer. Sci Rep. 2016;6:1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S3
Table S1