

Summary
Fedratinib, an oral Janus kinase‐2 (JAK2) inhibitor, reduces splenomegaly and improves symptom burden in patients with myelofibrosis. Regulatory approval of fedratinib 400‐mg daily was based on results of an updated analysis of the pivotal phase III, placebo‐controlled JAKARTA trial in patients with JAK‐inhibitor‐naïve myelofibrosis. At week 24, spleen volume response rate was 47% and symptom response rate was 40% with fedratinib 400 mg, versus 1% and 9% respectively, with placebo. Common adverse events were diarrhoea, nausea, anaemia, and vomiting. No Wernicke encephalopathy occurred in patients receiving fedratinib 400 mg/day. These updated data support use of first‐line fedratinib in patients with myelofibrosis.
Keywords: myelofibrosis, fedratinib, JAK inhibitor
Introduction
Myelofibrosis (MF) is characterised by anaemia, splenomegaly, debilitating constitutional symptoms, and shortened survival, often presenting with Janus kinase 2 (JAK2) mutations 1 , 2 that result in constitutively activated JAK2 proteins and activation of the JAK–signal transducer and activators of transcription (STAT) pathway, 2 thereby increasing levels of circulating pro‐inflammatory cytokines. 3 , 4 Fedratinib is an oral JAK2 inhibitor approved in the USA, European Union, UK, Canada, and elsewhere for use in adult patients with MF, as first‐line therapy or following prior ruxolitinib treatment.
The pivotal phase III, randomised, placebo‐controlled JAKARTA (NCT01437787) trial evaluated the efficacy and safety of fedratinib in patients with JAK‐inhibitor‐naïve MF. In 2013, fedratinib development was placed on clinical hold because of suspected cases of Wernicke encephalopathy, prompting closure of ongoing clinical trials including JAKARTA (initiated in 2011). The clinical hold was lifted in August 2017 after additional safety data were provided to the USA regulatory authorities. 5
Initial findings from JAKARTA were previously reported, showing a spleen volume response rate (SVRR) of 36% [with 4‐week magnetic resonance imaging/computed tomography (MRI/CT) confirmation scan] and a symptom response rate of 36% at the end of the six‐cycle randomised treatment period (RTP) [i.e. at 24 weeks/end of cycle six (EOC6)] for patients who received fedratinib 400 mg/day (recommended dose). 2 , 6 To prepare for USA regulatory approval, JAKARTA data were re‐analysed. Described are findings from that re‐analysis, focussing on outcomes for patients treated with the recommended fedratinib 400 mg/day dose or placebo during the six‐cycle RTP.
Methods
Detailed eligibility criteria and study design are reported elsewhere. 2 Briefly, adult patients with intermediate‐2 or high‐risk primary or secondary MF, Eastern Cooperative Oncology Group performance status scores of ≤2, and platelet counts ≥50 × 109/l were randomised 1:1:1 to fedratinib 400 mg, 500 mg, or placebo administered once daily in continuous 28‐day cycles during the six‐cycle RTP. Patients who showed clinical benefit or stable disease per modified International Working Group for Myelofibrosis Research and Treatment (IWG‐MRT) criteria 7 at EOC6 could continue treatment until progressive disease (PD) or unacceptable toxicity. Patients randomised to placebo who completed the RTP (or developed PD before EOC6) could crossover to fedratinib.
The primary end‐point was SVRR, the proportion of patients achieving ≥35% spleen volume reduction from baseline on MRI/CT at EOC6, confirmed on a follow‐up scan 4 weeks later. Duration of response (DOR; the time from documented response until PD or death) was estimated using Kaplan–Meier methods; patients ongoing at study termination were censored at last follow‐up visit. Because most patients were receiving treatment at study termination, DOR analysis was subject to extensive censoring.
The symptom response rate was the proportion of patients with ≥50% reduction from baseline in total symptom score (TSS) on the modified Myelofibrosis Symptom Assessment Form (MFSAF; version 2.0) at EOC6. The modified MFSAF was completed daily during the week before day 1 of each cycle and the week before EOC6. Patients rated the severity of six key MF symptoms (night sweats, abdominal discomfort, early satiety, pruritis, bone/muscle pain, pain below ribs on left side) on a scale from 0 (‘absent’) to 10 (‘worst imaginable’); the TSS is the sum of individual symptom scores (range 0–60). Evaluable patients had a non‐missing baseline TSS (completed 5/7 days before cycle 1 day 1) that was >0.
