

Summary
Admixture is a mechanism by which species of long‐lived plants may acquire novel alleles. However, the potential role of admixture in the origin and maintenance of tropical plant diversity is unclear. We ask whether admixture occurs in an ecologically important clade of Eschweilera (Parvifolia clade, Lecythidaceae), which includes some of the most widespread and abundant tree species in Amazonian forests.
Using target capture sequencing, we conducted a detailed phylogenomic investigation of 33 species in the Parvifolia clade and investigated specific hypotheses of admixture within a robust phylogenetic framework.
We found strong evidence of admixture among three ecologically dominant species, E. coriacea, E. wachenheimii and E. parviflora, but a lack of evidence for admixture among other lineages. Accepted species were largely distinguishable from one another, as was geographic structure within species.
We show that hybridization may play a role in the evolution of the most widespread and ecologically variable Amazonian tree species. While admixture occurs among some species of Eschweilera, it has not led to widespread erosion of most species’ genetic or morphological identities. Therefore, current morphological based species circumscriptions appear to provide a useful characterization of the clade's lineage diversity.
Keywords: adaptive introgression, Amazon basin, hybridization, hyperdominance, Lecythidaceae (Brazil nut family), Parvifolia clade, target enrichment sequencing, tropical diversity
Introduction
The extent to which hybridization and introgression (i.e. admixture) have affected the evolutionary history of tropical trees is only beginning to be understood. Admixture is expected to have various evolutionary consequences depending on the context of its occurrence, ranging from infrequent, localized production of hybrid offspring to the formation of new species (Rieseberg & Wendel, 1993). Adaptive introgression is a possible mechanism by which tropical tree populations may acquire favourable alleles, as has been demonstrated in various other plant clades (e.g. Martin et al., 2006; Whitney et al., 2010; Pease et al., 2016; Leroy et al., 2020) and may facilitate local adaptation beyond what might occur through selection acting on standing genetic variation and de novo mutations (Suarez‐Gonzalez et al., 2018).
Hybridization among tropical trees has historically been considered a relatively rare phenomenon, primarily because of the dearth of morphological intermediates in herbarium specimens of tropical tree floras (Ashton, 1969; Parnell et al., 2013). However, recent work using next generation sequencing methods has demonstrated evidence of hybridization in tropical trees including in Brownea (Fabaceae; Schley et al., 2020), Diospyros (Ebenaceae; Linan et al., 2020), Melicope (Rutaceae; Paetzold et al., 2019) and Metrosideros (Myrtaceae; Choi et al., 2020). Caron et al. (2019) found that across tree taxa at a site in northern French Guiana, chloroplast haplotype diversity was more frequent in species with a local congener than those without, which they attributed to introgression. However, direct evidence of hybridization remains elusive for most clades of tropical trees. Because tests for admixture are inherently comparative, such tests should ideally be nested within a robust and broadly inclusive phylogeny (Eaton et al., 2015). Such phylogenies are not yet available for many tropical clades, although phylogenomic datasets are becoming increasingly available (e.g. Prata et al., 2018; Couvreur et al., 2019; Loiseau et al., 2019; Linan et al., 2020; Christe et al., 2021). Investigations that characterize gene flow at well studied forest plots may also enhance our understanding of the role of admixture in tropical forests, because (1) gene pools can be delimited without having to consider the confounding effects of geographic variation (Linan et al., 2020; Schley et al., 2020), and (2) permanently tagged trees provide a kind of ‘living herbarium’ in which variation in field characteristics not evident in herbarium collections (e.g. branching architecture, microhabitat preferences, tree size) may be studied.
Target capture sequencing, also called target enrichment, is becoming increasingly popular for phylogenomic studies of nonmodel plants (Cronn et al., 2012; Baker et al., 2021) and often produces datasets with low missing data, even when the input DNA is partially degraded. The sizes of target loci vary, but generally range from hundreds to a few thousand base pairs (bp) in length. The number of targets also varies, but is frequently a few hundred loci, which is usually sufficient for phylogeny reconstruction, but is far fewer than is typically used for inferring admixture, especially compared with methods such as RADseq, which can recover tens of thousands of RAD loci (Eaton & Ree, 2013; Eaton et al., 2015; Johnson et al., 2018; Vargas et al., 2020). Gene tree‐based methods for inferring admixture using species networks can be used with several types of data, including target capture, although the resulting networks can include patterns of reticulate evolution that are sensitive to model parameters and gene tree quality (Morales‐Briones et al., 2020). Given this and the increasing use of target capture for studies of plant evolution comes the need to explore additional methods capable of identifying evidence of admixture.
Our study taxa are tree species in the Brazil nut family, Lecythidaceae (Ericales). Lecythidaceae are ecologically important in many Neotropical forests and several species in the genus Eschweilera are among the most abundant trees across the Amazon basin (ter Steege et al., 2013). The Parvifolia clade of Eschweilera comprises 66 described species, characterized by morphological features including a distinctive double‐coiled androecium (Fig. 1d) and lateral arils on their seeds (Mori et al., 2010 onward; Huang et al., 2015). Several members of the Parvifolia clade have been described as hyperdominant (i.e. species with disproportionate abundance across a large area of the Amazon; ter Steege et al., 2013). The most abundant species of Lecythidaceae, Eschweilera coriacea (DC.) S.A. Mori, ranks third in abundance out of the more than 16 000 estimated Amazonian tree species. It is ecologically variable, thriving in floodplains as well as upland terra firme (Mori et al., 2010 onward) and is the only tree species that attains ecological dominance in all geographic subregions of the Amazon basin (ter Steege et al., 2013, 2020).
Fig. 1.

Examples of the morphology of members of the Parvifolia clade. (a) Flower of Eschweilera parviflora. (b) Lateral view of a flower of Eschweilera wachenheimii. (c) Flower of Eschweilera coriacea. (d) Flower of Eschweilera collina with androecial hood sectioned. (e) Fruit bases, opercula and seeds of Eschweilera parviflora. (f) Fruits, operculum and seeds of Eschweilera coriacea. (g) Leaves and old fruit of Eschweilera atropetiolata. (h) Abaxial view of a leaf of Eschweilera coriacea. (i) Bark of Eschweilera tessmannii. (j) Bark of Eschweilera truncata. (k) Bark of Eschweilera sagotiana. (l) Bark of Eschweilera atropetiolata. Photograph attribution: (a, b, e, f, g, i, j, l) to Scott Alan Mori; (c, d, k) to Carol Ann Gracie; (h) to Xavier Cornejo. Reproduced under terms of the CC BY‐NC‐SA 3.0 license. Captions are adapted from Mori et al. (2010 onward).
As is the case for many clades of tropical trees, species boundaries in Lecythidaceae are not precisely understood, although the taxonomy of the family is relatively well studied (e.g. Prance & Mori, 1979; Mori & Prance, 1990; Mori et al., 2010 onward). Previous studies have found discordance between morphology and plastid‐based phylogenies, suggesting that chloroplast capture (i.e. the chloroplast of one species being introgressed into another) may be common in the group (Huang et al., 2015). However, hybridization followed by repeated directional backcrossing can result in chloroplast capture with little genetic or morphological evidence of nuclear admixture (Rieseberg & Wendel, 1993). A recent study using microsatellite DNA markers suggested that the nuclear genomes of Eschweilera may also conflict with morphological based species circumscriptions (Heuertz et al., 2020), although we are not aware of any previous studies that have shown explicit evidence of nuclear admixture in Lecythidaceae. We addressed the following questions:
Is there evidence of nuclear admixture among species of the Parvifolia clade of Eschweilera, including species that are among the most abundant and ecologically variable trees in the Neotropics?
To what extent do accepted species of Eschweilera represent monophyletic lineages that are distinguishable from one another using nuclear genomic data?
The answers to these questions may shed light on whether the hyperabundance of widespread species such as E. coriacea could be partly explained by a history of genetic introgression. We used a multifaceted sampling strategy and target capture sequencing to generate the largest phylogenomic dataset for the family to date. Our methods included the implementation of an explicit test for admixture suitable for target capture data, which may prove useful for other phylogenomic datasets.
Materials and Methods
Focal study site and sampling strategy
We conducted sampling using two approaches. First, we sampled 12 focal species of the Parvifolia clade (Table 1) that co‐occur at a single 100‐ha forest plot in which all individuals of Lecythidaceae ≥ 10 cm diameter at 1.3 m height have been tagged and identified by specialists beginning in the late 1980s (see Supporting Information Notes S1; Mori & Lepsch‐Cunha, 1995; Mori et al., 2001). The ‘Lecythidaceae plot’ lies within Reserve 1501, also known as Km 41, of the Biological Dynamics of Forest Fragments Project (BDFFP) located c. 70 km north of Manaus, Brazil (2°24′54″S, 59°50′39″W). The plot was established to study the Lecythidaceae of the central Amazon, a geographic center of diversity for the clade, but an area in which its taxonomy was poorly characterized (Mori & Lepsch‐Cunha, 1995). By pairing ecological studies with alpha taxonomy, the investigators sought to characterize nuanced morphological differences among species across population samples and, in doing so, identify new species and their ecological differences (Mori & Lepsch‐Cunha, 1995; Mori et al., 2001). Flowers and fruits are produced only sporadically in many species of Lecythidaceae, but species determinations for each tree in the plot were made using fertile material whenever possible (Mori et al., 2001). The site was re‐censused in 2019, which showed there to be 6741 trees from 36 described species of Lecythidaceae (T. Milton et al., unpublished). Here, we refer to this 100‐ha Lecythidaceae plot as Reserve 1501.
Table 1.
Summary of the number of samples before and after making redeterminations.
| Group or focal species | Number of samples (based on morphology in parentheses) | Named spp. represented |
|---|---|---|
| E. atropetiolata S.A. Mori | 5 (5) | 1 |
| E. bracteosa (Poepp. ex O. Berg) Miers | 4 (6) | 1 |
| E. collina Eyma | 5 (5) | 1 |
| E. coriacea (DC.) S.A. Mori | 12 (13) | 1 |
| E. cyathiformis S.A. Mori | 5 (4) | 1 |
| E. laevicarpa S.A. Mori | 7 (6) | 1 |
| E. micrantha (O. Berg) Miers | 2 (6) | 1 |
| E. pedicellata (Rich.) S.A. Mori | 6 (7) | 1 |
| E. pseudodecolorans S.A. Mori | 5 (4) | 1 |
| E. rankiniae S.A. Mori | 4 (4) | 1 |
| E. truncata A.C. Sm. | 10 (9) | 1 |
| E. wachenheimii (Benoist) Sandwith | 4 (6) | 1 |
| Focal species or admixed from Reserve 1501 | 58 (60) | 12 |
| Admixed within Parvifolia clade | 4 (3) | n.a. |
| Parvifolia clade | 109 (107) | 33 |
| Lecythidaceae | 240 (240) | 127 |
We chose focal species that were among the most abundant and most closely related species of Lecythidaceae at Reserve 1501 (Mori & Lepsch‐Cunha, 1995; Huang et al., 2015). Whenever possible, we sampled four to six tagged trees of each focal species and observed a minimum of at least 100 m between conspecifics to reduce the chances of sampling immediate relatives. Our field collections relied on prior tree identifications of S. Mori et al. and we prioritized collection of three individual trees that seemed to have intermediate morphology, including in branching architecture (Notes S2). For each field‐collected sample, leaf tissue was desiccated in silica gel and a voucher was deposited at the BDFFP collection at the National Institute of Amazonian Research (INPA), in Manaus, Brazil. In total, our sampling included 60 individuals collected at Reserve 1501 that were identified as a focal species or suspected hybrid based on morphology (Table 1).
Our second sampling approach aimed for wider phylogenetic and geographic breadth and used herbarium material and existing forest inventory vouchers. For this broader sampling, the New York Botanical Garden Herbarium (NY) provided about half of our samples, which also included several nonfocal species collected at Reserve 1501 and the surrounding area. Our overall sampling included 240 individuals from 127 of the 230 described species of Neotropical Lecythidaceae and seven outgroup species from Paleotropical genera. This included 109 individuals of the Parvifolia clade from 33 described species as well as several species that have not yet been formally described (Table S1). A full analysis of the relationships among all major clades, as well as a revised taxonomy of Lecythidaceae utilizing this sampling is forthcoming (O. Vargas et al., unpublished).
Sequencing and assembly
We performed DNA extractions using the NucleoSpin Plant Mini Kit II (Macherey‐Nagel, Düren, Germany) following the manufacturer's protocol, but we extended the digestion step to one h and added 5 μl of proteinase K (20 mg ml−1; Qiagen, Hilden, Germany). Preparation of unenriched libraries for genome skimming and target‐enriched libraries followed by 150‐bp paired‐end sequencing on an Illumina HiSeq4000 machine (Illumina Inc., San Diego, CA, USA) was performed by Rapid Genomics (Gainesville, FL, USA). The probes used to enrich libraries were designed to capture 344 nuclear genes previously inferred to be low or single copy and genetically variable in Lecythidaceae (Vargas et al., 2019). Raw reads were processed with seqyclean (Zhbannikov et al., 2017) to trim sequencing adapters, filter out low‐quality reads and trim reads at regions with a Phred score < 20 using a window of 10 bp. Trimmed reads were checked with fastqc v.0.11.3 (Andrews, 2010). Target loci were assembled using hybpiper v.1.3.1 (Johnson et al., 2016) with default settings and a target file that included DNA sequences based on complete cDNA targets (Vargas et al., 2019). The Hybpiper pipeline uses exonerate (Slater & Birney, 2005), blast+ (Camacho et al., 2009), biopython (Cock et al., 2009), bwa (Li & Durbin, 2009), samtools (Li et al., 2009), GNU parallel (Tange, 2011) and spades (Bankevich et al., 2012).
Paralog filtering and alignment
When using target capture, paralogs can be enriched during library preparation and recovered in locus assemblies. While evidence suggests all or most Lecythidaceae are diploid (Heuertz et al., 2020), the lineage is thought to have experienced a whole genome duplication that occurred near the time of the most recent common ancestor of Ericales (Larson et al., 2020). Given that paralogs from gene duplications can confound many phylogenetic analyses, we used a tree‐based pruning approach meant to reduce misidentified orthologs and assembly errors (Yang & Smith 2014). The parameters used in this trimming procedure were derived based a priori knowledge of the Lecythidaceae phylogeny and inspection of hundreds of amino acid phylogenies (Methods S1; Mori et al., 2015; Rose et al., 2018; Larson et al., 2020). The procedure included multiple sequence alignment with mafft v.7.271 (Katoh et al., 2002; Katoh & Standley, 2013) followed by amino acid tree estimation with raxml v.8.2.11 (Stamatakis, 2014) and was meant to reduce nonorthologous sequences in the orthogroup alignments, while minimizing loss of phylogenetic information for taxa in the Parvifolia clade (Methods S1). We use the term orthogroup to denote groups of sequences that appear to be reciprocally orthologous based on sequence similarity, regardless of their present function in individual species.
Preliminary phylogenetic investigation
To identify clades of closely related individuals, check determinations for specimens and to verify which individuals were nested within the Parvifolia clade, a phylogenetic tree (here referred to as the preliminary phylogeny) was estimated with the assembled sequences from all 240 samples after the paralog filtering procedure described above. The preliminary phylogeny was estimated using raxml v.8.2.11 and a separate gtrcat model partition for each of the exon and intron alignments of each orthogroup (Stamatakis, 2014). To assess support for clades in the preliminary phylogeny, rapid bootstrapping with 200 replicates was conducted. The results were visualized with figtree (https://github.com/rambaut/figtree/).
Genotyping and SNP analysis
To investigate the genetic structure of Parvifolia species and identify potentially admixed individuals, we called single nucleotide polymorphisms (SNPs) for each individual using gatk v.4.1.0.0 (McKenna et al., 2010). The exon sequences for one individual for which we recovered 343 target loci with a combined length of 836 403 bp were used as a reference assembly (Methods S2). Genomic variants were called for each individual following gatk best practices, with modifications when necessary to accommodate the available genomic resources for these nonmodel species (Methods S2; DePristo et al., 2011; Li et al., 2009; Li, 2013; Van der Auwera et al., 2013; Poplin et al., 2017; Hanlon et al., 2019). Several clades were identified based on the preliminary phylogeny and a clade‐specific SNP dataset in approximate linkage equilibrium was generated for each (Methods S2; Table S2; Purcell et al., 2007). We used structure v.2.3.4 (Pritchard et al., 2000) to investigate genetic clustering of individuals within each clade and determined the most appropriate number of populations (K) for each subset of taxa by comparing the estimated posterior probability of the data for multiple values of K in conjunction with a priori taxonomic information (Methods S2; Table S2). In cases when an individual showed strong evidence of clustering with a species other than that to which it was identified based on morphology, the identity of the individual was further investigated, and its determination was updated to reflect taxonomic uncertainty and all available evidence (Fig. S1; Methods S3). Special consideration was given to E. roseocalyx, which appeared to be nested within the broadly distributed species E. coriacea based on preliminary results (Methods S3; Batista et al., 2017). To further explore patterns of genetic variation within E. coriacea, we used the gdsfmt and snprelate packages (Zheng et al., 2012) in R v.3.6.0 (R Core Team, 2019) to produce an additional SNP dataset and conducted a genetic principal component analysis (PCA), which was visualized with a custom R script that utilized the plotly.js library (Sievert, 2020).
Verifying admixture with rooted triple tests
To corroborate the admixed ancestry of individuals identified using structure and test for evidence of ancestral introgression among closely related species, we implemented a test capable of inferring admixture from a set of gene trees using rooted triplets (RT; i.e. gene trees consisting of three ingroup individuals and an outgroup; Fig. 2), which we conducted using the novel script Run_RT_tests.py (see Data Availability Statement). A version of this test has been proposed previously (Hudson et al., 2005), but we are not aware of any previous studies that have used it to investigate admixture in target capture datasets. The RT tests were conducted by subsetting each orthogroup alignment to include the four individuals of interest and estimating a gene tree with branch lengths for each subalignment using iq‐tree v.1.6.3 (Nguyen et al., 2014; Chernomor et al., 2016). This obviated the need to re‐align sequences for each test and allowed the sequence data from all 240 samples to inform the subalignment, which may have helped alleviate alignment issues due to missing data. Then, the topologies of the resulting trees were summarized to assess whether the data were compatible with a scenario of no admixture, using the same theoretical framework as the D‐statistic (Green et al., 2010). However, unlike most implementations of the D‐statistic that count patterns in multiple sequence alignments or SNP datasets, our test is based on gene trees and can therefore readily be used with phylogenomic datasets consisting of relatively large gene regions in which all sites within a region are assumed to share the same phylogenetic history.
Fig. 2.
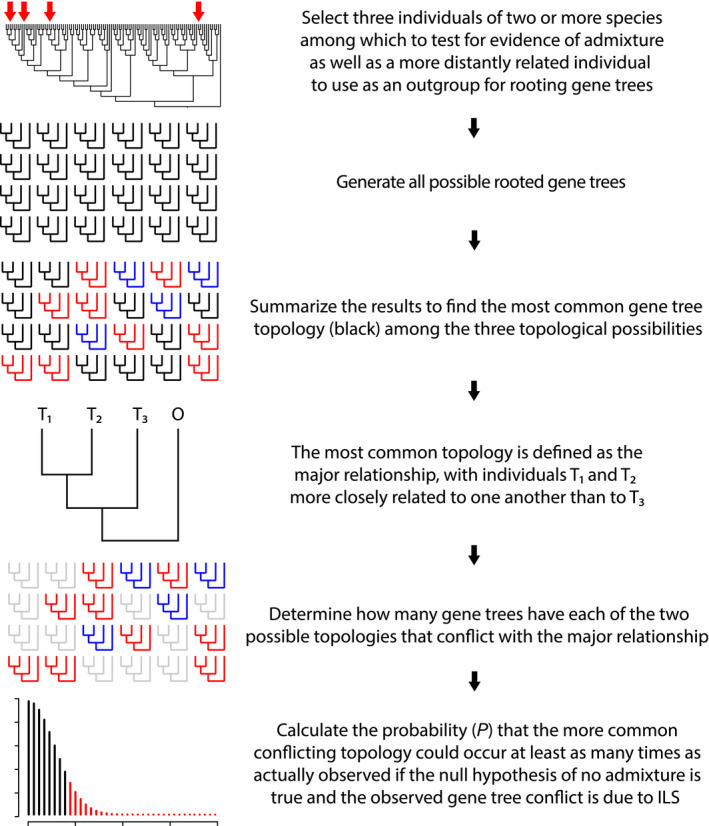
Schematic of the rooted triplet test for assessing evidence of admixture. Red arrows indicate four hypothetical samples selected for the test. The test assumes that the outgroup diverges first in all gene trees and at least two species are represented in the ingroup. Blue and red phylogenies represent the two possible topologies that conflict with the most common topology after all possible gene trees have been generated. Any statistically significant deviation from equal numbers of the two conflicting topologies, where P is the probability of a result at least as unequal as the observed frequencies using a binomial test, is considered evidence that the assumptions of the multispecies coalescent have been violated by admixture among species.
When all four‐taxon gene trees are rooted on a known outgroup, the result is a set of rooted triplets. For each gene tree in the set, there is a single tree bipartition that contains the topological information for the ingroup, since two individuals will be sister to the exclusion of the third. For a set of rooted triplets consisting of ingroup taxa A, B and C, the three possible ingroup bipartitions are (AB|C), (AC|B) and (BC|A). We define the most frequent of the three possibilities as the ‘major relationship’ and the other two possibilities as ‘conflicting relationships’. The two individuals that form the major relationship are inferred to be the two that are most closely related and are here referred to as T1 and T2 (Fig. 2). T1 and T2 are assumed to share a most recent common ancestor (MRCA) that occurred more recently than the MRCA of all three ingroup individuals, whether or not there is ongoing gene flow between/within the population(s) to which T1 and T2 belong (i.e. they can be the same or different species). As long as there is a null expectation of no gene flow with the populations to which the third ingroup (T3) or the outgroup (O) individuals belong (i.e. T3 and O are different species from one another as well as from T1 and T2) and it can be assumed that for each gene tree, O has the earliest diverging sequence, then in the absence of gene flow between the lineages represented by T3 and T1 and/or T2, the number of gene tree with each of the two possible conflicting topologies should be statistically equal, because each is equally likely to occur due to incomplete lineage sorting (ILS; Bryant & Hahn, 2020).
Any statistically significant deviation from equality can be considered evidence that the assumptions of the multispecies coalescent model have been violated by gene flow between the lineages to which T3 and T1 and/or T2 belong. We calculate P as the probability of a result at least as unequal as the observed frequencies using a binomial test in which each gene tree that conflicts with the major relationship represents a trial and the probability of either conflicting relationship is equal to 0.5.
To correct for multiple comparisons, we used the Holm–Bonferroni method with α = 0.01 to adjust our critical value for rejecting the null hypothesis (Holm, 1979; Eaton et al., 2015). The statistical power of each RT test is affected by the number of gene trees that conflict with the major relationship, which is expected to vary based on the time since the MCRA of the relevant individuals. The type II error rate (i.e. failing to reject the null hypothesis of no admixture when in fact there has been admixture) of this type of RT test may be relatively high for many target capture datasets, due to the relatively low number of independent trials available compared with some other tests for admixture using RADseq or whole genome assemblies. Because of this, our results may represent a conservative estimate of admixture among our sampled species, especially for cases of historical introgression involving small proportions of the genome. However, our statically significant results provide strong evidence of admixture.
It should be noted that because we utilized coding sequences and the introns adjacent to them, each locus is subject to natural selection. However, it is unlikely that selection would generally lead to a systematic bias for one conflicting gene tree topology over the other for a large enough number of independent loci to significantly increase the type I error rate (i.e. rejecting the null hypothesis of no admixture, when in fact no admixture has occurred). It is also important to note that the test as implemented does not explicitly account for heterozygosity, as each locus is represented by a single consensus sequence per sample, as is typical in most phylogenomic datasets. The effect that differing consensus‐calling approaches during sequence assembly might have on phylogeny‐based inferences of admixture warrants future study.
Parvifolia clade phylogeny
To build a robust phylogenetic hypothesis for the Parvifolia clade, we conducted additional analyses without individuals with evidence of recent admixture. We used additional tree‐based paralog pruning and generated two supermatrices, one that included data from introns and another that did not (Methods S4). For clarity, we refer to the best‐scoring tree for the dataset that included both intron and exon sequences as the ‘Parvifolia phylogeny’ and the best‐scoring tree for the other supermatrix as the ‘exon‐only Parvifolia phylogeny’. For visualization, a version of each phylogeny was produced by trimming tips to include a single representative of each accepted species (Table S1) using the pxrmt function in phyx (Brown et al., 2017). Conflict between the reduced‐representation phylogenies was visualized using the phytools package in R (Revell, 2012). A version of the Parvifolia phylogeny with all tips, as well as an analysis of topological conflict with the untrimmed exon‐only Parvifolia phylogeny, generated using the pxbp function in phyx, is also reported.
Summaries of collection records and phenology for selected species
To visualize the extent of known range overlap among hyperdominant species E. coriacea, E. parviflora, E. truncata and E. wachenheimii, we used a dataset curated by Mori et al. (2017) comprizing available species occurrence records for these taxa (Vargas & Dick, 2020). All records for each species were plotted with qgis v.3.16.3 (https://github.com/qgis). We used a river shapefile available from the World Bank (https://datacatalog.worldbank.org/dataset/major‐rivers‐world, CC‐BY 4.0 license), the World Borders Dataset (http://thematicmapping.org, CC‐BY‐SA 3.0 license) and the digital elevation model of Lehner & Grill (2013). We plotted individual occurrences, rather than range summaries, to more clearly show the available data and corresponding gaps in existing collection records. To investigate flowering times of E. coriacea, E. parviflora and E. wachenheimii, we used the C.V. Starr Virtual Herbarium (http://sweetgum.nybg.org/science/vh/) to examine all collections from Amazonas, Brazil housed at NY. We identified specimens with flowers or flower buds at time of collection and verified the collection date and determination for each based on the specimen label. The results were plotted as box plots and dot plots for each species in R using ggplot2 (Wickham, 2016) after removing duplicate collections made from the same tree on the same day.
Results
Admixture among species of the Parvifolia clade
Our SNP‐calling approach identified 148 310 polymorphic sites among 109 individuals in the Parvifolia clade. Both structure analyses and RT tests support evidence of admixture among two species pairs in our sampling. Two individuals collected at Reserve 1501 were supported as having near equal ancestry of E. coriacea and E. wachenheimii (Fig. 3). These individuals were not recovered as sister to one another in the preliminary phylogeny (Fig. S2) and RT tests showed significant evidence of admixture for separate tests that included these individuals (Tables 2, S3). Two additional individuals were supported as genetic intermediates between E. wachenheimii (c. 70–75% ancestry) and E. parviflora (c. 25–30% ancestry) in structure, with RT tests also supporting evidence of admixture (Fig. 3; Tables 2, S3). This second pair of individuals was recovered as sister to one another in the preliminary phylogeny (Fig. S2).
Fig. 3.
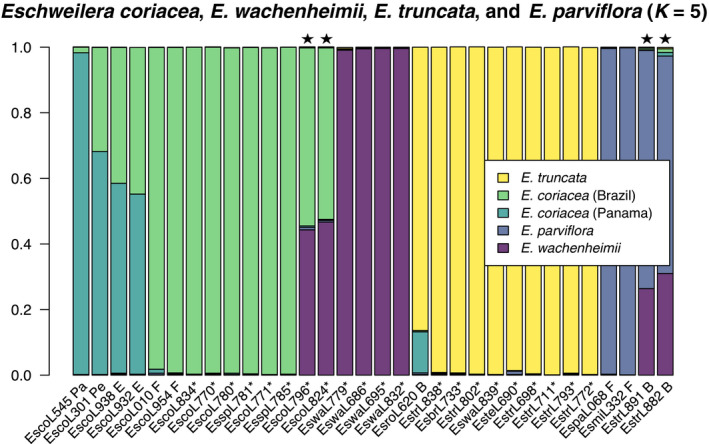
Population structure (K = 5) of all samples in the clade that included E. coriacea, E. wachenheimii, E. truncata and E. parviflora. Each bar represents the ancestry of an individual inferred with structure. Each individual is labelled with a unique code used throughout all analyses and asterisks indicate samples from focal species collected at Reserve 1501. Collection locations outside Reserve 1501 are indicated as follows: Pa – Panama, Pe – Peru, E – Ecuador, F – French Guiana, B – Brazil. Black stars above bars indicate individuals with significant evidence of admixture based on an RT test.
Table 2.
Summary of 15 rooted triplet tests, ranked in order of increasing P‐value.
| Samples forming major relationship | Third ingroup | Major relationship count | Conflict 1 count | Conflict 2 count | P‐value | Corrected crit. value | Reject H‐null |
|---|---|---|---|---|---|---|---|
| EswaL779, EscoL796 | EscoL834 | 141 | 124 | 53 | 4.87E‐08 | 2.22E‐04 | Yes |
| EstrL882, EswaL779 | EspaL068 | 134 | 124 | 56 | 2.18E‐07 | 2.27E‐04 | Yes |
| EswaL779, EscoL824 | EscoL834 | 129 | 126 | 59 | 4.69E‐07 | 2.33E‐04 | Yes |
| EstrL891, EspaL068 | EswaL779 | 131 | 113 | 64 | 1.42E‐04 | 2.38E‐04 | Yes |
| EsmiL332, EspaL068 | EswaL779 | 208 | 69 | 40 | 3.52E‐03 | 2.44E‐04 | No |
| EscoL885, EslaL783 | EsbrL794 | 147 | 101 | 69 | 8.58E‐03 | 2.50E‐04 | No |
| EstrL838, EscoL834 | EssaL335 | 140 | 104 | 73 | 0.0119 | 2.56E‐04 | No |
| EsroL664, EsamL886 | EsmiL823 | 120 | 111 | 83 | 0.0261 | 2.63E‐04 | No |
| EscoL241, EscoL828 | EscoL885 | 224 | 50 | 36 | 0.0803 | 2.70E‐04 | No |
| EscoL771, EswaL839 | EstrL711 | 212 | 55 | 42 | 0.111 | 2.78E‐04 | No |
| EsteL690, EstrL772 | EspaL068 | 222 | 50 | 38 | 0.120 | 2.86E‐04 | No |
| EstrL838, EswaL779 | EssaL335 | 170 | 80 | 66 | 0.141 | 2.94E‐04 | No |
| EspaL386, EspaL868 | EsteL704 | 221 | 49 | 39 | 0.169 | 3.03E‐04 | No |
| EscyL797, EsrhL578 | EsatL643 | 164 | 77 | 65 | 0.178 | 3.13E‐04 | No |
| EstrL838, EswaL779 | EscoL834 | 118 | 107 | 93 | 0.179 | 3.23E‐04 | No |
We also tested for evidence of more ancient introgression among lineages using RT tests with three ingroup individuals representing three different species determinations or structure clusters (in cases in which the individual’s identity was unclear). Individuals whose determination contained an affinis modifier were considered to be their own lineage for this purpose. We conducted 25 such tests, selecting one individual per lineage and excluding individuals with evidence of recent admixture in structure analyses. We did not find significant evidence of admixture in any of these tests (Figs 4, S3; Tables 2, S3), although three resulted in an uncorrected P < 0.05 but that was not significant at the level of α = 0.01 after correcting for multiple tests with the Holm–Bonferroni method (Tables 2, S3). One such test included individuals determined as E. parviflora, E. aff. parviflora and E. wachenheimii in which 63.3% of conflicting gene trees supported one alternative (P = 3.52 × 10−3). Another test included individuals of E. laevicarpa, E. bracteosa and an individual determined as E. aff. laevicarpa: for this test 59.4% of conflicting gene trees supporting one alternative (P = 8.58 × 10−3). The third test, which included individuals of E. truncata, E. coriacea and E. sagotiana resulted in 58.8% of conflicting gene trees supporting one alternative (P = 1.19 × 10−2; Tables 2, S3).
Fig. 4.
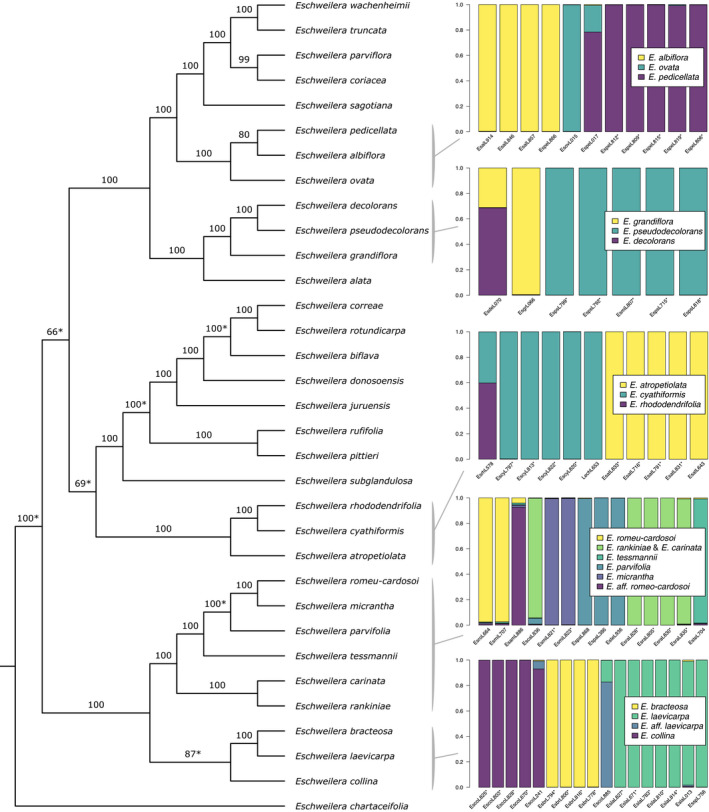
Phylogeny of the Parvifolia clade visualized using a single representative per accepted species. Branch labels are iq‐tree ultrafast bootstrap support. Asterisks on branch labels indicate relationships that conflict with the best‐scoring maximum likelihood topology recovered with an exon‐only supermatrix. The results of structure analyses are shown for SNP datasets that included all individuals within the corresponding clades indicated on the phylogeny. Each individual is labelled with a unique code used throughout all analyses and asterisks on these labels indicate samples from focal species collected at Reserve 1501. The legend for each subplot indicates the one or more species that most closely corresponded to each cluster based on accepted taxonomy. The individuals in these clades generally clustered along morphologically defined species boundaries and there was no significant evidence of admixture for these taxa based on rooted triplet tests.
Monophyly of described species in the Parvifolia clade
In structure analyses, individuals collected at Reserve 1501 were consistently assigned ancestry corresponding almost exclusively (i.e. > 95%) to a single cluster, with notable expectations for two individuals with evidence of admixture (Figs 3, 4, S4). There did not appear to be admixture within several clades based on samples collected at Reserve 1501 including (1) E. collina, E. bracteosa and E. laevicarpa, (2) E. atropetiolata and E. cyathiformis, or (3) E. micrantha and E. rankiniae (Fig. 4). When considering individuals from these species collected outside our focal plot, some were inferred to have ancestry corresponding to multiple species. However, this appeared to be the result of intraspecific variation due to geographic structure, as there was no evidence of admixture in relevant RT tests (Table S3). Intraspecific variation could have caused ancestry to be assigned to a second cluster due to the parameterization of the analysis or uneven sampling across subpopulations (e.g. several individuals sampled from Reserve 1501, one individual from another locality). Indeed, the tendency for structure to assign mixed ancestry in the presence of isolation by distance (Pritchard et al., 2010) or when sampling is uneven across hierarchical levels of population structure (Puechmaille, 2016) has been well documented. Alternatively, this signal could represent admixture that RT tests failed to detect.
Overall, most individuals had morphological determinations that agreed with genetic evidence. There were 60 individuals collected at Reserve 1501 with morphological determinations as one of our focal species or suspected hybrids. Of these, seven (11.7%) were shown to require redeterminations based on genetic evidence and two were shown to be admixed (Table S1). There were 51 individuals in our broader sampling of the Parvifolia clade that did not meet both of the following criteria: (1) determined to be a focal species based on morphology; and (2) collected at Reserve 1501. Of these 51, there were 11 (21.6%) that required redeterminations and two that showed evidence of admixture. Seven could be redetermined to species and four were assigned a putative species determination with an affinis modifier to reflect uncertainty (Fig. S1; Table S1).
Geographic structure in E. coriacea
There was strong evidence of geographic structure among 12 samples of E. coriacea with no evidence of recent admixture. In a PCA of SNP data, the first, second and third principal components explained 15.14%, 11.24% and 10.82% of the total variance, respectively, and individuals with the same country of origin clustered together (Fig. S5). In phylogenetic analyses, collections from Brazil formed a clade which was strongly supported as sister to collections from French Guiana (Table S1; Fig. S6). The single individual collected in Panama was sister to an individual collected at Los Amigos field station at Madre de Dios, Peru, with those sister to a clade of two individuals collected at Yasuní National Park in Ecuador; those four individuals were also inferred to have varying amounts of ancestry corresponding to a second cluster in structure analyses, while individuals from Brazil and French Guiana had inferred ancestry almost exclusively corresponding to a single cluster (Figs 3, S4).
Phylogenetic relationships in the Parvifolia clade
Our target capture approach resolved most of the phylogenetic relationships among sampled species of the Parvifolia clade, although for some, support was dataset dependent (Figs 4, S6, S7; Table S4). Seven relationships among accepted species differed between the Parvifolia phylogeny (i.e. intron and exon data) and the exon‐only Parvifolia phylogeny (Fig. S7). Inferred relationships among individuals within a species tended to vary more than relationships among species across datasets (Fig. S6). Regardless of whether intron data were included, E. truncata and E. wachenheimii were inferred to be sister taxa, as were E. coriacea and E. parviflora. Those four species formed a clade with E. sagotiana, with that clade of five species sister to a clade consisting of E. pedicellata, E. ovata and E. albiflora (Fig. 4).
Summary of collection records and phenology of selected species
Existing collection records showed broad overlap in the geographic ranges of the four species we investigated (Fig. 5). Our survey of phenology yielded 63 unique collections in flower from Amazonas, Brazil (Table S6). Collection date ranges and interquartile ranges for each of the three species overlapped, with the medians for each falling within 3 wk of one another during the dry season (Fig. S8).
Fig. 5.
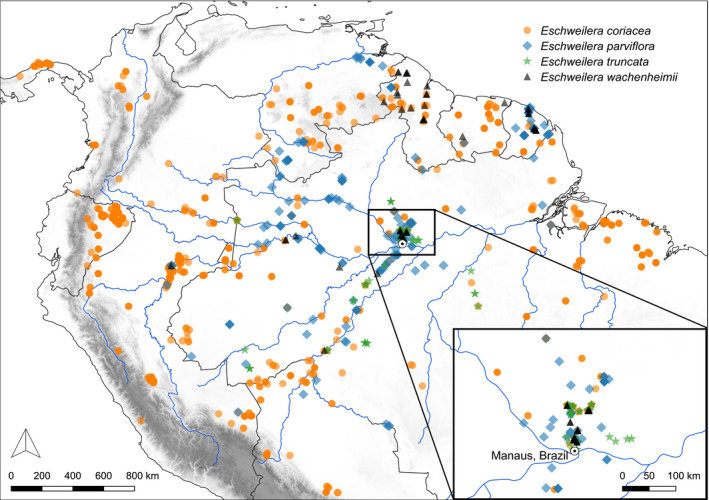
Occurrence records across the Neotropics for four closely related species in the Parvifolia clade, three of which show evidence of admixture in this study. Few or no records are available for these species across much of the Amazon basin as most come from collections made in permanent plots. Because of this, there is uncertainty regarding the true extent of range overlap among these and other species of Lecythidaceae as is the case for many other clades of Neotropical trees.
Discussion
Admixture in the Parvifolia clade
Our results add to the small, but growing, body of evidence regarding admixture among tropical trees and are, to our knowledge, the first examples of nuclear admixture among hyperdominant Amazonian species. Our sampling included all accepted species of the Parvifolia clade known to occur in the intensively studied Reserve 1501 Lecythidaceae plot (Table S1; Mori & Lepsch‐Cunha, 1995). All individuals of our 12 focal species collected at Reserve 1501 could be assigned robust species determinations based on structure analyses and tree‐based phylogenomic inference (Figs 3, 4, S2–S4; Table S1). Our results provide robust evidence of admixture between two of our focal species, E. coriacea and E. wachenheimii. The two E. coriacea × wachenheimii individuals were recovered as successively sister to all E. wachenheimii individuals in our preliminary phylogeny, consistent with each sharing a high degree of genetic similarity with E. wachenheimii while also harbouring genetic dissimilarities with E. wachenheimii and with one another (Fig. S2). In addition, there was significant evidence for rejecting the null hypothesis of no admixture for RT tests that included one E. wachenheimii, one E. coriacea and either putative E. coriacea × wachenheimii individual (Tables 2,S3).
Our results also strongly support hypotheses of admixture between E. wachenheimii and E. parviflora (Fig. 3; Table 2). The E. parviflora × wachenheimii individuals were inferred to have unequal ancestry from the two parent species, suggesting that hybridization followed by backcrossing may have occurred (Figs 3, S4). We note that only a single individual of E. parviflora has ever been recorded at Reserve 1501 and therefore was not among our focal species; the collections of these admixed individuals were made within the BR‐319 plot network, south of Reserve 1501 (https://ppbio.inpa.gov.br/sitios/br319; Table S1). Both sampled individuals with >95% ancestry corresponding to E. parviflora in structure analyses were collected in French Guiana. However, our results were not consistent with geographic structure: the relevant RT tests provided evidence to reject null hypotheses of no admixture for triplets consisting of one E. wachenheimii, one E. parviflora and either putative E. parviflora × wachenheimii intermediate (Tables 2, S3).
All three species with evidence of admixture, E. coriacea, E. wachenheimii and E. parviflora, have been described as hyperdominant: members of a group of 217 tree species that comprise c. 50% of the tree numbers and biomass of Amazon forests (ter Steege et al., 2013). Eschweilera coriacea is the third most abundant tree species across an Amazon‐wide network of forest inventory plots, with an estimated census population size of between four and five billion individuals (ter Steege et al., 2013, 2020) and is the only tree species to be considered hyperdominant in both the Amazon basin and Guiana Shield regions (ter Steege et al., 2013).
Is admixture widespread among species of Eschweilera?
Given the sizable gaps in available data on hyperdominant species of Eschweilera, additional research is clearly needed to reveal the full extent of admixture among them. We found admixture between two species pairs of hyperdominant Eschweilera at two different localities, despite sampling 12 or fewer individuals for any species (Table 1). Of the three individuals suspected to be hybrids based on morphology, only one showed evidence of admixture, while three other individuals, one originally determined as E. coriacea and two as E. truncata, were also found to be admixed (Table S1). This suggests that trees with or without obvious morphological signs of hybridity may have admixed genomes. Data on the phenology of these species is quite limited but indicates that broad overlap in flowering times during the dry season cannot be ruled out based on existing data (Fig. S8) and evidence of admixture clearly demonstrates that phenological overlap can occur in the Amazon basin.
Given the current evidence, the large population sizes of these species, their large and frequently overlapping ranges (Fig. 5; Mori et al., 2017) and the prevalence of gene tree conflict in our results (Tables 2, S3), we argue that admixture among E. coriacea, E. wachenheimii and E. parviflora may be extensive and that future efforts are likely to reveal further evidence that admixture has played a role in the evolution of these, and possibly other, species of Eschweilera. However, deeper sampling is necessary to determine the extent of admixture and whether additional species admix. The results of several RT tests showed patterns of gene tree conflict suggestive of ancestral evolutionary reticulations, but that failed to meet our criteria for statistical significance (Tables 2, S3). Future work that implements explicit tests for admixture with more independent loci may provide stronger evidence regarding whether ancient evolutionary reticulations have occurred in Eschweilera. Future sampling efforts with a larger geographic focus could also produce quantitative estimates of gene flow among lineages across the Neotropics and investigate whether entire populations, rather than individuals, bear genomic signatures of admixture.
Biological implications of admixture among dominant tropical lineages
If admixture is widespread, interspecific gene flow may be an important factor in the evolution of the Parvifolia clade and could shape their reproductive biology, local adaptation and ecological interactions. Hybridization and introgression can have various outcomes including increasing genetic diversity, sharing of adaptive alleles, and either increasing or decreasing the strength of reproductive isolation barriers (Rieseberg & Wendel, 1993). In some cases, a complete breakdown of reproductive isolation barriers can cause ‘lineage collapse’ or ‘speciation reversal’, resulting in a new lineage with a mosaic genome (Kearns et al., 2018). Alternatively, if hybrids are inviable, prezygotic isolation barriers may evolve (i.e. reinforcement) or there may be little or no lasting population level effects of hybridization. For Eschweilera, current evidence suggests that chloroplast capture may be quite common (Huang et al., 2015; Caron et al., 2019; O. Vargas et al., unpublished), indicating that at least some hybrids are capable of backcrossing with their parent species.
Our results show that morphologically defined species largely correspond to distinctive gene pools in our focal species, even in those that admix. The continued genetic cohesion of admixing species could be due to several factors, including hybrid inviability or divergent selection acting on suites of traits that differ among these species. Unfortunately, data about the reproductive biology and ecology of the species found to admix are limited. All three species most often occur in nonflooded forests, although E. coriacea appears to tolerate flooding more readily than the other two (Mori & Lepsch‐Cunha, 1995; Mori et al., 2010 onward). Eschweilera coriacea frequently reach the canopy while E. wachenheimii are typically smaller and occupy the understory. Eschweilera parviflora are most often found in the understory, but can also reach the canopy (Mori et al., 2010 onward). All three species differ somewhat in floral morphology (Fig. 1a–c; Table S5) and may attract different pollinators, although observations of floral visitors are lacking for these species (Mori et al., 2010 onward). A better understanding of the nuanced ecological differences among these species may help shed light the selective forces that maintain their genetic separation.
A group of taxa that remains largely distinct despite incomplete reproductive barriers is sometimes called a syngameon (Lotsy, 1925; Suarez‐Gonzalez et al., 2018). Several of the best studied examples of syngameons in trees are found within the oaks (Quercus), which hybridize prodigiously (e.g. Eaton et al., 2015; Hipp et al., 2020), yet largely retain their cohesion as species (Hardin, 1975; Cavender‐Bares, 2019; Kremer & Hipp, 2020) and have likely facilitated one another’s ecological success through introgression (Leroy et al., 2020). Our results suggest that some members of the Parviflora clade including E. coriacea, E. wachenheimii and E. parviflora could represent a syngameon, which have been hypothesized to be common in tropical trees (Cannon & Lerdau, 2015; Schmitt et al., 2021), but have not often been documented with genomic evidence. Exchanging genes with other species might facilitate local adaptation across the broad ranges of species such as E. coriacea (Fig. 5), but further investigation is needed to test for evidence of a relationship between admixture, species abundances and ecological amplitude.
Population structure
We found evidence of geographic structure within the hyperdominant species E. coriacea (Figs 3, S4, S5). In structure analyses, runs with the best posterior probability consistently inferred individuals of E. coriacea to correspond to two clusters, with individuals assigned varying proportions of the two clusters depending on where the specimen was collected, in a gradient from Panama to Ecuador and Peru to French Guiana and Brazil (Figs 3, S4). We also found evidence to suggest population structure in other species in the Parvifolia clade, including E. truncata (Figs 3, S4), E. sagotiana (Fig. S4), E. collina (Fig. 4) and E. pedicellata (Fig. 4), although we note our sampling was not designed to make inferences on geographic structure in these species. Phylogeographic structure is expected within broadly distributed Neotropical trees (Dick & Pennington, 2019) and has previously been uncovered in several species (e.g. Dick & Heuertz, 2008; Nazareno et al., 2019).
Implications for the taxonomy of Amazonian trees
While a reassessment of species limits is outside the scope of this work, our results suggest that our focal species can be robustly identified with the methods we used (Figs 2, 3, S3, S4). Despite the occurrence of admixture in some species, most individuals identified as a focal species clustered with other individuals with the same morphological species identification (Table S1). Our results therefore suggest that admixture has not led to the widespread erosion of species boundaries within the clade and, therefore, morphology can be used to reliably distinguish among most co‐occurring species of Lecythidaceae. However, cases in which genomic evidence did not match existing determinations suggest that refined taxonomic and genetic studies may be warranted for some species including E. coriacea and E. micrantha (Table S1).
Our results show that morphological determinations for specimens collected outside Reserve 1501 more frequently conflicted with genomic evidence than did determinations for specimens from the intensively studied plot (Table S1). Intraspecific morphological variability, identification errors, as well as admixture may have contributed to this discordance. Many species in the Parvifolia clade, including the three species for which we find evidence of admixture, have similar vegetative characteristics (Table S5), overlapping phenology (Fig. S8; Table S6) and are broadly distributed across the Neotropics (Fig. 5; Mori et al., 2010 onward; Mori et al., 2017). Our results suggest that the methodology used here might be useful for investigating species delimitation in relation to the geography of broadly distributed tropical tree species. To better characterize species boundaries in tropical trees, studies should explicitly investigate morphological characters in conjunction with genomic evidence, including for admixed individuals and/or populations.
Utility of target capture for studying tropical tree populations
There are several methods available to detect evidence of admixture, each with its own benefits and assumptions. The target capture protocol used here, with probes specifically designed to recover low copy number, genetically variable loci in Lecythidaceae (Vargas et al., 2019), allowed us to investigate evolutionary history using phylogenetic and Bayesian clustering approaches. Our inferences were based on highly variable coding regions, which may be under natural selection. The effect that targeting such regions has on studies of admixture and species delimitation warrants further study. While using neutral markers or a larger number of loci may have led to different estimates of ancestry, our dataset allowed us to identify admixed individuals and distinguish intraspecific geographic variation from admixture using an explicit test. There are drawbacks to using target capture at infraspecific phylogenetic scales, including the relatively high per sample cost compared with other reduced‐representation genome sequencing approaches such as RADseq. However, RADseq protocols often require relatively high‐molecular‐weight input DNA (Graham et al., 2015), while target capture can more readily allow researchers to include samples from partially degraded herbarium specimens (Brewer et al., 2019). We recovered sequences from 343 loci, far fewer than often recovered with RADseq, but far more than most studies that use microsatellites. However, unlike for many RADseq datasets, we recovered sequence data for nearly all target loci for most samples (average 339.6 of 344 loci per individual), which enabled gene tree‐based methods for explicitly testing hypotheses of admixture. Our results suggest that target capture can be used to study admixture in topical trees and may be especially useful for studies that wish to include herbarium specimens.
Author contributions
DAL and CWD conceived the study. AV hosted the field work, obtained collection and export permits and provided access to existing BDFFP collections. DAL and CWD conducted the field work to obtain new collections. DAL conducted the laboratory work for focal species and designed and performed the analyses. OMV led the sampling and laboratory work for the broader phylogeny. OMV and DAL mapped collection records. The figures and tables were prepared by DAL. The manuscript was written by DAL with editing by CWD and input from all authors. All authors contributed to and approved the final version of the manuscript.
Supporting information
Fig. S1 Schematic of the process for making redeterminations based on all available evidence.
Fig. S2 The preliminary phylogeny of the Parvifolia clade estimated from a supermatrix of intron and exon target capture data.
Fig. S3 Results of several structure analyses with alternative values of K.
Fig. S4 Results of structure analyses using a SNP dataset for the clade that included E. coriacea, E. wachenheimii, E. sagotiana, E. truncata and E. parviflora.
Fig. S6 The Parvifolia phylogeny without reduced representation, produced using a supermatrix of intron and exon target capture data.
Fig. S7 Comparison of the reduced‐representation Parvifolia phylogenies recovered with two supermatrices.
Fig. S8 Boxplots overlayed with dot plots showing the day of the year that collections were made for specimens at the New York Botanical Garden Herbarium.
Methods S1 Paralog filtering and alignment.
Methods S2 Genotyping and SNP dataset analyses.
Methods S3 Redetermination of individuals based on all available evidence.
Methods S4 Parvifolia phylogeny supermatrix construction and phylogeny estimation.
Notes S1 Note on species of the Parvifolia clade at Reserve 1501.
Notes S2 Notes on sampling at Reserve 1501 and prioritization of morphological intermediates.
Fig. S5 Three‐dimensional scatterplot for a genetic principal component analysis showing geographical structuring in samples of E. coriacea.
Table S1 Voucher and accession information for samples used in the study.
Table S2 Summary statistics for all SNP datasets and estimated probability of the data for all structure analyses for differing values of K.
Table S3 Summary of all rooted triplet tests conducted.
Table S4 Results of tree searches and likelihood recalculations for the Parvifolia phylogenies, ordered by increasing Akaike information criterion (AIC) score.
Table S5 Summaries of morphological and ecological traits of species inferred to engage in admixture.
Table S6 Information for flowering specimens collected in Amazonas, Brazil and housed at the New York Botanical Garden Herbarium.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Acknowledgements
We dedicate this work to the memory of two of our collaborators. Scott A. Mori (1941–2020) was the Lecythidaceae specialist who co‐founded the Reserve 1501 Lecythidaceae plot and kindly and generously supported our work. Paulo A. C. L. Assunção (1956–2021) was a field technician who trained with Scott on the Lecythidaceae plot in the 1980s, became a renowned field botanist in his own right and trained us in Lecythidaceae field identification. We thank Priscila Souza, Juvenal Batista, Michel Ribeiro, Bruno Garcia Luize and Xavier Cornejo for contributing collections for our broader sampling. We thank Priscila Souza, Tamara Milton, Nicolli Cabello and BDFFP Director José Luís Camargo for their help in Brazil and Joseph F. Walker for several helpful discussions about theory. We thank Stephen Smith, Deise Gonçalves, Hannah Marx, Hector Figueroa, Tamara Milton and three anonymous reviewers for their comments on a previous version of the manuscript. Financial support came from NSF FESD 1338694 to DAL and OMV and NSF DEB 1240869 to CWD. Field work by DAL was also supported by the Global Fellowship in Agricultural Development within CA&ES at the University of California, Davis. Samples from Brazil were part of a collaboration (CNPq AEX 01300.006387/2017‐42) between the University of Michigan and the National Institute of Amazonian Research (INPA) and were registered in SISGEN‐Brazil (no. AA0B72D) by AV. Collecting at BDFFP reserves was conducted under ICMBio permit no. 58484‐1. This is study number 819 of the Technical Series of the Biological Dynamics of Forest Fragments Project (BDFFP – INPA/STRI).
Data availability
The data, novel scripts and output files that support the findings of this study are openly available from Dryad at https://doi.org/10.5061/dryad.fj6q573t4. Raw sequence reads are available from NCBI BioProject PRJNA641333.
