

Summary
Background
Infliximab is a chimeric monoclonal antibody against tumour necrosis factor‐alpha for the treatment of Crohn's disease (CD) and ulcerative colitis (UC). Recently, a subcutaneous formulation of CT‐P13, an infliximab biosimilar, was approved for clinical use.
Aims
To characterise CT‐P13 pharmacokinetics (PK) and its clinically relevant determinants after subcutaneous administration through population PK modelling.
Methods
Data from a two‐part Phase I study with intravenous (5 mg/kg) and variable maintenance subcutaneous dosing of CT‐P13 with frequent PK sampling in patients with CD or UC were used. Population PK analysis was conducted by non‐linear mixed effects modelling. Covariates affecting PK parameters were chosen based on their clinical relevance (effect size of ≥20%) using a full fixed‐effect modelling approach.
Results
CT‐P13 PK was described by a two‐compartment model with linear elimination. The half‐life in a typical 70 kg patient with serum albumin of 44 g/L was 10.8 days. The typical value for clearance was 0.355 L/d, absorption constant 0.273/d, bioavailability 79.1%, central volume of distribution 3.10 L and peripheral volume of distribution 1.93 L. Clinically relevant covariates affecting clearance were body weight (+43.2% from 70 to 120 kg), the presence of anti‐drug antibodies (+39%) and serum albumin concentration (+30.1% from 44 to 32 g/L). Simulated drug exposure was comparable between routes of administration for patients weighing 50 or 70 kg, but lower with subcutaneous dosing in patients weighing 120 kg.
Conclusions
This first population PK model for subcutaneous CT‐P13 supports fixed subcutaneous maintenance dosing, although heavy patients had lower cumulative drug exposure.
For the same cumulative drug exposure (area under the curve), trough concentrations are numerically higher with subcutaneous dosing. Heavier patients may be underexposed with subcutaneous dosing. Drug clearance increases with body weight, lower serum albumin concentrations, and in the presence of anti‐drug antibodies.

1. INTRODUCTION
Crohn's disease (CD) and ulcerative colitis (UC) are chronic inflammatory bowel diseases (IBD) that can result in bowel damage and disability. 1 , 2 Biologic treatment is indicated for patients failing conventional therapy with 5‐aminosalicylates, corticosteroids and/or immunosuppressants. Infliximab, a chimeric immunoglobulin G1κ monoclonal antibody against tumour necrosis factor (TNF)‐α, was the first biologic drug approved for the treatment of IBD. 3
As patents for infliximab reference products have expired, several biosimilars, such as CT‐P13, have accessed the market. 4 , 5 Recently, a subcutaneous formulation of CT‐P13 has been approved for clinical use in IBD after demonstration of pharmacokinetic (PK) non‐inferiority compared to intravenous CT‐P13. 6 After weight‐based intravenous induction at weeks 0 and 2, 120 mg of CT‐P13 is administered subcutaneously every 2 weeks regardless of body weight. Subcutaneous administration has the prospect of increased convenience for patients, reduced demand for hospital resources and potentially a lower risk for exposure to COVID‐19. 7 , 8 , 9 , 10
Compared to intravenous administration, subcutaneous administration of monoclonal antibodies is characterised by slower absorption, incomplete bioavailability and lower peak concentrations. 11 The pharmacokinetics of intravenous infliximab has been described across the range of its indications. 12 , 13 , 14 , 15 , 16 Broadly speaking, the clearance is mainly dependent on body weight, serum albumin concentration and the presence of anti‐drug antibodies. The pharmacokinetics of the subcutaneous formulation remain uncharacterised. A better understanding of the PK determinants for subcutaneous CT‐P13 is a prerequisite for potential optimisation of drug dosing regimens.
Despite heterogeneity between studies and special patient groups (eg perianal fistulising CD), a serum infliximab concentration at trough >3 mg/L during maintenance treatment with intravenous infliximab is generally accepted as the therapeutic target. 17 In general, the relationship between trough concentrations and cumulative drug exposure as expressed with the area under the curve may differ by route of administration. Consequently, it remains unclear how trough thresholds from intravenous infliximab translate to those for subcutaneous CT‐P13.
We aimed to characterise the pharmacokinetics of subcutaneous CT‐P13 and identify clinically relevant PK determinants in patients with IBD, based on a population PK analysis of subcutaneous infliximab CT‐P13 in patients with CD and UC from a two‐part randomised phase 1 study with rich PK sampling. Monte Carlo simulations were performed to evaluate drug exposure with fixed‐dose subcutaneous maintenance treatment.
2. METHODS
2.1. Clinical study design and patients
We used patient data from the CT‐P13 1.6 study (NCT02883452), a Phase I study to evaluate the pharmacokinetics, efficacy and safety of subcutaneous CT‐P13 in patients with active CD and UC. 6
Part 1 of the study was designed to find the optimal dose of subcutaneous CT‐P13. It enrolled 44 patients with moderate‐to‐severe CD (CD activity index scores 220‐450) with at least 3 months of disease duration, insufficient response or intolerance to corticosteroids and/or immunosuppressants and no previous exposure to biological treatment. All patients received 5 mg/kg CT‐P13 intravenously at weeks 0 and 2 and were randomised to one of four regimens at week 6: intravenous CT‐P13 at week 6 and every 8 weeks thereafter until week 54 (cohort 1; n = 13), 120 mg CT‐P13 subcutaneously (cohort 2; n = 11), 180 mg CT‐P13 subcutaneously (cohort 3; n = 12) or 240 mg CT‐P13 subcutaneously (cohort 4; n = 8) at week 6 and every 2 weeks up to week 54. The PK sampling scheme is shown in Table S1.
Part 2 of the study was designed to demonstrate non‐inferiority in pharmacokinetics between subcutaneous and intravenous CT‐P13. It enrolled 131 patients (53 CD, 78 UC). In addition to inclusion criteria listed above, patients with CD were required to have at least one of the following at screening: C‐reactive protein (CRP) >5 mg/L, faecal calprotectin (FC) >100 mg/kg, Simple endoscopic score for CD (SES‐CD) of ≥6 for ileocolonic disease or ≥4 including the presence of ulcers from at least one segment for ileal or colonic CD. Patients with UC had a total Mayo score between 6 and 12 points with an endoscopic subscore ≥2. In the intravenous arm, patients (n = 65; CD 25, UC 40) received 5 mg/kg CT‐P13 intravenously at weeks 0, 2, 6, 14 and 22, followed by CT‐P13 subcutaneously at week 30 and every 2 weeks up to week 54, with weight‐based subcutaneous doses (≤80 kg: 120 mg; >80 kg: 240 mg). In the subcutaneous arm, patients (n = 66; CD 28, UC 38) received 5 mg/kg CT‐P13 intravenously at weeks 0 and 2, followed by subcutaneous weight‐based doses at week 6 and every 2 weeks up to week 54. The PK sample scheme is given in Table S1.