Safety was evaluated by adverse event (AE) reporting and laboratory assessments during the RTP. In the present updated analysis, laboratory values that worsened during treatment were considered AEs; the original analysis reported all abnormal laboratory values on‐study as AEs, even if present at baseline. 2 Safety was assessed in patients who received one or more study drug dose.
Results
Overall, 96 patients were randomised to fedratinib 400 mg/day and 96 to placebo. One placebo‐randomised patient died before receiving study drug and was excluded from safety analyses. The most common reason for discontinuation of fedratinib was study termination (63·5%) and of placebo was crossover to fedratinib (74%). At baseline, the median (range) age was 63 (39–86) years in the fedratinib 400‐mg arm and 66 (27–85) years in the placebo arm, and median (range) time since MF diagnosis was longer in the fedratinib arm, at 43 (0·8–311) months versus 28 (0·2–412) months. Similar proportions of patients in the fedratinib and placebo groups had primary MF (65% and 60% respectively), and median (range) spleen volume at baseline was comparable at 2652 (316–6430) ml and 2660 (662–7911) ml. The median treatment exposure to fedratinib 400 mg overall was 15·5 months in the fedratinib 400‐mg arm and to placebo was 6 months. In these analyses, the median drug exposure was 24 weeks in both treatment arms during the six‐cycle RTP.
The SVRR at EOC6 (with 4‐week confirmation) was significantly higher with fedratinib 400 mg/day compared with placebo: 37% [95% confidence interval (CI) 27–46] versus 1% (95% CI 0–3) (P < 0·0001). Without 4‐week confirmation scan (consistent with other large clinical trials of JAK inhibitors for MF) 7 , 8 , 9 , 10 , 11 , SVRR at EOC6 was 47% (95% CI 37–57) with fedratinib 400 mg and 1% (95% CI 0–3) with placebo (P < 0·0001). Of 75 patients in the fedratinib 400‐mg arm who completed the RTP and had spleen volume data available at both baseline and EOC6, all but three (96%) experienced a spleen volume reduction at EOC6. Conversely, spleen volume was increased at EOC6 for most patients in the placebo arm [44/58 (76%)] (Fig 1). For patients who achieved a spleen volume response, estimated median DOR in the fedratinib 400‐mg arm was 18·2 months, but as mentioned this analysis was subject to extensive censoring due to study termination; only six of 54 responding patients (11%) progressed or died during the study.
Fig 1.
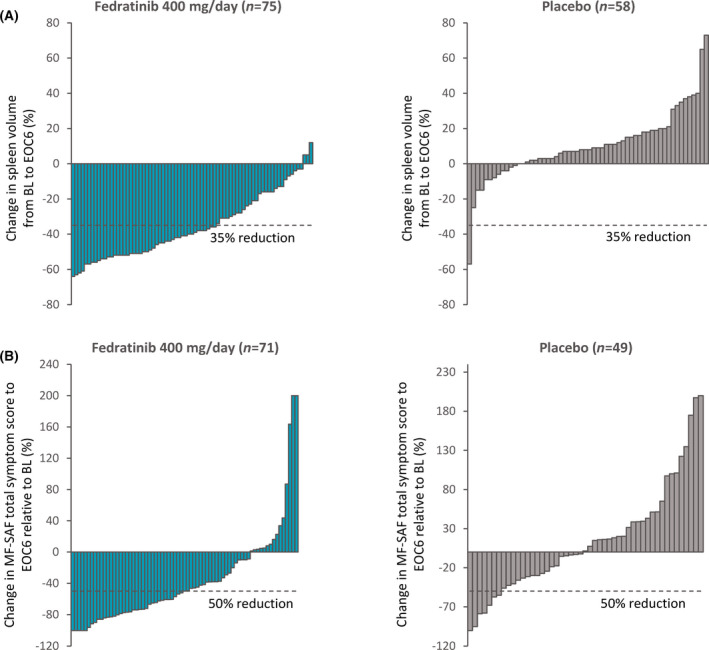
(A) Individual changes in spleen volume from baseline to end of cycle six (EOC6) for patients with data available at both time‐points; (B) Individual changes in total symptoms scores from baseline at EOC6 for patients with data available at both time‐points. [Colour figure can be viewed at wileyonlinelibrary.com]
Symptom scores for MFSAF‐evaluable patients suggested mild‐to‐moderate MF symptoms at baseline: mean (SD)] TSS was 18·0 (13.4) in the fedratinib 400‐mg arm (n = 89) and 15·4 (11.8) in the placebo arm (n = 81). On‐study, fedratinib was associated with a significant reduction in MF symptom burden from baseline and relative to placebo. At EOC6, the symptom response rate was 40% (95% CI 30–51) in the fedratinib 400‐mg/day group vs 9% (95% CI 3–15) with placebo (P < 0·0001).
The most common AEs with fedratinib during the RTP were diarrhoea, nausea, anaemia, and vomiting (Table I). Gastrointestinal events were most frequent during early treatment and decreased in incidence as treatment continued. Anaemia was the most common Grade ≥3 event (30%) and no other Grade ≥3 AEs occurred in >5% of patients. The most common Grade ≥3 laboratory abnormalities were anaemia (34%) and thrombocytopenia (12%). No treatment‐related Grade ≥3 bleeding events occurred. Common any grade biochemistry abnormalities were elevations in creatinine (59%), alanine aminotransferase (ALT, 43%), and aspartate aminotransferase (AST, 40%); Grade ≥3 elevations were uncommon (Table I). Grade ≥3 lipase increases occurred in 10% of fedratinib‐treated patients. During the RTP, 21% of patients had fedratinib treatment interrupted because of an AE, most commonly diarrhoea (5%) and nausea (4%); and 19% of patients had a fedratinib dose reduction because of an AE, most often anaemia (6%), diarrhoea (3%), vomiting (3%), and thrombocytopenia (2%). The frequency of Grade 3–4 AEs leading to treatment interruption or dose‐reduction during the RTP was 15% in the fedratinib arm and 13% in the placebo arm. AEs led to discontinuation of fedratinib 400 mg for 14% of patients (13/96); those occurring in more than one patient were cardiac failure (three patients), and (two each) thrombocytopenia, myocardial ischaemia, diarrhoea, and increased blood creatinine. In the placebo arm, 8% of patients (eight of 95) discontinued because of an AE, including cardiac failure in two patients. No patient receiving fedratinib 400 mg/day experienced Wernicke encephalopathy.
Table I.
Adverse events reported in ≥5% of patients treated with fedratinib 400 mg, with a difference between arms of >5% during randomised treatment; and selected laboratory abnormalities that worsened from baseline in ≥20% of fedratinib‐treated patients and with a difference between arms of >10%. 6
| Fedratinib 400 mg (n = 96) | Placebo (n = 95*) | |||
| All grades | Grade ≥3† | All grades | Grade ≥3 | |
| % | ||||
| Adverse events‡ | ||||
| Diarrhoea | 66 | 5 | 16 | 0 |
| Nausea | 62 | 0 | 15 | 0 |
| Anaemia | 40 | 30 | 14 | 7 |
| Vomiting | 39 | 3·1 | 5 | 0 |
| Fatigue or asthenia | 19 | 5 | 16 | 1·1 |
| Muscle spasms | 12 | 0 | 1·1 | 0 |
| Blood creatinine increased | 10 | 1 | 1·1 | 0 |
| Pain in extremity | 10 | 0 | 4·2 | 0 |
| ALT increased | 9 | 0 | 1·1 | 0 |
| Headache | 9 | 0 | 1·1 | 0 |
| Weight increased | 9 | 0 | 4·2 | 0 |
| Dizziness | 8 | 0 | 3·2 | 0 |
| Bone pain | 8 | 0 | 2·1 | 0 |
| Urinary tract infection§ | 6 | 0 | 1·1 | 0 |
| Dysuria | 6 | 0 | 0 | 0 |
| AST increased | 5 | 0 | 1·1 | 0 |
| Laboratory parameters | ||||
| Haematology | ||||
| Anaemia | 74 | 34 | 32 | 10 |
| Thrombocytopenia | 47 | 12 | 26 | 10 |
| Neutropenia | 23 | 5 | 13 | 3·3 |
| Biochemistry | ||||
| Creatinine increased | 59 | 3·1 | 19 | 1·1 |
| ALT increased | 43 | 1 | 14 | 0 |
| AST increased | 40 | 0 | 16 | 1·1 |
| Lipase increased | 35 | 10 | 7 | 2·2 |
| Hyponatremia | 26 | 5 | 11 | 4·3 |
| Amylase increased‖ | 24 | 2·1 | 5 | 0 |
ALT, alanine aminotransferase; AST, aspartate aminotransferase.
