

Abstract
Pharmacokinetics‐matched digital electrocardiogram data (n = 503 measurements from 180 patients) collected in a first‐in‐human, multi‐part, dose‐escalation (from 80 to 800 mg) and dose expansion (at 480 mg) phase 1 study in patients with advanced solid malignancies, were used to assess potential risk of QT prolongation associated with the AKT inhibitor capivasertib. The relationship between plasma drug concentrations and baseline‐adjusted Fridericia‐corrected QT (ΔQTcF) values was estimated using a prespecified linear mixed‐effects model. The model provided an unbiased reproduction of the experimental data set, estimating a small but positive correlation between capivasertib concentration and ΔQTcF. At the expected therapeutic dose (400 mg twice daily) the predicted mean ΔQTcF at the steady state maximum concentration was 3.97 ms with an upper limit of the 90% CI of 5.07 ms; below the 10 ms limit proposed by ICH E14 guidance. This analysis suggests that capivasertib is not expected to present a clinically significant risk for QT prolongation that is associated with pro‐arrhythmic effects.
Keywords: AKT, arrhythmia, breast cancer, capivasertib, concentration‐QT modelling, prostate cancer
What is already known about this subject
Capivasertib is a potent, selective inhibitor of the serine/threonine AKT1, AKT2 and AKT3 kinases currently being investigated for clinical use in various cancers, including breast and prostate cancer.
No clinically relevant cardiovascular safety issues, including serious electrocardiogram changes, have been identified to date in AstraZeneca‐sponsored clinical studies with capivasertib.
What this study adds
This systematic study, using concentration‐QT modelling of clinical data, indicates that capivasertib is not predicted to produce clinically significant prolongation of the QT interval in patients at the expected therapeutic dosage (400 mg twice daily, 4 days on/3 days off).
1. INTRODUCTION
The AKT serine/threonine protein kinases are downstream effectors of the PI3K/AKT/mTOR pathway, mediating cell proliferation and resistance to apoptosis. 1 , 2 AKT activation, commonly occurring due to upstream loss of PTEN function or activating mutations in PIK3CA or AKT1, occurs in a wide range of solid tumors. 3 , 4 , 5 Capivasertib, an oral, potent, selective inhibitor of AKT1, AKT2 and AKT3, has demonstrated effective inhibition of growth in preclinical cancer cell lines, 6 , 7 particularly those expressing PIK3CA or PTEN mutations, 8 and is under clinical investigation for therapeutic indications including triple‐negative breast cancer (TNBC), estrogen receptor‐positive/human epidermal growth factor receptor 2‐negative (ER+/HER2−) breast cancer and prostate cancer. 9 , 10 , 11 , 12 , 13 , 14 , 15
In the phase 2 FAKTION study, 15 patients with advanced/metastatic ER+/HER2− breast cancer receiving capivasertib (at a dosage of 400 mg twice daily, 4 days on/3 days off) in combination with fulvestrant had significantly longer progression‐free survival (PFS) compared with patients receiving fulvestrant plus placebo; in addition, there was a trend towards improved overall survival (OS) although the data were immature at the time of primary analysis. In the PAKT study, 12 this capivasertib dosing regimen, when combined with paclitaxel, also produced significantly longer PFS and OS in patients with advanced/metastatic TNBC compared with patients receiving paclitaxel alone. The benefit–risk profile of capivasertib is now being investigated in phase 3 studies, including the CAPItello‐290 (NCT03997123), CAPItello‐291 (NCT04305496) and CAPItello‐281 (NCT04493853) trials. Available clinical data from AstraZeneca‐sponsored studies with capivasertib reveal that there have been no reports of sudden death, torsades de pointes, seizures or electrocardiogram (ECG) changes that were considered serious by the investigator. 9 , 10 , 11 , 13 , 14 This work describes the assessment of the effect of capivasertib on the corrected QT (QTc) interval by categorical analysis and modelling of the concentration‐QTc relationship in patients with solid tumours.
