

Abstract
Centronuclear myopathy (CNM) is a genetically heterogeneous congenital myopathy characterized by muscle weakness, atrophy, and variable degrees of cardiorespiratory involvement. The clinical severity is largely explained by genotype (DNM2, MTM1, RYR1, BIN1, TTN, and other rarer genetic backgrounds), specific mutation(s), and age of the patient. The histopathological hallmark of CNM is the presence of internal centralized nuclei on muscle biopsy. Information on the phenotypical spectrum, subtype prevalence, and phenotype–genotype correlations is limited. To characterize CNM more comprehensively, we retrospectively assessed a national cohort of 48 CNM patients (mean age = 32 ± 24 years, range 0–80, 54% males) from the Netherlands clinically, histologically, and genetically. All information was extracted from entries in the patient's medical records, between 2000 and 2020. Frequent clinical features in addition to muscle weakness and hypotonia were fatigue and exercise intolerance in more mildly affected cases. Genetic analysis showed variants in four genes (18 DNM2, 14 MTM1, 9 RYR1, and 7 BIN1), including 16 novel variants. In addition to central nuclei, histologic examination revealed a large variability of myopathic features in the different genotypes. The identification and characterization of these patients contribute to trial readiness.
Keywords: BIN1, centronuclear myopathy, cohort, DNM2, MTM1, Netherlands, RYR1
1. INTRODUCTION
Centronuclear myopathies (CNM) are a group of congenital myopathies named after the abnormal localization of cell nuclei in the center rather than their normal location at the periphery of skeletal muscle cells. 1 CNMs are genetically heterogeneous and have been associated with autosomal‐dominant (AD), autosomal‐recessive (AR), and X‐linked inheritance. 2 Over the past three decades, different genetic causes of CNM have been identified, including variants in MTM1, DNM2, BIN1, RYR1, and TTN and—less frequently—SPEG1, MYF6, MAP3K20 (ZAK). 1 , 3 , 4 , 5 MTM1, DNM2, and BIN1, the genes originally implicated in CNM, all encode proteins involved in membrane remodeling and trafficking, while more recently implicated genes such as RYR1 and TTN play important roles in excitation‐contraction coupling and sarcomere assembly, respectively. 5
As with other congenital myopathies, the most prominent clinical manifestations of CNM are early‐onset muscle weakness, hypotonia, and associated disabilities. 2 There is substantial variability in the course and degree of functional impairment among the various CNMs. 6 , 7 , 8 Patients may present within the spectrum of the floppy infant syndrome, or with variable degrees of weakness with delayed gross motor milestones, respiratory and/or bulbar involvement. 7 , 9 , 10 , 11 Presentation is predominantly in infancy and childhood, but some patients do not present until their teens or adolescence with reduced exercise tolerance and mild ptosis, and often remain ambulatory throughout adult life.
The X‐linked form (XL‐MTM) due to MTM1 gene variants usually gives rise to a severe phenotype in males presenting at birth with marked weakness and hypotonia, external ophthalmoplegia and respiratory failure. 7 , 12 , 13 Patients may never crawl or walk and remain wheelchair‐dependent. XL‐MTM is often lethal in childhood or the teenage years. 14 Carriers of XL‐MTM are generally considered not to be affected, but several manifesting carriers have been reported in recent years. 15 , 16
As a group, patients with DNM2‐, BIN1‐, and RYR1‐ variants are generally more mildly affected, but occasionally more severely affected male patients with RYR1‐related CNM may mimic XL‐MTM. 2
Recently, two large studies on MTM1‐ and DNM2‐related CNM have provided essential data concerning their natural history, thus contributing to trial readiness. 13 , 17 Also based on these studies, the protocols for current clinical trials in XL‐MTM and DNM2‐CNM have included several clinical outcome measures considered to be most discriminative for and/or responsive to change. Another important step towards trial readiness is patient identification and epidemiological data concerning the different genetic subtypes.
To date, CNM epidemiological reports provide limited incidence and prevalence data. A recent integrated model utilizing available literature has been proposed to obtain a better estimate of overall CNM patient numbers by age, causative gene, severity, and geographic region. 18 This model calculated a CNM incidence higher than the current estimates. Therefore, knowledge on the actual prevalence in a geographically defined region is essential. Our aim therefore was to obtain epidemiological information regarding the Dutch CNM cohort, and to report their clinical, genetic, and histological features. This could also facilitate CNM trial recruitment in the future.
2. METHODS
This retrospective, cross‐sectional study was conducted at the Radboudumc Neuromuscular Centre, Nijmegen, in collaboration with the Dutch Neuromuscular Centre. All CNM patients had been referred to our center between 2000 and 2020. The study was approved by the local ethics committee (Protocol 2017‐4022), and all participants or, as appropriate, their parents provided informed consent.
2.1. Patients
Inclusion criteria were (1) a (likely) pathogeneous mutation in one of the genes associated with CNM: MTM1, DNM2, BIN1, and RYR1, and a clinical phenotype of a myopathy; or (2) a clinical or histopathological diagnosis of CNM and genetic confirmation of first degree affected family member. In the first group, histological confirmation was not required since histopathological confirmation is not performed in all cases anymore. We included subjects without genetic confirmation in the second group since some XL‐MTM patients had passed away before the diagnostic availability of genetic testing. Patients were divided into groups per genotype, including a distinction between male patients and female manifesting carriers with an MTM1 variant.
2.2. Data collection
CNM patients of all ages were identified through four routes: (1) the (Paediatric) Neurology Outpatient Clinic at the Radboudumc; (2) the Genetics Department at the Radboudumc; (3) the Dutch Patient Organization Spierziekten Nederland; and (4) (Paediatric) Neurologists of the Dutch Neuromuscular Centre. This is estimated to provide a high coverage (>80%) since the Radboudumc is the national referral center for congenital myopathies and is acknowledged as such by the other Dutch Neuromuscular Centres. Hence, CNM patients are generally referred to the Radboudumc at least once as part of clinical management, for registration and for participation in studies. Clinical data from the patients were stored in our electronic patient file system, and systematically extracted by the researchers (S.R. and D.Z.). Data were pseudonymized and stored in a Castor database.
2.3. Data collection, clinical features
We collected information regarding family history, medical history, clinical features, and ancillary investigations. Clinical features were grouped into motor symptoms (signs of delayed gross motor development, muscle weakness, muscle atrophy, and hypotonia), myalgia and cramps (myalgia, muscle cramps, and stiffness), facial and bulbar symptoms (facial weakness, abnormal ocular movements, dysphagia, and dysarthria), respiratory symptoms and cardiac involvement. Age at onset was retrieved from the medical file or estimated based on the history (congenital: 0 years; early childhood: ± 3 years; childhood: ±5 years). Age at diagnosis was determined by the time point where either a histological or suspected genetic diagnosis of CNM was made. Reference values for CK (in IU/L) used in our medical center are ≤710 for neonates, ≤295 for infants, ≤230 for children, ≤270 and ≤ 123 for male and female adolescents, ≤170 and ≤ 145 for men and women.
2.4. Data collection, genetic findings
Results of genetic testing previously performed as part of the diagnostic procedure were retrieved from the medical files. In most patients, Sanger sequencing was performed until the introduction of whole‐exome sequencing with muscle panel analysis in 2013. 19 The variants were classified as pathogenic, likely pathogenic, variant of uncertain significance (VOUS), likely benign or benign, according to the ACMG classification. 20
2.5. Histologic features
Results of muscle biopsy were retrieved from the medical files. Muscle biopsy samples were frozen and stored at −80°C, specimens were processed for routine histological procedures. The majority of samples were processed with several enzyme histochemical staining, including hematoxylin and phloxin (HPhlox), nicotinamide adenosine dinucleotide (NADH), succinate dehydrogenase (SDH), cytochrome C oxidase (COX), Gömöri trichrome, and ATPase 4.2, ATPase 4.6, and ATPase 10.3. All available muscle biopsy slides were reviewed by the pathologist at our center (B.K.) to confirm and/or amend the findings described in the clinical report. We paid particular attention to the following histological features: increased fiber size variability, type I fiber predominance, increased internal and central nuclei (>5%), fatty or connective tissue, nuclear clumps, and radial sarcoplasmic strands (RSS).
2.6. Statistical analysis
Data were analyzed using IBM SPSS Statistics software (version 25. Armonk, NY: IBM Corp.). Descriptive statistics used were mean with SD (n ± SD) and frequencies with percentages (n[%]).
3. RESULTS
3.1. Patients
We identified 50 patients with a CNM diagnosis in the Netherlands. Two patients with an additional diagnosis of nemaline myopathy were excluded. We retained two patients with a DNM2 variant and a mixed myopathy—neuropathy phenotype (neurophysiologically and histologically classified).
The most common genotype was DNM2 (18/48, 37%, 11 families), followed by MTM1 (14/48, 29%, 9 families) and RYR1 (9/48, 19%, 8 families). Variants in BIN1 were least frequent (7/48, 15%, 1 family). There were 10 male MTM1 patients (10/48, 21%) and four female manifesting carriers (4/48, 8%). Genotype prevalence is shown in Figure 1A.
FIGURE 1.
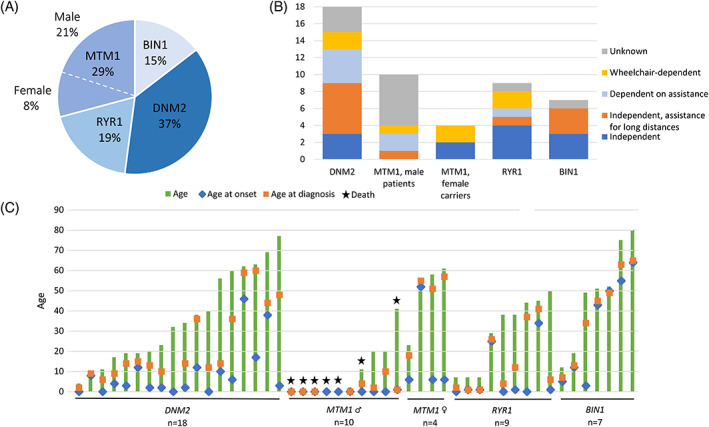
(A) Prevalence of CNM genotypes in the Netherlands. (B) Ambulatory status per genotype. (C) Ages of all patients, including ages at onset and diagnosis [Colour figure can be viewed at wileyonlinelibrary.com]
Seven of the 10 male XL‐MTM patients had passed away (30% survival, mean age at death was 7 ± 15 years). Five out of seven XL‐MTM patients died shortly after birth because of respiratory failure. One patient died at the age of 11 due to respiratory failure after recurrent respiratory tract infections and pneumothorax. The oldest XL‐MTM patient (41 years) passed away because of sudden cardiac failure, this patient was reported previously (III.3). 21 Survival in the other genotypes was 100%. Of the DNM2‐CNM patients, 61% had family members with a diagnosed myopathy; in MTM1 and RYR1 patients this was the case in 50% and 44%, respectively. In 13% of the patients stillbirths in the family were reported; these were mainly MTM1 families. Percentages per genotype are given in Table 1.
TABLE 1.
Patient characteristics per genotype
| DNM2 (n = 18) | MTM1, male patients (n = 10) | MTM1, female carriers (n = 4) | RYR1 (n = 9) | BIN1 (n = 7) | Overall (n = 48) | |
|---|---|---|---|---|---|---|
| Male sex (%) | 8 (44) | 10 (100) | 0 | 5 (56) | 3 (43) | 26 (54) |
| Age (range), y | 36 ± 23 (4–77) | 9 ± 14 (0–41) | 50 ± 18 (23–61) | 29 ± 18(7–50) | 48 ± 26 (12–80) | 32 ± 24 (0–80) |
| Age onset, y | 9 ± 13 | 0 ± 0 | 17 ± 24 | 7 ± 13 | 33 ± 26 | 11 ± 18 |
| Age diagnosis, y | 24 ± 19 (n = 17) | 2 ± 3 (n = 8) | 45 ± 18 | 14 ± 16 | 39 ± 23 | 22 ± 21 (n = 45) |
| Age at death, y | ‐ | 7 ± 15(n = 7) | ‐ | ‐ | ‐ | 7 ± 15(n = 7) |
| Delay between onset and diagnosis, y | 14 ± 14 (n = 17) | 2 ± 4 (n = 8) | 29 ± 24 | 7 ± 12 | 6 ± 11 | 11 ± 14 (n = 45) |
| Family members with neuromuscular disease/symptoms (%) | 11 (61) | 5 (50) | 2 (50) | 4 (44) | 7 (100) | 29 (60) |
| Stillbirth in family members (%) | 1 (6) | 2 (20) | 3 (75) | 0 | 0 | 6 (13) |
| Creatine kinase level, IU/L | 199 ± 265 (n = 11) | 199 ± 144 (n = 4) | 428 ± 385 (n = 4) | 117 ± 160 (n = 6) | 261 ± 218 (n = 4) | 222 ± 249 (n = 29) |
Note: Values are presented as means with SD (n ± SD) or counts with percentages (n[%]).
Abbreviations: IU/L, international units per liter; y, year.
3.2. Clinical features
Patient characteristics are summarized in Table 1. Overall mean age was 32 ± 24 years, ranging from 0 to 80 years. All male MTM1 patients had congenital onset, most RYR1 and DNM2 patients had onset in childhood. Age at onset was highly variable for BIN1 patients. Age at onset in the two DNM2 patients with a mixed phenotype was 17 and 46 years, respectively. The delay between age at onset and age at diagnosis was only short for male MTM1 patients (2 ± 4 years) and longest for female MTM1 carriers (29 ± 24 years). Patient age, including the age at onset and diagnosis, is depicted in Figure 1C.
Clinical features for each genotype are illustrated in Figure 2, more detailed information is listed in Table 2. Ambulatory status was highly variable (Figure 1B). None of the male MTM1 patients achieved independent ambulation, and none of the BIN1 patients were dependent on assistance or a wheelchair. Two patients used disease modifying medicine (nutritional supplements); one BIN1 patient and one MTM1 patient used pyridostigmine. In both patients, this had no effect. Two RYR1 patients used acetylcysteine, but without significant effects. All CNM patients had at least one motor symptom (signs of delayed gross motor development, muscle weakness, muscle atrophy, and hypotonia), except for two BIN1 patients whose main clinical features were myalgia and muscle cramps. Myalgia and cramps were reported by many BIN1 patients and female MTM1 carriers, while fatigue and exercise intolerance were common in all groups of CNM. These symptoms were less prevalent in male MTM1 and only reported by older XL‐MTM patients. Furthermore, a predominance of facial and bulbar symptoms was reported in RYR1, DNM2, and male MTM1 patients, with bulbar symptoms being most prominent in RYR1 patients. Respiratory insufficiency was most frequently observed in male MTM1 patients, but occurred also in the other subgroups of CNM except for BIN1 patients. The prevalence of cardiac involvement was 6 to 14% in DNM2, MTM1, and BIN1 patients. Abnormal ocular movement was reported in 12 (mainly RYR1) patients. Ptosis was reported in 16 (33%) patients, most frequently in DNM2 patients (n = 9). Disturbed vision due to strabismus or diplopia was reported in 7 patients (15%), mainly in RYR1 and MTM1 patients.
FIGURE 2.
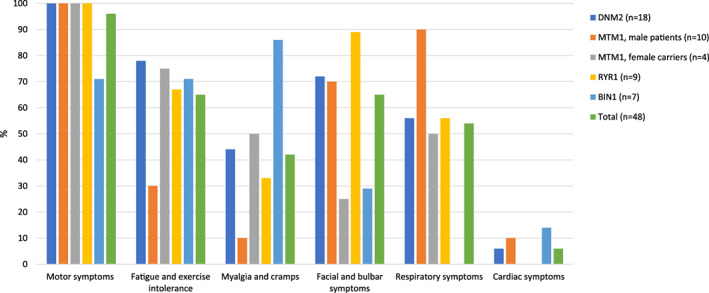
Clinical features per genotype [Colour figure can be viewed at wileyonlinelibrary.com]
TABLE 2.
Detailed clinical features per genotype
| DNM2 (n = 18) | MTM1, male patients (n = 10) | MTM1, female carriers (n = 4) | RYR1 (n = 9) | BIN1 (n = 7) | Overall (n = 48) | |
|---|---|---|---|---|---|---|
| Any symptoms | 18 (100) | 10 (100) | 4 (100) | 9 (100) | 7 (100) | 48 (100) |
| Motor symptoms | 18 (100) | 10 (100) | 4 (100) | 9 (100) | 5 (71) | 46 (96) |
| Delayed gross motor development | 12/12 | 3/3 | 1/2 | 6/6 | 2/3 | 24/26 |
| Muscle weakness | 18/18 | 7/7 | 4/4 | 8/9 | 5/6 | 42/44 |
| Muscle atrophy | 6/11 | 4/4 | 2/3 | 5/7 | 3/6 | 20 /31 |
| Hypotonia | 3/4 | 8/8 | 0/1 | 6/6 | 0/1 | 17/20 |
| Fatigue and exercise intolerance | 14 (78) | 3 (30) | 3 (75) | 6 (67) | 5 (71) | 31 (65) |
| Fatigue | 14/14 | 3/4 | 3/3 | 4/5 | 4/4 | 27/29 |
| Exercise intolerance | 10/10 | 2/2 | 3/3 | 3/4 | 4/4 | 22/23 |
| Myalgia and cramps | 8 (44) | 1 (10) | 2 (50) | 3 (33) | 6 (86) | 20 (42) |
| Myalgia | 7/9 | 0/2 | 1/2 | 2/5 | 4/4 | 14/22 |
| Muscle cramps | 3/5 | 0/1 | 2/2 | 0/1 | 2/2 | 7/11 |
| Muscle stiffness | 1/2 | 1/1 | 0/0 | 1/1 | 3/3 | 6/7 |
| Facial and bulbar symptoms | 13 (72) | 7 (70) | 1 (25) | 8 (89) | 2 (29) | 31 (65) |
| Facial weakness | 11/14 | 7/7 | 1/2 | 8/9 | 1/4 | 28/36 |
| Abnormal ocular movement | 3/11 | 3/4 | 1/3 | 5/7 | 0/4 | 12/29 |
| Dysphagia | 4/13 | 3/5 | 0/1 | 4/7 | 0/3 | 11/29 |
| Dysarthria | 2/8 | 1/2 | 1/2 | 3/7 | 2/3 | 9/22 |
| Respiratory symptoms | 10 (56) | 9 (90) | 2 (50) | 5 (56) | 0 | 26 (54) |
| Respiratory insufficiency | 10/15 | 9/10 | 2/3 | 5/6 | 0/1 | 26/35 |
| Cardiac symptoms | 1 (6) | 1 (10) | 0 | 0 | 1 (14) | 3 (6) |
| Cardiac involvement | 1/12 | 1/4 | 0/2 | 0/4 | 1/2 | 3/24 |
| Ambulation | ||||||
| ‐Independent | 3/15 | 0/4 | 2/4 | 4/8 | 3/6 | 12/37 |
| ‐Independent, assistance for long distances | 6/15 | 1/4 | 0/4 | 1/8 | 3/6 | 11/37 |
| ‐Dependent on assistance | 4/15 | 2/4 | 0/4 | 1/8 | 0/6 | 7/37 |
| Wheelchair‐dependent | 2/15 | 1/4 | 2/4 | 2/8 | 0/6 | 7/37 |
Note: The blue rows represent a group of clinical features and are a summary of the symptoms listed below. The total counts are shown with percentages (n[%]) per genotype. The white rows represent the individual symptoms, the counts are shown as number/total number without missing data.
Other frequently reported symptoms in our cohort of CNM patients were limited range of joint motion in 16 (33%) patients and joint hypermobility in 5 (10%, mainly RYR1) patients. Limited range of motion was in 7 (39%) patients due to contractures, in MTM1, DNM2, and RYR1 patients. Shortening of tibialis anterior, Achilles tendons or hamstrings were mainly reported by DNM2 patients. The remaining patients with a limited range of motion had severe muscle weakness. Four patients (8%) reported paraesthesia. Scoliosis (13/48, 27%) was reported in all genotypes. Reduced reflexes were another frequent feature (27/48, 56%), occurring with all genotypes. In addition to the well‐known clinical myopathy symptoms, bladder complaints including micturition difficulties, recurrent urinary tract infections, and the use of a urine catheter were reported throughout the different subgroups.
3.3. Creatine kinase
Creatine kinase (CK) levels were available in 29 CNM patients. Mean CK values are shown in Table 1; 261 ± 218 IU/L in BIN1, 117 ± 160 IU/L in RYR1, 199 ± 265 IU/L in DNM2, 199 ± 144 IU/L in MTM1 patients, and 428 ± 385 IU/L in female MTM1 carriers. Of the 29 patients whose CK levels were known, 12 patients had an elevated CK level; 4 DNM2 patients, 3 MTM1 carriers, 3 BIN1 patients, 1 RYR1 and 1 MTM1 patient.
3.4. Genetic findings
Variants in CNM‐related genes were detected in 45 of 48 participants (94%). Thirteen patients (27%) had a confirmed pathogenic variant, and 16 patients (33%) had a likely pathogenic variant according to the ACMG classification. Three patients (classified as 1 BIN1 and 2 MTM1 patients) could not be tested, but were deemed likely to carry a familial pathogenic variant, due to the presence of clinical and histopathological CNM features very similar to those of a genetically confirmed relative. The two MTM1 patients died postnatally and postmortem genetic testing had not been performed. Only after the death of the third male child in this family, XL‐MTM was diagnosed. The BIN1 patient was part of the large family, to which all BIN1 patients in this cohort belong.
Thirty‐six distinct genetic variants were identified in the families (n = 29). Twenty of the variants have previously been reported and 16 variants are novel. Genetic variants and their classification identified in the participants are summarized in Table 3, and more detail is listed in Table S1. Ten out of eleven DNM2 variants were missense variants and one in‐frame deletion was reported. Two DNM2 variants were identified as de novo after segregation analysis (c.596G > A and c.1105C > T) and are associated with a more severe phenotype than the other DNM2 variants in this study. One DNM2‐CNM patient had a somatic mosaicism for the DNM2 variant (c.1666G > A). Nine variants in MTM1 were reported, including nonsense, missense, and splice‐site variants, as well as one whole gene deletion. Thirteen different RYR1 variants were identified, most were occurring in combination with one or two other RYR1 variants. Three patients only had one RYR1 variant identified, which we included in this cohort because of evident clinical and/or histological features consistent with CNM. One BIN1 variant was identified in one family with an autosomal dominant inheritance pattern.
TABLE 3.
Genetic variants
| Patient | Variant DNA | Protein | Variant type | ACMG classification | Reference |
|---|---|---|---|---|---|
| DNM2 (NM_004945.3) | |||||
| 1 | c.596G > A a | p.(Arg199Gln) | Missense | Likely pathogenic | ‐ |
| 2 b | c.1058C > G | p.(Thr353Ser) | Missense | Uncertain significance | ‐ |
| 3 | c.1102G > A | p.(Glu368Lys) | Missense | Pathogenic | 3 |
| 4–6 | c.1105C > T a | p.(Arg369Trp) | Missense | Pathogenic | 3 |
| 7–9 | c.1393C > T | p.(Arg465Trp) | Missense | Pathogenic | 3 |
| 10, 11 | c.1553G > A | p.(Arg518His) | Missense | Likely pathogenic | 18 |
| 12 | c.1666G > A | p.(Glu556Lys) | Missense | Likely pathogenic | 19 |
| 13–15 | c.1832G > T | p.(Ser611Ile) | Missense | Uncertain significance | ‐ |
| 16 | c.1840G > A | p.(Ala614Thr) | Missense | Uncertain significance | 20 |
| 17 | c.1931_1933del | p.(Gln644del) | In‐frame deletion | Uncertain significance | ‐ |
| 18 b | c.2245G > A | p.(Asp749Asn) | Missense | Uncertain significance | ‐ |
| MTM1 (NM_000252.2) | |||||
| 19 | c.85C > T | p.(Arg29*) | Nonsense | Likely pathogenic | 21, 22 |
| 20–22 | c.686C > A | p.(Ser229*) | Nonsense | Likely pathogenic | ‐ |
| 23, 24 | c.1210G > A | p.(Glu404Lys) | Missense | Uncertain significance | 23 |
| 25 | c.1233G > C | p.(Trp411Cys) | Missense | Uncertain significance | 24 |
| 26 | c.1260 + 2 T > C | r.spl | Splice‐site (in frame) | Uncertain significance | ‐ |
| 27 | c.1261C > T | p.(Arg421*) | Nonsense | Pathogenic | 23 |
| 28, 29 | c.1354‐2A > T | r.spl | Splice‐site (in frame) | Uncertain significance | ‐ |
| 30 | c.1496G > T | p.(Trp499Leu) | Missense | Likely pathogenic | 25 |
| 31 | c‐76‐?_*1548del | p.0 | Entire gene deletion | Likely pathogenic | 26 |
| RYR1 (NM_000540.2) | |||||
| 32 | c.325C > T | p.(Arg109Trp) | Missense | Uncertain significance |
‐ |
| c.5815‐16G > A | r.(spl?) | Splice‐site | Uncertain significance | ||
| 33 | c.1100G > T | p.(Arg367Leu) | Missense | Uncertain significance | 28 |
| 34 | c.2653C > T | p.(Arg885Cys) | Missense | Uncertain significance | ‐ |
| c.2671_2786 + 34del | p.(Thr891fs) | Frameshift | Likely pathogenic | ‐ | |
| c.4405C > T | p.(Arg1469Trp) | Missense | Uncertain significance | 5 | |
| 35 | c.10616G > A | p.(Arg3539His) | Missense | Uncertain significance | |
| c.2870 + 1G > A | r.spl | Splice‐site (in‐frame) | Uncertain significance | ||
| 36 | c.10616G > A | p.(Arg3539His) | Missense | Uncertain significance |
‐ |
| c.13033_13067del | p.(Ala4345fs) | Frameshift | Likely pathogenic | ||
| 37 | c.10616G > A | p.(Arg3539His) | Missense | Uncertain significance | |
| c.14804‐1G > A | r.spl | Splice‐site | Likely pathogenic | ||
| 38 | c.4454G > A | r.(spl?)/p.(Ser1485Asn) | Splice‐site | Uncertain significance | ‐ |
| c.9103G > C | p.(Glu3035Gln) | Missense | Likely pathogenic | ‐ | |
| 39, 40 | c.12083C > T | p.(Ser4028Leu) | Missense | Likely pathogenic | 32 |
| BIN1 (NM_139343.2) | |||||
| 41–45 | c.53 T > A | p.(Val18Glu) | Missense | Pathogenic | 33 |
Abbreviations: ACMG, American College of Medical Geneticists; VOUS, variants of unknown significance.
De novo variants.
Patients with a mixed CNM/CMT phenotype.
3.5. Muscle histology
Muscle biopsy had been performed in 31 (65%) of 48 patients as part of the routine diagnostic process (M = 16, F = 15). The majority of biopsies were taken from the quadriceps femoris (n = 19, 61%), one was taken from the tibialis anterior (2%); the remaining 11 muscle biopsy sites were unknown. Age at biopsy was known for 29 of the patients, with a mean of 29 ± 23 (range 0–66) years. Histological information was extracted from the muscle biopsy reports. Twenty‐three biopsies were reviewed prospectively. Histologic examination revealed frequent internal and central nuclei, in 71% of the muscle biopsies. Increased fiber size variability (17/31, 55%) and type I fiber predominance (13/31, 42%) were also common, although the latter was not observed in female MTM1 carriers. Fatty or connective tissue was observed in 26% of all muscle biopsies, but not in BIN1 patients. Nuclear clumps were only reported in DNM2 and BIN1 patients and female MTM1 carriers. Furthermore, radial sarcoplasmic strands (RSS) were frequently present in DNM2 patients (54%), but only sporadically seen in RYR1‐CNM, BIN1‐CNM, and female MTM1 carriers. Core‐like structures were observed in six muscle biopsies, mainly in patients with a RYR1 variant. In one female MTM1 carrier, necklace fibers were observed. Typical histopathological features seen in our cohort are displayed in Table 4 and shown in Figure 3.
TABLE 4.
Histologic findings per genotype
| DNM2 (n = 11) | MTM1, male patients (n = 5) | MTM1, female carriers (n = 3) | RYR1 (n = 7) | BIN1 (n = 5) | Overall (n = 31) | |
|---|---|---|---|---|---|---|
| Increased fiber size variability | 6 (55) | 1 (20) | 3 (100) | 5 (71) | 2 (40) | 17 (55) |
| Type I fiber predominance | 4 (36) | 2 (40) | ‐ | 5 (71) | 2 (40) | 13 (42) |
| Increased internal nuclei | 7 (64) | 5(100) | 3 (100) | 6 (86) | 4 (80) | 22 (71) |
| Increased central nuclei | 7 (64) | 5 (100) | 1 (33) | 6 (86) | 3 (60) | 22 (71) |
| Fatty/connective tissue | 4 (36) | 1 (20) | 2 (67) | 1 (14) | ‐ | 8 (26) |
| Nuclear clumps | 2 (18) | ‐ | 3 (100) | ‐ | 2 (40) | 7 (23) |
| RSS | ||||||
| ‐ High degree | 3 (27) | ‐ | ‐ | ‐ | ‐ | 3 (10) |
| ‐ Low degree | 3 (27) | ‐ | 1 (33) | 1 (14) | 2 (40) | 7 (23) |
Note: Numbers are presented as counts with percentages (n[%]).
Abbreviation: RSS, radial sarcoplasmic strands.
FIGURE 3.
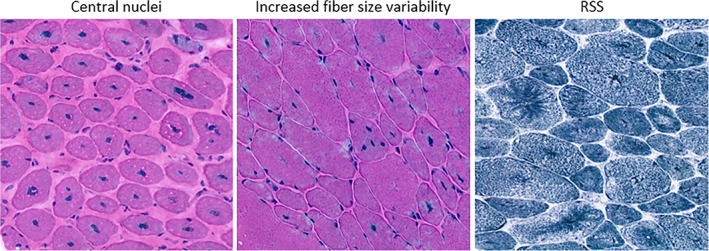
Histopathological features observed in our cohort; central nuclei, increased fiber size variability, and RSS [Colour figure can be viewed at wileyonlinelibrary.com]
4. DISCUSSION
This study describes the clinical, genetic, and histopathological features of a Dutch CNM cohort (n = 48). A majority of 37% had a DNM2 gene variant (n = 18, 11 families), 29% had a MTM1 variant (n = 14, 9 families), 19% a RYR1 variant (n = 9, 8 families), and 15% a BIN1 variant (n = 7, 1 family). This nationwide cohort study underlined the wide range of disease severity among the different genotypes and includes data of pediatric and adult patients throughout the life span (0–80 years). Most prominent consistent clinical findings next to muscle weakness were hypotonia, fatigue, and exercise intolerance. Thirty‐six unique variants were identified, including 16 novel variants. Most prominent histological features were by definition frequent internal and central nuclei. Our results detail the clinical, genetic, and histological features of this rare group of congenital diseases in the Netherlands, and give some insights into their prevalence as a basis for future trial readiness.
This is the first study to report the entire cohort of CNM patients in the Netherlands. Similar studies have been previously performed in Denmark and Italy. 22 , 23 In these and our current cohort, DNM2 was the most common genotype. In contrast to the Italian cohort, we did not identify TTN mutations. This might be related to the only relatively recent introduction of TTN sequencing into clinical diagnostics, and the considerable challenges of variant interpretation in the giant TTN gene. Furthermore, we identified a family of seven patients with autosomal dominant BIN1‐CNM, which was not observed in the other European cohorts.
The prevalence of CNM in this Dutch cohort differs from the estimated prevalence of CNM in Europe, predicted by the model of Vandersmissen et al. 18 : MTM1 variants were considered most prevalent (56%), followed by DNM2 and RYR1 variants (both 12%), and, least commonly, autosomal dominant BIN1 variants (4%). 18 The high prevalence of DNM2‐CNM in our cohort might be related to the inclusion of several large DNM2 families; we included 18 DNM2‐CNM patients from 11 families. The wide availability of genetic and prenatal testing in the Netherlands might have contributed to the lower XL‐MTM prevalence, 24 Dutch policy has a lot of influence on this. Because of the routine ultrasound examination around 20 weeks of pregnancy, termination of pregnancy is more common. 25 In addition, euthanasia for neonates has been allowed in the Netherlands since 2005. 26 Moreover, we have performed a retrospective study most likely confounded by selection bias. Therefore, we might not have reported the totality of prevalent patients but rather the majority of them, reaching an estimate of the prevalence in the Netherlands.
We will discuss the most important findings for each genotype in the following section.
4.1. DNM2‐CNM
Most patients had symptom onset in childhood or adolescence (1–17 years). The three patients with de novo variants had an early onset and respiratory insufficiency, as reported in some other patients with de novo DNM2 variants. 27 Some DNM2 variants have been described to be more frequent in late‐onset phenotypes, 23 however, this was not observed in our cohort. The delay between onset and diagnosis was long (14 ± 14 years), probably related to the late onset, relatively mild symptoms, and slow progression, 9 and the limited diagnostic options before the era of exome sequencing.
Two patients (patient 2 and 18) were diagnosed with a mixed phenotype of a congenital myopathy and a polyneuropathy, confirmed by electromyography (EMG) and nerve conduction velocity (NCV) studies. In one patient, NCV showed a reduced CMAP amplitude of tibial nerve (0.2 mV). EMG of distal muscles in arms and legs showed signs of denervation, proximal muscles showed signs of both denervation and reinnervation. In the second patient, NCV showed reduced SMAP amplitude of median, ulnar, peroneal, and tibial nerve (2.2; 2.8; 0.8; 0.2 mV). SNAP amplitudes of median and ulnar nerves were also reduced (3.6; 3.8 mV). EMG of distal muscles in arms and legs showed signs of denervation. The genetic variants of these patients are listed in Table 3. DNM2 plays an important role in mutated cytoskeleton and membrane proteins, both involved in CNM and Charcot–Marie–Tooth disease (CMT). In the CNM phenotype it affects mainly skeletal muscles and in CMT it mainly affects peripheral nerves. 28 These mixed phenotypes, although not common, have been well‐recognized in DNM2‐CNM and might be considered for inclusion in future CNM trials. 29 Mosaicism detected in one patient has only been reported once, 30 also with a mild phenotype (no hypotonia, respiratory or feeding difficulties at birth). Two of 11 mutations were de novo, which is not uncommon in DNM2‐CNM. 27 , 30
Connective tissue replacement was the most prevalent muscle biopsy finding in DNM2 patients. This could be an age effect, since four DNM2 patients had their muscle biopsy at ages >35 years. Variable degrees of radial sarcoplasmic strands were present in six patients; both features have been described previously. 9 , 31
4.2. XL‐MTM male patients and female carriers
Male XL‐MTM patients mostly have congenital onset and are severely affected. Cardiac involvement was reported in one male patient in our cohort and not in female carriers, although cardiomyopathies have been previously also described in MTM1 carriers. 32 There is also one report of a mildly affected male with XL‐MTM who developed a cardiomyopathy in early adulthood. 33 Seven patients had passed away during the retrospective study window of 20 years. The severe phenotype in males may be the reason for the only short delay between onset and diagnosis (2 ± 3 years). Fatigue and exercise intolerance were common in all groups of CNM, except for the male MTM1 patients (30%). This is probably biased by the early death of half of these patients, and also because of their severely reduced mobility. Female manifesting carriers generally have a later onset and a less severe phenotype. This, and the fact that disease manifestation in carriers has been neglected for a long time, has probably contributed to the long diagnostic delay (29 ± 24 years). Our cohort included four affected XL‐MTM carriers, one of them previously published by Biancalana et al. 16 , 34 The female phenotype covers almost the full disease spectrum in males, with wheelchair dependency (50%), respiratory insufficiency (50%), and facial and bulbar symptoms (25%, including extraocular muscle involvement).
Nine variants in the MTM1 gene were identified, including one entire gene deletion which has been reported before. 35 Type I fiber predominance in muscle biopsies was not reported in female MTM1 carriers, and nuclear clumps did not occur in XL‐MTM males. In one female MTM1 carrier, necklace fibers were observed in the biopsy.
4.3. RYR1‐CNM
Most RYR1 patients had onset in childhood or adolescence. Findings in the patients with RYR1‐CNM were similar to those previously reported in the literature, including joint hypermobility, 36 facial and bulbar symptoms, 5 , 10 absence of cardiac involvement, 5 , 10 and normal CK levels. 37 Thirteen different RYR1 variants were reported, mainly occurring in combination with one or two other variants. Three patients only had one RYR1 variant identified. In five patients, additional core‐like structures, which are a common feature in RYR1‐related myopathies, were observed in the muscle biopsies. 10 Nuclear clumps were not observed in any of the RYR1 patients, RSS only in one patient with a low degree.
4.4. BIN1‐CNM
All BIN1 patients in our cohort belonged to one previously reported family with a dominant inheritance pattern. 38 Family members had a mild phenotype with variable age at onset, with the longest diagnostic delay (31 years) in the index patient. After the first family member was diagnosed, the others soon followed. This might give a biased representation of the diagnostic delay in this patient group. Two genetically confirmed patients in this pedigree did not have any motor symptoms, but had only myalgia and muscle cramps. None of the patients in our cohort was reported to have respiratory insufficiency. The mild phenotype is in line with other dominant cases, 11 , 39 whereas recessive BIN1 cases are generally more severe. 11
Fatty or connective tissue was not observed in BIN1 biopsies. In a case report of a severely affected BIN1 patient, there was an increased amount of connective tissue. 40 However, this is not known as a typical feature in BIN1‐CNM patients.
We included patients with either a histopathological diagnosis of CNM, or suspected genetic diagnosis of mutations in a gene implicated in CNM. Muscle biopsy had been performed in 65% of all patients as part of the routine diagnostic process. The other 35% of patients had a mutation in one of the CNM‐related genes and a clinical phenotype consistent with CNM and/or family members with the same diagnosis and histopathological CNM features. This illustrates how the diagnosis of congenital myopathies is gradually changing with the increasing availability of genetic testing. We included patients with variants of uncertain significance and patients with only one RYR1 variant identified in our cohort, because we assume all‐encompassing (clinical diagnosis, family history/diagnosis, and possibly biopsy) that this is the cause of the clinical manifestation.
Central nuclei would be expected in all CNM muscle biopsies. However, the muscle biopsy of several patients with a pathogenic variant in one of the CNM genes (n = 9) showed no centralized nuclei at all. This is likely to be related to our current sequence of ancillary investigations—first genetic testing and subsequently muscle biopsy 4 , 41 —in contrast to the historical diagnostic approach where genetic testing was mainly prompted by suggestive features on muscle biopsy. Fatty or connective tissue and nuclear clumps were less common features in our CNM cohort. Nuclear clumps can occur in long‐standing neurogenic or myopathic conditions, but also in other genetic muscle disorders such as myotonic dystrophy type 2 (DM2). 42 , 43 Fibrosis and increases in fatty tissue have previously been reported in DNM2‐ and RYR1‐CNM with varying frequency and severity. In our cohort, fibrosis was observed in 36% of our DNM2 patients. 30 , 41 Although RSS were described in previous studies focusing on CNM patients with mainly autosomal‐dominant inheritance, we found this feature in only 10 of our biopsies, including six biopsies from patients with a DNM2 mutation. 41 The range of patient age when the muscle biopsy was performed was wide (0–66 years), with a likely effect on the observed variability of features, as some abnormalities in the muscle are not always visible at a younger age and may only develop over time. Other features, for example type I fiber predominance and the presence of connective tissue that are known to increase as part of the aging process, 44 may be more prominent in biopsies taken at an older age.
A limitation of this study is the retrospective study design. Data were collected by medical chart review, preventing a more detailed description of the phenotype of this cohort. Another constraint is the small size of the different genetic subgroups, resulting in difficulties in making comparisons between the different genotypes and with regards to the wider applicability of our findings. In addition, previously reported genotype–phenotype variability and intrafamilial variability have to be taken into account. 38 , 45 , 46 The next step will be to assess these patients prospectively to collect natural history data. A recent natural history study in Belgium and France focusing on CNM patients with a DNM2 mutation has provided reliable natural history data and sensitive outcome measures. 17 Results of this have contributed to the design of the ongoing phase I/II trial (ClinicalTrials.gov Identifier: NCT04033159), investigating a new medicine named DYN101 in patients with DNM2 and MTM1 mutations. 47
In conclusion, to our knowledge, this is the first detailed study in the Netherlands to report the complete identified Dutch CNM cohort. The identification and characterization of these patients contributes to trial readiness.
CONFLICT OF INTEREST
The authors declare to have no conflicts of interest.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/cge.14054.
Supporting information
Appendix S1: Supplementary Information
ACKNOWLEDGMENTS
We would like to thank all neuromuscular neurologists and clinical geneticists who contributed patients to our study cohort. Furthermore, we thank Spierziekten Nederland for dissemination of information about this study. We also thank Daan Zegers for his contribution in data collection, and Stéphanie Hoffmann, Chris Freitag, and Leen Thielemans from Dynacure Inc. for contributing to the discussion on future studies concerning CNM. Several authors of this paper are members of the Netherlands Neuromuscular Center (NL‐NMD), and the European Reference Network for rare neuromuscular diseases (EURO‐NMD).
Reumers SFI, Erasmus CE, Bouman K, et al. Clinical, genetic, and histological features of centronuclear myopathy in the Netherlands. Clinical Genetics. 2021;100(6):692-702. doi: 10.1111/cge.14054
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Gonorazky HD, Bönnemann CG, Dowling JJ. Chapter 36 ‐ the genetics of congenital myopathies. In: Geschwind DH, Paulson HL, Klein C, eds. Handbook of Clinical Neurology. Elsevier; 2018:549‐564. [DOI] [PubMed] [Google Scholar]
- 2. Jungbluth H, Treves S, Zorzato F, et al. Congenital myopathies: disorders of excitation‐contraction coupling and muscle contraction. Nat Rev Neurol. 2018;14:151‐167. [DOI] [PubMed] [Google Scholar]
- 3. Bitoun M, Maugenre S, Jeannet PY, et al. Mutations in dynamin 2 cause dominant centronuclear myopathy. Nat Genet. 2005;37:1207‐1209. [DOI] [PubMed] [Google Scholar]
- 4. Nicot AS, Toussaint A, Tosch V, et al. Mutations in amphiphysin 2 (BIN1) disrupt interaction with dynamin 2 and cause autosomal recessive centronuclear myopathy. Nat Genet. 2007;39:1134‐1139. [DOI] [PubMed] [Google Scholar]
- 5. Wilmshurst JM, Lillis S, Zhou H, et al. RYR1 mutations are a common cause of congenital myopathies with central nuclei. Ann Neurol. 2010;68:717‐726. [DOI] [PubMed] [Google Scholar]
- 6. Jungbluth H, Voermans NC. Congenital myopathies: not only a paediatric topic. Curr Opin Neurol. 2016;29:642‐650. [DOI] [PubMed] [Google Scholar]
- 7. Jungbluth H, Gautel M. Pathogenic mechanisms in centronuclear myopathies. Front Aging Neurosci. 2014;6:339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nance JR, Dowling JJ, Gibbs EM, Bönnemann CG. Congenital myopathies: an update. Curr Neurol Neurosci Rep. 2012;12:165‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Abath Neto O, Martins Cde A, Carvalho M, et al. DNM2 mutations in a cohort of sporadic patients with centronuclear myopathy. Genet Mol Biol. 2015;38:147‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Abath Neto O, Moreno CAM, Malfatti E, et al. Common and variable clinical, histological, and imaging findings of recessive RYR1‐related centronuclear myopathy patients. Neuromuscul Disord. 2017;27:975‐985. [DOI] [PubMed] [Google Scholar]
- 11. Böhm J, Biancalana V, Malfatti E, et al. Adult‐onset autosomal dominant centronuclear myopathy due to BIN1 mutations. Brain. 2014;137:3160‐3170. [DOI] [PubMed] [Google Scholar]
- 12. Annoussamy M, Lilien C, Gidaro T, et al. X‐linked myotubular myopathy: a prospective international natural history study. Neurology. 2019;92:e1852‐e1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Amburgey K, Tsuchiya E, de Chastonay S, et al. A natural history study of X‐linked myotubular myopathy. Neurology. 2017;89:1355‐1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Graham RJ, Muntoni F, Hughes I, et al. Mortality and respiratory support in X‐linked myotubular myopathy: a RECENSUS retrospective analysis. Arch Dis Child. 2020;105:332‐338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Savarese M, Musumeci O, Giugliano T, et al. Novel findings associated with MTM1 suggest a higher number of female symptomatic carriers. Neuromuscul Disord. 2016;26:292‐299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Biancalana V, Scheidecker S, Miguet M, et al. Affected female carriers of MTM1 mutations display a wide spectrum of clinical and pathological involvement: delineating diagnostic clues. Acta Neuropathol. 2017;134:889‐904. [DOI] [PubMed] [Google Scholar]
- 17. Annoussamy M, Baets J, De Ridder W, et al. Clinical changes over time in patients with centronuclear myopathy due to mutations in DNM2 gene enrolled in a European prospective natural history study. Unreviewed Poster Published. Neuromuscul Disord. 2019;29:S79‐S80. [Google Scholar]
- 18. Vandersmissen I, Biancalana V, Servais L, et al. An integrated modelling methodology for estimating the prevalence of centronuclear myopathy. Neuromuscul Disord. 2018;28:766‐777. [DOI] [PubMed] [Google Scholar]
- 19. Westra D, Schouten MI, Stunnenberg BC, et al. Panel‐based exome sequencing for neuromuscular disorders as a diagnostic service. J Neuromuscul Dis. 2019;6:241‐258. [DOI] [PubMed] [Google Scholar]
- 20. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. van Wijngaarden GK, Fleury P, Bethlem J, Meijer AEFH. Familial 'myotubular' myopathy. Neurology. 1969;19:901‐908. [DOI] [PubMed] [Google Scholar]
- 22. Werlauff U, Petri H, Witting N, Vissing J. Frequency and phenotype of Myotubular myopathy amongst Danish patients with congenital myopathy older than 5 years. J Neuromuscul Dis. 2015;2:167‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fattori F, Maggi L, Bruno C, et al. Centronuclear myopathies: genotype‐phenotype correlation and frequency of defined genetic forms in an Italian cohort. J Neurol. 2015;262:1728‐1740. [DOI] [PubMed] [Google Scholar]
- 24. Lord J, McMullan DJ, Eberhardt RY, et al. Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): a cohort study. Lancet. 2019;393:747‐757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. ten Cate K, van de Vathorst S, Onwuteaka‐Philipsen BD, van der Heide A. End‐of‐life decisions for children under 1 year of age in the Netherlands: decreased frequency of administration of drugs to deliberately hasten death. J Med Ethics. 2015;41:795‐798. [DOI] [PubMed] [Google Scholar]
- 26. Koper JF, Bos AF, Janvier A, Verhagen AAE. Dutch neonatologists have adopted a more interventionist approach to neonatal care. Acta Paediatr. 2015;104:888‐893. [DOI] [PubMed] [Google Scholar]
- 27. Jungbluth H, Cullup T, Lillis S, et al. Centronuclear myopathy with cataracts due to a novel dynamin 2 (DNM2) mutation. Neuromuscul Disord. 2010;20:49‐52. [DOI] [PubMed] [Google Scholar]
- 28. Koutsopoulos OS, Koch C, Tosch V, Böhm J, North KN, Laporte J. Mild functional differences of dynamin 2 mutations associated to centronuclear myopathy and Charcot‐Marie tooth peripheral neuropathy. PLoS One. 2011;6:e27498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhao M, Maani N, Dowling JJ. Dynamin 2 (DNM2) as cause of, and modifier for, human neuromuscular disease. Neurotherapeutics. 2018;15:966‐975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Catteruccia M, Fattori F, Codemo V, et al. Centronuclear myopathy related to dynamin 2 mutations: clinical, morphological, muscle imaging and genetic features of an Italian cohort. Neuromuscul Disord. 2013;23:229‐238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bitoun M, Bevilacqua JA, Prudhon B, et al. Dynamin 2 mutations cause sporadic centronuclear myopathy with neonatal onset. Ann Neurol. 2007;62:666‐670. [DOI] [PubMed] [Google Scholar]
- 32. García‐García J, Fernández‐García MA, Blanco‐Arias P, et al. Non‐compaction cardiomyopathy and early respiratory failure in an adult symptomatic female carrier of centronuclear myopathy caused by a MTM1 mutation. Neuromuscul Disord. 2018;28:952‐955. [DOI] [PubMed] [Google Scholar]
- 33. Yu S, Manson J, White S, et al. X‐linked myotubular myopathy in a family with three adult survivors. Clin Genet. 2003;64:148‐152. [DOI] [PubMed] [Google Scholar]
- 34. Cocanougher BT, Flynn L, Yun P, et al. Adult MTM1‐related myopathy carriers: classification based on deep phenotyping. Neurology. 2019;93:e1535‐e1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dahl N, Hu LJ, Chery M, et al. Myotubular myopathy in a girl with a deletion at Xq27‐q28 and unbalanced X inactivation assigns the MTM1 gene to a 600‐kb region. Am J Hum Genet. 1995;56:1108‐1115. [PMC free article] [PubMed] [Google Scholar]
- 36. Donkervoort S, Bonnemann CG, Loeys B, Jungbluth H, Voermans NC. The neuromuscular differential diagnosis of joint hypermobility. Am J Med Genet C Semin Med Genet. 2015;169c:23‐42. [DOI] [PubMed] [Google Scholar]
- 37. Amburgey K, Bailey A, Hwang JH, et al. Genotype‐phenotype correlations in recessive RYR1‐related myopathies. Orphanet J Rare Dis. 2013;8:117‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kouwenberg C, Bohm J, Erasmus C, et al. Dominant Centronuclear myopathy with early childhood onset due to a novel mutation in BIN1. J Neuromuscul Dis. 2017;4:349‐355. [DOI] [PubMed] [Google Scholar]
- 39. Prokic I, Cowling BS, Laporte J. Amphiphysin 2 (BIN1) in physiology and diseases. J Mol Med. 2014;92:453‐463. [DOI] [PubMed] [Google Scholar]
- 40. Mejaddam AY, Nennesmo I, Sejersen T. Severe phenotype of a patient with autosomal recessive centronuclear myopathy due to a BIN1 mutation. Acta Myol. 2009;28:91‐93. [PMC free article] [PubMed] [Google Scholar]
- 41. Jeannet PY, Bassez G, Eymard B, et al. Clinical and histologic findings in autosomal centronuclear myopathy. Neurology. 2004;62:1484‐1490. [DOI] [PubMed] [Google Scholar]
- 42. