

Abstract
Eukaryote symbionts of animals are major drivers of ecosystems not only because of their diversity and host interactions from variable pathogenicity but also through different key roles such as commensalism and to different types of interdependence. However, molecular investigations of metazoan eukaryomes require minimising coamplification of homologous host genes. In this study we (1) identified a previously published “antimetazoan” reverse primer to theoretically enable amplification of a wider range of microeukaryotic symbionts, including more evolutionarily divergent sequence types, (2) evaluated in silico several antimetazoan primer combinations, and (3) optimised the application of the best performing primer pair for high throughput sequencing (HTS) by comparing one‐step and two‐step PCR amplification approaches, testing different annealing temperatures and evaluating the taxonomic profiles produced by HTS and data analysis. The primer combination 574*F – UNonMet_DB tested in silico showed the largest diversity of nonmetazoan sequence types in the SILVA database and was also the shortest available primer combination for broadly‐targeting antimetazoan amplification across the 18S rRNA gene V4 region. We demonstrate that the one‐step PCR approach used for library preparation produces significantly lower proportions of metazoan reads, and a more comprehensive coverage of host‐associated microeukaryote reads than the two‐step approach. Using higher PCR annealing temperatures further increased the proportion of nonmetazoan reads in all sample types tested. The resulting V4 region amplicons were taxonomically informative even when only the forward read is analysed. This region also revealed a diversity of known and putatively parasitic lineages and a wider diversity of host‐associated eukaryotes.
Keywords: antimetazoan, eDNA, eukaryome, metabarcoding, microeukaryote, symbiome
1. INTRODUCTION
It is increasingly recognised that larger organisms provide an environment for a range of other smaller organisms, including bacteria, viruses, and eukaryotes that are not exclusively microeukaryotes. Arising from this model is the holobiont concept—that is, the host and its microbiota (Rohwer et al., 2002; Skillings, 2016). However, the diversity of host‐associated organisms is greater than those involved in long‐term and/or “active” associations with the host, including transient and/or minimally interacting associations (for example a larger organism acting as a mechanical vector of a smaller one). Therefore, more recently the terms “symbiont” and “symbiome” have been used to describe the totality of host‐associated organisms (Bass & del Campo, 2020; Bass et al., 2019).
Host‐associated bacteria have received the most attention so far, in large part due to their importance for health and nutrition of vertebrates, for example human and ungulate gut microbiomes (Bull & Plummer, 2014; O’Hara et al., 2020). Bacterial diversity studies have also been carried out in environmental systems for several decades, so far culminating in large‐scale studies initiatives like the Earth Microbiome Project (Gilbert et al., 2010) and the Tara Oceans (Karsenti et al., 2011). Such studies are facilitated by the availability of a well‐structured molecular and analytical toolkit based on small‐subunit (16S) ribosomal RNA amplicon sequencing, which focuses the genetic screening effort onto a well understood and comprehensively sampled taxonomic marker gene that excludes other domains of life (Lozupone et al., 2013; Qin et al., 2010; Tringe & Hugenholtz, 2008). Following the bacterial lead, broadly‐targeted microeukaryotic small subunit (18S) gene surveys of environmental samples were initiated in the early 2000s targeting protists (reviewed in Amaral‐Zettler et al., 2009; Stoeck et al., 2009).
Although 16S amplicon approaches are well suited to investigate animal host‐associated bacteria, the equivalent approach for eukaryotes (18S) is generally unsuitable for this purpose as host 18S genes are coamplified, often completely obscuring signal from any other (host‐associated) eukaryotes. This is true for the study of both plant and animal eukaryomes, as microeukaryotes are paraphyletic with respect to both animals and plants. To obviate this issue for animal hosts, a “universal nonmetazoan” PCR system facilitating amplification of parasitic protistan 18S fragments (encompassing the hypervariable V4 region) from infected animal tissues was developed (Bower et al., 2004; Carnegie et al., 2003) and subsequently adapted for high throughput metabarcoding sequencing (del Campo et al., 2019). The latter is a two‐step method in which 18S V4 region amplicons are firstly generated by the universal nonmetazoan primer pair are then reamplified by the “universal” V4 primer pair E572F ‐ E1009R (Comeau et al., 2011) to generate amplicons of suitable length for high throughput sequencing (HTS) (e.g., Illumina MiSeq platform) (del Campo et al., 2019). This method significantly reduced metazoan signal both in silico and in vitro (Bass & del Campo, 2020; del Campo et al., 2019). However, the use of a two‐step amplification for diversity studies can increase PCR amplification biases leading to skewed taxonomic representations, and potentially increasing the sequence error rate (del Campo et al., 2019). These considerations also apply to any methodological decision relevant to metabarcoding studies in general.
Therefore it is necessary to determine whether generating sequence libraries in a single step, using fully‐formed fusion primers following Illumina's TruSeq genomic library construction protocols (forward and reverse primers containing the Illumina adapter sequence, the sample‐specific index, the pad and link sequences, and the gene‐specific primer; Caporaso et al., 2011; Kozich et al., 2013) is preferable to a two‐step approach: initial amplification with gene‐specific primers connected to overhang sequences, followed by a second amplification to add the Illumina adapter sequences, the sample‐specific index, and the pad and link sequences (Baetens et al., 2011; Cruaud et al., 2017). There is some debate over the choice of using fusion primers (one‐step) or the two‐step approach. The former is quicker and less prone to cross‐contamination (O’Donnell et al., 2016; Zizka et al., 2019), yet it can be more susceptible to PCR inhibition (Schnell et al., 2015) and more costly, since several primer versions carrying different in‐line tags will be needed (Elbrecht & Steinke, 2019). The two‐step is overall more laborious with extra cleaning steps that could render more contaminations (Schnell et al., 2015) and produce slightly lower taxa numbers (Zizka et al., 2019).
Primer choice greatly influences the taxonomic recovery from a given sample and despite primer optimization steps, PCR inhibition, template sequence variation at the primer sites, and other taxonomic biases, meaning that it is very unlikely that all taxa present will be retrieved. Nonetheless, to better match a wider range of microeukaryotic symbionts and parasites, including divergent lineages, the original nonmetazoan PCR primers were modified by Bass and del Campo (2020), using sequence alignment and in silico analyses to determine a more inclusive primer combination: the forward primer 574*F (Hugerth et al., 2014) and a revised version of the reverse primer 18S‐EUK‐1134‐R (Bower et al., 2004; Carnegie et al., 2003): UNonMet_DB (Bass & del Campo, 2020). When tested in silico, this combination amplified a greater diversity of nonmetazoan sequence types than other forward primers with UNonMet_DB, including theoretical amplification of 50% of all excavates and 78% of amoebozoans present in the reference database SILVA 138 RefNR (Bass & del Campo, 2020; Yilmaz et al., 2011). Interestingly and usefully, the primer pair also amplified 43% of myxozoans, highly specialised parasitic cnidarians (Atkinson et al., 2018). The antimetazoan primer strategy is different from the blocking primer approach since it can be used as a classic one‐step PCR reaction, does not require the addition of dideoxynucleotide‐labelled primers to inhibit the enzymatic elongation of metazoan reads (Seyama et al., 1992), and, as the size of the primers is constrained (forward: 17 bp; reverse: 20 bp), it is suitable for the addition of Illumina adapters, indexes, pad and link for high throughput sequencing.
Microeukaryotic pathogens/symbionts of larger organisms are continually being discovered, and a unified method of investigating these while avoiding nontarget amplification is a powerful tool (Bass & del Campo, 2020; Bass et al., 2015; Bower et al., 2004; del Campo et al., 2020). The more nuanced roles of a wider diversity of host‐associated eukaryotes in the “symbiotic continuum” (Bass & del Campo, 2020; del Campo et al., 2019), and in the pathobiome (Bass et al., 2019), further increase demand for a simple high throughput protocol for learning more about these hitherto underinvestigated symbionts.
The aim of this study was to determine whether a one‐step PCR amplification using the antimetazoan 574*F—UNonMet_DB (Antimet‐1) primer pair is preferable to a two‐step PCR (Antimet‐2) to generate an amplicon of suitable length for recovering paired‐end contigs with minimal metazoan/host reads amplification, using environmental and animal tissue samples. We compared the taxonomic profiles generated by one‐ and two‐step antimetazoan PCR approaches against the universal eukaryotic primer pair 574*F–1132r (Hugerth et al., 2014) to determine whether they behaved comparably, and if not, how the results would influence the balance of the advantages and disadvantages of each approach as outlined above. We also tested the effect of different annealing temperatures on the taxonomic outputs and evaluated in vitro the performance of the antimetazoan primer pair 574*F–UNonMet_DB.
2. MATERIALS AND METHODS
2.1. Samples
Four type of samples were used: (1) gills from an individual barb (Barbus sp.) infected with the myxozoan Myxobolus sp. (parasitic infection confirmed by pathological examination, PCR, and Sanger sequencing, data not shown), and (2) hepatopancreas from an individual whiteleg shrimp (Litopenaeus vannamei) infected with the microsporidian Enterocytozoon hepatopenaei (EHP, parasitic infection confirmed by pathological examination, PCR, and Sanger sequencing, data not shown) collected from two aquaculture ponds in Thailand (undisclosed locations); (3) unsorted plankton grab, dominated by the copepods Paracartia grani and Acartia spp, collected from water column with a WP2 net equipped with a 180 µm mesh cup and sieved through a 1 mm plankton mesh to remove debris and jellyfish; and (4) cephalothorax from an individual common prawn (Palaemon serratus) both collected from Agapollen (59°50′23.6″N 5°14′49.7″E, Norway). All samples were stored in 100% ethanol until further processing.