2. METHODS
2.1. Study design
Patients with advanced or metastatic solid tumours were enrolled to a phase 1, open‐label, multi‐part study (NCT01226316). Parts A and B were dose‐escalation and dose‐expansion phases, respectively; parts C and D were expansion cohorts of patients with qualifying PIK3CA or AKT1 mutations, respectively. Key exclusion criteria included clinically significant abnormalities of glucose metabolism; treatment with chemotherapy, immunotherapy, anticancer agents, cytochrome P450 (CYP) 3A4 inducers/inhibitors/substrates or CYP2D6 substrates; severe or uncontrolled systemic disease; and abnormal organ function. Patients were also excluded if they had any of the following cardiac criteria: mean resting QTc interval > 470 ms in three consecutive ECG measurements; any clinically important abnormalities in rhythm, conduction or morphology of resting ECG; any factors that increase the risk of QTc prolongation or arrhythmic events such as heart failure, hypokalaemia, congenital long QT syndrome, family history of long QT syndrome or unexplained sudden death under 40 years of age or any concomitant medication known to prolong the QT interval; any incidence of coronary artery bypass graft, angioplasty, vascular stent, myocardial infarction, angina pectoris, congestive heart failure, uncontrolled hypotension; left ventricular ejection fraction below lower limit of normal for site. In parts A and B, patients (n = 90) were given capivasertib either continuously (80–600 mg twice daily) or intermittently on a 4 days on/3 days off (480–640 mg twice daily) or 2 days on/5 days off (640–800 mg twice daily) schedule; in parts C and D, patients (n = 118) were given capivasertib 4 days on/3 days off (480 mg twice daily). Efficacy and safety results from these study parts have been reported previously. 9 , 16 Time matched pharmacokinetic (PK)‐ECG measurements were collected in parts A and B at baseline and at 1, 2, 6 and 24 hours after the first capivasertib dose; in parts C and D, measurements were taken at baseline and at 2 and 4–6 hours after the first dose. As no post‐dose data were available after repeated administration, only single dose data were evaluated. In total, 503 time‐matched measurements were available from 180 patients and were included in the analysis. Further details are reported in Table S1 in the Supporting Information.
2.2. ECG recording and processing
Data were collected using a 12‐lead digital ECG recorder after patients had been resting semi‐supine for a minimum of 10 minutes. For each time point, three ECG recordings were taken at approximately 5‐minute intervals and the arithmetic means of ECG intervals measured from these triplicates were used for the analysis. The data were transferred electronically for central analysis; heart rate (HR) and PR, RR, QRS and QT intervals were determined and reviewed by a central ECG core laboratory (eResearch Technology Limited). The QT values were corrected for HR using the Fridericia and Bazett correction formulae. 17 , 18 The optimal correction method was selected based on visual inspection of RR versus QT (uncorrected), Fridericia‐corrected (QTcF) and Bazett‐corrected (QTcB) QTc plots.
2.3. Exploratory analysis
Exploratory data analysis was performed in two steps according to a prespecified exposure–response analysis plan, as outlined by the International Conference on Harmonisation (ICH) E14 guidance, the ICH implementation working group questions and answers (R3), the concentration‐QT whitepaper and in line with model‐informed approaches to cardiovascular safety. 19 , 20 , 21 , 22 First, categorical evaluation of QTc, PR and QRS intervals was performed to identify patients with significant treatment‐emergent QTc, PR and QRS prolongation (QTc: ≥450, ≥480, and ≥500 ms; ΔQTc: ≥30 and ≥60 ms; PR: ≥200 ms; QRS: ≥110 ms). Second, exploratory graphical analysis was performed to check assumptions of a prespecified linear mixed‐effects (LME) model and justify application of the model to the given data set (Figures S1–S4 in the Supporting Information). These assumptions included lack of drug effect on HR, appropriateness of selected HR correction method, no time delay between drug concentration and ΔQTc values, and linearity of concentration‐QT relationship.
