

Abstract
The objective of this phase 1 study was to evaluate the pharmacokinetics, safety, and tolerability of baricitinib after single and multiple doses in healthy Chinese adults. Eligible subjects received a once‐daily dose of baricitinib 2, 4, or 10 mg or placebo on day 1 (single dose) and days 4 through 10 for 7 consecutive days (multiple doses). Plasma pharmacokinetic samples were collected up to 48 hours after dosing on days 1 and 10, with predose samples collected before dosing on day 1 and days 4 through 10. Safety and tolerability were also assessed. Baricitinib was rapidly absorbed, reaching peak plasma concentrations within 0.5 to 1 hour (median). Plasma concentrations declined rapidly following the attainment of peak concentrations, with a mean terminal half‐life of 5.7 to 7.3 hours. Steady‐state plasma concentrations of baricitinib were achieved after the second day of once‐daily dosing, with minimal accumulation of baricitinib in plasma (up to 10% increase in area under the plasma concentration–time curve). Single‐ and multiple‐dose mean values for area under the plasma concentration–time curve from time zero to infinity and maximum plasma concentration appeared to increase in an approximately dose‐proportional manner across the dose range. Single and multiple oral doses of once‐daily baricitinib up to 10 mg were well tolerated by healthy Chinese subjects.
Keywords: baricitinib, China, Janus kinase family of protein tyrosine kinases, pharmacokinetics, rheumatoid arthritis
The dysregulation of cytokines has been implicated in the pathogenesis of chronic inflammatory diseases including autoimmune diseases such as rheumatoid arthritis (RA). 1 , 2 The Janus kinase (JAK) family of protein tyrosine kinases plays an important role in the signaling of a number of cytokines implicated in the pathogenesis of RA via JAK‐signal transducers and activators of transcription pathways. 1 , 3 , 4 Inhibition of JAK‐signal transducers and activators of transcription signaling can target multiple RA‐associated cytokine pathways and thereby reduce inflammation, cellular activation, and proliferation of key immune cells. 1 , 4 , 5 Thus, selective JAK inhibitors are viewed as disease‐modifying anti‐inflammatory drugs for the treatment of RA. 1 , 4 , 5 The JAK family comprises 4 members: JAK1, JAK2, JAK3, and tyrosine kinase 2. Two JAK inhibitors, tofacitinib and baricitinib, have recently been approved for the treatment of RA, and many JAK inhibitors currently in development have a significant effect on JAK1. It is unclear whether JAK2, JAK3, and tyrosine kinase 2 are desirable therapeutic targets in RA. 6 The agents that selectively inhibit JAKs have received significant attention in recent years as potential new treatments for the RA. 7
Baricitinib (Figure 1), is an oral, potent, and selective inhibitor of the JAK family of protein tyrosine kinases, with high potency and selectivity for JAK1 and JAK2 and a lower potency for JAK3 and tyrosine kinase 2. 8 , 9 , 10 Baricitinib is efficacious in patients with moderately to severely active RA and in patients with inadequate response to methotrexate or intolerance to conventional disease‐modifying anti‐inflammatory drugs. 11 , 12 Baricitinib has a favorable safety profile in the context of demonstrated efficacy, with reversible, dose‐dependent decline in absolute neutrophil count (ANC) that has no correlation with increased infection risk. 13 Baricitinib has been approved in doses of 2 mg (United States, European Union, Japan) and 4 mg (European Union, Japan) once daily for the treatment of adult patients with moderate to severely active RA and authorized in more than 50 countries. 14
Figure 1.
Structure of baricitinib.
In healthy White volunteers, baricitinib was rapidly absorbed, usually attaining maximum plasma concentration (Cmax) within 1.5 hours after dosing, after which baricitinib plasma concentrations declined in an apparent biexponential fashion, with a geometric mean terminal‐phase disposition half‐life (t1/2) of about 8 hours. 13 The t1/2 of baricitinib in patients weighing <40 kg was substantially shorter than in adult populations, requiring the need for dosing up to 4 times daily. 15 Baricitinib demonstrates dose‐linear and time‐invariant pharmacokinetics (PK), with low oral‐dose clearance (17 L/h) and minimal systemic accumulation observed following repeat dosing. 13 Baricitinib is mainly excreted in the urine (∼75%) in an unchanged form with a mean renal clearance of ∼12 L/h2. 13 There is minimal oxidative metabolism of baricitinib, mediated by cytochrome P450 3A4, with <10% of the dose identified as undergoing biotransformation. 16 In vitro, baricitinib was a substrate for organic anion transporter 3, multidrug and toxin extrusion protein 2‐K, P‐glycoprotein, and breast cancer resistance protein. 17 It was observed that the area under the plasma concentration–time curve (AUC) from time zero to infinity (AUC0‐∞) of baricitinib increased by 2‐fold and renal clearance decreased to 69% of control in healthy subjects with the use of probenecid, a strong organic anion transporter 3 inhibitor. 17 Population PK/pharmacodynamic modeling suggested that baricitinib 2 mg once daily had potential as an efficacious dose. 18
The PK and safety of drugs can differ between populations, reflecting differences in the intrinsic and extrinsic factors that influence the PK 19 and safety profile 20 , 21 of a particular drug. PK and safety characteristics of baricitinib in the Chinese population were unknown. The present study was designed to evaluate the PK, safety, and tolerability of single and multiple doses of baricitinib in healthy Chinese subjects. Baricitinib 2 and 4 mg once daily were investigated in previous pivotal phase 3 studies for RA indication, whereas baricitinib 10 mg was investigated in previous global early‐phase studies. 13 Based on the above facts, the present ethnic bridging study selected baricitinib 2, 4, and 10 mg doses. Baricitinib 10 mg was selected in case there was potential for a higher dose in indications other than RA.