2.2. Serum infliximab and anti‐drug antibody concentrations
Serum infliximab levels were quantitatively measured using Meso Scale Discovery electrochemiluminescence (Meso Scale Diagnostics, LLC, Rockville, MD). The lower limit of quantification (LLOQ) was 0.1 mg/L, samples below that value were fixed at 0.05 mg/L. When consecutive samples were below the LLOQ, only the first sample was retained in the PK database. Anti‐drug antibodies were analysed using an electrochemiluminescence platform with an affinity capture elution step (Celltrion, Inc, Incheon, Republic of Korea). The assays were drug tolerant and could detect an anti‐drug antibody concentration of ≥25 ng/mL in the presence of 120 mg/L infliximab in serum.
2.3. Population pharmacokinetic model development
The base pharmacokinetic model was developed using the first‐order conditional estimation with η‐ε interaction (FOCE‐I). As PK of IFX has been demonstrated to be linear, models with one and two compartments with first‐order absorption with and without lag time and first‐order elimination, were tested. The models were parameterised in terms of clearance and volume of distribution. Since CT‐P13 was administered by both subcutaneous and intravenous route, bioavailability was estimated. Inter‐individual and inter‐occasion variability of the PK parameters was modelled assuming log‐normal distribution of the individual parameters. The residual error was modelled with a combination of an additive and proportional error.
After the development of the structural population PK model, associations between patient characteristics and PK parameters were evaluated. A full covariate modelling approach (full fixed‐effects modelling) was used. 18 Based on published literature and clinical interest, and mechanistic plausibility, covariates for testing were predefined (Table 1). The association of continuous covariates and PK parameters was described using power models. Categorical covariates entered the model as power functions with a separate dichotomous (0, 1) covariate serving as an on‐off switch for the effect.
TABLE 1.
Pre‐defined covariate‐parameter relations with a short description of the physiologic rationale
| Covariate | Parameter | Rationale |
|---|---|---|
| Weight (time‐dependent) | CL, V1, V2, Q, F | Weight‐based dosing |
| Age at diagnosis (time‐independent) | CL | Younger age at diagnosis could portend more aggressive disease with higher CL |
| Albumin (time‐dependent) | CL | Marker of disease activity, surrogate marker of antibody turnover |
| C‐reactive protein (time‐dependent) | CL | Marker of disease activity |
| White blood cell count (time‐dependent) | CL | Marker of disease activity |
| Platelet count (time‐dependent) | CL | Marker of disease activity |
| Calprotectin (time‐dependent) | CL | Marker of disease activity |
| Haemoglobin (time‐dependent) | CL | Marker of disease activity |
| Crohn's disease vs ulcerative colitis (time‐independent) | CL | More evidence for faecal drug loss in ulcerative colitis than in Crohn's disease |
| Anti‐drug antibodies (time‐dependent) | CL, F | Prior knowledge |
| Concomitant immunomodulator (time‐dependent) | CL | Potential additional effect on clearance beyond suppressing anti‐drug antibody formation |
| Concomitant corticosteroid (time‐dependent) | CL | Clinical interest |
| Subcutaneous dose (time‐independent) | F | Clinical interest |
Abbreviations: CL, clearance; F, bioavailability; Q, intercompartmental clearance; V1, central compartment volume of distribution; V2, peripheral compartment volume of distribution.
Correlations between covariates were assessed, variables with absolute correlation coefficient values greater than 0.30 were not simultaneously included as predictors in the model. The effect of a covariate was judged to be clinically relevant if the effect size was at least 20% of the normalised value of individual PK parameters. Non‐parametric 95% confidence intervals (CI) for effect sizes were derived from the variance‐covariance matrix with 1000 bootstrap replicates. For continuous covariates, the magnitude of effect was calculated for the minimum, median and maximum value present in the dataset. If the magnitude of a covariate effect size did not exceed 20% and its 95% CI was fully within this range, it was removed from the model irrespective of its statistical significance. Covariates with an effect size estimate within the range of clinical unimportance and 95% CI extending outside this range were evaluated on an individual basis, considering the precision of the estimate and the likelihood of encountering such a value in a clinical population of patients with IBD. Variables, whose effect on PK could not be uniquely estimated due to strong correlations, were evaluated post hoc in an exploratory analysis using the final population PK model.
2.4. Model evaluation
The fit of the model to data was evaluated on the basis of several criteria. Relative standard errors of the estimated parameters and standard goodness‐of‐fit plots were inspected. Shrinkage was calculated for each individual PK parameter value to evaluate possible regression to the mean. The final model was evaluated using predicted‐corrected visual predictive checks stratified for the route of administration and the dose. Bootstrapping was performed to obtain non‐parametric estimates of uncertainty for the final model (n = 2000 bootstraps).
2.5. Monte Carlo simulations
Monte Carlo simulations were performed to evaluate serum trough concentrations were above target trough concentrations identified for intravenous dosing during maintenance therapy with fixed subcutaneous dosing. A virtual population was simulated of 3000 patients with a body weight of 50, 70 or 120 kg (1000 per weight). Albumin was randomly sampled from the probability density function of albumin in the original dataset and the virtual patients were modelled as not having anti‐drug antibodies. At day 0 and day 14 an intravenous dose of 5 mg/kg was administered, followed by subcutaneous doses of 120 mg every 2 weeks starting at day 42. Conventional intravenous 5 mg/kg dosing every 8 weeks was also simulated for the 1000 virtual patients per weight stratum. The cumulative area under the curve over 8 weeks was calculated for every patient. Monte Carlo simulations were performed with the developed final population PK model, including inter‐individual and inter‐occasion variability.
2.6. Software
Data were imported in R (version 3.4.1, R‐core team, Vienna, Austria) for data wrangling and descriptive analyses and visualisation (packages dplyr, ggplot2, xpose4). The population analysis of the PK data was performed using non‐linear mixed effects modelling (NONMEM software v7.3.0, ICON Development Solutions, Ellicott City, Maryland, USA).
3. RESULTS
3.1. Study population
Data were available from 175 patients in the CT‐P13 1.6 study (part 1: n = 44; part 2: n = 131) (Table 2). A total of 2901 IFX serum concentrations were available; 127 samples (4.4%) were below the LLOQ. Of these, 36 were excluded as only the first sample in a series of consecutive samples below the LLOQ in an individual patient was retained, a further 50 samples were censored due to implausible increases or decreases in concentration. Three patients with concentrations below the LLOQ after subcutaneous administration were removed from the dataset (43 samples censored). These three patients were excluded from the analysis as the majority of their IFX concentrations after subcutaneous administration was below the LLOQ due to the presence of anti‐drug antibodies. Inclusion of these patients incorrectly inflated inter‐individual variability of bioavailability, while their inclusion did not change clearance‐related parameters. Eventually, a total of 2772 samples (95.6% of all available samples) were used for the population PK analysis.
TABLE 2.
Patient characteristics at baseline
| Women, n (%) | 95 (54) |
| Age, years, median (IQR) [range] | 36 (29‐48) [18‐70] |
| Age at diagnosis, years, median (IQR) [range] | 33 (22‐42) [3‐68] |
| Crohn's disease, n (%) | 97 (55) |
| Ulcerative colitis, n (%) | 78 (45) |
| Body weight, kg, median (IQR) [range] | 69 (60‐80) [43‐118] |
| Immunomodulators | |
| Pre‐treatment, n (%) | 147 (84) |
| During treatment, n (%) | 80 (46) |
| Corticosteroids | |
| Pre‐treatment, n (%) | 106 (61) |
| During treatment, n (%) | 71 (41) |
|
Crohn's disease activity index (n = 96), median (IQR) [range] Full Mayo clinic score (n = 73), median (IQR) [range] |
283 (249‐333) [222‐446] 8 (7‐9) [4‐11] |
| Haemoglobin, g/L, median (IQR) [range] | 128 (117‐139) [43‐170] |
| Platelet count, ×109/L, median (IQR) [range] | 295 (243‐366) [43‐698] |
| White blood cell count, ×109/L, median (IQR) [range] | 7.1 (5.6‐9.1) [2.7‐43] |
| Albumin, g/L, median (IQR) [range] | 44 (40‐46) [28‐54] |
| C‐reactive protein, mg/L, median (IQR) [range] | 3.2 (1.2‐7.9) [0.2‐89.4] |
| Faecal calprotectin, mg/kg, median (IQR) [range] | 749 (265‐1576) [10‐6913] |
| Neutralising antibodies during treatment, n (%) | 58 (33) |
Part 1 of the study (n = 44, all with Crohn's disease) was designed to find the optimal dose of subcutaneous CT‐P13. Part 2 (n = 131, 53 with Crohn's disease, 78 with ulcerative colitis) of the study was designed to demonstrate non‐inferiority in pharmacokinetics between subcutaneous and intravenous CT‐P13.
3.2. Population PK analysis
3.2.1. Base model
A two‐compartment model with first‐order absorption and first‐order elimination best described the data (Table 3, Figure S1). Inter‐individual variability in PK parameters could be estimated for the absorption constant, clearance, central compartment volume of distribution and bioavailability. Inter‐occasion variability was quantified for clearance with each dosing event defined as a new occasion. The residual error was described with a combined additive and proportional error model. In all the analyses, the value of the additive error was higher than 0.1 mg/L.
TABLE 3.
Base and final model parameter estimates
| Parameter | Estimate (%RSE) [%shrinkage] | Estimate (%RSE) [%shrinkage] | Bootstrapped estimate (95% CI) |
|---|---|---|---|
| Base model (OFV = 11 866.0) | Final model (OFV = 11 607.7) | ||
| CL (L/d) | 0.365 (3) | 0.355 (2) | 0.355 (0.338‐0.373) |
| Vc (L) | 3.12 (4) | 3.10 (3) | 3.10 (2.94‐3.26) |
| Vp (L) | 1.96 (2) | 1.93 (2) | 1.93 (1.71‐2.13) |
| F1 (%) | 77.3 (3) | 79.1 (3) | 79.1 (74.0‐84.3) |
| Ka (/d) | 0.273 (8) | 0.273 (8) | 0.272 (0.235‐0.309) |
| Q (L/d) | 0.599 (4) | 0.598 (4) | 0.607 (0.403‐0.836) |
| Covariate effects | |||
| Albumin on CL | — | −0.826 (11) | −0.821 (−1.128‐−0.564) |
| Body weight on CL | — | 0.666 (15) | 0.671 (0.447‐0.862) |
| ATI on CL | — | 1.39 (3) | 1.40 (1.28‐1.54) |
| Body weight on Vc | — | 0.385 (34) | 0.388 (0.167‐0.616) |
| Body weight on Vp | — | 1.08 (9) | 1.06 (0.662‐1.480) |
| Body weight on Q | — | 1.26 (15) | 1.24 (0.01‐2.56) |
| Inter‐individual variability | |||
| IIV on CL (%) | 38.9 (14) [4] | 27.7 (14) [6] | 27.5 (23.3‐31.7) |
| IIV on F1 (%) | 20.3 (13) [12] | 16.4 (15) [16] | 16.2 (12.3‐21.0) |
| IIV on Vc (%) | 25.4 (22) [16] | 21.4 (21) [18] | 21.2 (16.9‐26.1) |
| IIV on Ka (%) | 45.6 (45) [29] | 48.5 (45) [26] | 50.5 (34.4‐69.9) |
| Correlation between CL and F1 | −0.024 (36.3) | −0.013 (42.2) | −0.013 (−0.025‐−0.001) |
| Correlation between CL and Vc | 0.055 (24.4) | 0.028 (31.4) | 0.027 (0.014‐0.044) |
| Correlation between CL and Ka | −0.050 (54.6) | −0.046 (53.2) | −0.047 (−0.085‐−0.008) |
| Correlation between F1 and Vc | −0.002 (429) | 0.00008 (744) | 0.002 (−0.011‐0.013) |
| Correlation between F1 and Ka | −0.011 (147) | 0.003 (413) | 0.005 (−0.017‐0.036) |
| Correlation between Vc and Ka | −0.066 (47) | −0.069 (47) | −0.064 (−0.103‐−0.01) |
| Inter‐occasion variability | |||
| IOV on CL (%) | 17.2 (6) [21] | 17.5 (6) [25] | 17.5 (14.9‐19.9) |
| Residual error model | |||
| Additive error (mg/L) | 1.77 (3) | 1.66 (3) | 1.63 (1.21‐2.02) |
| Proportional error | 0.104 (2) | 0.102 (2) | 0.102 (0.089‐0.115) |
Equations for the main pharmacokinetic parameters; anti‐drug antibodies equal 1 if present and 0 if absent: . .
Bootstrap: A total of 1705 (85.3%) successfully minimised runs were used to calculate the medians and the 2.5th and 97.5th percentile.
Abbreviations: ATI, antibodies to infliximab; CI, confidence interval; CL, clearance; F1, bioavailability; IIV, inter‐individual variability; IOV, inter‐occasion variability; Ka, absorption constant; OFV, objective function value; Q, intercompartmental clearance; RSE, relative standard error; Vc, volume of distribution in the central compartment; Vp, volume of distribution in the peripheral compartment.
3.3. Final model
During the development of the covariate model, strong correlations were noted between the following variables: albumin–CRP (r = −0.55), albumin–haemoglobin (r = 0.43), albumin–calprotectin (r = −0.32), CRP–platelet count (r = 0.42), CRP–FC (r = 0.35) (Figure S2). These variables were therefore not simultaneously included in the full covariate model. Of these variables, albumin had the largest effect on clearance and therefore this was used for subsequent covariate model development (Table S2).
The effect sizes of individual covariates on PK parameters from the full covariate model are shown in Figure 1. Based on criteria of a clinically relevant effect on clearance, body weight, anti‐drug antibodies and serum albumin concentrations were included in the final model. Population PK parameter estimates for a typical patient with a body weight of 70 kg, serum albumin of 44 g/L and no anti‐drug antibodies were: clearance 0.355 L/d, absorption constant 0.273/d, bioavailability 79.1%, volume of distribution in the central compartment 3.10 L and volume of distribution in the peripheral compartment 1.93 L (Table 3). Clearance increased with body weight (+43.2% from 70 kg to 120 kg), the presence of anti‐drug antibodies (+39%) and decreasing serum albumin concentrations (+30.1% from 44 g/L to 32 g/L). The inclusion of covariates reduced the inter‐individual variability of clearance by 11.2 percentage points. In a post hoc exploratory analysis forcing CRP into the final model, the inter‐individual variability for clearance decreased by 0.7 percentage points.
FIGURE 1.
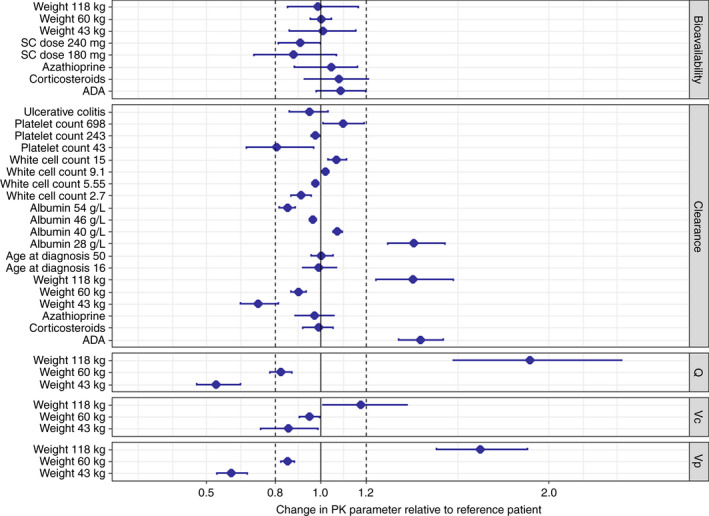
Covariate effects on pharmacokinetic parameters of infliximab (estimated with the full covariate model) relative to the reference subject (70 kg, 32 years at diagnosis, Crohn's disease, no concomitant corticosteroids or immunomodulators, albumin 44 g/L, white blood cell count 7.1 × 109/L, platelet count 295 × 109/L, no anti‐drug antibodies). An effect greater than ±20% was considered clinically important. The point estimate with 95% bootstrapped confidence intervals is depicted. Abbreviations: ADA, anti‐drug antibodies; Q, intercompartmental clearance; SC, subcutaneous; Vc, volume of distribution in the central compartment; Vp, volume of distribution in the peripheral compartment [Colour figure can be viewed at wileyonlinelibrary.com]
Body weight also had a clinically relevant effect on the volume of distribution in the central compartment, peripheral compartment and intercompartmental clearance. Inclusion of only body weight in the model explained 8.0% of inter‐individual variability of clearance (3.1 percentage points of 38.6%) and 13.0% of inter‐individual variability of the volume of distribution in the central compartment (3.3 percentage points of 25.4%).
The estimated parameters of the final population model were precise and the goodness‐of‐fit plots did not show any major deficiencies (Figure S3). The prediction‐corrected visual predictive checks stratified for the route of administration demonstrated that the final model adequately captured both the central tendency and the inter‐individual variability in drug exposure independent of the route or the administered dose (Figure 2).
FIGURE 2.
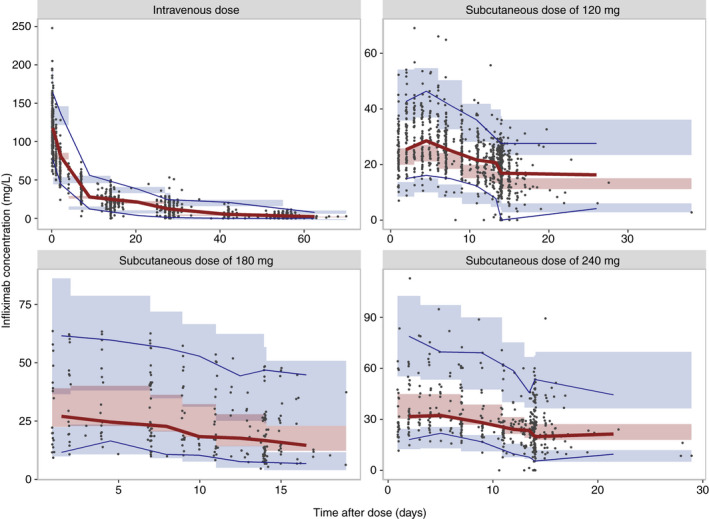
Prediction‐corrected visual predictive checks of the observed infliximab concentrations per route of administration and dose: intravenous 5 mg/kg (upper left), subcutaneous 120 mg (upper right), subcutaneous 180 mg (lower left), subcutaneous 240 mg (lower right). Observed infliximab concentrations are represented by dots, the solid red line connects the observed median prediction‐corrected infliximab concentrations. The blue lines connect the 5th and 95th percentiles of the prediction‐corrected observations. Shaded areas denote the 95% confidence interval of the median, 5th, and 95th percentile of the simulated values (n = 1000) [Colour figure can be viewed at wileyonlinelibrary.com]
3.4. Monte Carlo simulations
Simulated concentration‐time profiles of typical patients illustrate that fixed dosing of subcutaneous CT‐P13 during maintenance treatment maintains trough concentrations considered adequate for intravenous dosing across a range of body weights (Figure 3). The Monte Carlo simulations demonstrated that for patients with body weights of 50, 70 and 120 kg 96.4%, 90.3% and 71.9% of the serum trough concentrations were above 7 mg/L during maintenance treatment; 99%, 98.3% and 90.6% above 5 mg/L; 99.9%, 99.9% and 99.5% above 3 mg/L (Figure 3). The area under the curve was comparable regardless of dosing route in patients weighing 70 kg; higher with subcutaneous than intravenous dosing in patients weighing 50 kg, and lower with subcutaneous dosing in patients weighing 120 kg. For the same area under the curve, trough concentrations with subcutaneous dosing were higher than with intravenous dosing (Figure 4).
FIGURE 3.
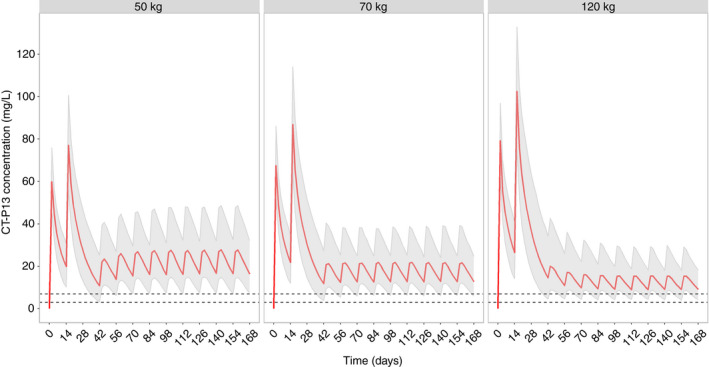
Simulated concentration‐time profiles of CT‐P13 with intravenous induction (5 mg/kg) at weeks 0 and 2 and subcutaneous maintenance dosing with 120 mg every 2 weeks for virtual patients with body weights of 50 kg, 70 kg and 120 kg (1000 per weight group). Albumin was randomly sampled from the probability density function of albumin in the original dataset; virtual patients had no anti‐drug antibodies. Medians, 5th and 95th percentile are indicated; dotted lines denote 3 and 7 mg/L [Colour figure can be viewed at wileyonlinelibrary.com]
FIGURE 4.
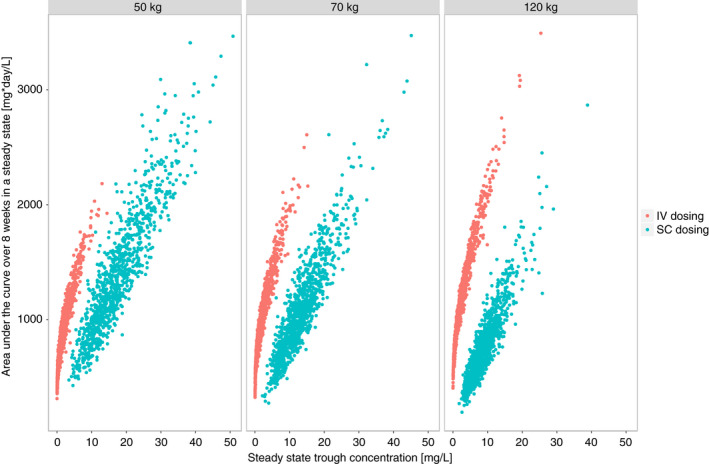
A comparison of cumulative drug exposure expressed as the area under the curve over 8 weeks during maintenance with trough concentrations at the end of the 8‐week period stratified by weight and route of administration. Results are based on simulations with 1000 patients per weight stratum per route of administration [Colour figure can be viewed at wileyonlinelibrary.com]
4. DISCUSSION
We developed the first population PK model for subcutaneous infliximab CT‐P13 in patients with CD and UC. Clearance increased to a clinically relevant extent with increasing body weight, the presence of anti‐drug antibodies and decreasing serum albumin concentrations. There were no relevant PK differences between CD and UC. Our findings support fixed dosing of CT‐P13 for subcutaneous maintenance treatment in most patients, although simulated patients weighing 120 kg had lower drug exposure with fixed subcutaneous compared to weight‐based intravenous dosing. The model adequately described the pharmacokinetics of CT‐P13 regardless of dose and route of administration.
The calculated half‐life of CT‐P13 for the typical patient, 10.8 days, was comparable to estimates from previous studies of infliximab pharmacokinetics in IBD. 12 , 13 , 19 The absence of other population PK models for subcutaneous infliximab, precludes further direct comparisons. The estimated bioavailability of subcutaneous CT‐P13 (79.1%) was higher than what was previously observed for other subcutaneously administered anti‐TNF monoclonal antibodies in IBD: adalimumab (64%), 20 golimumab (51%‐53%), 21 , 22 but lower than for ustekinumab (87%). 23 Due to variable sampling schedules, estimates of absorption constants from studies with sparse sampling may not be as reliable. Focusing only on studies with more intensive PK sampling, the absorption constant for CT‐P13 is comparable to adalimumab 24 and higher than for golimumab 21 or ustekinumab. 23
Mechanistic understanding of absorption of biologics after subcutaneous administration is evolving. Incomplete bioavailability may be the result of proteolysis at the injection site. 25 Catabolism is decreased by binding to the neonatal Fc receptor (FcRn), as has been shown in an animal model where IgG variants with higher affinity for FcRn had higher bioavailability. 26 Target‐mediated drug deposition is an additional potential mechanism of catabolism at the injection site. Transport from the interstitial space to the lymphatic system is the rate‐limiting step for absorption. 25 Concentration‐time profiles with subcutaneous administration are characterised by lower peak concentrations and smaller differences between peak and trough concentrations. Higher trough concentrations were observed with subcutaneous dosing, but overall drug exposure, expressed as the area under the curve, between the intravenous and subcutaneous arm was comparable. 6 The clinical implications of these differences are unclear. Trough concentrations were incorporated into therapeutic drug monitoring algorithms for intravenous infliximab for pragmatic reasons, even though it is not clear whether this is the optimal exposure metric to guide treatment decisions nor if it has the strongest association with therapeutic efficacy. 27 Available studies for subcutaneous CT‐P13 were not powered to assess differences in efficacy between the intravenous and subcutaneous route. Subcutaneous dosing could theoretically result in lower clinical efficacy if the effect of infliximab were dependent on peak concentrations. There is some indication that peak infliximab concentrations during induction are associated with subsequent remission, 28 but there is no evidence for this phenomenon during maintenance. In the published clinical study, rates of clinical remission (68.4% in the subcutaneous arm vs. 61.5% in the intravenous arm) and endoscopic improvement (55.3% in the subcutaneous arm vs. 56.4% in the intravenous arm) were comparable regardless of the route of administration for maintenance dosing. 6 A quartile‐based exposure‐response analysis of subcutaneous CT‐P13 trough concentrations at week 54 showed a plateau of clinical remission in the third quartile with trough concentrations ≥21.5 mg/L, whilst the proportion of patients with a FC ≤250 mg/kg increased up to the fourth quartile with trough concentrations ≥26.7 mg/L. 29
Clearance of CT‐P13 in our study increased with body weight, as has already been observed with intravenous infliximab. 12 , 13 It may therefore seem counterintuitive to suggest fixed dosing during subcutaneous maintenance treatment for a biologic which is dosed by weight when given intravenously. The significance of body weight for the pharmacokinetics of a monoclonal antibody is determined by the magnitude of effect in the PK model and the proportional contribution to interindividual variability of a given PK parameter. Wang et al simulated weight‐based and fixed dosing for 12 monoclonal antibodies. 30 In a power function covariate model, fixed dosing maintained consistent drug exposure across the range of body weights for exponents smaller than 0.32, while weight‐based had an advantage for exponents larger than 0.68. Neither approach could maintain drug exposure within ±20% for intermediate exponent values. In our model, the impact of body weight on clearance was in the intermediate range with an exponent of 0.666 where either approach would be acceptable, but fixed dosing provides greater simplicity and convenience. Body weight explained 8.0% and 13.0% of interindividual variability for clearance and volume of distribution in the central compartment respectively. Taken together, this suggests that weight‐based dosing would not provide an appreciable reduction in drug exposure variability.
