

Abstract
Background
Platelet‐derived protein disulfide isomerase 1 (PDIA1) regulates thrombus formation, but its role in the regulation of platelet function is not fully understood.
Aims
The aim of this study was to characterize the role of PDIA1 in human platelets.
Methods
Proteomic analysis of PDI isoforms in platelets was performed using liquid chromatography tandem mass spectometry, and the expression of PDIs on platelets in response to collagen, TRAP‐14, or ADP was measured with flow cytometry. The effects of bepristat, a selective PDIA1 inhibitor, on platelet aggregation, expression of platelet surface activation markers, thromboxane A2 (TxA2), and reactive oxygen species (ROS) generation were evaluated by optical aggregometry, flow cytometry, ELISA, and dihydrodichlorofluorescein diacetate‐based fluorescent assay, respectively.
Results
PDIA1 was less abundant compared with PDIA3 in resting platelets and platelets stimulated with TRAP‐14, collagen, or ADP. Collagen, but not ADP, induced a significant increase in PDIA1 expression. Bepristat potently inhibited the aggregation of washed platelets induced by collagen or convulxin, but only weakly inhibited platelet aggregation induced by TRAP‐14 or thrombin, and had the negligible effect on platelet aggregation induced by arachidonic acid. Inhibition of PDIA1 by bepristat resulted in the reduction of TxA2 and ROS production in collagen‐ or thrombin‐stimulated platelets. Furthermore, bepristat reduced the activation of αIIbβ3 integrin and expression of P‐selectin.
Conclusions
PDIA1 acts as an intraplatelet regulator of the ROS‐TxA2 pathway in collagen‐GP VI receptor‐mediated platelet activation that is a mechanistically distinct pathway from extracellular regulation of αIIbβ3 integrin by PDIA3.
Keywords: bepristat, flow cytometry, PDIs expression, platelet activation, proteomics
Essentials.
The role of PDIA1 in the regulation of platelet function has not been defined.
The platelet aggregation, reactive oxygen species and thromboxane A2 in platelets were measured.
PDIA1 blockade specifically inhibits platelet aggregation induced by GPVI receptor agonists.
PDIA1 regulates the reactive oxygen species‐thromboxane A2–dependent pathway in platelets.
1. INTRODUCTION
Antiplatelet therapy represents a central therapeutic strategy for preventing atherothrombotic events in patients with cardiovascular, cerebrovascular, and peripheral artery disease. 1 Platelets contain several members of the protein disulfide isomerase (PDI) family of enzymes, including PDIA1, PDIA3, PDIA4, and PDIA6, which are secreted and recruited to the platelet surface in response to platelet activation. 2 , 3 , 4 , 5 , 6 PDIA1 is required for platelet accumulation and tissue factor‐dependent fibrin formation at site of vascular injury in vivo. 7 , 8 Then, PDIA3, PDIA6, and PDIA4 were also found to regulate platelet function and thrombus formation in vivo. 2 , 3 , 9 , 10 Therefore, PDI isoforms have emerged as attractive targets for new antithrombotic agents. However, the specific roles of PDIA1, PDIA3, PDIA4, and PDIA6 have not been thoroughly explored.
Previous reports convincingly demonstrated that inhibition of extracellular PDIA3 with specific anti‐PDIA3 antibodies resulted in reduced arterial thrombosis at the site of vascular injury. 10 , 11 Further studies of platelet‐specific PDIA3 deficiency confirmed that platelet‐derived PDIA3 plays a crucial role in supporting thrombus formation in vivo 2 , 12 by regulating platelet adhesion, 10 aggregation, 2 and granule secretion. 10 , 11 It has been hypothesized that PDIA3 exposed on the surface of activated platelets controls the conformational changes of integrins, including the αIIbβ3 integrin receptor, from the inactive state to the ligand‐binding state based on the rearrangement of disulfide bonds within integrins or within other yet‐undefined targets of PDIA3. 2 , 10
Platelet surface PDIs, including not only PDIA3, 2 , 10 , 11 but also PDIA1, 13 , 14 , 15 PDIA4, 9 and PDIA6, 3 have been implicated in the regulation of platelet function. Although all of these thiol isomerases can bind to the αIIbβ3 receptor and mediate its activation and platelet aggregation, 2 , 3 , 9 , 16 they appear to have a distinct role in the regulation of platelet aggregation. Indeed, surprisingly, different PDIs were not able to compensate for each other in platelets, 9 , 15 , 17 and some reports indicated that various PDIs play a distinct role in platelet function. For example, several studies found that PDIA3 and PDIA4 regulated Ca2+ mobilization in platelets, 10 , 18 whereas PDIA1 and PDIA6 did not. 17 , 19 These results suggest that various PDIs differ among each other in their targeting of various mechanisms relevant for platelet activation.
Furthermore, PDIA1 and PDIA3 have been demonstrated to support thrombus formation in vivo through distinct mechanisms regulating coagulation and fibrin formation. 12 , 15 , 20 Fibrin formation at the site of vessel injury was shown to be regulated by PDIA1 released from endothelial cells, whereas platelet‐derived PDIA1 was primarily required for platelet accumulation in a growing thrombus. 15 , 17 , 20 Several postulated procoagulant mechanisms of PDIA1 include activation of a tissue factor, 8 platelet‐derived factor V, 21 and factor XI, 22 and binding of coagulation factors to the platelet surface. 6 These findings contrast with platelet‐derived PDIA3, which seems to have only a minor role in regulating fibrin deposition. 12
Altogether, the accumulated evidence suggests that PDIA3 and PDIA1 may have distinct roles in controlling thrombus formation and platelet activation; however, the specific role of PDIA1 in platelet activation has not been thoroughly evaluated. Currently, to the best of our knowledge, bepristat is the only available PDIA1 inhibitor that is relatively selective, but the antiplatelet effect of bepristat, although reported, has not been characterized in detail. 23
Hence, the aim of this study was to investigate PDI content in nonstimulated and stimulated platelets, surface expression of major PDI platelet isoforms and, particularly, the role of PDIA1 in platelet function using a selective PDIA1 inhibitor: bepristat. We evaluated the effects of bepristat on generation of thromboxane A2 (TxA2) and reactive oxygen species (ROS), which are both important mediators of platelet activation, compared with the effects of bepristat on platelet aggregation and expression of platelet surface activation markers.
2. MATERIALS AND METHODS
2.1. Reagents
Bepristat 2a hydrochloride (bepristat) and acetylsalicylic acid (ASA; aspirin) were obtained from Sigma‐Aldrich. Rutin was obtained from Merck. Recombinant PDI enzymes including PDIA1, PDIA3, and PDIA6 were purchased from MyBioSource. The αIIbβ3 receptor antagonist Integrilin (eptifibatide) was purchased from GlaxoSmithKline. A complete list of reagents is described in the Supporting Information.
2.2. Blood collection
All procedures on human blood samples were approved by the Bioethical Committee of the Jagiellonian University or Medical University of Lodz and were carried out in accordance with the Declaration of Helsinki. Venous blood was obtained from healthy and drug‐free male volunteers, and informed consent was given by the volunteers before blood withdrawal.
2.3. Preparation of washed platelets
Human washed platelets were prepared from blood collected into vacutainer tubes containing 3.2% sodium citrate solution at the volume ratio 9:1 (BD Vacutainer System), unless another anticoagulant was stated. Whole blood was centrifuged at 260g for 15 min at room temperature (RT) to obtain platelet‐rich plasma (PRP). PRP from at least two donors was pooled. To obtain washed platelets, a wash buffer consisting of Dulbecco's phosphate–buffered saline without calcium and magnesium ions (Gibco), 1 mg/mL glucose, and 1 mg/mL bovine serum albumin was used. PRP was centrifuged at 960g for 10 min at RT in the presence of 100 ng/mL prostacyclin. Platelets in the pellet were resuspended in wash buffer with 100 ng/mL prostacyclin and subsequently recentrifuged at 810g for 10 min at RT. The final pellet was resuspended in wash buffer to reach a platelet count required for the individual assays. The platelet suspension was allowed to rest for a minimum of 30 min at RT before measurements.