2.4. Model description
A predefined model structure adapted for placebo‐free oncology trials was used to characterize the concentration‐∆QTc relationship (Equation 1) 20 :
| (1) |
where ∆QTc i,k is the change from baseline in QTc for subject i at time k; θ0 is the population mean intercept; η0,i is the random effect associated with the intercept term θ0; θ1 is the population mean slope of the assumed linear association between concentration and ΔQTc i,k ; η1,i is the random effect associated with the slope θ1; C i,k is the concentration for subject i at time k; θ2 is the fixed effect associated with baseline QTc i,k = 0; is the overall mean of QTc i,k = 0 (i.e., the mean of all the baseline [= time 0] QTc values); and ɛ is the residual error. Parameter estimation was performed in R version 3.5.1 (R Foundation for Statistical Computing, Vienna, Austria; package nlme version 3.1–137); model predictions were performed using the lsmeans package (version 2.26–3). Model parameters were estimated using restricted maximum log‐likelihood assuming that random effects and residuals followed normal distributions. Quality of the experimental data reproduction by the final model was evaluated via analysis of goodness‐of‐fit plots.
2.5. Model‐based QT simulations
The verified model was used to calculate the mean and 90% CI for ΔQTc at expected therapeutic capivasertib exposure levels. The maximum plasma concentration (C max) of capivasertib was derived by non‐compartmental analysis and used as an independent variable for ΔQTc calculation. ΔQTc was predicted at geometric mean C max values for nominal doses at Days 8, 4 and 2 for continuous, 4 days on/3 days off, and 2 days on/5 days off dosing schedules, respectively.
2.6. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20. 23 , 24
3. RESULTS
3.1. Categorical analysis
Of the evaluated total of 180 patients (see Table S1 in the Supporting Information), eight patients (4.4%) had QTcF ≥ 450 ms (seven at 480 mg and one at 640 mg) and no patient had QTcF ≥ 480 ms. One patient (0.6%) had ΔQTcF ≥ 30 ms (at 640 mg) and no patient had ΔQTcF ≥ 60 ms. Twelve patients (6.7%) had PR ≥ 200 ms and nine patients (5.0%) had QRS ≥ 110 ms, all at doses ranging from 80 mg to 800 mg (Figure S1 in the Supporting Information).
3.2. Evaluation of model assumptions
Visual inspection of scatterplots and linear regression of RR vs QT, QTcF and QTcB intervals confirmed that the Fridericia correction method was most appropriate for this data set (Figure S2 in the Supporting Information), hence, ΔQTcF values were used for further analysis. The time course of changes in post‐dose plasma capivasertib concentration, ΔHR and ΔQTcF are presented in Figure 1. Dose‐dependent increases in drug concentration and C max, which was reached approximately 2 hours after dosing, were observed (Figure 1A). A small decrease in HR was detected at 1 hour; however, ΔHR was not dose‐dependent and did not exceed the predefined threshold of 10 beats per minute (Figure 1B). The time to maximum ΔQTcF was typically within 2 hours after administration (Figure 1C). No evidence of a time‐delay was observed in hysteresis plots (Figure S3 in the Supporting Information). A linear relationship between capivasertib concentration and ΔQTcF was shown to describe the data satisfactorily (Figure S4 in the Supporting Information). Consequently, as no violations of the model assumptions were detected, the prespecified model was used.
FIGURE 1.
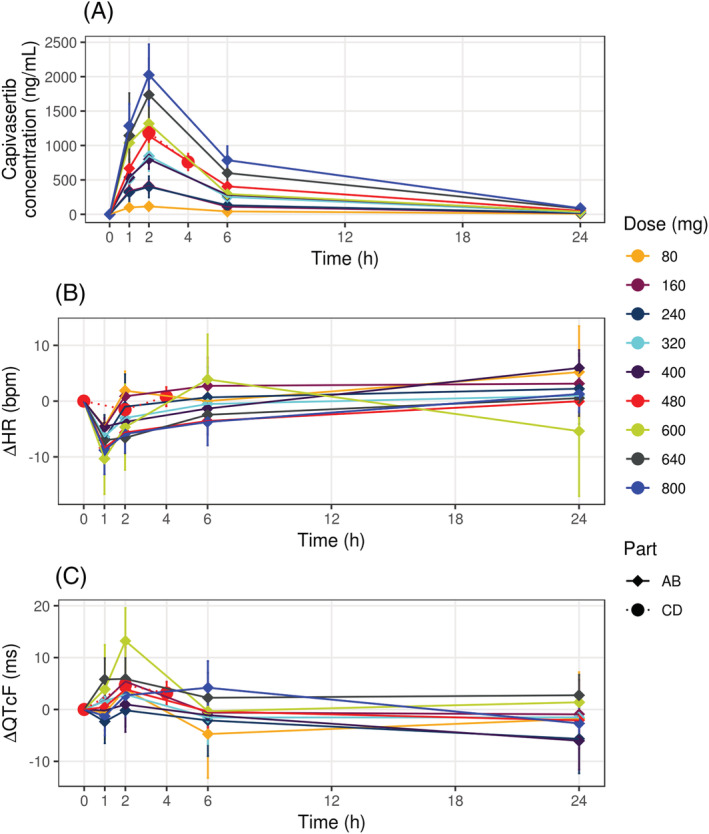
Time course of mean and 90% CI of (A) capivasertib concentration, (B) ΔHR and (C) ΔQTcF. Lines denote arithmetic mean values; error bars denote 90% CI for mean values. Data for different capivasertib doses are shown by colour.bpm, beats per minute; HR, heart rate; QTcF, Fridericia‐corrected QT interval
3.3. Description of the final concentration QTcF model
The model parameters and their precision are shown in Table 1. The slope parameter indicated a minor but statistically significant linear relationship between capivasertib concentration and ΔQTcF, with a value of 0.003 ms/(ng/mL) (95% CI: 0.002–0.004 ms/(ng/mL); P < .001). The model accurately matched experimentally observed ΔQTcF across all capivasertib concentrations (Figure 2). Evaluation of goodness‐of‐fit plots demonstrated unbiased reproduction of the experimental data by the final model (Figure S5 in the Supporting Information).
TABLE 1.
Linear mixed‐effects model parameters
| Parameter | Description | Estimate | RSE (%) | P‐value |
|---|---|---|---|---|
| θ0 | Intercept (ms) | −0.269 | 237.15 | 0.674 |
| η0 | Random effect for intercept (ms) | 5.350 | ||
| θ1 | Slope (ms/ (ng/mL)) | 0.00337 | 17.003 | <.001 |
| η1 | Random effect for slope (ms/(ng/mL)) | 2.271 | ||
| θ2 | Impact of baseline QTcF on ΔQTcF | −0.119 | 25.90 | <.001 |
| ɛ | Residual variability (ms) | 6.452 |
QTcF, Fridericia‐corrected QT interval; RSE, relative standard error.
FIGURE 2.
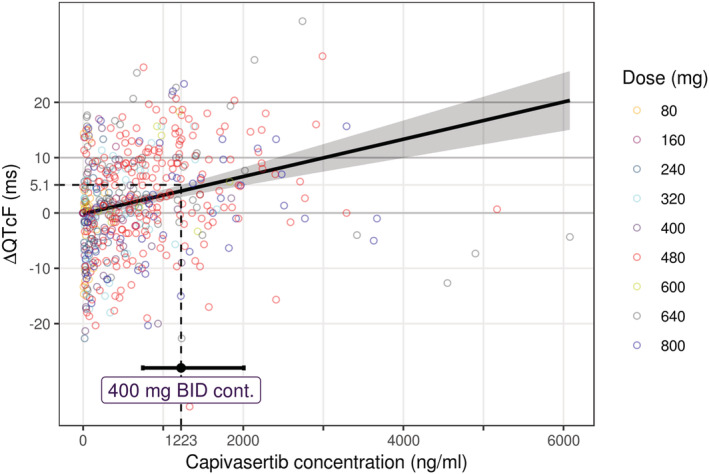
Observed and model‐derived ΔQTcF vs plasma capivasertib concentration. Solid black line and gray area denote the mean and 90% CI of the model prediction; circles denote individual patient data with capivasertib dosage indicated by colour; dashed black line denotes the upper 90% CI limit of the model derived ΔQTcF at the geometric mean steady state C max measured with the expected therapeutic dose; error bars denote 95% CI for the geometric mean C max; BID, twice daily; C max, maximum plasma concentration; cont., continuous; QTcF, Fridericia‐corrected QT interval
3.4. QT estimation at clinically relevant capivasertib concentrations
The geometric mean C max for each capivasertib dose and the corresponding predicted mean and 90% CI ΔQTcF are reported in Table S2 in the Supporting Information. In the absence of rich PK data for the anticipated therapeutic capivasertib dose regimen (400 mg twice daily, 4 days on/3 days off), Day 8 PK data from continuous 400 mg twice daily dosing was used as an estimate of the therapeutic steady‐state C max (1223 ng/mL). At this concentration, the predicted mean ΔQTcF was 3.97 ms (90% CI: 2.87–5.07) (Figure 2).