2.2. DNA extraction
Fifty to 100 mg of barb gills and shrimp hepatopancreas were homogenised with mortar and pestle in 500 µl of lysis buffer (50 mM TRIS pH 9, 0.1 M EDTA pH 8, 50 mM NaCl, 2% SDS, filter sterilised with a ⌀ 0.2 µm filter) containing 10 µl of proteinase K at 5 µg/ml, followed by a 30 min incubation at 65°C. Total DNA was immediately purified using a standard phenol‐chloroform extraction method (Green & Sambrook, 2012). Fifty to 100 mg of whole plankton and whole common prawn were homogenised with Lysing Matrix A FastPrep tubes in 800 µl of Lifton buffer (100 mM EDTA, 25 mM Tris‐HC1 pH 7.5, 1% SDS) containing 20 µl of proteinase K at 10 µg/ml, followed by overnight incubation at 56°C. Total DNA was immediately purified using a standard phenol‐chloroform extraction method (Nishiguchi et al., 2002). DNA concentrations were measured with the QuantiFluor ONE dsDNA System (Promega) fluorometric assay and samples diluted in molecular grade water to 5 ng/µl.
2.3. In silico primer evaluation
We selected the two most taxonomically inclusive antimetazoan primer combinations recently featured in the literature (574*F—UNonMet_DB [Bass & del Campo, 2020; Hugerth et al., 2014]; 18S‐EUK581‐F—18S‐EUK‐1134‐R [Bower et al., 2004; Carnegie et al., 2003]), and a “universal” eukaryote primer pair (574*F–1132r [Hugerth et al., 2014]). Primers sequences are provided in Appendix S1. Primer combinations were compared and evaluated in silico using TestPrime (Klindworth et al., 2013) against SILVA 138 RefNR (Yilmaz et al., 2011) database, with no mismatches allowed between primers and in silico template sequences, flanking a 574 bp of the 18S rRNA V4 gene region.
2.4. In vitro HTS primer design
Amplification of the 18S rRNA ~574 bp gene with one‐step PCR approaches was obtained using primer pair 574*F and 1132r (Hugerth et al., 2014), here referred as “universal”, and the antimetazoan primer pair 574*F and UNonMet_DB (Bass & del Campo, 2020), here referred as “Antimet‐1”. All forward and reverse primer combinations were designed to include the Illumina adapter (P5/P7), 8 bp indexes (i5/i7), pad and link, and 18S primers (Figure 1; Appendix S1).
FIGURE 1.
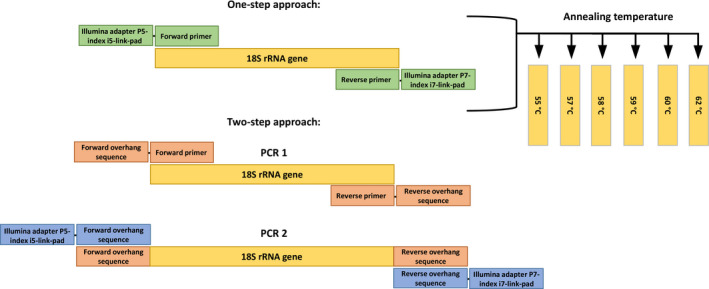
Experimental design scheme highlighting differences between the one‐ and two‐step antimetazoan PCR approaches tested in this study
The two‐step PCR approach (here referred as “Antimet‐2”) amplified the same V4 region as the Antimet‐1; however the PCR amplification was performed in two steps. For the first round PCR, all forward and reverse primers comprised the antimetazoan primer pair, each with an overhang sequence (Illumina TruSeq Read 1 and 2 sequences). For the second round PCR, all forward and reverse primer combinations included the Illumina adapter (P5/P7), 8 bp indexes (i5/i7), and primer overhang sequence (Figure 1).
2.5. PCR amplification and library preparation
The primer combinations, annealing temperatures and three PCR approaches (antimetazoan and “universal” one‐step and antimetazoan two‐step) were tested for each sample. All PCR approaches were set in triplicates with each replicate tagged by different combinations of i5 and i7 indexes to allow bioinformatic demultiplexing of each sample and replicate (Appendix S1).
PCR reactions for the Antimet‐1 and universal PCRs contained 12.5 µl of NEBNext PCR mix (New England BioLabs), 10 ng of DNA, 0.5 µM of each primer (final concentration), and molecular grade water up to a final volume of 25 µl. PCR cycling conditions consisted of 30 s of denaturation at 98°C, followed by 30 cycles of 10 s at 98°C, 30 s at 6 specific annealing temperatures (T a), 30 s at 72°C, and final extension for 2 min at 72°C. T as (Temp 1–6) were: Temp 1 = 55°C; Temp 2 = 57°C; Temp 3 = 58°C; Temp 4 = 59°C; Temp 5 = 60°C; Temp 6 = 62°C.
The first round of the Antimet‐2 PCR mix and cycling conditions were the same as the one‐step PCRs, except using 15 cycles instead of 30. PCR products were visualized by gel electrophoresis and purified with ProNex Size‐Selective Purification System (Promega). To add the Illumina adapters and indexes to the Antimet‐2 PCRs, the second round PCR mix contained 12.5 µl of NEBNext PCR mix (New England BioLabs), 0.5 µM of second round primers (comprising the sample‐specific index and Illumina adaptor; see Figure 1), 7 µl of purified PCR products and molecular grade water up to a final volume of 25 µl. PCR2 cycling conditions were as for the first round PCR, with annealing temperature set at 55°C for all samples and 15 cycles. For both one‐ and two‐step approaches, PCR products were visualized by gel electrophoresis and purified with ProNex Size‐Selective Purification System (Promega) (single band on gel) or with QIAquick Gel Extraction Kit (Qiagen) (multiple bands on gel). Purified samples were then quantified with the ProNex NGS Library Quant Kit (Promega). To minimise sequencing bias between amplicons, only samples with a concentration of at least 2 ng/µl were pooled in equimolar concentrations to create three separate libraries (Antimet‐1 library, “universal” library, and Antimet‐2 library). Each library was sequenced on a separate flow cell on an Illumina MiSeq, using v3 reagent kits (600‐cycle).
2.6. Bioinformatics and data processing
Raw sequences were processed using version 3.5.1 of the R statistical programming language and version 1.8.0 of the dada2 package (Callahan et al., 2016; R Core Team, 2020). The distribution of quality scores was visualised prior to trimming the raw sequencing data. Forward reads were trimmed to 200 bp in length approximately corresponding to the point at which the lower quartile fell below 20. Low quality reads were removed when estimated errors were greater than two and truncated if quality scores fell below two. Reverse reads were excluded from the analyses, as their overall quality was much lower than the forward reads. Samples comprising fewer than 100 reads after trimming were discarded. Data from each sequencing library was processed separately, including parameterisation of error rate models, dereplication of reads, and identification of unique sequence variants, using pooled data from across each sequencing run. After denoising, the resulting sequencing tables were merged. Chimeric sequences were identified using the consensus method defined for de novo detection of chimeric sequences in the dada2 package (removeBimeraDenovo function) in R and removed.
Taxonomies were assigned to the resulting amplicon sequence variants (ASVs) using the PR2 database (version 4.12) (Guillou et al., 2013) and the assignTaxonomy function in dada2. Classification followed the RDP Naïve Bayesian Classifier algorithm described in Wang et al. (2007) (kmer size: 8; bootstrap replicates:100; min. bootstrap support: 50). To avoid inflating the diversity when checking the efficiency of the different primer combinations, annealing temperatures and PCR approaches, singletons and ASVs with a count <10 and present in only one library were removed (trimmed data). Percentages and proportions of reads classified as metazoan or nonmetazoan were derived from the trimmed data. Initial comparison of PCR strategies (universal, Antimet‐1, and Antimet‐2) was performed on pooled reads obtained from each T as. Classification at genus level was chosen to determine the best individual annealing temperature for each sample, defined as the one amplifying the most diverse set of nonmetazoan sequences (number of phyla and genera) from the samples. To estimate alpha diversity and between‐treatments dissimilarity, Chao1 dissimilarity index was used on the untrimmed unannotated data set immediately after analysis with dada2. Chao1 is a nonparametric estimator of species richness that accounts for potential under sampling of a population by counting the number of species observed only once (singletons) or twice (doubletons) within the sample (Hughes et al., 2001). Therefore, it must be calculated using the untrimmed data to correctly estimate diversity and the associated standard error. As an alternative way of accounting for the different number of reads found in each sample, ASV richness was calculated after rarefying the untrimmed data using a sample size of 40,000 reads. To explore what proportion of the ASV richness was attributable to metazoan sequences and to show the nonmetazoan diversity detected across the temperature gradient, the rarefied data set was compared to a parallel data set from which metazoans ASV were removed after rarefaction at 40,000 reads. Chao1 and ASV richness were calculated for each sample type and at each T a of the Antimet‐1 library, with data from technical replicates combined prior to calculating diversity or rarefying the data.
Data compilation, alpha diversity, and ASV richness measures were performed using the r statistical programming language (version 3.5.1) (Ihaka & Gentleman, 1996). In addition, ggplot2 (version 3.3.2), stringr (version 1.3.1), biostrings (version 2.48.0), phyloseq (version 1.24.2), vegan (version 2.5.7), and Venn Diagrams (http://bioinformatics.psb.ugent.be/webtools/Venn/) were used to manipulate the data and produce images (McMurdie & Holmes, 2013; Oksanen et al., 2020; Pages et al., 2018; Wickham, 2016; Wickham & RStudio, 2018).
3. RESULTS
3.1. In silico PCR amplification
The two antimetazoan 18S V4 primer combinations performed largely similarly but the combination used in this study, 574*F—UNonMet_DB, retrieved a greater proportion of sequences for some groups, notably Amoebozoa (which are often biased against by 18S amplicon studies) and Discoba (Figure 2). The universal primer pair also “amplified” a large proportion of Discoba, as well as metamonads, which were underrepresented by both antimetazoan primer pairs. However, the universal primer pair also amplified >85% of metazoan sequences compared to the very low levels amplified by both antimetazoan combinations. Note that PCR performance in vitro is less constrained than the stringent 100% primer‐template match criterion used for this in silico comparison. The median amplicon lengths produced by the in silico PCRs were 574 bp.
FIGURE 2.
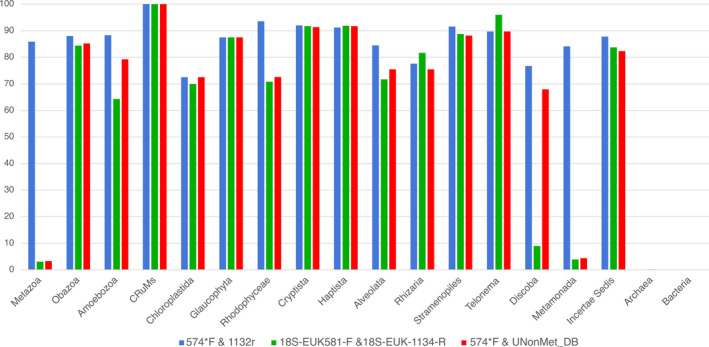
Comparison of in silico “PCR” amplification across a comprehensive set of eukaryote groups, between the universal primers 574*F—1132r (blue) (Hugerth et al., 2014), the antimetazoan primer combination 18S‐EUK581‐F—18S‐EUK‐1134‐R (green) (Carnegie et al., 2003), and 574*f—UNonMet_DB (red) (Bass & del Campo, 2020; Hugerth et al., 2014). The y‐axis represents the percentage of total database sequences amplified in each taxonomic group indicated on the x‐axis
3.2. In vitro PCR amplification
PCR products were successfully generated from all samples, primer strategies, and T a combinations, producing a single band of around 690 bp (Antimet‐2 first round PCR), and around 700 bp including tagged primers (Antimet‐1, universal, and Antimet‐2 second round PCR). Comparing equal volumes of PCR products loaded on a gel, the intensity of the bands was higher in the samples amplified at lower T as, fading towards higher T as (results not shown).
After PCR product purification, some replicates did not yield enough PCR product for library preparation: these were universal library barb Temp 6 (one replicate), plankton Temp 6 (two replicates), shrimp Temps 1, 5, and 6 (one each); Antimet‐1 library: prawn Temp 1 (one) and Temp 2 (two); Antimet‐2 library: barb Temp 3 (one), plankton Temps 3, 4, and 5 (one each); shrimp Temp 4 (one). Shrimp hepatopancreas samples amplified with Antimet‐1 and −2 had a second smaller band (490 bp) at all T as (Antimet‐1 PCR and second‐round of the Antimet‐2 PCR). After gel separation and purification of these bands individually, it was not possible to obtain enough PCR products from all T as to be sequenced, therefore sequencing was carried out for Temps 1 to 4. Sequencing of the smaller bands from Antimet‐1 PCR resulted in a large proportion (>99%) of microsporidian (Enterocytozoon spp.) reads and a very low proportion (<0.03%) of host reads. By contrast, the 700 bp bands comprised a higher proportion (ranging from 13% to 26%) of host reads and 73%–86% microsporidian reads (results not shown). As the taxonomic composition of both bands were similar, they were combined within each temperature and used in the Antimet‐1 analysis.
3.3. “Universal” approach amplified predominantly host 18S
The universal library produced a total of 3,214,228 sequences of which 1,755,896 (mean by sample: 438,974 ± 286,149) were retained and classified into 205 ASVs across 17 phyla for the plankton sample, 494 ASVs (four phyla) for barb, 30 ASVs (two phyla) for shrimp, and 33 ASVs (two phyla) for prawn (Table 1; Appendix S2). The universal library was dominated by metazoan sequences with 100% of sequences corresponding to the metazoan host for prawn and shrimp and >99% for barb (Table 1; Appendix S2).
TABLE 1.
Summary of reads percentages at phylum level for each sample and each amplification approach
| ASV taxonomic assignation | Barb | Common prawn | Plankton | Whiteleg shrimp | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Domain | Supergroup | Phylum | Antimet‐1 | Antimet‐2 | Universal | Antimet‐1 | Antimet‐2 | Universal | Antimet‐1 | Antimet‐2 | Universal | Antimet‐1 | Antimet‐2 | Universal |
| Eukaryota | Alveolata | Apicomplexa | – | – | – | – | – | – | 0.12 | 0.06 | – | – | – | – |
| Eukaryota | Alveolata | Ciliophora | 7.03 | 0.11 | 0.01 | 6.11 | 0.39 | – | 5.52 | 2.60 | 0.17 | – | – | – |
| Eukaryota | Alveolata | Dinoflagellata | 0.07 | 0.01 | – | <0.01 | – | – | 8.15 | 4.38 | 0.16 | – | – | – |
| Eukaryota | Alveolata | NA | – | – | <0.01 | – | – | – | 0.18 | 0.07 | <0.01 | – | – | – |
| Eukaryota | Alveolata | Perkinsea | – | – | – | – | – | – | 0.01 | <0.01 | – | – | – | – |
| Eukaryota | Amoebozoa | Amoebozoa_X | – | – | – | – | – | – | <0.01 | <0.01 | – | – | – | – |
| Eukaryota | Amoebozoa | Breviatea | – | – | – | – | – | – | <0.01 | <0.01 | – | – | – | – |
| Eukaryota | Amoebozoa | Conosa | <0.01 | – | – | – | – | – | 0.04 | 0.02 | – | – | <0.01 | – |
| Eukaryota | Amoebozoa | Lobosa | – | – | – | – | – | – | 0.18 | 0.07 | – | – | – | – |
| Eukaryota | Amoebozoa | NA | 0.01 | – | – | – | – | – | <0.01 | – | – | – | – | – |
| Eukaryota | Apusozoa | Apusomonadidae | <0.01 | – | – | – | – | – | 0.10 | 0.04 | – | – | – | – |
| Eukaryota | Apusozoa | Hilomonadea | – | – | – | – | – | – | 0.16 | 0.07 | – | – | – | – |
| Eukaryota | Apusozoa | Mantamonadidea | – | – | – | – | – | – | <0.01 | – | – | – | – | – |
| Eukaryota | Archaeplastida | Chlorophyta | 0.12 | <0.01 | – | 1.17 | 0.13 | – | 36.21 | 29.70 | 0.76 | 0.02 | – | – |
| Eukaryota | Archaeplastida | NA | – | – | – | – | – | – | <0.01 | – | – | 0.01 | – | – |
| Eukaryota | Archaeplastida | Rhodophyta | 0.14 | – | – | – | – | – | 0.26 | 0.12 | – | – | – | – |
| Eukaryota | Archaeplastida | Streptophyta | <0.01 | – | – | <0.01 | – | – | 8.87 | 6.51 | 0.15 | – | – | – |
| Eukaryota | Eukaryota_X | Eukaryota_XX | – | – | – | – | – | – | <0.01 | <0.01 | – | – | – | – |
| Eukaryota | Excavata | Discoba | 0.15 | 0.03 | – | – | – | – | 0.18 | 0.07 | – | – | – | – |
| Eukaryota | Excavata | Metamonada | – | – | – | – | – | – | 0.01 | – | – | – | – | – |
| Eukaryota | Excavata | NA | – | 0.01 | – | – | – | – | – | – | – | – | – | – |
| Eukaryota | Hacrobia | Centroheliozoa | 0.06 | <0.01 | – | – | – | – | 0.09 | 0.05 | 0.01 | – | – | – |
| Eukaryota | Hacrobia | Cryptophyta | 0.06 | <0.01 | – | – | – | – | 0.03 | 0.02 | – | – | – | – |
| Eukaryota | Hacrobia | Haptophyta | <0.01 | – | – | – | – | – | 0.42 | 0.19 | 0.01 | – | – | – |
| Eukaryota | Hacrobia | Katablepharidophyta | 0.01 | – | – | – | – | – | <0.01 | – | – | – | – | – |
| Eukaryota | Hacrobia | Picozoa | – | – | – | – | – | – | 0.01 | <0.01 | – | – | – | – |
| Eukaryota | Hacrobia | Telonemia | – | – | – | – | – | – | 0.05 | 0.03 | – | – | – | – |
| Eukaryota | NA | NA | 0.43 | 0.38 | – | 0.03 | 0.04 | – | 5.35 | 2.52 | 2.62 | – | 0.01 | – |
| Eukaryota | Opisthokonta | Choanoflagellida | – | – | – | – | – | – | 0.31 | 0.18 | <0.01 | – | – | – |
| Eukaryota | Opisthokonta | Fungi | 1.93 | <0.01 | – | 0.03 | 0.01 | – | 4.77 | 3.12 | 0.04 | 95.14 | 21.61 | <0.01 |
| Eukaryota | Opisthokonta | Mesomycetozoa | 0.03 | <0.01 | – | – | – | – | 1.09 | 0.39 | <0.01 | – | – | – |
| Eukaryota | Opisthokonta | Metazoa | 78.97 | 99.18 | 99.99 | 92.64 | 99.42 | 100.00 | 4.93 | 34.48 | 76.33 | 4.83 | 78.38 | 100.00 |
| Eukaryota | Opisthokonta | NA | 0.22 | <0.01 | – | – | <0.01 | <0.01 | 0.55 | 0.22 | 19.31 | – | – | – |
| Eukaryota | Rhizaria | Cercozoa | 0.22 | – | – | – | – | – | 1.66 | 0.74 | 0.21 | – | – | – |
| Eukaryota | Rhizaria | Foraminifera | <0.01 | – | – | – | – | – | – | – | – | – | – | – |
| Eukaryota | Rhizaria | NA | – | – | – | – | – | – | <0.01 | – | – | – | – | – |
| Eukaryota | Stramenopiles | NA | – | – | – | – | – | – | 0.11 | 0.03 | – | – | – | – |
| Eukaryota | Stramenopiles | Ochrophyta | 10.40 | 0.26 | 0.01 | 0.01 | 0.01 | – | 18.10 | 13.33 | 0.18 | <0.01 | – | – |
| Eukaryota | Stramenopiles | Opalozoa | 0.14 | – | – | – | – | – | 0.20 | 0.08 | – | – | – | – |
| Eukaryota | Stramenopiles | Pseudofungi | – | – | – | – | – | – | 1.56 | 0.59 | 0.02 | – | – | – |
| Eukaryota | Stramenopiles | Sagenista | – | – | – | – | – | – | 0.77 | 0.34 | 0.01 | – | – | – |
| Eukaryota | Stramenopiles | Stramenopiles_X | – | – | – | – | – | – | <0.01 | <0.01 | – | – | – | – |
NA, taxonomic assignation not available; –, ASVs not present in the library. The density of shaded grey, light to dark reflects increasing proportions of reads.