2.4. Identification and quantification of PDI enzymes in platelets using the liquid chromatography tandem mass spectometry (LC‐MS/MS) method and Exponentially Modified Protein Abundance Index (emPAI) calculation
Washed platelets were prepared from blood samples as described previously, using K2EDTA as an anticoagulant. For the proteomic analysis, platelets suspended at 10 × 108/mL in Dulbecco's phosphate–buffered saline (Gibco) with glucose (1 mg/mL) were supplemented with CaCl2 and MgCl2 to final concentrations of 1 and 2 mM, respectively; subsequently, the platelets were stimulated with 0.5 U/mL thrombin for 8 min at 37°C under static conditions or were allowed to remain nonstimulated for the same period. The platelets were then centrifuged at 700g for 10 min at RT. The supernatants, referred to as releasates (n = 3), and pellets (n = 6) were further processed following the protocol by Sitek et al., 24 with minor modifications. Briefly, pellets were lysed in lysis buffer containing 7 M urea, 2 M thiourea, MS safe protease and phosphatase inhibitor cocktail (1:100), and 30 mM Tris‐HCl (pH 8.0). The pellets were then sonicated on ice, centrifuged (16 000g, 15 min, 4°C), and the supernatants, referred to as lysates, were then collected. The protein concentration in the releasates and lysates was assessed using Bradford assay‐Quick Start Bradford 1x Dye Reagent. Proteomic identification of PDI isoforms was performed using the LC‐MS/MS technique as described in detail in the Supporting information. The levels of PDI isoforms were semiquantified using the emPAI calculation as described previously, 25 normalized to the total emPAI value, and compared as a percentage of the overall composition. 26
2.5. Platelet aggregation
Aggregation of washed platelets at 2 × 108/mL was measured using a two‐channel optical aggregometer (Lum–aggregometer Model 700, Chrono‐Log Corporation) at 37℃ with continuous stirring. Before the measurements, platelets were supplemented with CaCl2 and MgCl2 to final concentrations of 1 and 2 mM, respectively. For platelet aggregation assays, platelets were preincubated at 37℃ for 3 min under stirring conditions with bepristat (10–30 µM) or rutin (60–120 µM) and then stimulated with various agonists including collagen (2, 4, 10, or 20 µg/mL), convulxin (50 or 100 ng/mL), bovine thrombin (0.015, 0.025, or 0.1 U/mL), or TRAP‐14 (10 or 20 µM). Additionally, platelet aggregation in the presence of eptifibatide (1 µg/mL), ASA (500 µM), ML171, or apocynin was measured. To study the effect of bepristat on platelet aggregation in PRP or GPIb/IX/V receptor‐mediated agglutination, platelets (2 × 108/mL) in PRP were preincubated at 37℃ for 2 min under stirring conditions with bepristat at the concentrations of 50 to 150 µM and then treated with collagen (4 µg/mL), arachidonic acid (250 µM), or ristocetin (1.25 mg/mL). Aggregation or agglutination traces were recorded for 6 min.
Further methods and statistics are described in detail in the Supporting information.
3. RESULTS
3.1. PDIA1 content in human platelets
Proteomic analysis of lysates, which indicate platelet cargo, revealed that resting human platelets contained a variety of PDI isoforms, of which PDIA1, PDIA3, and PDIA6 were the most abundant. PDIA1 expression was comparable to that of PDIA6, but significantly lower than PDIA3 (Figure 1). In contrast, PDIA4, PDIA5, and PDIA9 were present at lower levels in platelets (Figure 1).
FIGURE 1.
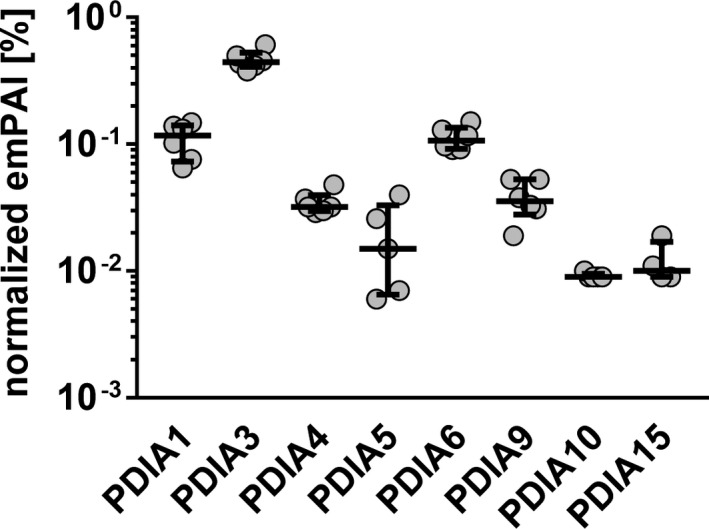
Levels of PDI enzymes in platelet lysates. Lysates of resting platelets were analyzed with LC‐MS/MS, and PDI isoforms were semiquantified using emPAI % calculation algorithm. Data represent individual values with medians and interquartile ranges, n = 4–6
Platelet activation with thrombin did not result in changes in PDIA1, PDIA3, and PDIA6 levels in platelets as demonstrated by analysis of platelet lysates. Similarly, in releasates representing the released secretory cargo of platelets, the level of PDIA1 and PDIA3 remained unchanged after platelet activation; however, PDIA6 was unexpectedly decreased (Table 1). In contrast to the unaltered PDIA1 content in activated platelets, platelet activation with thrombin resulted in the release of a variety of factors, including procoagulant (FV, PAI‐1), proadhesive (vWf, P‐sel, TSP‐1), and growth (TGF‐β1) factors, as indicated by decreased content of these factors in lysates and increased content in releasates after platelet activation (Table 1). These results may suggest that PDIs released from activated platelets are retained on the platelet surface.
TABLE 1.
The effect of platelet stimulation on the levels of PDI enzymes and other selected platelet‐secreted molecules
| Lysate | Releasate | |
|---|---|---|
| T granules | ||
| PDIA1 | No change | No change |
| PDIA3 | No change | No change |
| PDIA4 | No change | No change |
| PDIA5 | No change | No change |
| PDIA6 | No change | ↓ * |
| PDIA9 | No change | No change |
| PDIA10 | ↑ ** | No change |
| PDIA15 | No change | No change |
| α granules | ||
| P‐sel | No change | ↑ * |
| PBP | ↓ ** | No change |
| PF4 | ↓ * | ↑ ** |
| vWf | ↓ *** | No change |
| TGFβ1 | No change | ↑ * |
| FV | ↓ ** | No change |
| TSP−1 | ↓ *** | ↑ *** |
| Plg | ‐ | ↑ *** |
| PAI−1 | ‐ | ↑ * |
Lysates and releasates of platelets resting and stimulated with 0.5 U/mL thrombin were analyzed with the LC‐MS/MS method. PDI isoforms and selected platelet‐secreted molecules were semiquantified using emPAI % calculation. ↑ increase; ↓ decrease; *p < .05, **p < .01, ***p < .001; n = 3–6; Student's t test or Mann‐Whitney test. Detailed data are presented in Table S1 in the Supporting information.
Abbreviations: FV, factor V; PAI‐1. plasminogen activator inhibitor 1; P‐sel, P‐selectin; PBP, protein basic protein; PF4, platelet factor 4; Plg, plasminogen; TGFβ1, transforming growth factor β1; TSP‐1, thrombospondin 1; vWf, von Willebrand factor.
In fact, activation of platelets resulted in the upregulation of PDIA1 expression on the platelet surface as indicated by flow cytometry. Expression of PDIA1 was the highest after stimulation with collagen (13‐fold increase, Figure 2C), whereas the PDIA1 increase in response to TRAP‐14 was less pronounced (5.5‐fold, Figure 2A) and unchanged after ADP (Figure 2E). In contrast to PDIA1, expression of PDIA3 increased substantially after stimulation with TRAP‐14 (398‐fold increase, Figure 2A), but was increased to a lesser degree in response to collagen (74‐fold, Figure 2C) and ADP (37‐fold, Figure 2E). These results revealed that the increased expression of PDIA1 on the surface of activated platelets was significantly lower than expression of PDIA3, and it was specifically linked to collagen‐induced platelet activation.
FIGURE 2.
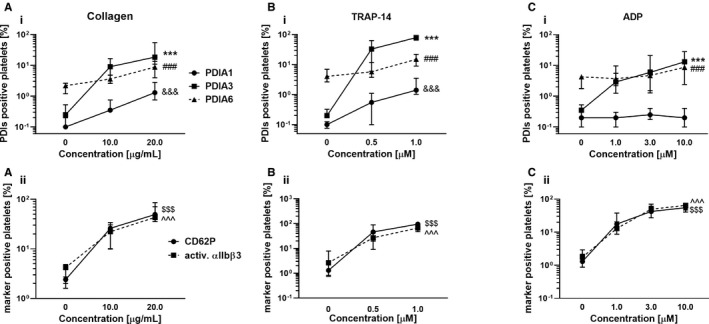
The expression of PDI enzymes on activated platelets compared with the expressions of platelet surface activation markers. Whole blood samples were stimulated with platelet agonists including (A i, A ii) collagen, (B i, B ii) TRAP‐14, or (C i, C ii) ADP. Expressions of PDIA1, PDIA3, PDIA6, P‐selectin, and the activated αIIbβ3 receptor were measured by flow cytometry. Data show medians and interquartile ranges, n = 10. The highest concentrations of agonists, &&&, ***, ###, $$$, or ^^^ symbols are relevant for PDIA1, PDIA3, PDIA6, P‐selectin, and the activated αIIbβ3 receptor, respectively; p < .001 vs. corresponding resting platelets; one‐way ANOVA with Dunnett's post hoc test
Interestingly, despite large differences in the level of PDIA3 expression promoted by TRAP‐14, collagen, and ADP, there were no differences in the degree of αIIbβ3 integrin activation induced by TRAP‐14, collagen, or ADP (Figure 2B, D and F).
3.2. Inhibition of PDIA1 by bepristat significantly affected platelet aggregation induced by collagen or convulxin compared with TRAP‐14 or thrombin
Rutin, a reference PDIA1 inhibitor, only slightly reduced aggregation of washed platelets in response to collagen (Figure 3A). The IC50 value for PDIA1 inhibition by rutin was almost 2‐fold higher compared with bepristat in an insulin reduction assay (4.0 vs. 2.1 µM, Table 2). In addition, rutin displayed non‐PDIA1‐related effects. 15 , 17 Accordingly, bepristat, but not rutin, was chosen as a tool to dissect the role of PDIA1 in platelet activation.
FIGURE 3.
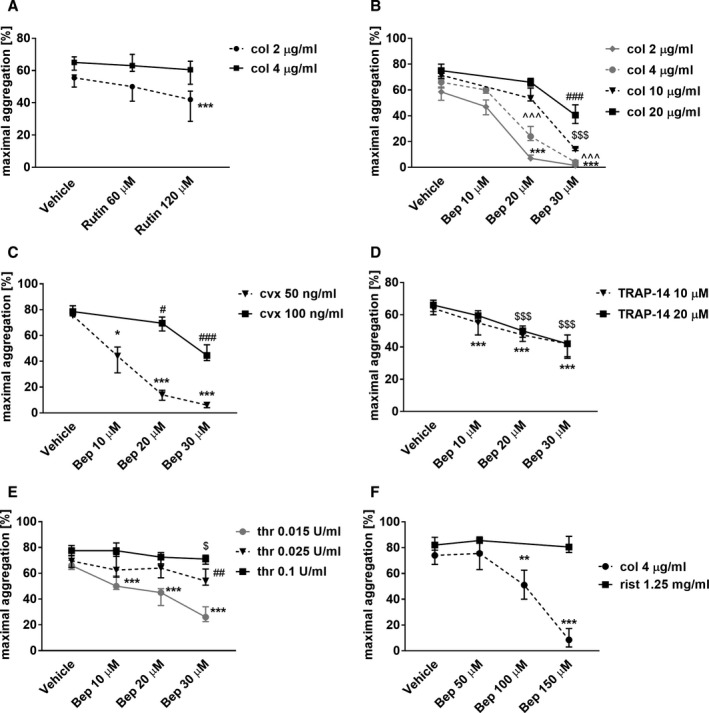
Effects of rutin and bepristat on platelet aggregation or agglutination. (A‐E) Washed human platelets (2 × 108/mL) were preincubated with rutin for 2 min and then stimulated with collagen (A), or preincubated with bepristat (Bep) and then stimulated with (B) collagen (col), (C) convulxin (cvx), (D) TRAP‐14, or (E) thrombin (thr) to induce aggregation. (F) Platelets in PRP (2 × 108/mL) were preincubated with bepristat (Bep) for 2 min and then stimulated with ristocetin (rist) or collagen (col) to induce agglutination and aggregation, respectively. Data are medians and interquartile ranges, n = 4–20. # or $ p < .05; ## or ** p < .01; ***, ### or $$$ p < .001 vs. corresponding vehicle control; one‐way ANOVA with Tukey's post hoc test or Kruskal‐Wallis test with Dunn's post‐hoc test
TABLE 2.
Comparison of the inhibitory effects of reference PDI inhibitors on the reductive activity of PDI isoforms
| PDIA1 | PDIA3 | PDIA6 | |
|---|---|---|---|
| Rutin, µM | 4.0 ± 0.8 | No effect | No effect |
| Bepristat, µM | 2.1 ± 0.5 | 127 ± 14 | No effect |
Recombinant PDI enzymes were incubated with inhibitors (0.005–200 µM) for 60 min at 37℃, and the activity of enzymes was then monitored with an insulin turbidimetric assay. Effects of the inhibition are expressed as the means ± SD of IC50 values, n = 3.
Abbreviation: PDI, protein disulfide isomerase.
Bepristat (10–30 µM) inhibited the aggregation of washed platelets induced by collagen, convulxin, TRAP‐14, or thrombin in a concentration‐dependent manner, and increasing concentrations of agonists were able to overcome the antiplatelet activity of bepristat 2a to some extent (Figure 3B‐E). Importantly, in the presence of bepristat, the inhibition of platelet aggregation induced by collagen or convulxin (Figure 3B,C) was more pronounced compared with aggregation initiated by TRAP‐14 or thrombin (Figure 3D,E). Platelet aggregation in response to collagen (2 or 4 µg/mL) or convulxin (50 ng/mL) was completely inhibited by 30 µM bepristat, whereas it was only partially inhibited when platelet aggregation was triggered by thrombin (0.015 U/mL) or TRAP‐14 (10 µM). These concentrations of thrombin and TRAP‐14 were chosen to induce submaximal platelet response (data not shown).
To test whether PDIA1 regulates the function of the glycoprotein Ib, ristocetin‐induced platelet agglutination (mediated by GPIb–vWf interactions) was measured in PRP in the presence of bepristat. Bepristat (150 µM) did not affect ristocetin‐induced platelet agglutination (Figure 3F), whereas it completely inhibited platelet aggregation in PRP in response to collagen (Figure 3F). These results confirmed that PDIA1 had no influence on the GPIb/IX/V receptor‐mediated response.
3.3. Inhibition of PDIA1 by bepristat decreased the expression of platelet surface activation markers
Bepristat (10–30 µM) decreased the expression of activated receptor αIIbβ3 (Figure 4A) and P‐selectin (Figure 4B) in gel‐filtered platelets in a concentration‐dependent manner. However, bepristat only slightly reduced the binding of vWf to platelet surfaces (Figure 4C).
FIGURE 4.
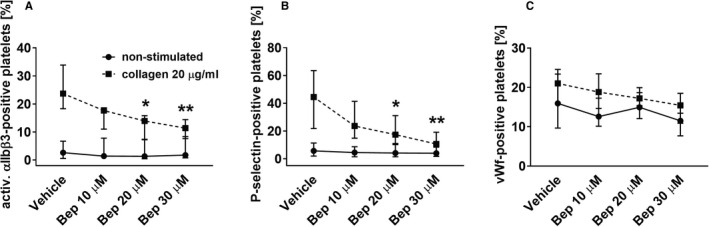
The effect of bepristat on expression of platelet surface activation markers. Gel‐filtered platelets at 2 × 108/mL were preincubated with bepristat (Bep) for 10 min, subsequently stimulated with collagen for 5 min, and then expression of activated αIIbβ3 receptor and P‐selectin, as well as the binding of von Willebrand factor were measured. Data are medians and interquartile ranges, n = 6. *p < .05, **p < .01 vs. corresponding vehicle control; one‐way ANOVA with Tukey's post‐hoc test
3.4. Inhibition of PDIA1 by bepristat reduced platelet TxA2 production
To demonstrate that PDIA1 regulates intraplatelet activation pathways, the production of thromboxane B2 (a stable metabolite of TxA2) by activated platelets treated with bepristat was measured. As shown in Figure 5A, bepristat (30 µM) strongly inhibited TxB2 production in platelets activated with collagen or thrombin (by approximately 85% and 70%, respectively). A high concentration of ASA (500 µM), used as a reference, inhibited TxB2 production completely. Despite the fact that bepristat or aspirin significantly inhibited platelet TxB2 production either in response to collagen (4 or 20 µg/mL, Figure 3B) or thrombin (0.1 U/mL, Figure 5B), bepristat or ASA did not inhibit platelet aggregation induced by thrombin at a concentration of 0.1 U/mL (Figure 5B). However, ASA partially diminished platelet aggregation triggered by a lower concentration of thrombin (0.015 U/mL, Figure S2 in Supporting Information), and this effect was similar to the effect of bepristat (Figure 3E). These results demonstrated that TxA2 is important for platelet aggregation caused by collagen or lower concentrations of thrombin, and that PDIA1 is specifically linked to the TxA2 generation pathway but not generally to the aggregation response.
FIGURE 5.
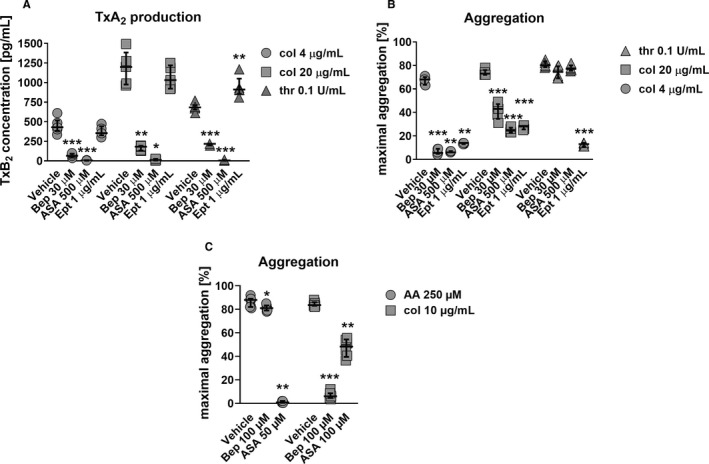
(A) The effect of bepristat on thromboxane A2 production in platelets compared with reference antiplatelet drugs. Washed platelets at 2 × 108/mL were preincubated with bepristat (Bep), aspirin (ASA), or eptifibatide (Ept) for 2 min and then stimulated for 6 min with collagen (col) or thrombin (thr). TxB2 (a stable metabolite of TxA2) was measured in the supernatant with an ELISA kit. Data are individual values with medians and interquartile ranges, n = 4–6. (B) The effect of bepristat on platelet aggregation compared with reference antiplatelet drugs. Washed human platelets (2 × 108/mL) were preincubated with bepristat (Bep), aspirin (ASA), or eptifibatide (Ept) for 2 min and then stimulated with collagen (col) or thrombin (thr) to induce aggregation. Data are individual values with medians and interquartile ranges, n = 5–6. (C) The effect of bepristat on platelet aggregation induced by arachidonic acid compared with collagen. Human platelets in PRP (2 × 108/mL) were preincubated with bepristat (Bep) or aspirin (ASA) for 2 min and then stimulated with arachidonic acid (AA) or collagen (col) to induce aggregation. Data are individual values with medians and interquartile ranges, n = 6–7. *p < .05, **p < .01, ***p < .001 vs. corresponding vehicle control; for A, B, and C Student's t‐test or Mann‐Whitney test
Additionally, to exclude the role of αIIbβ3 outside‐in signaling in the effect of bepristat on TxA2 generation, platelets were activated in the presence of an αIIbβ3 integrin antagonist, eptifibatide. Although eptifibatide abolished platelet aggregation induced by collagen or thrombin, blockade of the αIIbβ3 receptor did not affect or even slightly enhance collagen‐ or thrombin‐induced TxB2 production (Figure 5A,B). These results clearly demonstrated the distinct effects of αIIbβ3 receptor blockade by eptifibatide and PDIA1 blockade by bepristat on platelet function.
To test whether PDIA1 directly regulates TxA2 synthesis pathway in platelets, the platelet aggregation in response to arachidonic acid (AA, 250 µM) was measured. Bepristat (100 µM) had a negligible effect on AA‐induced platelet aggregation in PRP, whereas ASA (50 µM), even at a concentration of twice lower than bepristat, abolished platelet aggregation induced by AA (Figure 5C). In comparison, bepristat completely inhibited platelet aggregation caused by a relatively high concentration of collagen (10 µg/mL, Figure 5C). These results clearly indicate that PDIA1 inhibition affects GPVI‐mediated platelet activation mechanisms upstream of AA‐TxA2 synthesis pathway.
3.5. Inhibition of PDIA1 by bepristat reduced intraplatelet ROS production
To verify the role of PDIA1 in the regulation of ROS production in platelets, intraplatelet ROS production was measured in platelets activated with collagen in the presence or absence of bepristat. Bepristat reduced ROS generation in a concentration‐dependent manner, with the strongest effect visible at a concentration of 30 µM (Figure 6A). ASA (500 µM) also inhibited ROS production in response to collagen (20 µg/mL), but the effect of ASA was weaker compared with bepristat (30 µM) (Figure 6A). In contrast, ASA (500 µM) significantly inhibited platelet aggregation and TxB2 production induced by collagen (20 µg/mL) compared with bepristat (Figures 3A and 5A,B), suggesting distinct intraplatelet mechanisms of action for ASA and bepristat. Of note, bepristat (30 µM) suppressed also the production of ROS in platelets stimulated with thrombin as we show in Figure S1 in Supporting Information.
FIGURE 6.
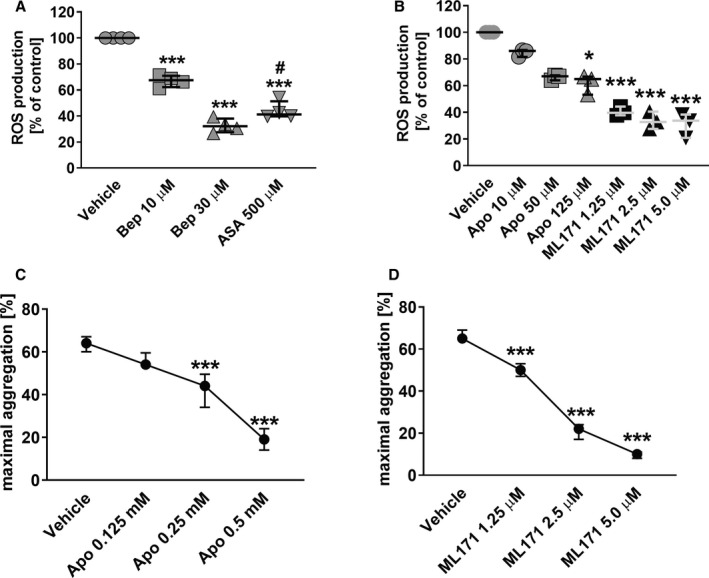
(A–B) Effects of bepristat on ROS production in platelets compared with aspirin, apocynin and ML171. Washed human platelets (2 × 108/mL) loaded with H2DCF‐DA were preincubated with (A) bepristat (Bep) or aspirin (ASA), and (B) apocynin (Apo) or ML171 for 5 min and then stimulated with collagen (20 μg/mL) to induce ROS generation. Data are individual values with medians and interquartile ranges, n = 3–4. *p < .05, ***p < .001 vs. corresponding vehicle control; #p < .05 vs. Bep 30; for A, one‐way ANOVA followed by Tukey's post hoc test; for B, Kruskal‐Wallis test with Dunn's post hoc test or one‐way ANOVA followed by Dunnett's post‐hoc test for apocynin and ML171, respectively. (C–D) Effects of apocynin and ML171 on platelet aggregation. Washed human platelets (2 × 108/mL) were preincubated with (C) apocynin (Apo) or (D) ML171 for 2 min and then stimulated with collagen (4 μg/mL) to induce aggregation. Data are medians and interquartile ranges, n = 6–9. ***p < .001 vs. corresponding vehicle control; for C and D, one‐way ANOVA followed by Tukey's post‐hoc test
3.6. Comparison of the effects of bepristat and Nox1 inhibition on intraplatelet ROS production and platelet aggregation
The effects of bepristat on ROS production in platelets was compared to the effects of the non‐selective NADPH oxidase (Nox) inhibitor apocynin or a specific Nox1 inhibitor ML171. As shown in Figure 6B, the inhibition of collagen‐induced ROS generation in platelets was more pronounced in the presence of ML171 compared with apocynin. Importantly, bepristat (30 µM) inhibited ROS production to a similar degree as ML171 (5 µM) (Figure 6A,B), and for both compounds, this inhibition was complete compared to the ROS level in nonstimulated platelets (data not shown). Apocynin (10–125 µM) inhibited collagen‐induced ROS generation in a concentration‐dependent manner (Figure 6B), but the inhibition was incomplete compared with the ROS level in resting platelets (data not shown). Furthermore, apocynin only moderately decreased platelet aggregation at the concentration of 125 µM, which caused maximal inhibition of ROS production. At higher concentrations (250–500 µM), herein, apocynin increased ROS signal (data not shown) as previously reported by others. 27 , 28 In contrast to apocynin, platelet treatment with ML171 (2.5 or 5 µM) displayed stronger inhibition of intraplatelet ROS production, which was associated with a profound inhibition of platelet aggregation (Figure 6B,D).
4. DISCUSSION
In the present work, to our knowledge we identified for the first time that PDIA1‐dependent regulation of platelet function is targeted to the intracellular ROS‐TxA2 pathway upstream of COX‐1. Therefore, PDIA1 has a distinct mechanism of action compared with PDIA3, which is known to regulate directly the activation of extracellular αIIbβ3 integrin on platelets. 2 Even though the expression of PDIA1 in platelets was lower compared with PDIA3, the blockade of PDIA1 activity by bepristat strongly inhibited platelet aggregation induced by collagen or convulxin, without a pronounced effects on thrombin‐ or TRAP‐14‐induced platelet aggregation but with visible effects of bepristat on ROS‐TxA2 pathway in platelets activated by thrombin. Importantly, bepristat potently reduced the production of ROS and TxA2 in platelets activated with collagen, which could be ascribed to the downregulation of Nox1 activation as PDIA1 was previously considered as a regulator of Nox1 activity. 29 Noteworthy is the effects of bepristat on ROS‐TxA2 pathway in platelets activated by thrombin that suggest a possible role of PDIA1 as a regulator of Nox2. 30 Finally, our findings demonstrate that PDIA1 supports the activation of the αIIbβ3 integrin by regulating the intraplatelet ROS‐TxA2 pathway, which is especially important for GPVI receptor‐mediated platelet activation (Figure S4 in Supporting Information). This mechanism does not involve the platelet response to thrombin and is distinct from extracellular controlling αIIbβ3 integrin activation by PDIA3. 2 Taken together, in the present work, we provide evidence that PDIA1 represents a novel intraplatelet target to inhibit the ROS‐TxA2 pathway, providing mechanistically distinct antiplatelet mechanisms of action to PDIA3‐mediated extracellular regulation of αIIbβ3 integrin.
In previous studies, the presence of PDIA1, PDIA3, PDIA4, or PDIA6 in platelets was demonstrated by specific antibody‐based western blotting, immunoprecipitation, flow cytometry, or fluorescence confocal microscopy. 2 , 3 , 4 , 5 , 6 , 9 , 19 Here, using a proteomic approach for semiquantitative analysis of PDI content in platelets we demonstrated that PDIA1, PDIA3, and PDIA6 were the most abundant PDI isoforms in resting human platelets compared with PDIA4, PDIA5, and PDIA9. Importantly, PDIA1 expression was comparable to PDIA6 but significantly lower than PDIA3. Following platelet activation, the levels of PDIA1, PDIA3, and PDIA6 in platelet lysates analyzed using a proteomic approach were unchanged, whereas the expression of these PDIs on the platelet surface characterized by flow cytometry was increased; this suggested that PDIs were released from platelets and retained on platelet surface. In contrast, platelet‐derived procoagulant (FV, PAI‐1), pro‐adhesive (vWf, P‐sel, TSP‐1) and growth (TGFβ1) factors, representing a specific cargo of platelet alpha granules, were released by activated platelets to surrounding milieu. Although PDIA1 was shown to be released from the endothelium, 31 our results suggested that PDIA1 localized in platelet T granules 32 was released and retained on the platelet surface in response to platelet activation. 5 , 6 Indeed, as we show in the present work, the pattern of PDI responses following platelet activation is consistent with previous reports showing that PDIA1, PDIA3, or PDIA6 secreted from platelets are exposed on platelet surface. 2 , 3 , 5 , 6 Furthermore, exogenous PDIs were shown to bind extensively to the surface of activated platelets compared to nonactivated platelets, 2 , 15 , 17 suggesting that platelet activation recruits platelet‐derived and extraplatelet PDIs to activate targets such as the αIIbβ3 integrin. 2 , 15 Notably, PDIA3 showed the most significant increase on the surface of activated platelets compared with PDIA1 or PDIA6 in our study, which is compatible with the role of PDIA3 in the extracellular regulation of αIIbβ3 integrin activation.
Interestingly, PDIA1 had the highest increase in the expression level on the surface of platelets treated with collagen compared with other platelet agonists, whereas PDIA3 showed the greatest increase in response to TRAP‐14 (Figure 2), suggesting important differences in PDIA1 and PDIA3 functions.
As previously reported, activation of platelets in the presence of exogenous PDIA1, PDIA3, or PDIA4 in the in vitro assays resulted in the enhanced binding of these PDI isoforms to platelet αIIbβ3 integrin. 2 , 9 , 15 It is likely that PDIs catalyze thiol disulfide exchange reactions within αIIbβ3 integrin, facilitating transition to the high affinity state of αIIbβ3 after an initiation of the inside‐out changes in αIIbβ3 in response to platelet activation. 33
Here, we specifically characterized the effects of PDIA1 inhibition on platelet aggregation. In previous reports, the role of platelet‐derived PDIA1 in regulating platelet function was shown with function‐blocking antibodies, PDIA1‐null platelets or small‐molecule inhibitors, such as PACMA‐31, a nonselective PDI inhibitor as well as rutin or bepristat, the selective PDIA1 inhibitors. 15 , 17 , 23 , 34 , 35 , 36 In our hands, rutin displayed a significantly weaker effect on collagen‐induced platelet aggregation compared with bepristat. Indeed, using an insulin reduction assay we showed that bepristat has a two‐fold lower IC50 value (Table 2) compared with rutin, which justified our choice of bepristat instead of rutin in our studies. Furthermore, a relatively lower cellular uptake of rutin compared with quercetin 37 may suggest that rutin might have displayed weaker effects on intraplatelet target than bepristat. On the other hand, our analysis revealed that bepristat was detected in platelets after incubation as evidenced by LC‐MS/MS–based measurements (Figure S3 in Supporting Information), supporting the notion that bepristat targets PDIA1 inside platelets.
Our study critically demonstrated that the inhibition of platelet PDIA1 by bepristat was more effective against platelet aggregation induced by GPVI receptor agonists such as collagen or convulxin than by thrombin or TRAP‐14 (Figure 3). We also demonstrated that bepristat strongly inhibited the production of TxA2 in platelets in response to either collagen or thrombin (Figure 5A). However, bepristat inhibition of collagen‐induced aggregation had negligible effects on thrombin‐induced platelet aggregation. Specifically, only at a lower concentration of thrombin bepristat reduced the platelet aggregation (Figure 3E), and importantly, this effect was similar to aspirin (Figure S2 in Supporting Information). These data show that PDIA1 targets platelet TxA2 production that is translated into the pronounced inhibition of collagen‐induced aggregation and weaker or absent effects on thrombin‐induced platelet aggregation. 38 , 39 Of note, the antiaggregatory effect of PDIA1 inhibition by bepristat was demonstrated for the first time in a study by Bekendam et al., 23 which used only the PAR‐1‐activating peptide SFLLRN to stimulate platelets, in contrast to our study, in which we used multiple platelets’ agonists and ascribing the antiaggregatory mechanisms of action of bepristat to the inhibition of TxA2 production.
GPVI‐dependent platelet activation as well as platelet stimulation by other agonists produce a variety of ROS 40 via nicotinamide adenine dinucleotide (phosphate) oxidase (Nox) including Nox1 and Nox2. Platelet‐derived ROS and TxA2 are important mediators required for full platelet activation. 30 , 40 , 41 , 42 Nox1 has been reported to play a larger role in platelet activation in response to collagen and mediate TxA2 production, whereas Nox2 mainly contributes to platelet activation by thrombin. 30 , 41 , 43 Accordingly, we hypothesized that PDIA1 inhibition in platelets would impair platelet aggregation via inhibition of the GPVI‐mediated ROS‐TxA2 pathway. Indeed, we demonstrated that inhibition of platelet PDIA1 by bepristat was associated with a substantial decrease in intraplatelet ROS production induced by collagen. These effects were comparable to those observed for a selective Nox1 inhibitor ML171 (Figure 6B). In contrast, apocynin, both a nonselective Nox inhibitor and a ROS scavenger, 44 inhibited intraplatelet ROS production to a lesser degree compared with ML171. The inhibition of ROS production by ML171 was related to the concentration‐dependent reduction in collagen‐induced platelet aggregation (Figure 6D), confirming that Nox1‐derived ROS are crucial for GPVI receptor‐mediated platelet aggregation. 41 , 43 These data imply that the blockade of PDIA1 by bepristat impairs platelet activation in response to collagen through the inhibition of Nox1‐mediated ROS production.
Although aspirin also limited ROS production in platelets, the effect of aspirin was weaker than bepristat (Figure 6A). However, aspirin inhibited TxA2 production to a greater degree than bepristat (Figure 5A). This suggests that the reduced ROS production caused by PDIA1 inhibition by bepristat was not a result of directly blocking TxA2 generation; instead, the inhibition of ROS production by bepristat reduced TxA2 generation. This may be further supported by results showing that bepristat displayed negligible inhibitory effect on platelet aggregation induced by AA in contrast to aspirin which completely abolished AA‐induced platelet aggregation (Figure 5C). On the other hand, TxA2 generated by activated platelets via interaction with TxA2‐receptors can induce Nox‐mediated ROS production to amplify platelet aggregation. 45 , 46 Additionally, COX may produce ROS 40 and peroxy compounds, which could oxidize H2DCF‐DA. 45
Given the recent evidence that the inhibition of peri/epicellular PDIA1 inhibited platelet aggregation in parallel with intracellular oxidative burst and mitochondrial respiration, 47 and that PDIA1 regulate Nox1 activity in vascular smooth muscle cells and in endothelial cells, 29 , 48 , 49 it is possible that PDIA1 in platelets also regulates Nox1‐mediated production of ROS. Gimenez et al. 29 demonstrated that in vascular smooth muscle cell PDIA1 increased phosphorylation of p47phox subunit in Nox1 activation. Interestingly, in parallel to our work, the role of PDIA1 in the regulation of platelet activation has been addressed in paper published recently by Gaspar et al. 50 These authors have proposed that Nox1 and PDIA1 cooperate with each other to regulate platelet activation; however, they control two distinct pathways downstream of GPVI receptor. Nox1 was suggested to be involved in the regulation of phosphoinositide 3‐kinase signaling pathway, whereas PDIA1 can regulate the activation of protein kinase C and mitogen‐activated protein kinases (MAPKs), which interact with p47phox to assemble the Nox1 complex. Superoxide generated by Nox1 may then activate protein kinase C and MAPKs in a positive feedback loop. 50 At the molecular level, the activation of TxA2 synthesis by Nox1‐derived ROS in response to GPVI receptor stimulation is mediated by the activation of p38 MAPK because the inhibition of p38 MAPK with the selective inhibitor SB202190 reduced TxA2 production following GPVI receptor stimulation. 41 These results and the findings reported by Gaspar et al., 50 showing that PDIA1 regulates the activation of MAPKs (ERK 1/2, p38) and subsequently Nox1 activity, support our concept that PDIA1 specifically controls GPVI receptor‐Nox1‐ROS and subsequent generation of TxA2 demonstrated here for the first time. In addition, the reduced production of Nox1‐derived ROS caused by PDIA1 inhibition may impair various intraplatelet redox mechanisms, including peroxynitrite‐mediated “peroxide tone” 51 or have also a direct effect on intraplatelet inside‐out αIIβ3 integrin activation (see Masselli et al. 40 for a recent review on intraplatelet redox regulations). This hypothesis is in line with the known inhibitory effects of ROS scavengers and Nox inhibitors on αIIbβ3 integrin activation and platelet aggregation. 45 , 46 We cannot exclude that PDIA1 regulates also Nox2‐ROS‐TxA2 pathway because the PDIA1 inhibition by bepristat suppressed ROS and TxA2 production in platelets activated with thrombin (Figure S1 in Supporting Information, Figure 5A), and as in the case of GPVI stimulation, p38 MAPK was shown to be involved in thrombin‐induced TxA2 synthesis. 52 The PDIA1‐dependent Nox2‐ROS‐TxA2 pathway can be important for activation of platelets by low concentrations of thrombin because aspirin inhibited platelet aggregation evoked by 0.015–0.02 U/mL of thrombin (Figure S2 in Supporting Information), and this effect was similar to bepristat (Figure 3E).
Apart from aggregation, PDIA1 has been found to also play a role in controlling other platelet functions such as platelet adhesion 13 or granule secretion. 15 , 17 In the present study, platelets pretreated with bepristat that were then stimulated with collagen displayed a reduced surface expression of P‐selectin (Figure 4B), thereby confirming that PDIA1 also controls the platelet secretion pathway. This regulation appears to be independent of its effects on the αIIbβ3 integrin, 15 but the mechanism involved needs to be identified.
PDIA1 has been reported to regulate the function of platelet glycoprotein GPIbα. 53 Here, we found that PDIA1 inhibition by bepristat did not modify ristocetin‐induced platelet agglutination mediated by GPIbα‐vWF binding in PRP (Figure 3F), whereas bepristat effectively inhibited platelet aggregation in PRP in response to collagen (Figure 3F). In contrast to a report by Li et al., 53 our data did not support a role for PDIA1 in regulating GPIbα function.
In previous studies, it was reported that the activation of αIIbβ3 integrin was impaired in PDIA1‐ or PDIA3‐null mouse platelets 2 , 15 , 17 ; however, αIIbβ3 integrin activation and platelet aggregation was recovered by exogenous active PDIA1 or PDIA3 enzymes. 2 , 15 , 17 Importantly, reduced aggregation of PDIA1‐ or PDIA3‐null platelets was recovered only by the addition of the PDI isoform that was missing, 9 suggesting that specific PDIs regulate αIIbβ3 integrin activation through distinct mechanisms. PDIA3 chiefly supports platelet aggregation through a direct interaction with extracellular αIIbβ3 integrin. In fact, our results confirmed that PDIA3 was more abundantly expressed than PDIA1 on the surface of activated platelets, which remained compatible with the regulation of platelet surface αIIbβ3 integrin activation. 2 Based on our results, we claimed that PDIA1‐dependent ROS‐TxA2 production represents a distinct mechanism that clearly differentiates the role of PDIA1 from PDIA3 in the regulation of platelet aggregation and explains why the reduced aggregation of PDIA1‐null platelets was recovered only by the addition of PDIA1 but not PDIA3.
To conclude, in the present work, we showed for the first time that PDIA1 can support platelet activation through regulating the intracellular Nox1‐ROS‐TxA2 pathway upstream of COX‐1, which plays an essential role in collagen‐induced activation of the αIIbβ3 integrin on the platelet surface. The PDIA1‐mediated mechanism of action seems to be distinct compared with the mechanism mediated by PDIA3 previously ascribed to αIIbβ3 integrin activation through the direct regulation of disulfide bond‐dependent conformational changes within the integrin. 2 , 10 The results of this present work indicate that PDIA1‐mediated Nox1‐ROS‐TxA2 production may be a promising and attractive target for inhibiting collagen‐induced platelet activation and thrombosis, without producing adverse effects on hemostasis. 43 This finding could be vital in preventing atherothrombosis, given the success of aspirin as a major antiplatelet drug limiting the production and release of TxA2. PDIA1‐based platelet pharmacology warrants future studies, given that recent studies highlighted the role of Nox1 function in platelets. 43 , 54 , 55
CONFLICT OF INTEREST
None of authors have a conflict of interest.
AUTHORS CONTRIBUTIONS
Kamil Przyborowski designed the study, performed the experiments, analyzed the data, and wrote the manuscript. Patrycja Kaczara, Anna Kurpinska, and Dagmara Wojkowska contributed to the study design, performed the experiments, and analyzed the data. Joanna Suraj‐Prazmowska, Kamil Karolczak, and Agnieszka Pelesz performed the experiments. Ivars Kalvins analyzed the data. Agata Malinowska and Anna Kurpinska performed the experiments and analyzed the data. Cezary Watala contributed to the study design and data analysis. Stefan Chlopicki designed the study and revised the manuscript. All authors approved the final version of the manuscript.
Supporting information
Supplementary Material
Przyborowski K, Kurpinska A, Wojkowska D, et al. Protein disulfide isomerase‐A1 regulates intraplatelet reactive oxygen species–thromboxane A2‐dependent pathway in human platelets. J Thromb Haemost. 2021;20:157–169. doi: 10.1111/jth.15539
Manuscript handled by: X. Long Zheng
Final decision: X. Long Zheng and 27‐Sep‐2021.
Funding information
This work was supported by funding from The National Centre for Research and Development (grant STRATEGMED1/233226/11/NCBR/2015 to S.Ch.), and partially by the Latvian National Fundamental and Applied Research Grant (grant No. Izp‐2018/1‐0143 to IK “Isoform selective PDI inhibitors: design, synthesis and SAR”). The open‐access publication of this article was funded by the Priority Research Area BioS under the program “Excellence Initiative – Research University” at the Jagiellonian University in Krakow. The equipment used for this work was sponsored in part by the Centre for Preclinical Research and Technology (CePT), a project cosponsored by the European Regional Development Fund and Innovative Economy, The National Cohesion Strategy of Poland.
Contributor Information
Kamil Przyborowski, Email: kamil.przyborowski@jcet.eu.
Stefan Chlopicki, Email: stefan.chlopicki@jcet.eu.
REFERENCES
- 1. Majithia A, Bhatt DL. Novel antiplatelet therapies for atherothrombotic diseases. Arterioscler Thromb Vasc Biol. 2019;39:546‐557. 10.1161/ATVBAHA.118.310955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang L, Wu Y, Zhou J, et al. Platelet‐derived ERp57 mediates platelet incorporation into a growing thrombus by regulation of the αIIbβ3 integrin. Blood. 2013;122:3642‐3650. 10.1182/blood-2013-06-506691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Passam FH, Lin L, Gopal S, et al. Both platelet‐ and endothelial cell‐derived ERp5 support thrombus formation in a laser‐induced mouse model of thrombosis. Blood. 2015;125:2276‐2285. 10.1182/blood-2013-12-547208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Holbrook L‐M, Watkins NA, Simmonds AD, Jones CI, Ouwehand WH, Gibbins JM. Platelets release novel thiol isomerase enzymes which are recruited to the cell surface following activation. Br J Haematol. 2010;148:627‐637. 10.1111/j.1365-2141.2009.07994.x [DOI] [PubMed] [Google Scholar]
- 5. Crescente M, Pluthero FG, Li L, et al. Intracellular trafficking, localization, and mobilization of platelet‐borne thiol isomerases. Arterioscler Thromb Vasc Biol. 2016;36:1164‐1173. 10.1161/atvbaha.116.307461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jurk K, Lahav J, Van aken H, Brodde MF, Nofer J‐R, Kehrel BE. Extracellular protein disulfide isomerase regulates feedback activation of platelet thrombin generation via modulation of coagulation factor binding. J Thromb Haemost. 2011;9:2278‐2290. 10.1111/j.1538-7836.2011.04509.x [DOI] [PubMed] [Google Scholar]
- 7. Cho J, Furie BC, Coughlin SR, Furie B. A critical role for extracellular protein disulfide isomerase during thrombus formation in mice. J Clin Invest. 2008;118:1123‐1131. 10.1172/jci34134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Reinhardt C, von Bruhl ML, Manukyan D, et al. Protein disulfide isomerase acts as an injury response signal that enhances fibrin generation via tissue factor activation. J Clin Invest. 2008;118:1110‐1122. 10.1172/jci32376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhou J, Wu Y, Chen F, et al. The disulfide isomerase ERp72 supports arterial thrombosis in mice. Blood. 2017;130:817‐828. 10.1182/blood-2016-12-755587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Holbrook LM, Sasikumar P, Stanley RG, Simmonds AD, Bicknell AB, Gibbins JM. The platelet‐surface thiol isomerase enzyme ERp57 modulates platelet function. J Thromb Haemost. 2012;10:278‐288. 10.1111/j.1538-7836.2011.04593.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wu Y, Ahmad SS, Zhou J, Wang L, Cully MP, Essex DW. The disulfide isomerase ERp57 mediates platelet aggregation, hemostasis, and thrombosis. Blood. 2012;119:1737‐1746. 10.1182/blood-2011-06-360685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhou J, Wu Y, Wang L, et al. The disulfide isomerase ERp57 is required for fibrin deposition in vivo. J Thromb Haemost. 2014;12:1890‐1897. 10.1111/jth.12709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lahav J, Wijnen EM, Hess O, et al. Enzymatically catalyzed disulfide exchange is required for platelet adhesion to collagen via integrin alpha2beta1. Blood. 2003;102:2085‐2092. 10.1182/blood-2002-06-1646 [DOI] [PubMed] [Google Scholar]
- 14. Wang L, Zhou J, Wang L, Wang CC, Essex DW. The b’ domain of protein disulfide isomerase cooperates with the a and a’ domains to functionally interact with platelets. J Thromb Haemost. 2019;17:371‐382. 10.1111/jth.14366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhou J, Wu Y, Wang L, et al. The C‐terminal CGHC motif of protein disulfide isomerase supports thrombosis. J Clin Invest. 2015;125:4391‐4406. 10.1172/jci80319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Qin RR, Zhu H, Wang F, et al. Platelet activation in diabetic mice models: the role of vascular endothelial cell‐derived protein disulfide isomerase‐mediated GP IIb/IIIa receptor activation. Aging. 2019;11(16):6358‐6370. 10.18632/aging.102192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim K, Hahm E, Li J, et al. Platelet protein disulfide isomerase is required for thrombus formation but not for hemostasis in mice. Blood. 2013;122:1052‐1061. 10.1182/blood-2013-03-492504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Holbrook LM, Sandhar GK, Sasikumar P, et al. A humanized monoclonal antibody that inhibits platelet‐surface ERp72 reveals a role for ERp72 in thrombosis. J Thromb Haemost. 2018;16:367‐377. 10.1111/jth.13878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jordan PA, Stevens JM, Hubbard GP, et al. A role for the thiol isomerase protein ERP5 in platelet function. Blood. 2005;105:1500‐1507. 10.1182/blood-2004-02-0608 [DOI] [PubMed] [Google Scholar]
- 20. Jasuja R, Furie B, Furie BC. Endothelium‐derived but not platelet‐derived protein disulfide isomerase is required for thrombus formation in vivo. Blood. 2010;116:4665‐4674. 10.1182/blood-2010-04-278184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stopa JD, Neuberg D, Puligandla M, Furie B, Flaumenhaft R, Zwicker JI. Protein disulfide isomerase inhibition blocks thrombin generation in humans by interfering with platelet factor V activation. JCI Insight. 2017;2:e89373. 10.1172/jci.insight.89373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zucker M, Seligsohn U, Yeheskel A, Mor‐Cohen R. An allosteric disulfide bond is involved in enhanced activation of factor XI by protein disulfide isomerase. J Thromb Haemost. 2016;14:2202‐2211. 10.1111/jth.13488 [DOI] [PubMed] [Google Scholar]
- 23. Bekendam RH, Bendapudi PK, Lin L, et al. A substrate‐driven allosteric switch that enhances PDI catalytic activity. Nat Commun. 2016;7:12579. 10.1038/ncomms12579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sitek B, Waldera‐Lupa DM, Poschmann G, Meyer HE, Stühler K. Application of label‐free proteomics for differential analysis of lung carcinoma cell line A549. In: Marcus K, editor. Quantitative Methods in Proteomics. Humana Press; 2012: 241‐248. [DOI] [PubMed] [Google Scholar]
- 25. Ishihama Y, Oda Y, Tabata T, et al. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol Cell Proteomics. 2005;4:1265‐1272. 10.1074/mcp.M500061-MCP200 [DOI] [PubMed] [Google Scholar]
- 26. Roy J, Wycislo KL, Pondenis H, Fan TM, Das A. Comparative proteomic investigation of metastatic and non‐metastatic osteosarcoma cells of human and canine origin. PLoS One. 2017;12:e0183930‐e. 10.1371/journal.pone.0183930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vejrazka M, Micek R, Stipek S. Apocynin inhibits NADPH oxidase in phagocytes but stimulates ROS production in non‐phagocytic cells. Biochim Biophys Acta. 2005;1722:143‐147. 10.1016/j.bbagen.2004.12.008 [DOI] [PubMed] [Google Scholar]