1 patient randomised to placebo died before receiving study drug and is excluded from safety reporting.
Only one Grade 4 adverse event (anaemia) was reported in the fedratinib 400‐mg arm.
Common Terminology Criteria for Adverse Events (CTCAE) version 4.03.
Includes cystitis.
Without clinical pancreatitis.
Discussion
Patients with intermediate‐2 or high‐risk MF treated with fedratinib 400 mg/day had significant improvements in spleen volume and symptom responses (both P < 0·0001) compared with placebo. In this re‐analysis, SVRR with fedratinib 400 mg at week 24 with confirmation 4 weeks later (37%), and without confirmation (47%), were similar to rates initially reported (36% and 47% respectively) 2 but symptom response rate was higher in this re‐analysis (40% vs. 36%).
In the initial JAKARTA report, laboratory abnormalities present at baseline that did not worsen during treatment were nonetheless considered AEs in safety assessments. 2 This re‐analysis employed the more common, well‐accepted approach of capturing only ‘new or worsening’ laboratory AEs. Safety outcomes reported here are more likely to reflect the actual frequency of treatment‐emergent laboratory abnormalities that might be expected during fedratinib 400‐mg treatment. Grade ≥3 laboratory abnormalities, including increases in liver enzymes and creatinine, were infrequent, and changes in pancreatic enzymes did not indicate pancreatitis. Renal function, liver enzymes, amylase, and lipase levels may require monitoring during fedratinib treatment, and patients taking strong cytochrome P450 family 3 subfamily A (CYP3A) inhibitors or with severe renal impairment require dose reductions. 6
Although diarrhoea, nausea, and anaemia were common during fedratinib treatment, they were managed mainly by dosing modifications, and infrequently required treatment discontinuation. These events may have been less frequent if prophylaxis had been required. The ongoing phase III FREEDOM (NCT03755518) and FREEDOM2 (NCT03952039) trials, which assess fedratinib clinical outcomes in patients with MF previously treated with ruxolitinib, include mitigation strategies to manage gastrointestinal events during fedratinib therapy. Preliminary results from the FREEDOM trial indicate rates of diarrhoea, nausea, and vomiting are markedly lower with these mitigation strategies. 12
These updated results of the JAKARTA study further support the clinical efficacy and safety of fedratinib 400‐mg daily in JAK‐inhibitor‐naïve patients with intermediate‐2 or high‐risk MF and may serve to inform expectations of therapeutic response and the potential toxicity of fedratinib in the clinic.
Author contributions
The sponsors collected and analysed data in conjunction with all authors. Animesh Pardanani wrote an early draft of the manuscript. All authors revised the manuscript and reviewed and approved the final version for submission.
Financial disclosures
Animesh Pardanani declares no conflicts. Ayalew Tefferi declares no conflicts. Tamás Masszi reports Advisory Committee participation for Abbvie, BMS, Janssen‐Cilag, Novartis, Pfizer, and Takeda. Elena Mishchenko declares no conflicts. Mark Drummond : Speakers’ Bureau: Novartis, BMS, Astellas, Jazz; Advisory Committee participation: Novartis, BMS, Pfizer; Research Funding: Novartis, BMS/Celgene. Eric Jourdan reports consulting fees and Advisory Committee participation for Novartis, BMS/Celgene, Astallas, and Abbvie. Alessandro Vannucchi holds positions on advisory committees for Novartis, Celgene (a Bristol‐Myers Squibb Company), Abbvie, Blueprint, and Incyte; and participates in speakers’ bureau for Novartis, Celgene (a Bristol‐Myers Squibb Company), and Abbvie. Mindaugas Jurgutis declares no conflicts. Vincent Ribrag reports ad board participation for Gilead, Infinity, MSD, BMS, Nanostring, Incyte, Roche, AstraZeneca; Research Funding from Astex, Argen‐X, and GSK. Alessandro Rambaldi reports personal fees from Celgene BMS, Sanofi, Novartis, Italfarmaco, Amgen, Pfizer, kite Gilead, Astellas, Jazz. Liang Piu Koh declares no conflicts. Shelonitda Rose and Jun Zhang report employment and equity ownership in BMS. Claire Harrison reports consultancy for Celgene, Novartis, CTI, Roche, Promedior, Galacteo, Sierra Oncology, Constellation, Janssen, and Geron; Honoraria from Celgene, Novartis, Roche, AOP Pharma; and Speakers’ Bureau participation for Celgene, Novartis, CTI, and Janssen.