4. DISCUSSION
Drugs that prolong the mean QTc interval by >20 ms have a substantially increased likelihood of being proarrhythmic. 19 Consequently, new chemical entities are expected to undergo a clinical ECG evaluation early in development, 19 for example by a ‘thorough QT/QTc’ (TQT) study at supratherapeutic doses, including placebo and a positive control, or by collecting ECG data in early studies that include a broad range of doses and performing concentration‐QTc modelling. 19 , 20 The latter approach has been applied across various therapeutic areas and has some advantages, particularly in the oncology setting where several challenges are encountered when conducting TQT studies in patients. These include the use of placebo or active controls (e.g. moxifloxacin) and concomitant medications (e.g., antidepressants, antiemetics, antibiotics), the high prevalence of risk factors for QT prolongations associated with side effects of cancer therapy (nausea and vomiting, dehydration followed by electrolyte imbalances) and other effects (e.g., kidney failure, liver dysfunction and poorly controlled diabetes). 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32
Indeed, like other anti‐cancer agents, capivasertib should not be administered to healthy volunteers at supratherapeutic doses. We therefore assessed the QTc prolongation risk for capivasertib, at the highest anticipated therapeutic exposure, in patients with solid tumours by applying a previously described prespecified LME model structure. 20 Our systematic analysis included more than 500 time‐matched digital concentration‐QTc assessments from 180 patients, who received single doses of capivasertib ranging from 80 mg to 800 mg.
The exploratory analysis supported the use of the prespecified model, enabling an unbiased reproduction of the experimental data. A statistically significant, but not clinically relevant, association between capivasertib concentration and ΔQTcF was demonstrated. At the estimated therapeutic steady state peak concentration, the predicted mean ΔQTcF was <5 ms and the upper limit of the 90% CI was <10 ms, below the limit proposed by ICH E14 guidance.
Although the model‐based analysis did not include steady state data, drug accumulation is less than twofold when capivasertib is given according to a 4 days on/3 days off intermittent schedule and no active metabolites have been identified. The single dose exposure from doses up to 800 mg is, therefore, representative of the therapeutic exposure from 400 mg. Supratherapeutic mean exposures have not been evaluated. However, the variability is anticipated to be representative of the target population, there have been no clinically relevant intrinsic or extrinsic factors affecting capivasertib PK identified to date, and the exposure that is predicted to cause QTcF prolongation of 20 ms is approximately four‐ to fivefold higher than the C max at the expected therapeutic dose. No patients in the data set reported here had a QTcF value of >480 ms or a ΔQTcF value of >60 ms.
In conclusion, phase 1 data from patients with advanced solid malignancies suggest that the recommended treatment regimen of capivasertib is not expected to present a clinically significant risk for QT prolongation associated with pro‐arrhythmic effects.
CONTRIBUTORS
M.C., S.Y.A.C., G.S., J.P., C.D. and D.R. were responsible for the study concept and design; V.V., P.D., S.Y.A.C. and M.C. analysed the data; V.V., P.D., S.Y.A.C. and D.R. performed the modelling; M.C. and V.V. wrote the manuscript; all authors reviewed and revised the manuscript.
COMPETING INTERESTS
M.C., J.P., G.S., C.D., B.M. and D.R. are employees of AstraZeneca and own stock or stock options. P.D. owns shares in AstraZeneca. S.Y.A.C. owns shares in AstraZeneca and is currently an employee of and owns shares in Certara. Certara received research funding from AstraZeneca. V.V. is an employee of M&S Decisions LLC. M&S Decisions received research funding from AstraZeneca.
Supporting information
TABLE S1 Summary of PK‐ECG data
TABLE S2 Observed C max and ΔQTcF predictions based on a prespecified concentration‐QTcF model
FIGURE S1 Scatterplots of capivasertib concentration and (A) PR interval and (B) QRS interval. Circles denote individual data with capivasertib dosage indicated by colour; solid black lines denote linear regression
FIGURE S2 Scatterplots of RR interval and (A) uncorrected, (B) Fridericia‐corrected and (C) Bazett‐corrected QT intervals. Circles denote individual data with capivasertib dosage indicated by colour; solid black lines denote linear regression
FIGURE S3 Evaluation of PK‐ΔQTcF hysteresis. Mean ΔQTcF and plasma capivasertib concentration connected in temporal order by dose (mg). Lines denote mean values; error bars denote 90% CI for mean values.
FIGURE S4 Scatterplot of paired ΔQTcF and plasma capivasertib concentration data. Circles denote individual data with capivasertib dosage indicated by colour; the red line denotes the second‐order loess smooth line; the dashed line denotes the linear regression; filled black circles denote the mean ΔQTcF (±SD) of each concentration bin. (A) Concentration on normal scale; (B) concentration on logarithmic scale
FIGURE S5 Goodness‐of‐fit plots: (A) Individual model predictions vs observed ∆QTcF; (B) quantile‐quantile plot of standardized residuals; (C) scatterplot of standardized residuals vs plasma capivasertib concentration; (D) scatterplot of standardized residuals vs centred baseline QTcF; (E) notched boxplot and scatterplot of the residuals, grouped by study parts
ACKNOWLEDGEMENTS
AZD5363 (capivasetib) was discovered by AstraZeneca subsequent to a collaboration with Astex Therapeutics (and its collaboration with the Institute of Cancer Research and Cancer Research Technology Limited). This work and study NCT01226316 were overseen and funded by AstraZeneca. Medical writing support was provided by Adam Errington, PhD, Oxford PharmaGenesis, Cardiff, UK and was funded by AstraZeneca.
Voronova V, Cullberg M, Delff P, et al. Concentration‐QT modelling shows no evidence of clinically significant QT interval prolongation with capivasertib at expected therapeutic concentrations. Br J Clin Pharmacol. 2022;88(2):858-864. 10.1111/bcp.15006
This manuscript describes a secondary investigation that addresses a specific scientific question with a modelling approach using previously collected clinical trial data. No new clinical data, requiring medical supervision, were collected for this manuscript. The Principal Investigator of the original study was an author on the primary publication but does not meet ICMJE criteria for authorship of this manuscript.
Funding information AstraZeneca, Grant/Award Number: NCT01226316; Institute of Cancer Research, Grant/Award Number: AZD5363
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Ortega MA, Fraile‐Martínez O, Asúnsolo Á, Buján J, García‐Honduvilla N, Coca S. Signal transduction pathways in breast cancer: the important role of PI3K/Akt/mTOR. J Oncol. 2020;2020:9258396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR signaling in cancer. Front Oncol. 2014;4:2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stemke‐Hale K, Gonzalez‐Angulo AM, Lluch A, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008;68(15):6084‐6091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Koboldt DC, Fulton R, McLellan M, et al. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4(12):988‐1004. [DOI] [PubMed] [Google Scholar]
- 6. Addie M, Ballard P, Buttar D, et al. Discovery of 4‐amino‐N‐[(1S)‐1‐(4‐chlorophenyl)‐3‐hydroxypropyl]‐1‐(7H‐pyrrolo[2,3‐d]pyrimidin‐4‐yl)piperidine‐4‐carboxamide (AZD5363), an orally bioavailable, potent inhibitor of Akt kinases. J Med Chem. 2013;56(5):2059‐2073. [DOI] [PubMed] [Google Scholar]