- 28. Kučera J, Binó L, Štefková K, et al. Apocynin and diphenyleneiodonium induce oxidative stress and modulate PI3K/Akt and MAPK/Erk activity in mouse embryonic stem cells. Oxid Med Cell Longev. 2016;2016:7409196. 10.1155/2016/7409196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gimenez M, Veríssimo‐Filho S, Wittig I, et al. Redox activation of Nox1 (NADPH oxidase 1) involves an intermolecular disulfide bond between protein disulfide isomerase and p47(phox) in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2019;39:224‐236. 10.1161/atvbaha.118.311038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vara D, Cifuentes‐Pagano E, Pagano PJ, Pula G. A novel combinatorial technique for simultaneous quantification of oxygen radicals and aggregation reveals unexpected redox patterns in the activation of platelets by different physiopathological stimuli. Haematologica. 2019;104:1879‐1891. 10.3324/haematol.2018.208819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sharda A, Kim SH, Jasuja R, et al. Defective PDI release from platelets and endothelial cells impairs thrombus formation in Hermansky‐Pudlak syndrome. Blood. 2015;125:1633‐1642. 10.1182/blood-2014-08-597419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thon JN, Peters CG, Machlus KR, et al. T granules in human platelets function in TLR9 organization and signaling. J Cell Biol. 2012;198:561‐574. 10.1083/jcb.201111136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wu Y, Essex DW. Vascular thiol isomerases in thrombosis: the yin and yang. J Thromb Haemost. 2020;18:2790‐2800. 10.1111/jth.15019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kung PH, Hsieh PW, Lin YT, Lee JH, Chen IH, Wu CC. HPW‐RX40 prevents human platelet activation by attenuating cell surface protein disulfide isomerases. Redox Biol. 2017;13:266‐277. 10.1016/j.redox.2017.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gaspar RS, da Silva SA, Stapleton J, et al. Myricetin, the main flavonoid in syzygium cumini leaf, is a novel inhibitor of platelet thiol isomerases PDI and ERp5. Front Pharmacol. 2019;10:1678. 10.3389/fphar.2019.01678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jasuja R, Passam FH, Kennedy DR, et al. Protein disulfide isomerase inhibitors constitute a new class of antithrombotic agents. J Clin Invest. 2012;122:2104‐2113. 10.1172/jci61228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang H, Hassan YI, Liu R, et al. Molecular mechanisms underlying the absorption of aglycone and glycosidic flavonoids in a Caco‐2 BBe1 cell model. ACS Omega. 2020;5:10782‐10793. 10.1021/acsomega.0c00379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rotondo S, Krauze‐Brzósko K, Manarini S, Evangelista V, Cerletti C. Licofelone, an inhibitor of cyclooxygenase and 5‐lipoxygenase, specifically inhibits cyclooxygenase‐1‐dependent platelet activation. Eur J Pharmacol. 2004;488:79‐83. 10.1016/j.ejphar.2004.02.006 [DOI] [PubMed] [Google Scholar]
- 39. Taylor ML, Ilton MK, Misso NL, Watkins DN, Hung J, Thompson PJ. The effect of aspirin on thrombin stimulated platelet adhesion receptor expression and the role of neutrophils. Br J Clin Pharmacol. 1998;46:139‐145. 10.1046/j.1365-2125.1998.00766.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Masselli E, Pozzi G, Vaccarezza M, et al. ROS in platelet biology: functional aspects and methodological insights. Int J Mol Sci. 2020;21:4866. 10.3390/ijms21144866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Walsh TG, Berndt MC, Carrim N, Cowman J, Kenny D, Metharom P. The role of Nox1 and Nox2 in GPVI‐dependent platelet activation and thrombus formation. Redox Biol. 2014;2:178‐186. 10.1016/j.redox.2013.12.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cho MJ, Liu J, Pestina TI, et al. The roles of αIIbβ3‐mediated outside‐in signal transduction, thromboxane A2, and adenosine diphosphate in collagen‐induced platelet aggregation. Blood. 2003;101:2646‐2651. 10.1182/blood-2002-05-1363 [DOI] [PubMed] [Google Scholar]
- 43. Vara D, Tarafdar A, Celikag M, et al. NADPH oxidase 1 is a novel pharmacological target for the development of an antiplatelet drug without bleeding side effects. FASEB J. 2020;34:13959‐13977. 10.1096/fj.202001086RRR [DOI] [PubMed] [Google Scholar]
- 44. Heumüller S, Wind S, Barbosa‐Sicard E, et al. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension. 2008;51:211‐217. 10.1161/hypertensionaha.107.100214 [DOI] [PubMed] [Google Scholar]
- 45. Begonja AJ, Gambaryan S, Geiger J, et al. Platelet NAD(P)H‐oxidase‐generated ROS production regulates alphaIIbbeta3‐integrin activation independent of the NO/cGMP pathway. Blood. 2005;106:2757‐2760. 10.1182/blood-2005-03-1047 [DOI] [PubMed] [Google Scholar]
- 46. Minuz P, Meneguzzi A, Fumagalli L, et al. Calcium‐dependent Src phosphorylation and reactive oxygen species generation are implicated in the activation of human platelet induced by thromboxane A2 analogs. Front Pharmacol. 2018;9:1‐15. 10.3389/fphar.2018.01081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gaspar RS, Mansilla S, Vieira VA, et al. The protein disulphide isomerase inhibitor CxxCpep modulates oxidative burst and mitochondrial function in platelets. Free Radic Biol Med. 2021;172:668‐674. 10.1016/j.freeradbiomed.2021.07.011 [DOI] [PubMed] [Google Scholar]
- 48. Soares Moretti AI, Martins Laurindo FR. Protein disulfide isomerases: redox connections in and out of the endoplasmic reticulum. Arch Biochem Biophys. 2017;617:106‐119. 10.1016/j.abb.2016.11.007 [DOI] [PubMed] [Google Scholar]
- 49. Fernandes DC, Wosniak J, Gonçalves RC, et al. PDIA1 acts as master organizer of NOX1/NOX4 balance and phenotype response in vascular smooth muscle. Free Radic Biol Med. 2021;162:603‐614. 10.1016/j.freeradbiomed.2020.11.020 [DOI] [PubMed] [Google Scholar]
- 50. Gaspar RS, Sage T, Little G, Kriek N, Pula G, Gibbins JM. Protein disulphide isomerase and NADPH oxidase 1 cooperate to control platelet function and are associated with cardiometabolic disease risk factors. Antioxidants. 2021;10:497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schildknecht S, van der Loo B, Weber K, Tiefenthaler K, Daiber A, Bachschmid MM. Endogenous peroxynitrite modulates PGHS‐1‐dependent thromboxane A2 formation and aggregation in human platelets. Free Radic Biol Med. 2008;45:512‐520. 10.1016/j.freeradbiomed.2008.04.042 [DOI] [PubMed] [Google Scholar]
- 52. Yacoub D, Théorêt J‐F, Villeneuve L, et al. Essential role of protein kinase C delta in platelet signaling, alpha IIb beta 3 activation, and thromboxane A2 release. J Biol Chem. 2006;281:30024‐30035. 10.1074/jbc.M604504200 [DOI] [PubMed] [Google Scholar]
- 53. Li J, Kim K, Jeong SY, et al. Platelet protein disulfide isomerase promotes glycoprotein Ibα‐mediated platelet‐neutrophil interactions under thromboinflammatory conditions. Circulation. 2019;139:1300‐1319. 10.1161/circulationaha.118.036323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vara D, Mailer RK, Tarafdar A, et al. NADPH oxidases are required for full platelet activation in vitro and thrombosis in vivo but dispensable for plasma coagulation and hemostasis. Arterioscler Thromb Vasc Biol. 2020;41:Atvbaha120315565. 10.1161/atvbaha.120.315565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gaspar RS, Ferreira PM, Mitchell JL, Pula G, Gibbins JM. Platelet‐derived extracellular vesicles express NADPH oxidase‐1 (Nox‐1), generate superoxide and modulate platelet function. Free Radic Biol Med. 2021;165:395‐400. 10.1016/j.freeradbiomed.2021.01.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material