Acknowledgments
We thank the patients, families, co‐investigators, and all study personnel who participated in this trial. The original study was supported by Sanofi S.A.; this re‐analysis was funded by Celgene, a Bristol‐Myers Squibb company. The authors received editorial assistance on an early draft from Brian Kaiser and Sheila Truten (Medical Communication Company, Inc.), sponsored by Bristol‐Myers Squibb. The authors are fully responsible for all content and editorial decisions, and all approved submission of the final manuscript.
References
- 1. Vannucchi AM, Lasho TL, Guglielmelli P, Biamonte F, Pardanani A, Pereira A, et al. Mutations and prognosis in primary myelofibrosis. Leukemia. 2013;27:1861–9. [DOI] [PubMed] [Google Scholar]
- 2. Pardanani A, Harrison C, Cortes JE, Cervantes F, Mesa RA, Milligan D, et al. Safety and efficacy of fedratinib in patients with primary or secondary myelofibrosis: a randomized clinical trial. JAMA Oncol. 2015;1:643–51. [DOI] [PubMed] [Google Scholar]
- 3. Bousoik E, Montazeri AH. "Do we know Jack" about JAK? A closer look at JAK/STAT signaling pathway. Front Oncol. 2018;8:287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kleppe M, Kwak M, Koppikar P, Riester M, Keller M, Bastian L, et al. JAK‐STAT pathway activation in malignant and nonmalignant cells contributes to MPN pathogenesis and therapeutic response. Cancer Discov. 2015;5:316–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Harrison CN, Schaap N, Vannucchi AM, Kiladjian JJ, Jourdan E, Silver RT, et al. Fedratinib in patients with myelofibrosis previously treated with ruxolitinib: An updated analysis of the JAKARTA2 study using stringent criteria for ruxolitinib failure. Am J Hematol. 2020;95:594–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. INREBIC® (fedratinib) prescribing information . Impact Biomedicines, Inc., Summit, NJ; Rev 08/2019.
- 7. Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. A double‐blind, placebo‐controlled trial of ruxolitinib for myelofibrosis. New Engl J Med. 2012;366:799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Harrison C, Kiladjian JJ, Al‐Ali HK, Gisslinger H, Waltzman R, Stalbovskaya V, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366:787–98. [DOI] [PubMed] [Google Scholar]
- 9. Mascarenhas J, Hoffman R, Talpaz M, Gerds AT, Stein B, Gupta V, et al. Pacritinib vs best available therapy, including ruxolitinib, in patients with myelofibrosis: A randomized clinical trial. JAMA Oncol. 2018;4:652–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mesa RA, Vannucchi AM, Mead A, Egyed M, Szoke A, Suvorov A, et al. Pacritinib versus best available therapy for the treatment of myelofibrosis irrespective of baseline cytopenias (PERSIST‐1): an international, randomised, phase 3 trial. Lancet Haematol. 2017;4:e225–e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Harrison CN, Vannucchi AM, Platzbecker U, Cervantes F, Gupta V, Lavie D, et al. Momelotinib versus best available therapy in patients with myelofibrosis previously treated with ruxolitinib (SIMPLIFY 2): a randomised, open‐label, phase 3 trial. Lancet Haematol. 2018;5:e73–e81. [DOI] [PubMed] [Google Scholar]
- 12. Gupta V, Yacoub A, Fazal S, Miller C, Verstovsek S, Mesa R, et al. Preliminary gastrointestinal safety and tolerability of fedratinib from the phase IIIb FREEDOM trial in patients with intermediate‐ or high‐risk myelofibrosis previously treated with ruxolitinib. Clin Lymphoma Myeloma Leuk. 2020;20(Suppl. 1):S187. [Google Scholar]