An individual participant pooled data analysis from key clinical trials of IFX (ACCENT‐I, ACT‐1, ACT‐2, SONIC) did not demonstrate significant differences in the odds of reaching clinical or endoscopic outcomes for obese (body mass index ≥30 kg/m2) compared to non‐obese patients, although trough concentrations were not compared. 31 In a recent meta‐analysis, of TNF antagonists, obesity was associated with a higher risk of treatment failure in UC, but not CD, for both weight‐based and dose‐fixed agents, suggesting that mechanisms other than pharmacokinetics may be responsible for these observations. 32 In contrast, a post hoc analysis of the IM‐UNITI trial of ustekinumab with fixed maintenance dosing showed that obese patients had significantly lower median maintenance trough concentrations (2.98 mg/L vs. 4.43 mg/L in patients with a normal or low body mass index). 33 The impact of body weight on ustekinumab clearance is in the intermediate range (0.641) where neither regimen can maintain consistent drug exposure. 23
The presence of anti‐drug antibodies measured with a drug‐tolerant assay increased clearance by 39%, which is a consistent observation in infliximab PK models with the magnitude of effect ranging from 29% to 72%. 12 , 13 , 19 , 34 Although the effect of immunosuppressants on clearance is presumed to occur through the decreased formation of anti‐drug antibodies, this relationship has not been universally identified in published PK models, including ours. 13 , 19 , 34 As this relationship is indirect, it is possible that our study was underpowered to detect it. Concomitant corticosteroids did not have a significant effect on clearance. Potential differences in immunogenicity between intravenously and subcutaneously administered CT‐P13 remain to be explored, although currently available data for monoclonal antibodies used outside IBD do not suggest an excess of anti‐drug antibody formation with subcutaneous administration. 35
Serum albumin concentrations had a clinically relevant effect on clearance, which is consistent with prior studies on infliximab, other anti‐TNF agents, 21 and non‐anti‐TNF monoclonal antibodies used in IBD. 23 , 36 This association has not been conclusively explained and at least two plausible hypotheses exist. First, the FcRn not only binds IgG, but also albumin and thus protects it from intracellular catabolism. 37 Higher serum albumin concentrations could reflect an increased number of FcRn and lower CT‐P13 clearance. Second, severe intestinal inflammation may result in loss of both albumin and monoclonal antibodies through the diseased mucosa. 38 Although a higher inflammatory burden is thought to result in accelerated clearance, none of the other measures of disease activity in our study (CRP, FC, white blood cell count, platelet count) had a clinically relevant effect on clearance beyond the effect of albumin, suggesting that serum albumin concentrations reflect multiple processes important for the pharmacokinetics of monoclonal antibodies and not merely disease activity. Finally, it should be noted that the inclusion of covariates in the model (body weight, anti‐drug antibodies, serum albumin concentration) explained 11.2 percentage points of interindividual variability for clearance, leaving 27.7 percentage points unexplained. Although this magnitude of reduction is at the higher end of PK models in IBD, the substantial residual interindividual variability highlights the necessity for Bayesian forecasting if using our model to inform precision dosing in future interventional studies.
Our results provide reassurance for practicing clinicians that fixed subcutaneous maintenance dosing provides drug exposure comparable with intravenous dosing for simulated patients weighing 50 or 70 kg. Nevertheless, exposure with subcutaneous dosing was lower for simulated patients weighing 120 kg. For a given area under the curve, trough concentrations with subcutaneous dosing were numerically higher than with intravenous dosing, which suggests that thresholds associated with clinical outcomes cannot be directly extrapolated from intravenous to subcutaneous dosing. Further clinical exposure‐response studies are required to identify concentration thresholds for relevant outcomes with subcutaneous dosing.
The strengths and limitations of our study should be acknowledged. Prospectively collected data in a randomised multi‐centre study with rich PK sampling enabled precise estimation of PK model parameters and IIV. A broad range of covariates permitted exploration of covariate relationships which were understudied in previous work on infliximab. While PK sampling at trough was performed on multiple occasions both during induction and maintenance treatment, rich sampling was concentrated between weeks 22 and 30. Despite the absence of evidence for convincingly different pharmacokinetics outside this sampling period, this element of study design should nonetheless be borne in mind. Furthermore, all patients included in the study were naïve to biological treatment, necessitating caution in directly extrapolating our findings to patients with previous exposure to biologics.
In conclusion, we successfully developed the first population PK model for subcutaneous CT‐P13 in patients with CD and UC. Key covariates influencing clearance were body weight, anti‐drug antibodies and serum albumin concentrations, which is consistent with previous observations for intravenous infliximab. Our analysis indicates fixed subcutaneous maintenance dosing is appropriate for most patients, but may result in underexposure in heavy patients.
AUTHORSHIP
Guarantor of the article: Ron A. A. Mathôt.
Authors Contributions: Data analysis: Jurij Hanzel, Laura Bukkems, Ron A. A. Mathôt. Study supervision: Ron A. A. Mathôt. Manuscript drafting: Jurij Hanzel, Laura Bukkems. Manuscript editing: Jurij Hanzel, Laura Bukkems, Krisztina B. Gecse, Geert R. D’Haens, Ron A. A. Mathôt. All authors approved the final version of the manuscript.