- 7. Davies BR, Greenwood H, Dudley P, et al. Preclinical pharmacology of AZD5363, an inhibitor of AKT: pharmacodynamics, antitumor activity, and correlation of monotherapy activity with genetic background. Mol Cancer Ther. 2012;11(4):873‐887. [DOI] [PubMed] [Google Scholar]
- 8. Chalhoub N, Baker SJ. PTEN and the PI3‐kinase pathway in cancer. Annu Rev Pathol. 2009;4(1):127‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Banerji U, Dean EJ, Pérez‐Fidalgo JA, et al. A phase I open‐label study to identify a dosing regimen of the pan‐AKT inhibitor AZD5363 for evaluation in solid tumors and in PIK3CA‐mutated breast and gynecologic cancers. Clin Cancer Res. 2018;24(9):2050‐2059. [DOI] [PubMed] [Google Scholar]
- 10. Tamura K, Hashimoto J, Tanabe Y, et al. Safety and tolerability of AZD5363 in Japanese patients with advanced solid tumors. Cancer Chemother Pharmacol. 2016;77(4):787‐795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Turner NC, Alarcón E, Armstrong AC, et al. BEECH: a dose‐finding run‐in followed by a randomised phase II study assessing the efficacy of AKT inhibitor capivasertib (AZD5363) combined with paclitaxel in patients with estrogen receptor‐positive advanced or metastatic breast cancer, and in a PIK3CA mutant sub‐population. Ann Oncol. 2019;30(5):774‐780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schmid P, Abraham J, Chan S, et al. Capivasertib plus paclitaxel versus placebo plus paclitaxel as first‐line therapy for metastatic triple‐negative breast cancer: the PAKT trial. J Clin Oncol. 2020;38(5):423‐433. [DOI] [PubMed] [Google Scholar]
- 13. Dean E, Banerji U, Schellens JHM, et al. A phase 1, open‐label, multicentre study to compare the capsule and tablet formulations of AZD5363 and explore the effect of food on the pharmacokinetic exposure, safety and tolerability of AZD5363 in patients with advanced solid malignancies: OAK. Cancer Chemother Pharmacol. 2018;81(5):873‐883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Crabb SJ, Birtle AJ, Martin K, et al. ProCAID: a phase I clinical trial to combine the AKT inhibitor AZD5363 with docetaxel and prednisolone chemotherapy for metastatic castration resistant prostate cancer. Invest New Drugs. 2017;35(5):599‐607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jones RH, Casbard A, Carucci M, et al. Fulvestrant plus capivasertib versus placebo after relapse or progression on an aromatase inhibitor in metastatic, oestrogen receptor‐positive breast cancer (FAKTION): a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 2020;21(3):345‐357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hyman DM, Smyth LM, Donoghue MTA, et al. AKT inhibition in solid tumors with AKT1 mutations. J Clin Oncol. 2017;35(20):2251‐2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fridericia LS. Die Systolendauer im Elektrokardiogramm bei normalen Menschen und bei Herzkranken. Acta Med Scand. 2009;53(1):469‐486. [Google Scholar]
- 18. Bazett HC. An analysis of the time‐relations of electrocardiograms. Ann Noninvasive Electrocardiol. 1997;2(2):177‐194. [Google Scholar]
- 19. European Medicines Agency . ICH: E14: The clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non‐antiarrhythmic drugs—step 5. 2005. https://www.ema.europa.eu/en/ich-e14-clinical-evaluation-qtqtc-interval-prolongation-proarrhythmic-potential-non-antiarrhythmic. Accessed March 5, 2021.
- 20. Garnett C, Bonate PL, Dang Q, et al. Scientific white paper on concentration‐QTc modeling. J Pharmacokinet Pharmacodyn. 2018;45(3):383‐397. [DOI] [PubMed] [Google Scholar]
- 21. International Council for Harmonisation E14 Implementation Working Group . ICH E14 guideline: the clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non‐antiarrhythmic drugs. Questions and answers (R3). 2015. https://database.ich.org/sites/default/files/E14_Q%26As_R3_Q%26As.pdf. Accessed March 5, 2021.