3.4. One‐step antimetazoan PCR is preferable to the two‐step approach
The Antimet‐2 library yielded a total of 8,020,614 sequences of which 6,561,040 (mean by sample: 1,640,260 ± 1,135,397) were retained and classified in 1611 ASVs for the plankton sample (34 phyla), 345 ASVs for barb (13 phyla), 18 ASVs for shrimp (four phyla), and 43 ASVs for prawn (seven phyla) (Table 1; Appendix S3). Although the Antimet‐2 approach increased the proportion of nonmetazoan sequences compared to the universal strategy (Figure 3a; Table 1), metazoan sequence representation remained unacceptably high (>99% in barb and prawn).
FIGURE 3.
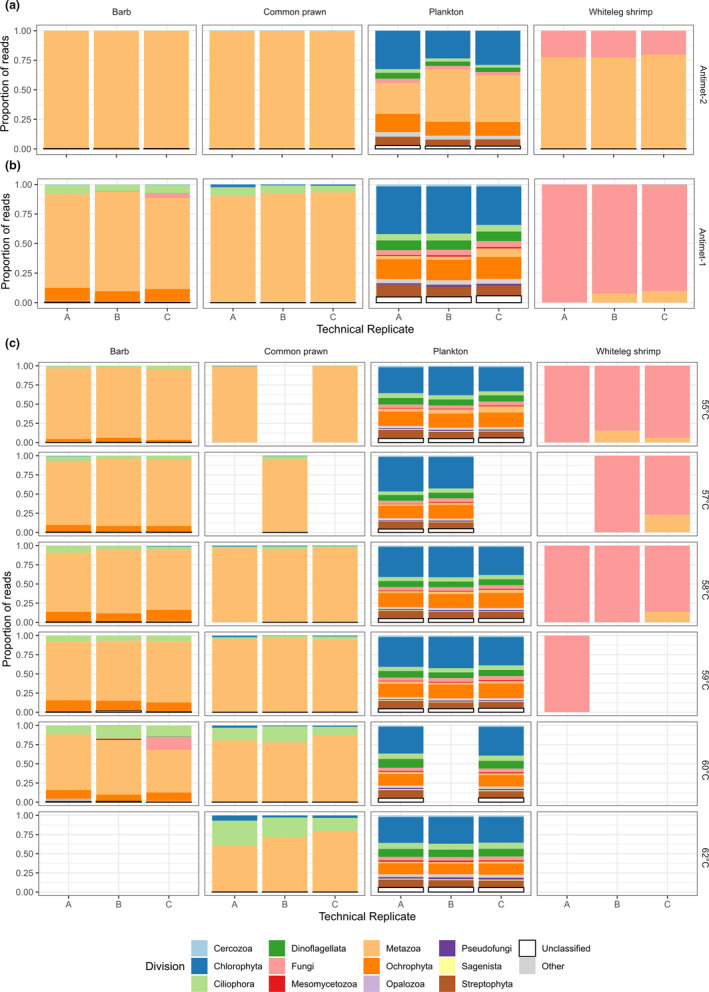
Proportion of sequences for each Phylum amplified by the one‐step and two‐step approaches with the antimetazoan primer pair by sample type. Technical replicates representing the sequences obtained by combining all T as for (a) Antimet‐2 and (b) Antimet‐1 libraries, showing a lower proportion of metazoan reads (peach colour) in the Antimet‐1 libraries. (c) Antimet‐1 sequences proportion for each T a and technical replicate, showing lower abundance of metazoan reads (colour peach) at higher T as
The Antimet‐1 library returned a total of 7,832,338 sequences of which 5,760,532 (mean by sample: 1,440,133 ± 840,250) were retained and classified in 3,546 ASVs for the plankton sample (40 phyla), 676 ASVs for barb (22 phyla), 43 ASVs for shrimp (five phyla), and 79 ASVs for prawn (eight phyla) (Table 1; Appendix S4). The Antimet‐1 library amplified a total of 41 phyla, in contrast to the universal (17 phyla) and Antimet‐2 libraries (35 phyla) (Table 1). The majority of retrieved phyla were shared between the three amplification approaches, with Antimet‐1 amplifying an extra 7 phyla (Figure 4a). The phylum unique to the Antimet‐2 approach was classified as Domain Eukaryota Supergroup Excavata Phylum “NA” (Table 1; Appendix [Link], [Link] and [Link], [Link]). The other two phyla belonging to the Excavata were classified as Discoba, present in both Antimet‐1 and Antimet‐2 libraries (Table 1; Appendix S3–4), and Metamonada, present only in the Antimet‐1 library (Table 1; Appendix S4). At the genus level, most genera were shared by the two Antimet approaches, with Antimet‐1 amplifying an extra 254 genera (Figure 4b). The genera shared only by the universal and the Antimet‐2 included five metazoan genera and the protistan genus Marteilia (present in plankton samples from Norway). Unique taxa present only in the Antimet‐2 and the universal libraries, and the ones shared between these two libraries are detailed in Appendix S5.
FIGURE 4.
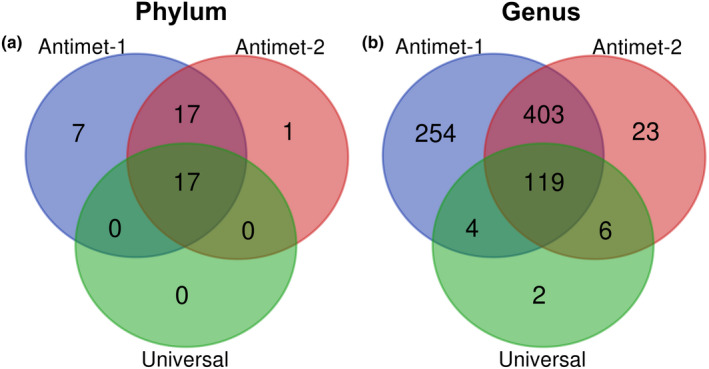
Number of shared and unique taxa at genus and phylum level amplified by the three approaches tested in this study. Antimet‐1 (blue), Antiemt‐2 (red), universal (blue)
The percentages of reads classified as metazoan in the Antimet‐1 library were considerably lower compared to both universal and Antimet‐2 libraries (Table 1; Figure 3b). Therefore, the “universal” and Antimet‐2 approaches were not investigated further. Antimet‐1 was then tested at different PCR annealing temperatures (T as) to determine the effect of such temperatures on the proportion of metazoan versus nonmetazoan reads.