Subjects and Methods
Subjects
Healthy male and female Chinese subjects of nonchildbearing potential between the ages of 18 and 60 years with a body mass index of 19.0 to 24.0 kg/m2 were screened based on medical history, physical examination, clinical laboratory profile, and electrocardiogram (ECG) evaluation. Additionally, only those whose biological parents and grandparents were exclusively of Chinese descent and were born in China were included. Subjects were excluded from the study if they had (1) used or intended to use over‐the‐counter medication, prescription medication, or Chinese herbal preparations within 14 days before dosing and during the study, or had previously received the investigational product; (2) a history of adverse drug reactions, or disorders capable of significantly altering the absorption, metabolism, or elimination of drugs; (3) stomach or intestinal surgery; (4) medical history of tuberculosis (active or latent); (5) herpes zoster; (6) hepatitis A virus immunoglobulin M, hepatitis B surface antigen, hepatitis C virus antibody, or human immunodeficiency virus antibody; or (7) screening ANC values <2000 cells/μL (2 × 109/L).
Study Design
This was a single‐site, subject‐ and investigator‐blinded, placebo‐controlled, randomized study (ClinicalTrials.gov identifier: NCT02758613). The study was conducted in 3 periods, which included a screening period, a treatment period (single and multiple dosing), and a follow‐up period. A total of 33 subjects were randomized to receive baricitinib (2, 4, or 10 mg) or placebo. Subjects received a single, oral dose of baricitinib or placebo on day 1 and multiple, oral doses of baricitinib or placebo on days 4 through 10 (once daily dosing for 7 days). The subjects received a fixed dose during the single‐ and multiple‐dose periods. Subject disposition is presented in Figure S1. The study protocol and informed consent were approved by the Institutional Review Board of Peking University First Hospital, Beijing. Informed consent was obtained from all subjects or legal representatives before performing any study‐related procedures. The study was conducted at the Clinical Research Unit of Peking University First Hospital in accordance with International Conference on Harmonization Good Clinical Practice guidelines, applicable laws, and regulations, the Declaration of Helsinki and its subsequent revisions, and the Council for International Organizations of Medical Sciences International Ethical Guidelines.
Study Drug Administration
Baricitinib and matching placebo tablets were supplied by Eli Lilly and Company, Indianapolis, Indiana. Following an overnight fast of at least 10 hours, study drugs were administered orally with 200 mL of room temperature water. Subjects were provided meals 3 hours after administration of the study drug.
PK Analyses
The investigators collected venous blood samples to evaluate plasma concentration of baricitinib in all subjects who received at least 1 dose of the study drug. Plasma PK samples were collected up to 48 hours (0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 16, 24, 36, and 48 hours) after dosing on days 1 and 10, with predose samples collected on day 1 (single‐dose period) and days 4 through 10 (multiple‐dose period). PK parameters for baricitinib were estimated using standard noncompartmental methods, which for the single‐dose period included AUC0‐∞, AUC from time zero to time t, AUC from time 0 to 24 hours after dosing, Cmax, time to maximum plasma concentration (tmax), t½, and apparent clearance (CL/F), and for the multiple‐dose period included AUC during 1 dosing interval at steady state (AUCτ,ss), Cmax during a dosing interval at steady state (Cmax,ss), tmax during 1 dosing interval at steady state, t½, CL/F, and accumulation ratio. A predose PK sample was collected from day 4 to day 10 to provide information on steady‐state achievement.
Safety and Tolerability Evaluations
Safety and tolerability evaluations included monitoring of adverse effects (AEs) and their frequency, physical examinations, and clinical laboratory tests throughout the course of the study. The incidence and severity of AEs were assessed for each study treatment. The causality of the AE with the investigational product was also evaluated based on the investigator's perception. AEs reported before the first dose were distinguished from those reported as new or those that increased in severity during the study. Each AE was classified by the most suitable term from the Medical Dictionary for Regulatory Activities. Single ECGs and supine/standing blood pressure and pulse rate were assessed.