- 22. Cheung SYA, Parkinson J, Wählby‐Hamrén U, et al. A tutorial on model informed approaches to cardiovascular safety with focus on cardiac repolarisation. J Pharmacokinet Pharmacodyn. 2018;45(3):365‐381. [DOI] [PubMed] [Google Scholar]
- 23. Alexander SPH, Fabbro D, Kelly E, et al. The Concise Guide to PHARMACOLOGY 2019/20: Enzymes. Br J Pharmacol. 2019;176:S297‐S396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Alexander SPH, Cidlowski JA, Kelly E, et al. The Concise Guide to PHARMACOLOGY 2019/20: Nuclear hormone receptors. Br J Pharmacol. 2019;176:S229‐S246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lu J, Li J, Helmlinger G, Al‐Huniti N. Assessing QT/QTc interval prolongation with concentration‐QT modeling for Phase I studies: impact of computational platforms, model structures and confidence interval calculation methods. J Pharmacokinet Pharmacodyn. 2018;45(3):469‐482. [DOI] [PubMed] [Google Scholar]
- 26. Kang D, Lin K, Ludwig E, Yin O. Concentration‐QT analysis of quizartinib in patients with relapsed/refractory AML. Presented at the Annual Meeting of the American Conference on Pharmacometrics; October 7–10, 2018; San Diego, California.
- 27. Jolling K, Äbelö A, Luyckx N, et al. Concentration‐QT modeling following inhalation of the novel inhaled phosphodiesterase‐4 inhibitor CHF6001 in healthy volunteers shows an absence of QT prolongation. CPT Pharmacometrics Syst Pharmacol. 2019;8(7):460‐468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hibma JE, Kantarjian HM, DeAngelo DJ, Boni JP. Effect of inotuzumab ozogamicin on the QT interval in patients with haematologic malignancies using QTc‐concentration modelling. Br J Clin Pharmacol. 2019;85(3):590‐600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tolcher AW, Chi KN, Shore ND, et al. Effect of abiraterone acetate plus prednisone on the QT interval in patients with metastatic castration‐resistant prostate cancer. Cancer Chemother Pharmacol. 2012;70(2):305‐313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nebot N, Arkenau HT, Infante JR, et al. Evaluation of the effect of dabrafenib and metabolites on QTc interval in patients with BRAF V600‐mutant tumours. Br J Clin Pharmacol. 2018;84(4):764‐775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Britto MR, Sarapa N. Clinical QTc assessment in oncology. In: Bonate PL, Howard DR, eds. Pharmacokinetics in Drug Development: Problems and Challenges in Oncology, Volume 4. Cham: Springer International; 2016:77‐106. [Google Scholar]
- 32. Coppola C, Rienzo A, Piscopo G, Barbieri A, Arra C, Maurea N. Management of QT prolongation induced by anti‐cancer drugs: target therapy and old agents. Different algorithms for different drugs. Cancer Treat Rev. 2018;63:135‐143. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1 Summary of PK‐ECG data
TABLE S2 Observed C max and ΔQTcF predictions based on a prespecified concentration‐QTcF model
FIGURE S1 Scatterplots of capivasertib concentration and (A) PR interval and (B) QRS interval. Circles denote individual data with capivasertib dosage indicated by colour; solid black lines denote linear regression
FIGURE S2 Scatterplots of RR interval and (A) uncorrected, (B) Fridericia‐corrected and (C) Bazett‐corrected QT intervals. Circles denote individual data with capivasertib dosage indicated by colour; solid black lines denote linear regression
FIGURE S3 Evaluation of PK‐ΔQTcF hysteresis. Mean ΔQTcF and plasma capivasertib concentration connected in temporal order by dose (mg). Lines denote mean values; error bars denote 90% CI for mean values.
FIGURE S4 Scatterplot of paired ΔQTcF and plasma capivasertib concentration data. Circles denote individual data with capivasertib dosage indicated by colour; the red line denotes the second‐order loess smooth line; the dashed line denotes the linear regression; filled black circles denote the mean ΔQTcF (±SD) of each concentration bin. (A) Concentration on normal scale; (B) concentration on logarithmic scale
FIGURE S5 Goodness‐of‐fit plots: (A) Individual model predictions vs observed ∆QTcF; (B) quantile‐quantile plot of standardized residuals; (C) scatterplot of standardized residuals vs plasma capivasertib concentration; (D) scatterplot of standardized residuals vs centred baseline QTcF; (E) notched boxplot and scatterplot of the residuals, grouped by study parts
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.