3.5. Higher annealing temperatures reduce the proportion of metazoan reads amplified
Taxonomic profiles obtained from each T a with the Antimet‐1 approach were analysed at the phylum level to establish the proportions of metazoan sequences amplified by each T a and at the genus level to determine whether different T as resulted in different taxonomic profiles or levels of diversity (Table 2). For the barb sample all three replicates from Temp 6 were discarded as fewer than 100 reads were retained after trimming. This was also the case for one plankton replicate at each of Temps 2 and 5. A summary of the retained reads across each individual sample type and T as is provided in Table 2.
TABLE 2.
Summary of taxonomic assignments for each sample, with detected genera separated by metazoan and nonmetazoan affiliations and retained reads at each annealing temperature (T a) for the Antimet‐1 library
| Barb | Domain | Supergroup | Phylum | Class | Order | Family | Genus | Temp 1 | Temp 2 | Temp 3 | Temp 4 | Temp 5 | Temp 6 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 55°C | 57°C | 58°C | 59°C | 60°C | 62°C | ||||||||
| Total taxonomic assignments † | 1 | 10 | 22 | 45 | 56 | 66 | 83 | ||||||
| Detected genera | 49 | 47 | 52 | 43 | 47 | ‐ | |||||||
| Metazoan genera | 10 | 10 | 11 | 10 | 8 | ‐ | |||||||
| Nonmetazoan genera | 39 | 37 | 41 | 33 | 39 | ‐ | |||||||
| Metazoan reads % | 92.13 | 85.99 | 78.30 | 76.37 | 62.72 | ‐ | |||||||
| Reads count* | 98,647 (3) | 147,496 (3) | 200,590 (3) | 118,515 (3) | 131,811 (3) | ‐ | |||||||
| Common prawn | |||||||||||||
| Total taxonomic assignments † | 1 | 5 | 8 | 14 | 17 | 21 | 28 | ||||||
| Detected genera | 7 | 8 | 16 | 19 | 18 | 11 | |||||||
| Metazoan genera | 3 | 3 | 3 | 3 | 3 | 2 | |||||||
| Nonmetazoan genera | 4 | 5 | 13 | 16 | 15 | 9 | |||||||
| Metazoan reads % | 99.40 | 95.56 | 96.68 | 95.49 | 84.22 | 70.55 | |||||||
| Reads count* | 143,398 (2) | 48,497 (1) | 499,953 (3) | 1,173,078 (3) | 494,820 (3) | 103,604 (3) | |||||||
| Plankton | |||||||||||||
| Total taxonomic assignments † | 1 | 11 | 40 | 128 | 223 | 378 | 738 | ||||||
| Detected genera | 619 | 622 | 599 | 569 | 498 | 551 | |||||||
| Metazoan genera | 21 | 18 | 15 | 18 | 12 | 13 | |||||||
| Nonmetazoan genera | 598 | 604 | 584 | 551 | 486 | 538 | |||||||
| Metazoan reads % | 7.72 | 2.43 | 3.32 | 2.67 | 2.32 | 1.99 | |||||||
| Reads count* | 819,826 (3) | 351,911 (2) | 177,207 (3) | 237,997 (3) | 62,335 (2) | 140,524 (3) | |||||||
| Whiteleg shrimp | |||||||||||||
| Total taxonomic assignments † | 1 | 3 | 5 | 10 | 10 | 11 | 13 | ||||||
| Detected genera | 12 | 3 | 7 | 3 | ‐ | ‐ | |||||||
| Metazoan genera | 6 | 2 | 5 | 1 | ‐ | ‐ | |||||||
| Nonmetazoan genera | 6 | 1 | 2 | 2 | ‐ | ‐ | |||||||
| Metazoan reads % | 6.63 | 4.65 | 1.95 | 0.03 | ‐ | ‐ | |||||||
| Reads count* | 453,179 (3) | 98,101 (2) | 232,449 (3) | 26,594 (1) | ‐ | ‐ | |||||||
Abbreviations: (), number of replicates/T a *, sum of all reads obtained in each T a; ‐, library missing due to low number of reads retained after trimming (barb) or low PCR product yield after PCR purification; T a for each sample type resulting in the detection of most nonmetazoan genera are highlighted in grey and bold. †, total taxonomic assignments considering all T as. Ta for each sample type resulting in the detection of most non‐metazoan genera are highlighted in grey and bold.
In all sample types, raising the T a reduced the proportion of metazoan reads amplified (Table 2; Figure 3c). This effect is particularly pronounced in the barb and prawn samples in which the overall proportion of metazoan reads decreased by ~30% with rising T a, while less noticeable in plankton and shrimp (reduction of ~5%–6%) (Table 2; Figure 3c). The barb sample showed an initial increase of host associated reads at Temp 2, followed by a steady decline and a steady rise of nonhost metazoan fraction (Supporting material S6). This latter fraction was represented by a high proportion of Myxobolus (Cnidaria, Myxozoa) reads (ranging from 35.14% to 20.24% from Temps 1 to 5, respectively) (Appendix S6), a parasitic infection previously observed in the gills of the barb sampled for this study (data not shown).
Temperatures 1 to 4 amplified the most nonmetazoan genera in shrimp, plankton, barb, and prawn samples, respectively (Table 2; Appendix S6). In the samples tested, one temperature was not sufficient to amplify the full range of genera detected considering all combined T as (Table 2). For example, considering all annealing temperatures, the total number of nonmetazoan ASVs genera detected in prawn was 22 (Appendix S6), however the highest number of nonmetazoan ASVs genera detected by a single T a was 16 (Table 2). A similar situation was observed for the other sample types (Table 2; Appendix S6). Appendix S7 provides taxonomic inventories at the phylum level for barb, prawn, plankton, and shrimp, respectively.
Chao1 diversity estimates calculated on the untrimmed data varied with temperature across the four sample types (Figure 5a). In the barb samples, the values were similar across all T as, with higher values for Temp 1. Prawn diversity peaked in the mid‐range of T as, while the whiteleg shrimp diversity was higher in Temp 1, and the values for plankton were highest in the lower temperature range (Figure 5a). ASV richness estimates varied with temperatures across the sample types (Figure 5b, c). After rarefying, the shrimp sample amplified at Temp 4 was removed from the analysis as fewer than 40’000 reads were present. Plotted ASV richness estimates on the untrimmed rarefied data (Figure 5b) and rarefied data without metazoan ASVs (Figure 5c) showed that the metazoan fraction of the ASVs has an influence on the richness values, which were lower in the absence of metazoan ASVs (Figure 5c). However, when comparing the ASV richness plots (Figure 5b, c) for each sample type and T a, the shapes of the richness distribution across temperatures are similar.
FIGURE 5.
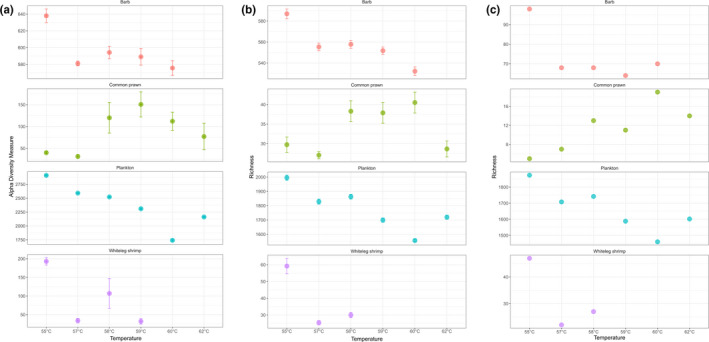
Alpha diversity (Chao1 index, a) and species richness estimates (b, c) of each sample type at each T a. (a) Chao1 indices were calculated on the untrimmed data sets; (b) species richness calculated on the untrimmed data sets, with read rarefaction set at 40’000; (c) as (b) but with metazoan ASVs removed after rarefaction. Shrimp sample amplified at 59°C was removed from the rarefied data due to low number of reads. For both indices, replicates were merged before estimations
4. DISCUSSION
The 18S V4 region primers 18S‐EUK581‐F and 18S‐EUK‐1134‐R were devised by Carnegie et al. (2003) to selectively amplify nonmetazoan genes from animal hosts infected with protistan parasites. This combination of “universal” forward and reverse primers excluding a mostly metazoan‐specific signature has shown to successfully reduce metazoan 18S amplification from material infected with parasites (Bower et al., 2004), and for revealing the eukaryome of coral and human samples (del Campo et al., 2019). In this experiment we used modified set of antimetazoan primers to optimise an Illumina metabarcoding approach producing the lowest proportion of metazoan/host reads, and the highest diversity of nonmetazoan reads.
4.1. Comparison of PCR strategies
When amplified with the broadly‐targeted primers 574*F–1132r (Hugerth et al., 2014), the barb, prawn, and shrimp tissue samples were overwhelmingly dominated by host/metazoan reads, and were not suitable for investigating eukaryotic symbionts. The plankton sample, being an environmental sample (i.e., a mix of whole zooplanktonic organisms coated by seawater and including their epibionts) was also dominated by planktonic metazoan reads (76.3%), but protists were also amplified, including parasitic lineages (e.g., oomycetes, syndinians).