References
- Andrews S. 2010. FastQC: a quality control tool for high throughput sequence data. [WWW document] URL http://www.bioinformatics.babraham.ac.uk/projects/fastqc [accessed 6 November 2018]. [Google Scholar]
- Ashton PS. 1969. Speciation among tropical forest trees: some deductions in the light of recent evidence. Biological Journal of the Linnaean Society 1: 155–196. [Google Scholar]
- Baker WJ, Bailey P, Barber V, Barker A, Bellot S, Bishop D, Botigué LR, Brewer G, Carruthers T, Clarkson JJ et al. 2021. A comprehensive phylogenomic platform for exploring the angiosperm tree of life. Systematic Biology. doi: 10.1093/sysbio/syab035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD et al. 2012. SPAdes: a new genome assembly algorithm and its appplications to single‐sell sequencing. Journal of Computational Biology 19: 455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista GJE, Mori SA, Harrison JS. 2017. New species of Eschweilera and a first record of Cariniana (Lecythidaceae) from Panama. Phytoneuron 2017–62: 1–16. [Google Scholar]
- Brewer GE, Clarkson JJ, Maurin O, Zuntini AR, Barber V, Bellot S, Biggs N, Cowan RS, Davies NMJ, Dodsworth S et al. 2019. Factors affecting targeted sequencing of 353 nuclear genes from herbarium specimens spanning the diversity of Angiosperms. Frontiers in Plant Science 10: 1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JW, Walker JF, Smith SA. 2017. Phyx: phylogenetic tools for unix. Bioinformatics 33: 1886–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant D, Hahn MW. 2020. The concatenation question. In: Delsuc F, Galtier N, Scornavacca C, eds. Phylogenetics in the Genomic Era. No commercial publisher. URL https://hal.inria.fr/PGE/. [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10: 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon CH, Lerdau M. 2015. Variable mating behaviors and the maintenance of tropical biodiversity. Frontiers in Genetics 6: 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron H, Molino J‐F, Sabatier D, Léger P, Chaumeil P, Scotti‐Saintagne C, Frigério J‐M, Scotti I, Franc A, Petit RJ. 2019. Chloroplast DNA variation in a hyperdiverse tropical tree community. Ecology and Evolution 9: 4897–4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavender‐Bares J. 2019. Diversification, adaptation, and community assembly of the American oaks (Quercus), a model clade for integrating ecology and evolution. New Phytologist 221: 669–692. [DOI] [PubMed] [Google Scholar]
- Chernomor O, von Haeseler A, Minh BQ. 2016. Terrace aware data structure for phylogenomic inference from supermatrices. Systematic Biology 65: 997–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JY, Purugganan M, Stacy EA. 2020. Divergent selection and primary gene flow shape incipient speciation of a riparian tree on Hawaii Island. Molecular Biology and Evolution 37: 695–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christe C, Boluda CG, Koubínová D, Gautier L, Naciri Y. 2021. New genetic markers for Sapotaceae phylogenomics: more than 600 nuclear genes applicable from family to population levels. Molecular Phylogenetics and Evolution 160: 107123. [DOI] [PubMed] [Google Scholar]
- Cock PJA, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B et al. 2009. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25: 1422–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couvreur TLP, Helmstetter AJ, Koenen EJM, Bethune K, Brandão RD, Little SA, Sauquet H, Erkens RHJ. 2019. Phylogenomics of the major tropical plant family Annonaceae using targeted enrichment of nuclear genes. Frontiers in Plant Science 9: 1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronn R, Knaus BJ, Liston A, Maughan PJ, Parks M, Syring JV, Udall J. 2012. Targeted enrichment strategies for next‐generation plant biology. American Journal of Botany 99: 291–311. [DOI] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M et al. 2011. A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nature Genetics 43: 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick CW, Heuertz M. 2008. The complex biogeographic history of a widespread tropical tree species. Evolution 62: 2760–2774. [DOI] [PubMed] [Google Scholar]
- Dick CW, Pennington RT. 2019. History and geography of Neotropical tree diversity. Annual Review of Ecology, Evolution, and Systematics 50: 279–301. [Google Scholar]
- Eaton DAR, Hipp AL, González‐Rodríguez A, Cavender‐Bares J. 2015. Historical introgression among the American live oaks and the comparative nature of tests for introgression. Evolution 69: 2587–2601. [DOI] [PubMed] [Google Scholar]
- Eaton DAR, Ree RH. 2013. Inferring phylogeny and introgression using RADseq data: an example from flowering plants (Pedicularis: Orobanchaceae). Systematic Biology 62: 689–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham CF, Glenn TC, McArthur AG, Boreham DR, Kieran T, Lance S, Manzon RG, Martino JA, Pierson T, Rogers SM et al. 2015. Impacts of degraded DNA on restriction enzyme associated DNA sequencing (RADSeq). Molecular Ecology Resources 15: 1304–1315. [DOI] [PubMed] [Google Scholar]
- Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, Patterson N, Li H, Zhai W, Fritz MH‐Y et al. 2010. A draft sequence of the neandertal genome. Science 328: 710–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanlon VC, Otto SP, Aitken SN. 2019. Somatic mutations substantially increase the per‐generation mutation rate in the conifer Picea sitchensis . Evolution Letters 3: 348–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardin JW. 1975. Hybridization and introgression in Quercus Alba . Journal of the Arnold Arboretum 56: 336–363. [Google Scholar]
- Heuertz M, Caron H, Scotti‐Saintagne C, Pétronelli P, Engel J, Tysklind N, Miloudi S, Gaiotto FA, Chave J, Molino J‐F. 2020. The hyperdominant tropical tree Eschweilera coriacea (Lecythidaceae) shows higher genetic heterogeneity than sympatric Eschweilera species in French Guiana. Plant Ecology and Evolution 153: 67–81. [Google Scholar]
- Hipp AL, Manos PS, Hahn M, Avishai M, Bodénès C, Cavender‐Bares J, Crowl AA, Deng M, Denk T, Fitz‐Gibbon S et al. 2020. Genomic landscape of the global oak phylogeny. New Phytologist 226: 1198–1212. [DOI] [PubMed] [Google Scholar]
- Holm S. 1979. A simple sequentially rejective multiple test procedure. Scandinavian Journal of Statistics 6: 65–70. [Google Scholar]
- Huang Y‐Y, Mori SA, Kelly LM. 2015. Toward a phylogenetic‐based generic classification of Neotropical Lecythidaceae—I. Status of Bertholletia, Corythophora, Eschweilera and Lecythis . Phytotaxa 203: 85–121. [Google Scholar]
- Huson DH, Klöpper T, Lockhart PJ, Steel MA. 2005. Reconstruction of reticulate networks from gene trees. In: Miyano S , Mesirov J, Kasif S, Istrail S, Pevzner PA, Waterman M, eds. Research in computational molecular biology. Berlin/Heidelberg: Springer, 233–249. [Google Scholar]
- Johnson MG, Gardner EM, Liu Y, Medina R, Goffinet B, Shaw AJ, Zerega NJC, Wickett NJ. 2016. HybPiper: extracting coding sequence and introns for phylogenetics from high‐throughput sequencing reads using target enrichment. Applications in Plant Sciences 4: 1600016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MG, Pokorny L, Dodsworth S, Botigué LR, Cowan RS, Devault A, Eiserhardt WL, Epitawalage N, Forest F, Kim JT et al. 2018. A universal probe set for targeted sequencing of 353 nuclear genes from any flowering plant designed using k‐medoids clustering. Systematic Biology 68: 594–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Research 30: 3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution 30: 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearns AM, Restani M, Szabo I, Schrøder‐Nielsen A, Kim JA, Richardson HM, Marzluff JM, Fleischer RC, Johnsen A, Omland KE. 2018. Genomic evidence of speciation reversal in ravens. Nature Communications 9: doi: 10.1038/s41467-018-03294-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer A, Hipp AL. 2020. Oaks: an evolutionary success story. New Phytologist 226: 987–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson DA, Walker JF, Vargas OM, Smith SA. 2020. A consensus phylogenomic approach highlights paleopolyploid and rapid radiation in the history of Ericales. American Journal of Botany 107: 773–789. [DOI] [PubMed] [Google Scholar]
- Lehner B, Grill G. 2013. Global river hydrography and network routing: baseline data and new approaches to study the world’s large river systems. Hydrological Processes 27: 2171–2186. [Google Scholar]
- Leroy T, Louvet J‐M, Lalanne C, Le Provost G, Labadie K, Aury J‐M, Delzon S, Plomion C, Kremer A. 2020. Adaptive introgression as a driver of local adaptation to climate in European white oaks. New Phytologist 226: 1171–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM. arXiv preprint arXiv:1303.3997. [WWW document] URL https://arxiv.org/abs/1303.3997.
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R ; 1000 Genome Project Data Processing Subgroup . 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linan AG, Lowry PP, Miller AJ, Schatz GE, Sevathian J‐C, Edwards CE. 2020. RAD‐sequencing reveals patterns of diversification and hybridization, and the accumulation of reproductive isolation in a clade of partially sympatric, tropical island trees. Molecular Ecology. doi: 10.1111/mec.15736. [DOI] [PubMed] [Google Scholar]
- Loiseau O, Olivares I, Paris M, de La Harpe M, Weigand A, Koubínová D, Rolland J, Bacon CD, Balslev H, Borchsenius F et al. 2019. Targeted capture of hundreds of nuclear genes unravels phylogenetic relationships of the diverse Neotropical palm tribe Geonomateae. Frontiers in Plant Science 10: 864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotsy JP. 1925. Species or linneon. Genetica 7: 487–506. [Google Scholar]
- Martin NH, Bouck AC, Arnold ML. 2006. Detecting adaptive trait introgression between Iris fulva and I. brevicaulis in highly selective field conditions. Genetics 172: 2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M. 2010. The genome analysis toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Research 20: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales‐Briones DF, Kadereit G, Tefarikis DT, Moore MJ, Smith SA, Brockington SF, Timoneda A, Yim WC, Cushman JC, Yang Y. 2020. Disentangling sources of gene tree discordance in phylogenomic data sets: testing ancient hybridizations in Amaranthaceae s.l. Systematic Biology 70: 219–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori SA, Becker P, Kincaid D. 2001. Lecythidaceae of a central Amazonian lowland forest: implications for conservation. In: Bierregaard RO, Gascon C, Lovejoy TE, Mesquita R, eds. Lessons from Amazonia: the ecology and conservation of a fragmented forest. New Haven, CT, USA: Yale University Press, 55–67. [Google Scholar]
- Mori SA, Kiernan EA, Smith NP, Kelly LM, Huang Y‐Y, Prance GT, Thiers BM. 2017. Observations on the phytogeography of the Lecythidaceae clade (Brazil nut family). Phytoneuron 30: 1–85. [Google Scholar]
- Mori SA, Lepsch‐Cunha N. 1995. The Lecythidaceae of a central Amazonian moist forest. Bronx, NY, USA: New York Botanical Garden Press. [Google Scholar]
- Mori SA, Prance GT. 1990. Lecythidaceae, Part 2. The zygomorphic‐flowered New World genera (Couroupita, Corythophora, Bertholletia, Couratari, Eschweilera, & Lecythis). New York, NY, USA: New York Botanical Garden Press. [Google Scholar]
- Mori SA, Smith NP, Cornejo X, Prance GT. 2010. The Lecythidaceae pages. The Lecythidaceae pages. [WWW document] URL http://sweetgum.nybg.org/science/projects/lp/ [accessed 9 July 2020]. [Google Scholar]
- Mori SA, Smith NP, Huang Y‐Y, Prance GT, Kelly LM, Matos CC. 2015. Toward a phylogenetic‐based generic classification of Neotropical Lecythidaceae—II. Status of Allantoma, Cariniana, Couratari, Couroupita, Grias and Gustavia . Phytotaxa 203: 122–137. [Google Scholar]
- Nazareno AG, Dick CW, Lohmann LG. 2019. A biogeographic barrier test reveals a strong genetic structure for a canopy‐emergent Amazon tree species. Scientific Reports 9: 18602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L‐T, Schmidt HA, von Haeseler A, Minh BQ. 2014. IQ‐TREE: a fast and effective stochastic algorithm for estimating maximum‐likelihood phylogenies. Molecular Biology and Evolution 32: 268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paetzold C, Wood KR, Eaton DAR, Wagner WL, Appelhans MS. 2019. Phylogeny of Hawaiian Melicope (Rutaceae): RAD‐seq resolves species relationships and reveals ancient introgression. Frontiers in Plant Science 10: 1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnell J, Pedersen H, Hodkinson T, Balslev H, Welzen PC, Simpson D, Middleton D, Esser H‐J, Pooma R, Utteridge T et al. 2013. Hybrids and the flora of Thailand. Thai Forest Bulletin 41: 1–9. [Google Scholar]
- Pease JB, Haak DC, Hahn MW, Moyle LC. 2016. Phylogenomics reveals three sources of adaptive variation during a rapid radiation. PLoS Biology 14: e1002379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poplin R, Ruano‐Rubio V, DePristo MA, Fennell TJ, Carneiro MO, Van der Auwera GA, Kling DE, Gauthier LD, Levy‐Moonshine A, Roazen D et al. 2017. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv. doi: 10.1101/201178 [DOI] [Google Scholar]
- Prance GT, Mori SA. 1979. Lecythidaceae: Part I: The actinomorphic‐flowered New World Lecythidaceae (Asteranthos, Gustavia, Grias, Allantoma, & Cariniana). Bronx, NY, USA: New York Botanical Garden Press. [Google Scholar]
- Prata EMB, Sass C, Rodrigues DP, Domingos FMCB, Specht CD, Damasco G, Ribas CC, Fine PVA, Vicentini A. 2018. Towards integrative taxonomy in Neotropical botany: disentangling the Pagamea guianensis species complex (Rubiaceae). Botanical Journal of the Linnean Society 188: 213–231. [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. 2000. Inference of population structure using multilocus genotype data. Genetics 155: 945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Wen W, Falush D. 2010. Documentation for STRUCTURE software: v.2.3. Chicago, IL, USA: University of Chicago. [Google Scholar]
- Puechmaille SJ. 2016. The program structure does not reliably recover the correct population structure when sampling is uneven: subsampling and new estimators alleviate the problem. Molecular Ecology Resources 16: 608–627. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd‐Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ et al. 2007. PLINK: a tool set for whole‐genome association and population‐based linkage analyses. American Journal of Human Genetics 81: 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . 2019. R: a language and environment for statistical computing, v.3.6.0. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- Revell LJ. 2012. phytools: an R package for phylogenetic comparative biology (and other things). Methods in Ecology and Evolution 3: 217–223. [Google Scholar]
- Rieseberg LH, Wendel JF. 1993. Introgression and its consequences in plants. In: Harrison RG, ed. Hybrid zones and the evolutionary process. New York, NY, USA: Oxford University Press, 70–109. [Google Scholar]
- Rose JP, Kleist TJ, Löfstrand SD, Drew BT, Schönenberger J, Sytsma KJ. 2018. Phylogeny, historical biogeography, and diversification of angiosperm order Ericales suggest ancient Neotropical and East Asian connections. Molecular Phylogenetics and Evolution 122: 59–79. [DOI] [PubMed] [Google Scholar]
- Schley RJ, Pennington RT, Pérez‐Escobar OA, Helmstetter AJ, de la Estrella M, Larridon I, Sabino Kikuchi IAB, Barraclough TG, Forest F, Klitgård B. 2020. Introgression across evolutionary scales suggests reticulation contributes to Amazonian tree diversity. Molecular Ecology 29: 4170–4185. [DOI] [PubMed] [Google Scholar]
- Schmitt S, Tysklind N, Derroire G, Heuertz M, Hérault B. 2021. Topography shapes the local coexistence of tree species within species complexes of Neotropical forests. Oecologia 196: 389–398. [DOI] [PubMed] [Google Scholar]
- Sievert C. 2020. Interactive web‐based data visualization with R, plotly, and shiny. Boca Raton, FL, USA: CRC Press. [Google Scholar]
- Slater GSC, Birney E. 2005. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics 30: 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ter Steege H, Pitman NCA, Sabatier D, Baraloto C, Salomão RP, Guevara JE, Phillips OL, Castilho CV, Magnusson WE, Molino J‐F et al. 2013. Hyperdominance in the Amazonian tree flora. Science 342: 1243092. [DOI] [PubMed] [Google Scholar]
- ter Steege H, Prado PI, de Lima RAF, Pos E, de Souza CL, de Andrade Lima Filho D, Salomão RP, Amaral IL, de Almeida Matos FD, Castilho CV et al. 2020. Biased‐corrected richness estimates for the Amazonian tree flora. Scientific Reports 10: 10130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez‐Gonzalez A, Lexer C, Cronk QCB. 2018. Adaptive introgression: a plant perspective. Biology Letters 14: 20170688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tange O. 2011. Gnu parallel‐the command‐line power tool. USENIX Magazine 36: 42–47. [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, del Angel G, Levy‐Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J et al. 2013. From FastQ data to high‐confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Current Protocols in Bioinformatics 43: 11.10.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas OM, Dick CW. 2020. Diversification history of Neotropical Lecythidaceae, an ecologically dominant tree family of Amazon rain forest. In: Rull V, Carnaval AC, eds. Neotropical diversification: patterns and processes. Cham. Switzerland: Springer International, 791–809. [Google Scholar]
- Vargas OM, Goldston B, Grossenbacher DL, Kay KM. 2020. Patterns of speciation are similar across mountainous and lowland regions for a Neotropical plant radiation (Costaceae: Costus). Evolution 74: 2644–2661. [DOI] [PubMed] [Google Scholar]
- Vargas OM, Heuertz M, Smith SA, Dick CW. 2019. Target sequence capture in the Brazil nut family (Lecythidaceae): marker selection and in silico capture from genome skimming data. Molecular Phylogenetics and Evolution 135: 98–104. [DOI] [PubMed] [Google Scholar]
- Whitney KD, Randell RA, Rieseberg LH. 2010. Adaptive introgression of abiotic tolerance traits in the sunflower Helianthus annuus . New Phytologist 187: 230–239. [DOI] [PubMed] [Google Scholar]
- Wickham H. 2016. ggplot2: elegant graphics for data analysis. New York, NY, USA: Springer‐Verlag. [Google Scholar]
- Yang Y, Smith SA. 2014. Orthology inference in nonmodel organisms using transcriptomes and low‐coverage genomes: improving accuracy and matrix occupancy for phylogenomics. Molecular Biology and Evolution 31: 3081–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhbannikov IY, Hunter SS, Foster JA, Settles ML. 2017. SeqyClean: a pipeline for high‐throughput sequence data preprocessing. In: Proceedings of the 8th ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics. New York, NY, USA: ACM, 407–416. [Google Scholar]
- Zheng X, Levine D, Shen J, Gogarten SM, Laurie C, Weir BS. 2012. A high‐performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 28: 3326–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Schematic of the process for making redeterminations based on all available evidence.
Fig. S2 The preliminary phylogeny of the Parvifolia clade estimated from a supermatrix of intron and exon target capture data.
Fig. S3 Results of several structure analyses with alternative values of K.
Fig. S4 Results of structure analyses using a SNP dataset for the clade that included E. coriacea, E. wachenheimii, E. sagotiana, E. truncata and E. parviflora.
Fig. S6 The Parvifolia phylogeny without reduced representation, produced using a supermatrix of intron and exon target capture data.
Fig. S7 Comparison of the reduced‐representation Parvifolia phylogenies recovered with two supermatrices.
Fig. S8 Boxplots overlayed with dot plots showing the day of the year that collections were made for specimens at the New York Botanical Garden Herbarium.
Methods S1 Paralog filtering and alignment.
Methods S2 Genotyping and SNP dataset analyses.
Methods S3 Redetermination of individuals based on all available evidence.
Methods S4 Parvifolia phylogeny supermatrix construction and phylogeny estimation.
Notes S1 Note on species of the Parvifolia clade at Reserve 1501.
Notes S2 Notes on sampling at Reserve 1501 and prioritization of morphological intermediates.
Fig. S5 Three‐dimensional scatterplot for a genetic principal component analysis showing geographical structuring in samples of E. coriacea.
Table S1 Voucher and accession information for samples used in the study.
Table S2 Summary statistics for all SNP datasets and estimated probability of the data for all structure analyses for differing values of K.
Table S3 Summary of all rooted triplet tests conducted.
Table S4 Results of tree searches and likelihood recalculations for the Parvifolia phylogenies, ordered by increasing Akaike information criterion (AIC) score.
Table S5 Summaries of morphological and ecological traits of species inferred to engage in admixture.
Table S6 Information for flowering specimens collected in Amazonas, Brazil and housed at the New York Botanical Garden Herbarium.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Data Availability Statement
The data, novel scripts and output files that support the findings of this study are openly available from Dryad at https://doi.org/10.5061/dryad.fj6q573t4. Raw sequence reads are available from NCBI BioProject PRJNA641333.