Supporting information
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank CelltrionHC for generously providing the clinical data. The study results and discussion presented in this article are solely the personal views of respective authors.
Declaration of personal interests: JH has received speaker's fees from Abbvie, Janssen and Takeda; consulting fees from Alimentiv Inc LHB has no conflicts of interest. KBG has received grants from Pfizer Inc and Celltrion; consultancy fees from AbbVie, Arena Pharmaceuticals, Galapagos, Gilead, Immunic Therapeutics, Janssen Pharmaceuticals, Novartis, Pfizer Inc, Samsung Bioepis and Takeda and speaker's honoraria from Celltrion, Ferring, Janssen Pharmaceuticals, Novartis, Pfizer Inc, Samsung Bioepis, Takeda and Tillotts. GRD has served as an advisor for Abbvie, Ablynx, Alimentiv, Allergan, Amakem, Amgen, AM Pharma, Arena Pharmaceuticals, AstraZeneca, Avaxia, Biogen, Bristol Meiers Squibb, Boerhinger Ingelheim, Celgene/Receptos, Celltrion, Cosmo, Covidien/Medtronics, Ferring, DrFALK Pharma, Eli Lilly, Engene, Galapagos, Genentech/Roche, Gilead, Glaxo Smith Kline, Hospira/Pfizer, Immunic, Johnson and Johnson, Lycera, Medimetrics, Millennium/Takeda, Mitsubishi Pharma, Merck Sharp Dome, Mundipharma, Nextbiotics, Novonordisk, Otsuka, Pfizer/Hospira, Photopill, Prometheus laboratories/Nestle, Progenity, Protagonist, Salix, Samsung Bioepis, Sandoz, Seres/Nestle, Setpoint, Shire, Teva, Tigenix, Tillotts, Topivert, Versant and Vifor; received speaker fees from Abbvie, Biogen, Ferring, Johnson and Johnson, Merck Sharp Dohme, Mundipharma, Norgine, Pfizer, Samsung Bioepis, Shire, Millenium/Takeda, Tillotts and Vifor. RAAM has received grants from governmental and societal research institutes such as NWO, ZonMW, Kidney Foundation and Innovation Fund; unrestricted investigator research grants from Baxter/Baxalta/Shire/Takeda, Bayer, CSL Behring, Sobi and CelltrionHC; and has served as an advisor for Bayer, CSL Behring, Merck Sharp & Dohme, Baxter/Baxalta/Shire/Takeda. (All grants and fees were paid to the institution).
Hanzel J, Bukkems LH, Gecse KB, D’Haens GR, Mathôt RAA. Population pharmacokinetics of subcutaneous infliximab CT‐P13 in Crohn’s disease and ulcerative colitis. Aliment Pharmacol Ther. 2021;54:1309–1319. 10.1111/apt.16609
The Handling Editor for this article was Professor Richard Gearry, and it was accepted for publication after full peer‐review.
DATA AVAILABILITY STATEMENT
Original clinical data are the property of Celltrion HC. Data pertaining to the pharmacokinetic model and simulation studies are available from the corresponding author upon reasonable request and approval of Celltrion HC.
REFERENCES
- 1. Torres J, Mehandru S, Colombel JF, Peyrin‐Biroulet L. Crohn's disease. Lancet. 2017;389:1741‐1755. [DOI] [PubMed] [Google Scholar]
- 2. Ungaro R, Mehandru S, Allen PB, Peyrin‐Biroulet L, Colombel J‐F. Ulcerative colitis. Lancet. 2017;389:1756‐1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. D'Haens GR, van Deventer S. 25 years of anti‐TNF treatment for inflammatory bowel disease: lessons from the past and a look to the future. Gut. 2021;70:1396‐1405. [DOI] [PubMed] [Google Scholar]
- 4. Ye BD, Pesegova M, Alexeeva O, et al. Efficacy and safety of biosimilar CT‐P13 compared with originator infliximab in patients with active Crohn's disease: an international, randomised, double‐blind, phase 3 non‐inferiority study. Lancet. 2019;393:1699‐1707. [DOI] [PubMed] [Google Scholar]
- 5. Kim HoUng, Alten R, Avedano L, et al. The future of biosimilars: maximizing benefits across immune‐mediated inflammatory diseases. Drugs. 2020;80:99‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schreiber S, Ben‐Horin S, Leszczyszyn J, et al. Randomized controlled trial: subcutaneous vs intravenous infliximab CT‐P13 maintenance in inflammatory bowel disease. Gastroenterology. 2021;160:2340‐2353. [DOI] [PubMed] [Google Scholar]
- 7. Allen PB, Lindsay H, Tham TC. How do patients with inflammatory bowel disease want their biological therapy administered? BMC Gastroenterol. 2010;10:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Verma AM, Patel A, Subramanian S, Smith PJ. From intravenous to subcutaneous infliximab in patients with inflammatory bowel disease: a pandemic‐driven initiative. Lancet Gastroenterol Hepatol. 2021;6:88‐89. [DOI] [PubMed] [Google Scholar]
- 9. Stoner KL, Harder H, Fallowfield LJ, Jenkins VA. Intravenous versus subcutaneous drug administration. Which do patients prefer? A systematic review. Patient. 2015;8:145‐153. [DOI] [PubMed] [Google Scholar]
- 10. Vavricka SR, Bentele N, Scharl M, et al. Systematic assessment of factors influencing preferences of Crohn's disease patients in selecting an anti‐tumor necrosis factor agent (CHOOSE TNF TRIAL). Inflamm Bowel Dis. 2012;18:1523‐1530. [DOI] [PubMed] [Google Scholar]
- 11. Bittner B, Richter W, Schmidt J. Subcutaneous administration of biotherapeutics: an overview of current challenges and opportunities. BioDrugs. 2018;32:425‐440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fasanmade AA, Adedokun OJ, Blank M, Zhou H, Davis HM. Pharmacokinetic properties of infliximab in children and adults with Crohn's disease: a retrospective analysis of data from 2 phase III clinical trials. Clin Ther. 2011;33:946‐964. [DOI] [PubMed] [Google Scholar]
- 13. Fasanmade AA, Adedokun OJ, Ford J, et al. Population pharmacokinetic analysis of infliximab in patients with ulcerative colitis. Eur J Clin Pharmacol. 2009;65:1211‐1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ternant D, Ducourau E, Perdriger A, et al. Relationship between inflammation and infliximab pharmacokinetics in rheumatoid arthritis. Br J Clin Pharmacol. 2014;78:118‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xu Z, Seitz K, Fasanmade A, et al. Population pharmacokinetics of infliximab in patients with ankylosing spondylitis. J Clin Pharmacol. 2008;48:681‐695. [DOI] [PubMed] [Google Scholar]
- 16. Gottlieb AB, Masud S, Ramamurthi R, et al. Pharmacodynamic and pharmacokinetic response to anti‐tumor necrosis factor‐alpha monoclonal antibody (infliximab) treatment of moderate to severe psoriasis vulgaris. J Am Acad Dermatol. 2003;48:68‐75. [DOI] [PubMed] [Google Scholar]
- 17. Feuerstein JD, Nguyen GC, Kupfer SS, et al. American gastroenterological association institute clinical guidelines C. American gastroenterological association institute guideline on therapeutic drug monitoring in inflammatory bowel disease. Gastroenterology. 2017;153:827‐834. [DOI] [PubMed] [Google Scholar]
- 18. Gastonguay M. Full covariate models as an alternative to methods relying on statistical significance for inferences about covariate effects: a review of methodology and 42 case studies. PAGE Conference; June 2011, 2011.