Bioanalytical Methodology
Baricitinib plasma concentrations were quantified using a validated, liquid chromatography with tandem mass spectrometry bioanalytical method at WuXi App Tech laboratories in China. Baricitinib was extracted from K2EDTA human plasma. The analyte was extracted using liquid‐liquid extraction, and the reconstituted sample residue was analyzed by liquid chromatography with tandem mass spectrometry under gradient conditions (3%‐97%) using mobile phase A (10 mM ammonium formate in water) and mobile phase B (0.1% formic acid in acetonitrile). The high‐performance liquid chromatography column was a Waters Atlantis HILIC Silica column (3 μm, 2.1 × 50 mm) (Waters Corporation, Milford, Massachusetts). Baricitinib was detected using a Sciex API 4000 mass spectrometer (Applied Biosystems, Foster City, California) with positive ionization and multiple reaction monitoring (baricitinib m/z 372.3/251.3 and 13C‐baricitinib as the internal standard m/z 377.3/253.3). The extraction efficiencies of both baricitinib and the internal standard were between 77.0% and 96.3%. The assay was validated over a range of 0.200 to 200.00 ng/mL. The interassay precision and accuracy ranged from 0.5% to 2.0% and 5.1% to 10.8%, respectively.
Statistical Methods
Statistical Evaluation of Safety
All AEs, treatment‐emergent AEs (TEAEs), and serious AEs (SAEs) were listed or tabulated. A TEAE was defined as an AE that occurred after dosing or was present before dosing and worsened after dosing. TEAEs were summarized by treatment and by relationship to the study drug. The frequency (number of TEAEs, number of subjects experiencing at least 1 TEAE, and percentage of subjects experiencing at least 1 TEAE) of TEAEs was summarized by treatment and by day of onset. Moreover, the severity (mild, moderate, or severe) of AEs and any required concomitant medication for AEs were recorded. Clinical and laboratory parameters, vital signs, and ECG parameters were listed and summarized by treatment.
PK Statistical Methodology
Descriptive statistics were used to summarize PK parameters and PK parameters were calculated by standard noncompartmental methods of analysis (Phoenix WinNonlin Version 6.4). A dose proportionality assessment was conducted based on the PK parameters AUC0‐∞ and Cmax after a single dose, and AUCτ and Cmax during multiple dosing, using a power model:
where i represents the ith subject, PK represents a PK parameter (AUC or Cmax), and ε i is a residual error term.
Rdnm is the model‐predicted ratio of dose‐normalized means for the highest dose relative to the lowest dose, and the corresponding 90% confidence intervals (CIs) were reported:
where h and l correspond to the highest and lowest doses.
Results
Demographics and Subject Disposition
Of the 33 healthy male subjects enrolled, 30 completed the study in accordance with the protocol. Two subjects discontinued the study on day 1 at their own discretion after receiving a single dose of 4 mg baricitinib or placebo, and 1 subject who received placebo was withdrawn based on the physician's and sponsor's decision due to ECG abnormalities (Figure S1). A total of 25 subjects (baricitinib 2 mg: 8 subjects; baricitinib 4 mg: 9 subjects; and baricitinib 10 mg: 8 subjects) were included in PK analyses. For the subject from the 4‐mg group withdrawn from the study after dosing on day 1, blood samples for PK analyses were only collected up to 6 hours after dosing; thus, only Cmax and tmax (N = 9) were evaluated for this subject. Other PK parameters were evaluated in 8 subjects in the 4‐mg group. The demographic characteristics, mean age, and weight were comparable across the 3 groups and placebo regimens (Table 1).
Table 1.
Subject Demographics and Characteristics
| Placebo (N = 8) | 2‐mg Baricitinib (N = 8) | 4‐mg Baricitinib (N = 9) | 10 mg Baricitinib (N = 8) | Overall (N = 33) | |
|---|---|---|---|---|---|
| Age, y | 26.6 (5.4) | 30.8 (7.3) | 26.6 (3.5) | 27.6 (6.7) | 27.8 (5.8) |
| Male, n (%) | 8 (100.0) | 8 (100.0) | 9 (100.0) | 8 (100.0) | 33 (100.0) |
| Weight, kg | 65.23 (8.05) | 63.44 (6.23) | 63.78 (5.44) | 61.94 (1.93) | 63.60 (5.68) |
| BMI, kg/m2 | 22.34 (1.21) | 22.15 (1.75) | 21.87 (1.28) | 21.68 (1.39) | 22.01 (1.38) |
BMI, body mass index; N, number of subjects; SD, standard deviation.
Values shown are for mean (SD) unless otherwise noted.
Pharmacokinetic Evaluations
Single‐Dose PK of Baricitinib
Following the single‐dose administration of 2‐, 4‐, or 10‐mg baricitinib, peak plasma concentrations were reached at similar times across all dose ranges, with median tmax values of 0.75 to 1.0 hour after dosing on day 1 (Figure 2, Table 2). Plasma concentrations declined rapidly following attainment of peak concentrations, with a mean t1/2 of 5.9 to 7.4 hours across the 2‐ to 10‐mg dose range (Table 2), and individual values ranging from 4.5 to 11.8 hours. There were no significant dose‐related trends in CL/F following single doses of 2‐, 4‐, or 10‐mg baricitinib, with mean CL/F values ranging from 12.9 to 15.0 L/h across all dose levels (Table 2). Mean values for AUC0‐∞ and Cmax increased in an approximately dose‐proportional manner across the dose range. The results of statistical analysis showed a dose‐proportional increase in Cmax, with the 90%CI for the ratio of dose‐normalized geometric means including unity; however, the upper 90%CI was high in the 10‐mg group (1.58). For AUC0‐∞, the 90%CI narrowly excluded unity (Table 3).
Figure 2.
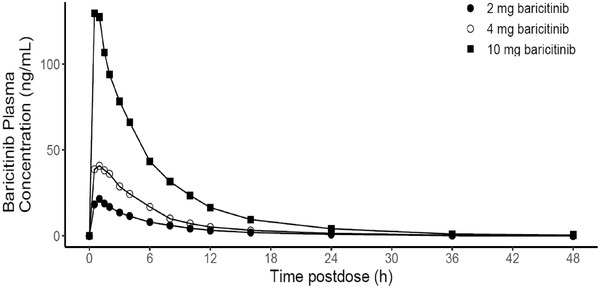
Arithmetic mean of plasma concentration versus time profiles of baricitinib following single oral dosing of 2 to 10 mg on day 1 in healthy male Chinese subjects (linear scale).
Table 2.
Summary of Plasma Pharmacokinetic Parameters of Baricitinib Following Single Oral Doses of 2 to 10 mg on Day 1 in Healthy Chinese Subjects
| Arithmetic Mean (SD) | |||
|---|---|---|---|
| 2‐mg Baricitinib | 4‐mg Baricitinib | 10‐mg Baricitinib | |
| PK Parameters | (N = 8) | (N = 9) | (N = 8) |
| AUC0‐24, ng · h/mL | 133 (13.7) | 256 (40.9) a | 740 (75.6) |
| AUC0‐t last, ng · h/mL | 134 (14.6) | 269 (46.2) a | 775 (72.9) |
| AUC0‐∞, ng · h/mL | 140 (15.3) | 274 (45.2) a | 780 (71.3) |
| Cmax, ng/mL | 24.6 (4.11) | 51.3 (20.4) | 152.0 (42.5) |
| tmax, b h | 1.00 (0.50‐1.50) | 1.00 (0.50‐2.03) | 0.75 (0.50‐1.50) |
| t1/2, h | 5.85 (0.37) | 7.36 (2.25) a | 7.23 (1.62) |
| CL/F, L/h | 14.5 (1.76) | 15.0 (2.48) a | 12.9 (1.24) |
AUC0‐24, area under the plasma concentration–time curve from time zero to 24 hours after dosing; AUC0‐∞, area under the plasma concentration–time curve from time zero to infinity; AUC0‐t last, area under the plasma concentration–time curve from time zero to time t, where t is the last time point with a measurable concentration; CL/F, apparent clearance; Cmax, maximum observed drug concentration; t½, apparent terminal half‐life; tmax, time to maximum concentration.
Values shown are for mean (SD) unless otherwise noted.
N = 8
Median (range).
Table 3.
Dose Proportionality Analysis of AUC and Cmax for Baricitinib Following Single Oral Doses of 2 to 10 mg on Day 1 and Multiple Oral Doses of 2 to 10 mg on Day 10 in Healthy Chinese Subjects
| Single Oral Doses of 2 to 10 mg on Day 1 | |||||
|---|---|---|---|---|---|
| Parameter | Power Model Equation | 90%CI of Exponent | Baricitinib Dose (mg) | Predicted Dose‐Normalized Geometric Means | Ratio of Dose‐Normalized Geometric Means (10 or 4 mg vs 2 mg) (90%CI) |
| AUC0‐∞, ng · h/mL | (e4.16):(Dose1.08) | 1.01‐1.14 | 2 | 67.43 | |
| 4 | 71.03 | 1.05 (1.00‐1.11) | |||
| 10 | 76.09 | 1.13 (1.01‐1.26) | |||
| Cmax, ng/mL | (e2.37):(Dose1.12) | 0.96‐1.28 | 2 | 11.67 | |
| 4 | 12.71 | 1.09 (0.97‐1.22) | |||
| 10 | 14.23 | 1.22 (0.94‐1.58) | |||
| Multiple oral doses of 2 to 10 mg on Day 10 | |||||
| AUCτ,ss, ng . h/mL | (e4.21):(Dose1.04) | 0.96‐1.13) | 2 | 69.44 | |
| 4 | 71.60 | 1.03 (0.97‐1.09) | |||
| 10 | 74.56 | 1.07 (0.94‐1.23) | |||
| Cmax,ss, ng/mL | (e2.59):(Dose0.99) | 0.87‐1.22 | 2 | 13.25 | |
| 4 | 13.17 | 0.99 (0.91‐1.08) | |||
| 10 | 13.07 | 0.99 (0.81‐1.20) | |||
AUC, area under the plasma concentration–time curve; AUC0‐∞, area under the plasma concentration–time curve from time zero to infinity; AUCτ,ss, area under the plasma concentration–time curve during one dosing interval at steady state; CI, confidence interval; Cmax, maximum observed drug concentration; Cmax,ss, maximum observed drug concentration at steady state.
Model: Log (Parameter) = Log (Dose) + Random Error.
Multiple‐Dose PK of Baricitinib
Similar to the single‐dose administration, the multiple dose of 2‐ to 10‐mg baricitinib was rapidly absorbed. The Cmax,ss of baricitinib occurred at similar times across all dose ranges, with median tmax values of 0.5 to 1.0 hour after dosing on day 10 (Figure 3). Similarly, the mean t1/2 of baricitinib was comparable across all dose ranges, with mean values ranging from of 5.7 to 7.6 hours after dosing on day 10 (Table 4). Steady‐state plasma concentrations of baricitinib were achieved after the second once‐daily dose of baricitinib (day 6 morning). Minimal accumulation of baricitinib was observed upon multiple dosing, with mean accumulation ratio values of 1.05 to 1.10 in plasma (up to 10%) during the 7 days of QD doses across dose ranges (2 to 10 mg) (Table 4). Following multiple doses of 2‐ to 10‐mg baricitinib, mean CL/F was comparable to estimations following single doses, with values ranging from 13.1 to 15.3 L/h across dose levels on day 10 (Table 4). Mean values for AUCτ,ss and Cmax,ss appeared to increase in a dose‐proportional manner over the multiple‐dose range of 2 to 10 mg (Table 4). The results of statistical analysis showed a dose‐proportional increase in AUCτ,ss and Cmax,ss, with the 90%CI for the ratio of the dose‐normalized geometric means of 10 mg vs 2 mg and 10 mg vs 4 mg including unity (Table 3).
Figure 3.
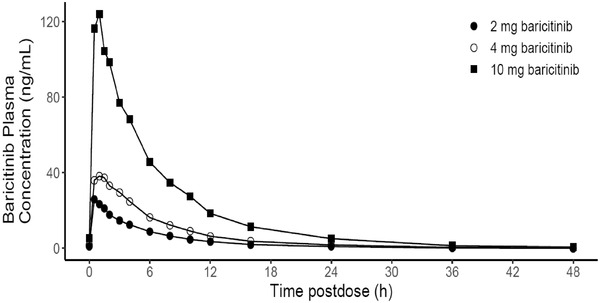
Arithmetic mean of plasma concentration vs time profiles of baricitinib on day 10 following multiple oral dosing of 2 to 10 mg in healthy male Chinese subjects (linear scale).
Table 4.
Summary of the Plasma Pharmacokinetic Parameters of Baricitinib on Day 10 Following Multiple Oral Doses of 2 to 10 mg in Healthy Chinese Subjects
| Arithmetic Mean (SD) | |||
|---|---|---|---|
| 2‐mg Baricitinib | 4‐mg Baricitinib | 10‐mg Baricitinib | |
| PK Parameters | (N = 8) | (N = 8) | (N = 8) |
| AUCτ,ss, ng · h/mL | 146 (17.0) | 268 (48.2) | 778 (101.0) |
| Cmax,ss, ng/mL | 28.6 (6.95) | 49.6 (14.4) | 138.0 (22.8) |
| Tmax,ss, a h | 0.50 (0.50‐1.50) | 1.00 (0.50‐1.50) | 1.00 (0.50‐1.50) |
| t½, h | 5.68 (0.63) | 7.55 (1.88) | 7.39 (2.48) |
| CL/F, L/h | 13.9 (1.66) | 15.3 (2.64) | 13.1 (1.95) |
| RA | 1.11 (0.11) | 1.06 (0.20) | 1.05 (0.07) |
AUCτ,ss, area under the plasma concentration–time curve during 1 dosing interval at steady state; CI, confidence interval; CL/F, apparent clearance; Cmax,ss, maximum observed drug concentration at steady state; CV, coefficient of variation; PK, pharmacokinetics; RA, accumulation ratio; t½, apparent terminal half‐life; tmax,ss, time to maximum concentration at steady state.
Data displayed are arithmetic mean (SD), unless otherwise stated.
Median (range).
Safety and Tolerability
All 33 subjects who entered the study were analyzed for AEs. Eleven subjects (33.3%) reported 21 TEAEs during the study (Table 5). All TEAEs reported were mild in severity, with no SAEs reported, and no subjects administered baricitinib withdrew from the study due to AEs. There was an apparent increase in the incidence of drug‐related TEAEs in the 2‐ to 10‐mg dose range, but the incidence of drug‐related TEAEs that occurred at the highest dose of 10‐mg baricitinib was comparable to that of placebo.
Table 5.
Summary of TEAEs
| Baricitinib | |||||
|---|---|---|---|---|---|
| Placebo (N = 8) | 2 mg (N = 8) | 4 mg (N = 9) | 10 mg (N = 8) | Overall (N = 33) | |
| All causalities | |||||
| Number of subjects (%) with ≥1 AE | 4 (50.0) | 1 (12.5) | 2 (22.2) | 4 (50.0) | 11 (33.3) |
| Total number of TEAEs (all severities were “mild”), n | 10 | 2 | 3 | 6 | 21 |
| Drug related (number of TEAEs [number of subjects with TEAEs]) | |||||
| ECG abnormal | 6 (2) | 0 | 0 | 0 | 6 (2) |
| ALT increased | 1 (1) | 0 | 1 (1) | 2 (2) | 4 (4) |
| AST increased | 1 (1) | 0 | 1 (1) | 1 (1) | 3 (3) |
| Bilirubin increased | 0 | 0 | 0 | 1 (1) | 1 (1) |
| ANC decreased | 0 | 0 | 1 (1) | 0 | 1 (1) |
| Hypotension | 1 (1) | 0 | 0 | 0 | 1 (1) |
| Orthostatic hypotension, n | 0 | 0 | 0 | 1 (1) | 1 (1) |
| Overall | 9 (4) | 0 (0) | 3 (2) | 5 (4) | 17 (10) |
AEs, adverse events; ALT, alanine aminotransferase; ANC, absolute neutrophil count; AST, aspartate aminotransferase; ECG, electrocardiogram; TEAEs, treatment‐emergent adverse events.
In the baricitinib groups, the most frequently reported TEAEs included an increase in alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels in subjects who received 4 and 10 mg of baricitinib (Table 5). The TEAEs of increase in ALT and AST in the baricitinib group were of mild severity and resolved: ALT increased from 10 to 39 U/L (predose) to 56 to 105 U/L (day 10), and AST increased from 14 to 21 U/L (predose) to 56 to 58 U/L (day 10). One subject who received placebo also reported TEAEs of increased ALT and AST (Table 5). Following single and multiple doses of baricitinib, there was a transient decrease in ANC, while the absolute leukocyte counts (ALCs) increased briefly. The magnitude of treatment‐related decrease from baseline in mean ANC on day 1 in the single‐dose group was dose related, with maximum mean decreases at 8 hours after dosing of 0.616, 0.808, and 1.706 GI/L at the 2‐, 4‐, and 10‐mg dose levels, respectively. Similarly, in the multiple‐dose groups, on day 10, similar dose‐related decreases in ANC were observed, with maximum mean decreases at 8 hours after dosing of 0.096, 0.706, and 1.443 GI/L at the 2‐, 4‐, and 10‐mg dose levels, respectively. The maximum decrease in ANC was observed at 8 hours, while the increase in ALC was noted at 16 hours after dosing. ANC and ALC values returned to baseline by 24 hours postdose on days 1 and 10. The changes in ANC and ALC were comparable between single (day 1) and multiple (day 10) doses. Only 1 drug‐related TEAE of decreased ANC was reported in the baricitinib 4‐mg dose group.
In summary, the regimens tested were generally well tolerated by the 33 subjects in this study. No clinically significant abnormal vital signs or other laboratory measurements were observed during the course of the study (Table 5); moreover, there were no clinically significant dose‐dependent ECG changes in the 12‐lead ECGs following administration of baricitinib.
Discussion
To our knowledge, this was the first study conducted in healthy Chinese subjects to characterize the PK, safety, and tolerability of baricitinib (2‐10 mg) following single and multiple doses. This phase 1 study revealed baricitinib is rapidly absorbed after oral administration and is rapidly eliminated, with minimal accumulation after multiple dosing, and demonstrated dose‐proportional PK and a well‐tolerated safety profile in healthy Chinese subjects. The baricitinib PK profile following multiple dosing was similar to that of single dosing in healthy Chinese volunteers. The increase in AUC and Cmax over the single and multiple doses of baricitinib over a 2‐ to 10‐mg dose range was dose proportional. This indicates that baricitinib exhibited dose‐linear PK characteristics.
The PK of baricitinib in Chinese subjects in this study is comparable to that of healthy White subjects in previous studies conducted outside China. 11 The mean terminal‐phase t1/2 following single and multiple doses of baricitinib is comparable to that reported previously in White subjects, which is approximately 8 hours in healthy subjects using a noncompartmental PK method. 11 Similarly, the AUC and Cmax are comparable between healthy Chinese and White subjects, 11 suggesting low ethnic sensitivity. Moreover, the PK characteristics of baricitinib in both Chinese and White subjects were linear and dose proportional. The lack of differences in the PK profile of baricitinib between Chinese and White subjects is consistent with the elimination mechanism of baricitinib.
The safety analysis showed single and multiple doses of 2‐ to 10‐mg baricitinib were generally well tolerated among healthy Chinese volunteers. The reported incidence of TEAEs was about 33% during the study, and all TEAEs were mild in intensity, with no SAEs or deaths reported. No subjects administered baricitinib were withdrawn due to AEs. Common AEs reported included an increase in ALT and AST. Although there was an apparent increase in the incidence of drug‐related TEAEs over the 2‐ to 10‐mg dose range, the incidence of TEAEs with the 10‐mg dose was comparable with that of placebo. In this study, 10 mg was the highest dose of baricitinib administered, which was well tolerated in healthy Chinese subjects, a result similar to that reported in the first‐in‐human global study. 11 The transient and dose‐dependent decrease in ANC and increase in ALC was observed after single and multiple doses of baricitinib; however, ANC returned to near baseline after 24 hours of dosing on days 1 and 10. In addition, at each dose level, the maximum changes in ANC and ALC were comparable between single (day 1) and multiple (day 10) doses. No infection‐related AEs were reported in this study. The transient changes in ANC and ALC observed in this study were mild in severity and consistent with previous findings demonstrated in White populations. 11 The changes in ANC after dosing were unlikely to represent bone marrow suppression as ANC returned to baseline by 24 hours after dosing, reflecting neutrophil margination. Baricitinib (up to 10 mg) appeared to be safe and well tolerated with an overall safety profile supporting its further clinical development in Chinese patients with RA.
Our study has several limitations. First, the study duration was short; hence, there are chances of the underreporting of AEs compared with previous studies. Second, after screening both male and female subjects, only male subjects were found eligible and subsequently enrolled in the present study for evaluation; therefore, the present results are based on male subjects only. Third, the sampling frequency of 0.5 hour after dosing during the absorption phase can be further increased due to the rapid absorption of baricitinib observed in previous studies. Although the PK profile of baricitinib in healthy Chinese subjects observed in this study was comparable to that observed in healthy White subjects, PK similarity between healthy White and Chinese subjects may not translate into similar efficacy in patients.
Conclusions
The JAK1/2 inhibitor baricitinib was well tolerated and safe in healthy Chinese subjects. Moreover, the PK parameters were comparable with a previous study of White subjects, suggesting a low ethnic sensitivity of baricitinib. Therefore, ethnic‐specific dose adjustment of baricitinib due to exposure difference is not considered necessary for the Chinese population.
Conflicts of Interest
F.W., C.D.P., and X.Z. are employees of Eli Lilly and Company; X.Z., X.Y.S., and Y.M.C. declare no conflicts of interest.
Funding
This study was funded and supported by Eli Lilly and Company, China.
Author Contributions
All authors met the International Committee of Medical Journal Editors criteria for authorship. X.Y.S. performed the experiments. C.D.P. contributed to the study design and concept. Z.X. contributed to data acquisition, and all authors contributed to analysis and interpretation. All authors were involved in the preparation and review of the article and approved the final version to be submitted.
Supporting information
Supplementary information
Acknowledgments
The authors thank Dr Rakesh Ojha, PhD, from Syneos Health, Pune, India for medical writing support, and Dana Schamberger, MA, from Syneos Health, USA, for editorial support in the preparation of this manuscript. The authors also thank Li Li Bian and Chu Yue Zeng for assistance in the publication project management, and Fei Ji for statistical review of the article (all of Eli Lilly and Company, China); and Ellen Cannady (Eli Lilly and Company, China) for providing consultation and expert review of the absorption, distribution, metabolism, and excretion part of the article.
Trial registration number: NCT02758613
[The copyright line for this article was changed on 7 December 2021 after original online publication.]
References
- 1. Nakayamada S, Kubo S, Iwata S, Tanaka Y. Recent progress in JAK inhibitors for the treatment of rheumatoid arthritis. BioDrugs. 2016;30(5):407‐419. [DOI] [PubMed] [Google Scholar]
- 2. Chen L, Deng H, Cui H, et al. Inflammatory responses and inflammation‐associated diseases in organs. Oncotarget. 2018;9(6):7204‐7218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ghoreschi K, Laurence A, O'Shea JJ. Janus kinases in immune cell signaling. Immunol Rev. 2009;228(1):273‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Choy EHS, Miceli‐Richard C, González‐Gay MA, et al. The effect of JAK1/JAK2 inhibition in rheumatoid arthritis: efficacy and safety of baricitinib. Clin Exp Rheumatol. 2019;37(4):694‐704. [PubMed] [Google Scholar]
- 5. Norman P. Selective JAK inhibitors in development for rheumatoid arthritis. Expert Opin Investig Drugs. 2014;23(8):1067‐1077. [DOI] [PubMed] [Google Scholar]
- 6. Choy EH. Clinical significance of Janus kinase inhibitor selectivity. Rheumatology (Oxford). 2019;58(6):953‐962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Taylor PC. Clinical efficacy of launched JAK inhibitors in rheumatoid arthritis. Rheumatology (Oxford). 2019;58(suppl 1):i17‐i26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fridman JS, Sherle PA, Collins R, et al. Selective inhibition of JAK1 and JAK2 is efficacious in rodent models of arthritis: preclinical characterization of INCB028050. J Immunol. 2010;184(9);5298‐5307. [DOI] [PubMed] [Google Scholar]
- 9. Al‐Salama ZT, Scott LJ. Baricitinib: a review in rheumatoid arthritis. Drugs. 2018;78(7):761‐772. [DOI] [PubMed] [Google Scholar]
- 10. Mayence A, Vanden Eynde JJ. Baricitinib: a 2018 novel FDA‐approved small molecule inhibiting Janus kinases. Pharmaceuticals (Basel). 2019;12(1):37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Keystone EC, Taylor PC, Drescher E, et al. Safety and efficacy of baricitinib at 24 weeks in patients with rheumatoid arthritis who have had an inadequate response to methotrexate. Ann Rheum Dis. 2015;74(2):333‐340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dougados M, van der Heijde D, Chen YC, et al. Baricitinib in patients with inadequate response or intolerance to conventional synthetic DMARDs: results from the RA‐BUILD study. Ann Rheum Dis. 2017;76(1):88‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shi JG, Chen X, Lee F, et al. The pharmacokinetics, pharmacodynamics, and safety of baricitinib, an oral JAK 1/2 inhibitor, in healthy volunteers. J Clin Pharmacol. 2014;54(12):1354‐1361. [DOI] [PubMed] [Google Scholar]
- 14. Schlueter M, Rouse P, Pitcher A, et al. A modeling framework for the economic evaluation of baricitinib in moderate‐to‐severe rheumatoid arthritis. Expert Rev Pharmacoecon Outcomes Res. 2020;20(2):221‐228. [DOI] [PubMed] [Google Scholar]
- 15. Kim H, Brooks KM, Tang CC, et al. Pharmacokinetics, pharmacodynamics, and proposed dosing of the oral JAK1 and JAK2 inhibitor baricitinib in pediatric and young adult CANDLE and SAVI patients. Clin Pharmacol Ther. 2018;104(2):364‐373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kawalec P, Śladowska K, Malinowska‐Lipień I, Brzostek T, Kózka M. New alternative in the treatment of rheumatoid arthritis: clinical utility of baricitinib. Ther Clin Risk Manag. 2019;15:275‐284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Posada MM, Cannady EA, Payne CD, et al. Prediction of transporter‐mediated drug‐drug interactions for baricitinib. Clin Transl Sci. 2017;10(6):509‐519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang X, Chua L, Ernest 2nd C, Macias W, Rooney T, Tham LS. Dose/exposure‐response modeling to support dosing recommendation for phase III development of baricitinib in patients with rheumatoid arthritis. CPT Pharmacometrics Syst Pharmacol. 2017;6(12):804‐813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bjornsson TD, Wagner JA, Donahue SR, et al. A review and assessment of potential sources of ethnic differences in drug responsiveness. J Clin Pharmacol. 2003;43(9):943‐967. [DOI] [PubMed] [Google Scholar]
- 20. Eliasson E. Ethnicity and adverse drug reactions. BMJ. 2006;332(7551):1163‐1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Baehr A, Peña JC, Hu DJ. Racial and ethnic disparities in adverse drug events: a systematic review of the literature. J Racial Ethn Health Disparities. 2015;2(4):527‐536. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information