The one‐step antimetazoan approach (Antimet‐1) performed best both in terms of minimising the proportion of metazoan/host reads from all sample types tested and retrieving a higher number of taxa. The sequence libraries from two of the most host material‐dominated samples, barb and prawn, were dominated (>97%) by host reads in the Antimet‐2 approach (two‐step), but reduced to 57.3% and 90.4% respectively in Antimet‐1 (one‐step). Metazoan reads in the plankton samples were reduced to as low as 5%. The shrimp sample was dominated by the microsporidian Enterocytozoon hepatopenaei (EHP), which is directly linked to its heavy infection levels but also to the shorter 18S V4 region of microsporidians resulting in the parasite ASVs dominating the Antimet‐1 libraries (usually, highly divergent microsporidian 18S amplicons are rarely amplified by broadly‐targeted eukaryotic primers [Bass et al., 2015]). Additionally, the antimetazoan primer pair amplified the myxozoan Myxobolus sp. from the barb sample, which corresponded to 21.4% of the Antimet‐1 library. Myxozoans are highly specialised parasitic cnidarians (Metazoa), with a diversity of animal hosts (Fiala et al., 2015), which are fortuitously amplified, at least to some extent, by 574*F –UNonMet_DB.
The results of our comparison of one‐step versus two‐step PCR amplification for nonmetazoans contrast interestingly with those of Zizka et al. (2019), who made a similar comparison for amplification of metazoan COI fragment primers. They found that the two‐step approach was more successful than the one‐step approach, and only slightly less successful than using an Illumina TruSeq kit. Possible explanations for our results consistently favouring the one‐step approach include the use of different primers, targeting a different gene, and the nature of the taxonomic targets. A useful conclusion here is that any particular library construction and sequencing approach is not inherent preferable in all cases, but we provide empirical evidence of its superior suitability for the present antimetazoan system.
4.2. Optimal combination of antimetazoan primers
The antimetazoan primer pair amplifying the largest diversity of nonmetazoan sequence types in silico was 574*F combined with our revised selective reverse primer UNonMet_DB. The in silico performances of other frequently used forward primers in 18S V4 metabarcoding studies were tested in del Campo et al. (2019) and Bass and del Campo (2020) Based on the latter, and confirmed by our own in silico experiments, we selected the 574*F primer for the present study.
The 574*F—UNonMet_DB primer pair can be used in a one‐step process without subsequent amplification by a different primer set because the forward primer is immediately adjacent to the hypervariable V4 region, which is taxonomically highly informative, even with the conservative approach we took here of using only the forward reads and cropping their length to 200 bp (from a maximum of 300 bp, thereby avoiding a possible drop in sequencing quality towards the 3’ end of the sequence). However, it is still possible to create “pseudocontigs” (e.g., using the mergePairs function in dada2), comprising paired forward and reverse reads with Ns padding an indeterminate length of missing sequence in the middle, which provide (discontinuous) sequences of at least 400 bp, enabling more robust phylogenetic analyses than are possible with the forward read alone (Liu et al., 2020; Werner et al., 2012). The average length of the 574*F—UNonMet_DB amplified region of 580 bp, which is unavoidable if the antimetazoan selection is required, precludes any attempt to recover overlapping paired‐end contigs from this amplicon with Illumina MiSeq technologies. Other forward primers with theoretically good taxonomic coverage that would amplify slightly shorter V4 amplicons are available for example, 616*f (Vaulot et al., 2021), but their utility for producing paired‐end contigs will still be affected by the variable length of the V4 region across microeukaryote taxa, and the quality of individual sequencing runs.
4.3. PCR annealing temperature
We did not necessarily expect to see a direct and consistent relationship between annealing temperatures and detection of nonmetazoan taxa in this experiment. We used alpha diversity measures (Chao1) on the untrimmed data and rarefied data to try to account for the different read numbers found per sample. However, there was no predictable relationship between nonmetazoan diversity detected and T a and only for the prawn sample did higher annealing temperatures result in higher nonmetazoan diversity. After removing metazoan ASVs from the untrimmed rarefied data set (Figure 5c), the ASV richness‐temperature relationship plots were generally similar in shape to those including metazoan ASVs (Figure 5b), the main predictable difference being that the richness values are lower in the absence of Metazoa. In the case of the whiteleg shrimp, the lowest temperature (55°C) did in fact result in a significantly greater diversity of nonmetazoan ASVs. In other cases, the differences in richness in the trimmed data between annealing temperatures were very low, and cumulatively detected more diversity than any one temperature considered alone.
The relationship between annealing temperature and diversity detected is complex, depending on the level and nature of diversity present in the sample, and conflicting PCR amplification trends: (1) decreasing proportion of metazoan reads with increasing T a, facilitating amplification of other taxa (if they are present in the sample), and (2) decreasing likelihood of amplification of some lineages with increasing annealing temperatures (particularly at higher temperatures), for example due to primer‐template mismatches. It should also be noted that the diversity of all taxa is artificially inflated by small‐scale differences between ASV sequences that do not represent real diversity ‐ due to PCR/sequencing errors and intragenomic polymorphism. However, as this “inflation” should apply randomly, the shape of the plots across time should reflect the underlying “real” diversity, just with inflated values due to these artefacts.
Our inference from the results presented here is that different eukaryotic assemblages amplify differently at different temperatures. This could be explained by several reasons: (1) different eukaryotic sample types are represented by diverse proportions of amplifiable DNA and amplify differently at different temperatures; (2) the relationships between T as and nonmetazoan taxa amplified are inevitably influenced by the level of nonmetazoan diversity in any particular sample, and by the taxonomic level at which these are assessed (phylum and genus levels in our case); (3) more replicates at each temperature would be required for more realistic diversity estimates.
Above a certain T a, the amount of amplicons generated by PCR from different samples was too low for HTS, such was the case for the shrimp sample at Temp 2 (55°C, one replicate), Temp 4 (59°C, two replicates), Temp 5 and Temp 6 (60 and 62°C, all replicates). Moreover, the samples used in this study showed optimal nonmetazoan genera amplification at different T as (55°C, shrimp; 57°C, plankton; 58°C, barb; 59°C, common prawn). Therefore, our current recommendation to other researchers (until any more generalised patterns emerge) is to amplify samples across a T a range of 55–60°C, in order to account for unpredictable and unquantifiable differences between different sample types, and to capture the maximum diversity of nonmetazoan taxa while simultaneously including amplifications that minimise metazoan amplification.
As relatively rare taxa will sometimes be of prime interest in antimetazoan metabarcoding studies, factors potentially creating biases against these should be mitigated as far as possible (Fonseca, 2018). Other more general considerations for optimal and replicable diversity characterisation of biodiverse samples should also be borne in mind, for example the importance of PCR reagents and conditions (Alberdi et al., 2018; Bista et al., 2018), chimera formation and secondary structures at lower annealing temperatures (Fonseca et al., 2012), artefactual diversity created by incorporation of PCR and sequencing errors, and intragenomic polymorphism between multicopy genes in a single cell (Jurburg et al., 2021).
4.4. Future perspectives
Amplicon sequencing to investigate microbial diversity (metabarcoding) using relatively short amplicons remains popular mainly because it is cost‐effective, relatively quick and can target simultaneously multiple species. The resulting data sets per sample are large enough to comprehensively, if not exhaustively, capture the diversity in each sample, and the large numbers of samples that can be produced per sequencing run are very suitable for robust comparisons and statistical analyses. The perspective of developing sequencing technologies that can accommodate longer amplicons are very attractive because of the much greater taxonomic and phylogenetic information that such amplicons contain (Jamy et al., 2020). To fully make use of its potential it is key to design general and group‐specific (or ‐excluding) primers across the whole ribosomal RNA gene array, and other genomic regions (e.g., mtDNA). Nonetheless the success of such approaches relies on the expansion and curation of reference databases, on high throughput standard protocols that could easily be reproducible in the laboratory as well as analytical procedures to streamline the data sets, at least for specific target taxa or sample types (e.g., eDNA, bulk samples, marine, freshwater, etc). With respect to antimetazoan eukaryotic amplification, other selective primer sites should be sought, to add to and/or complement the useful nonmetazoan signature in UNonMet_DB.
AUTHOR CONTRIBUTIONS
Conceptualisation, David Bass; in silico analysis, Javier del Campo; in vitro methods development, David Bass, Diana Minardi, Vera G. Fonseca, Alberto Pallavicini; sample collection for whiteleg shrimp and barb, Diana Minardi; plankton and common prawn samples, Stein Mortensen; DNA extractions from whiteleg shrimp, barb, and common prawn, Diana Minardi; DNA extraction from plankton, Rose Kerr; library preparation, sequencing, and data collection, Diana Minardi; bioinformatic analysis, David Ryder; data analysis and paper writing, David Bass, Diana Minardi, David Ryder, Javier del Campo; review editing, David Bass, Diana Minardi, David Ryder, Javier del Campo, Vera G. Fonseca. All authors contributed to the final draft and gave their approval for its publication.
Supporting information
Appendix S1
Appendix S2
Appendix S3
Appendix S4
Appendix S5
Appendix S6
Appendix S7
Appendix S8
Appendix S9
Appendix S10
ACKNOWLEDGEMENTS
This study was conceived and developed in its early stages within the European Project H2020 VIVALDI, which received funding from the European Union H2020 research and innovation programme under grant agreement no. 678589. The work was supported by funding from the UK Department of Environment, Food and Rural Affairs (Defra) under contract FB002 to D.M., D.R., and D.B; Institute of Marine Research (IMR), Aquaculture programme to S.M. In addition, the authors would like to thank Chantelle Hooper for advice regarding sequencing runs.
Minardi, D. , Ryder, D. , del Campo, J. , Garcia Fonseca, V. , Kerr, R. , Mortensen, S. , Pallavicini, A. , & Bass, D. (2022). Improved high throughput protocol for targeting eukaryotic symbionts in metazoan and eDNA samples. Molecular Ecology Resources, 22, 664–678. 10.1111/1755-0998.13509
DATA AVAILABILITY STATEMENT
The data from this study can be found in the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA694647 and will be made accessible upon publication of this article.
REFERENCES
- Alberdi, A. , Aizpurua, O. , Gilbert, M. T. P. , & Bohmann, K. (2018). Scrutinizing key steps for reliable metabarcoding of environmental samples. Methods in Ecology and Evolution, 9(1), 134–147. 10.1111/2041-210X.12849 [DOI] [Google Scholar]
- Amaral‐Zettler, L. A. , McCliment, E. A. , Ducklow, H. W. , & Huse, S. M. (2009). A method for studying protistan diversity using massively parallel sequencing of V9 hypervariable regions of small‐subunit ribosomal RNA genes. PLoS One, 4(7), e6372. 10.1371/journal.pone.0006372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson, S. D. , Bartholomew, J. L. , & Lotan, T. (2018). Myxozoans: Ancient metazoan parasites find a home in phylum Cnidaria. Zoology, 129, 66–68. 10.1016/j.zool.2018.06.005 [DOI] [PubMed] [Google Scholar]
- Baetens, M. , Van Laer, L. , De Leeneer, K. , Hellemans, J. , De Schrijver, J. , Van De Voorde, H. , Renard, M. , Dietz, H. , Lacro, R. V. , Menten, B. , Van Criekinge, W. , De Backer, J. , De Paepe, A. , Loeys, B. , & Coucke, P. J. (2011). Applying massive parallel sequencing to molecular diagnosis of Marfan and Loeys‐Dietz syndromes. Human Mutation, 32(9), 1053–1062. 10.1002/humu.21525 [DOI] [PubMed] [Google Scholar]
- Bass, D. , & del Campo, J. (2020). Microeukaryotes in animal and plant microbiomes: Ecologies of disease? European Journal of Protistology, 76, 125719. 10.1016/j.ejop.2020.125719 [DOI] [PubMed] [Google Scholar]
- Bass, D. , Stentiford, G. D. , Littlewood, D. T. J. , & Hartikainen, H. (2015). Diverse applications of environmental DNA methods in parasitology. Trends in Parasitology, 31(10), 499–513. 10.1016/j.pt.2015.06.013 [DOI] [PubMed] [Google Scholar]
- Bass, D. , Stentiford, G. D. , Wang, H.‐C. , Koskella, B. , & Tyler, C. R. (2019). The Pathobiome in animal and plant diseases. Trends in Ecology & Evolution, 34(11), 996–1008. 10.1016/j.tree.2019.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bista, I. , Carvalho, G. R. , Tang, M. , Walsh, K. , Zhou, X. , Hajibabaei, M. , Shokralla, S. , Seymour, M. , Bradley, D. , Liu, S. , Christmas, M. , & Creer, S. (2018). Performance of amplicon and shotgun sequencing for accurate biomass estimation in invertebrate community samples. Molecular Ecology Resources, 18(5), 1020–1034. 10.1111/1755-0998.12888 [DOI] [PubMed] [Google Scholar]
- Bower, S. M. , Carnegie, R. B. , Goh, B. , Jones, S. R. , Lowe, G. J. , & Mak, M. W. (2004). Preferential PCR amplification of parasitic protistan small subunit rDNA from metazoan tissues. The Journal of Eukaryotic Microbiology, 51(3), 325–332. 10.1111/j.1550-7408.2004.tb00574.x [DOI] [PubMed] [Google Scholar]
- Bull, M. J. , & Plummer, N. T. (2014). Part 1: The human gut microbiome in health and disease. Integrative Medicine (Encinitas, Calif.), 13(6), 17–22. [PMC free article] [PubMed] [Google Scholar]
- Callahan, B. J. , McMurdie, P. J. , Rosen, M. J. , Han, A. W. , Johnson, A. J. A. , & Holmes, S. P. (2016). DADA2: High‐resolution sample inference from Illumina amplicon data. Nature Methods, 13(7), 581–583. 10.1038/nmeth.3869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso, J. G. , Lauber, C. L. , Walters, W. A. , Berg‐Lyons, D. , Lozupone, C. A. , Turnbaugh, P. J. , Fierer, N. , & Knight, R. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proceedings of the National Academy of Sciences, 108(Supplement 1), 4516–4522. 10.1073/pnas.1000080107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnegie, R. B. , Meyer, G. R. , Blackbourn, J. , Cochennec‐Laureau, N. , Berthe, F. C. J. , & Bower, S. M. (2003). Molecular detection of the oyster parasite Mikrocytos mackini, and a preliminary phylogenetic analysis. Diseases of Aquatic Organisms, 54(3), 219–227. 10.3354/dao054219 [DOI] [PubMed] [Google Scholar]
- Comeau, A. M. , Li, W. K. W. , Tremblay, J.‐É. , Carmack, E. C. , & Lovejoy, C. (2011). Arctic Ocean microbial community structure before and after the 2007 record sea ice minimum. PLoS One, 6(11), e27492. 10.1371/journal.pone.0027492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruaud, P. , Rasplus, J.‐Y. , Rodriguez, L. J. , & Cruaud, A. (2017). High‐throughput sequencing of multiple amplicons for barcoding and integrative taxonomy. Scientific Reports, 7, 41948. 10.1038/srep41948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Campo, J. , Bass, D. , & Keeling, P. J. (2020). The eukaryome: Diversity and role of microeukaryotic organisms associated with animal hosts. Functional Ecology, 34(10), 2045–2054. 10.1111/1365-2435.13490 [DOI] [Google Scholar]
- del Campo, J. , Pons, M. J. , Herranz, M. , Wakeman, K. C. , del Valle, J. , Vermeij, M. J. A. , Leander, B. S. , & Keeling, P. J. (2019). Validation of a universal set of primers to study animal‐associated microeukaryotic communities. Environmental Microbiology, 21(10), 3855–3861. 10.1111/1462-2920.14733 [DOI] [PubMed] [Google Scholar]
- Elbrecht, V. , & Steinke, D. (2019). Scaling up DNA metabarcoding for freshwater macrozoobenthos monitoring. Freshwater Biology, 64(2), 380–387. 10.1111/fwb.13220 [DOI] [Google Scholar]
- Fiala, I. , Bartošová‐Sojková, P. , & Whipps, C. M. (2015). Classification and phylogenetics of Myxozoa. In Okamura B., Gruhl A., & Bartholomew J. L. (Eds.), Myxozoan evolution, ecology and development (pp. 85–110). Springer International Publishing. 10.1007/978-3-319-14753-6_5 [DOI] [Google Scholar]
- Fonseca, V. G. (2018). Pitfalls in relative abundance estimation using eDNA metabarcoding. Molecular Ecology Resources, 18(5), 923–926. 10.1111/1755-0998.12902 [DOI] [Google Scholar]
- Fonseca, V. G. , Nichols, B. , Lallias, D. , Quince, C. , Carvalho, G. R. , Power, D. M. , & Creer, S. (2012). Sample richness and genetic diversity as drivers of chimera formation in nSSU metagenetic analyses. Nucleic Acids Research, 40(9), e66. 10.1093/nar/gks002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert, J. A. , Meyer, F. , Antonopoulos, D. , Balaji, P. , Brown, C. T. , Brown, C. T. , Desai, N. , Eisen, J. A. , Evers, D. , Field, D. , Feng, W. U. , Huson, D. , Jansson, J. , Knight, R. , Knight, J. , Kolker, E. , Konstantindis, K. , Kostka, J. , Kyrpides, N. , … Stevens, R. (2010). Meeting report: the terabase metagenomics workshop and the vision of an Earth microbiome project. Standards in Genomic Sciences, 3(3), 243–248. 10.4056/sigs.1433550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green, M. R. , & Sambrook, J. (2012). Molecular Cloning: A Laboratory Manual, 4th ed. Cold Spring Harbor Laboratory Press; [Google Scholar]
- Guillou, L. , Bachar, D. , Audic, S. , Bass, D. , Berney, C. , Bittner, L. , Boutte, C. , Burgaud, G. , de Vargas, C. , Decelle, J. , del Campo, J. , Dolan, J. R. , Dunthorn, M. , Edvardsen, B. , Holzmann, M. , Kooistra, W. H. C. F. , Lara, E. , Le Bescot, N. , Logares, R. , … Christen, R. (2013). The Protist Ribosomal Reference database (PR2): a catalog of unicellular eukaryote small sub‐unit rRNA sequences with curated taxonomy. Nucleic Acids Research, 41, 597–604. 10.1093/nar/gks1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugerth, L. W. , Muller, E. E. L. , Hu, Y. O. O. , Lebrun, L. A. M. , Roume, H. , Lundin, D. , Wilmes, P. , & Andersson, A. F. (2014). Systematic design of 18S rRNA gene primers for determining eukaryotic diversity in microbial consortia. PLoS One, 9(4), e95567. 10.1371/journal.pone.0095567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, J. B. , Hellmann, J. J. , Ricketts, T. H. , & Bohannan, B. J. (2001). Counting the uncountable: statistical approaches to estimating microbial diversity. Applied and Environmental Microbiology, 67(10), 4399–4406. 10.1128/aem.67.10.4399-4406.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihaka, R. , & Gentleman, R. (1996). R: A language for data analysis and graphics. Journal of Computational and Graphical Statistics, 5(3), 299–314. 10.1080/10618600.1996.10474713 [DOI] [Google Scholar]
- Jamy, M. , Foster, R. , Barbera, P. , Czech, L. , Kozlov, A. , Stamatakis, A. , Bending, G. , Hilton, S. , Bass, D. , & Burki, F. (2020). Long‐read metabarcoding of the eukaryotic rDNA operon to phylogenetically and taxonomically resolve environmental diversity. Molecular Ecology Resources, 20(2), 429–443. 10.1111/1755-0998.13117 [DOI] [PubMed] [Google Scholar]
- Jurburg, S. D. , Keil, P. , Singh, B. K. , & Chase, J. M. (2021). All together now: Limitations and recommendations for the simultaneous analysis of all eukaryotic soil sequences. Molecular Ecology Resources, 21(6), 1759–1771. 10.1111/1755-0998.13401 [DOI] [PubMed] [Google Scholar]
- Karsenti, E. , Acinas, S. G. , Bork, P. , Bowler, C. , De Vargas, C. , Raes, J. , Sullivan, M. , Arendt, D. , Benzoni, F. , Claverie, J.‐M. , Follows, M. , Gorsky, G. , Hingamp, P. , Iudicone, D. , Jaillon, O. , Kandels‐Lewis, S. , Krzic, U. , Not, F. , Ogata, H. ,., … Wincker, P. (2011). A holistic approach to marine eco‐systems biology. PLOS Biology, 9(10), e1001177. 10.1371/journal.pbio.1001177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klindworth, A. , Pruesse, E. , Schweer, T. , Peplies, J. , Quast, C. , Horn, M. , & Glöckner, F. O. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next‐generation sequencing‐based diversity studies. Nucleic Acids Research, 41(1), e1. 10.1093/nar/gks808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozich, J. J. , Westcott, S. L. , Baxter, N. T. , Highlander, S. K. , & Schloss, P. D. (2013). Development of a dual‐index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Applied and Environmental Microbiology, 79(17), 5112–5120. 10.1128/AEM.01043-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, T. , Chen, C.‐Y. , Chen‐Deng, A. , Chen, Y.‐L. , Wang, J.‐Y. , Hou, Y.‐I. , & Lin, M.‐C. (2020). Joining Illumina paired‐end reads for classifying phylogenetic marker sequences. BMC Bioinformatics, 21(1), 105. 10.1186/s12859-020-3445-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone, C. A. , Stombaugh, J. , Gonzalez, A. , Ackermann, G. , Wendel, D. , Vazquez‐Baeza, Y. , Jansson, J. K. , Gordon, J. I. , & Knight, R. (2013). Meta‐analyses of studies of the human microbiota. Genome Research, 23(10), 1704–1714. 10.1101/gr.151803.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurdie, P. J. , & Holmes, S. (2013). phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One, 8(4), e61217. 10.1371/journal.pone.0061217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiguchi, M. K. , Doukakis, P. , Egan, M. , Kizirian, D. , Phillips, A. , Prendini, L. , & Giribet, G. (2002). DNA Isolation Procedures. In DeSalle R., Giribet G., & Wheeler W. (Eds.), Techniques in Molecular Systematics and Evolution (pp. 249–287). Birkhäuser Basel. 10.1007/978-3-0348-8125-8_12 [DOI] [Google Scholar]
- O’Donnell, J. L. , Kelly, R. P. , Lowell, N. C. , & Port, J. A. (2016). Indexed PCR primers induce template‐specific bias in large‐scale DNA sequencing studies. PLoS One, 11(3), e0148698. 10.1371/journal.pone.0148698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hara, E. , Neves, A. L. A. , Song, Y. , & Guan, L. L. (2020). The role of the gut microbiome in cattle production and health: Driver or passenger? Annual Review of Animal Biosciences, 8(1), 199–220. 10.1146/annurev-animal-021419-083952 [DOI] [PubMed] [Google Scholar]
- Oksanen, J. , Guillaume Blanchet, F. , Friendly, M. , Kindt, R. , Legendre, P. , McGlinn, D. , & Wagner, H. (2020). vegan: Community Ecology Package. Retrieved from https://cran.r‐project.org/package=vegan%0A [Google Scholar]
- Pages, H. , Aboyoun, P. , Gentleman, R. , DebRoy, S. (2018). Biostrings: Efficient manipulation of biological strings. Retrieved from https://bioconductor.org/packages/Biostrings
- Qin, J. , Li, R. , Raes, J. , Arumugam, M. , Burgdorf, K. S. , Manichanh, C. , Nielsen, T. , Pons, N. , Levenez, F. , Yamada, T. , Mende, D. R. , Li, J. , Xu, J. , Li, S. , Li, D. , Cao, J. , Wang, B. , Liang, H. , Zheng, H. , Xie, Y., … Wang, J. (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature, 464(7285), 59–65. 10.1038/nature08821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . (2020). R: A language and environment for statistical computing. R Foundation for Statistical Computing. Retrieved from http://www.r‐project.org/index.html [Google Scholar]
- Rohwer, F. , Seguritan, V. , Azam, F. , & Knowlton, N. (2002). Diversity and distribution of coral‐associated bacteria. Marine Ecology Progress Series, 243, 1–10. 10.3354/meps243001 [DOI] [Google Scholar]
- Schnell, I. B. , Bohmann, K. , & Gilbert, M. T. P. (2015). Tag jumps illuminated – reducing sequence‐to‐sample misidentifications in metabarcoding studies. Molecular Ecology Resources, 15(6), 1289–1303. 10.1111/1755-0998.12402 [DOI] [PubMed] [Google Scholar]
- Seyama, T. , Ito, T. , Hayashi, T. , Mizuno, T. , Nakamura, N. , & Akiyama, M. (1992). A novel blocker‐PCR method for detection of rare mutant alleles in the presence of an excess amount of normal DNA. Nucleic Acids Research, 20(10), 2493–2496. 10.1093/nar/20.10.2493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skillings, D. (2016). Holobionts and the ecology of organisms: Multi‐species communities or integrated individuals? Biology and Philosophy, 331(6), 875–892. 10.1007/s10539-016-9544-0 [DOI] [Google Scholar]
- Stoeck, T. , Behnke, A. , Christen, R. , Amaral‐Zettler, L. , Rodriguez‐Mora, M. J. , Chistoserdov, A. , Orsi, W. , & Edgcomb, V. P. (2009). Massively parallel tag sequencing reveals the complexity of anaerobic marine protistan communities. BMC Biology, 7, 72. 10.1186/1741-7007-7-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tringe, S. G. , & Hugenholtz, P. (2008). A renaissance for the pioneering 16S rRNA gene. Current Opinion in Microbiology, 11(5), 442–446. 10.1016/j.mib.2008.09.011 [DOI] [PubMed] [Google Scholar]
- Vaulot, D. , Geisen, S. , Mahé, F. , & Bass, D. (2021). pr2‐primers: An 18S rRNA primer database for protists. Molecular Ecology Resources, 1–12. 10.1111/1755-0998.13465 [DOI] [PubMed] [Google Scholar]
- Wang, Q. , Garrity, G. M. , Tiedje, J. M. , & Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and Environmental Microbiology, 73(16), 5261–5267. 10.1128/AEM.00062-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner, J. J. , Zhou, D. , Caporaso, J. G. , Knight, R. , & Angenent, L. T. (2012). Comparison of Illumina paired‐end and single‐direction sequencing for microbial 16S rRNA gene amplicon surveys. The ISME Journal, 6(7), 1273–1276. 10.1038/ismej.2011.186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham, H. (2016). ggplot2: Elegant Graphics for Data Analysis. 2nd ed. Springer. 10.1007/978-0-387-98141-3 [DOI] [Google Scholar]
- Wickham, H. , & RStudio . (2018). stringr: Simple, Consistent Wrappers for Common String. Retrieved from http://stringr.tidyverse.org [Google Scholar]
- Yilmaz, P. , Gilbert, J. A. , Knight, R. , Amaral‐Zettler, L. , Karsch‐Mizrachi, I. , Cochrane, G. , Nakamura, Y. , Sansone, S.‐A. , Glöckner, F. O. , & Field, D. (2011). The genomic standards consortium: bringing standards to life for microbial ecology. The ISME Journal, 5(10), 1565–1567. 10.1038/ismej.2011.39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zizka, V. M. A. , Elbrecht, V. , Macher, J.‐N. , & Leese, F. (2019). Assessing the influence of sample tagging and library preparation on DNA metabarcoding. Molecular Ecology Resources, 19(4), 893–899. 10.1111/1755-0998.13018 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Appendix S2
Appendix S3
Appendix S4
Appendix S5
Appendix S6
Appendix S7
Appendix S8
Appendix S9
Appendix S10
Data Availability Statement
The data from this study can be found in the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA694647 and will be made accessible upon publication of this article.