- 19. Dotan I, Ron Y, Yanai H, et al. Patient factors that increase infliximab clearance and shorten half‐life in inflammatory bowel disease: a population pharmacokinetic study. Inflamm Bowel Dis. 2014;20:2247‐2259. [DOI] [PubMed] [Google Scholar]
- 20. EMA . Humira (Adalimumab). Summary of product characteristics. European Medicines Agency (EMA); 2017. [Google Scholar]
- 21. Adedokun OJ, Xu Z, Liao S, et al. Population pharmacokinetics and exposure‐response modeling of golimumab in adults with moderately to severely active ulcerative colitis. Clin Ther. 2020;42:157‐174 e154. [DOI] [PubMed] [Google Scholar]
- 22. Xu Z, Wang Q, Zhuang Y, et al. Subcutaneous bioavailability of golimumab at 3 different injection sites in healthy subjects. J Clin Pharmacol. 2010;50:276‐284. [DOI] [PubMed] [Google Scholar]
- 23. Xu Y, Hu C, Chen Y, et al. Population pharmacokinetics and exposure‐response modeling analyses of ustekinumab in adults with moderately to severely active ulcerative colitis. J Clin Pharmacol. 2020;60:889‐902. [DOI] [PubMed] [Google Scholar]
- 24. Vande Casteele N, Baert F, Bian S, et al. Subcutaneous absorption contributes to observed interindividual variability in adalimumab serum concentrations in Crohn's disease: a prospective multicentre study. J Crohn's Colitis. 2019;13:1248‐1256. [DOI] [PubMed] [Google Scholar]
- 25. Richter WF, Jacobsen B. Subcutaneous absorption of biotherapeutics: knowns and unknowns. Drug Metab Dispos. 2014;42:1881‐1889. [DOI] [PubMed] [Google Scholar]
- 26. Deng R, Meng YG, Hoyte K, et al. Subcutaneous bioavailability of therapeutic antibodies as a function of FcRn binding affinity in mice. MAbs. 2012;4:101‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vermeire S, Dreesen E, Papamichael K, Dubinsky MC. How, when, and for whom should we perform therapeutic drug monitoring? Clin Gastroenterol Hepatol. 2020;18:1291‐1299. [DOI] [PubMed] [Google Scholar]
- 28. Liefferinckx C, Bottieau J, Toubeau JF, et al. Collecting new peak and intermediate infliximab levels to predict remission in inflammatory bowel diseases. Inflamm Bowel Dis. 2021. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 29. Ye BD, Leszczyszyn J, Dudkowiak R, et al. Exposure‐response relationship of subcutaneous infliximab (CT‐P13 SC) in patients with active Crohn’s disease and ulcerative colitis: analysis from a multicenter, randomized controlled pivotal trial. United European Gastroenterol J. 2020;8:385‐386. [Google Scholar]
- 30. Wang DD, Zhang S, Zhao H, Men AY, Parivar K. Fixed dosing versus body size‐based dosing of monoclonal antibodies in adult clinical trials. J Clin Pharmacol. 2009;49:1012‐1024. [DOI] [PubMed] [Google Scholar]
- 31. Singh S, Proudfoot J, Xu R, Sandborn WJ. Obesity and response to infliximab in patients with inflammatory bowel diseases: pooled analysis of individual participant data from clinical trials. Am J Gastroenterol. 2018;113:883‐889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dai ZH, Xu XT, Ran ZH. Associations between obesity and the effectiveness of anti‐tumor necrosis factor‐alpha agents in inflammatory bowel disease patients: a literature review and meta‐analysis. Ann Pharmacother. 2020;54:729‐741. [DOI] [PubMed] [Google Scholar]
- 33. Wong ECL, Marshall JK, Reinisch W, Narula N. Body mass index does not impact clinical efficacy of ustekinumab in Crohn's disease: a post hoc analysis of the IM‐UNITI trial. Inflamm Bowel Dis. 2021;27:848‐854. [DOI] [PubMed] [Google Scholar]
- 34. Buurman DJ, Maurer JM, Keizer RJ, Kosterink JG, Dijkstra G. Population pharmacokinetics of infliximab in patients with inflammatory bowel disease: potential implications for dosing in clinical practice. Aliment Pharmacol Ther. 2015;42:529‐539. [DOI] [PubMed] [Google Scholar]
- 35. Hamuro L, Kijanka G, Kinderman F, et al. Perspectives on subcutaneous route of administration as an immunogenicity risk factor for therapeutic proteins. J Pharm Sci. 2017;106:2946‐2954. [DOI] [PubMed] [Google Scholar]
- 36. Rosario M, Dirks NL, Gastonguay MR, et al. Population pharmacokinetics‐pharmacodynamics of vedolizumab in patients with ulcerative colitis and Crohn's disease. Aliment Pharmacol Ther. 2015;42:188‐202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim J, Hayton WL, Robinson JM, Anderson CL. Kinetics of FcRn‐mediated recycling of IgG and albumin in human: pathophysiology and therapeutic implications using a simplified mechanism‐based model. Clin Immunol. 2007;122:146‐155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brandse JF, van den Brink GR, Wildenberg ME, et al. Loss of infliximab into feces is associated with lack of response to therapy in patients with severe ulcerative colitis. Gastroenterology. 2015;149:350‐355.e352. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
Original clinical data are the property of Celltrion HC. Data pertaining to the pharmacokinetic model and simulation studies are available from the corresponding author upon reasonable request and approval of Celltrion HC.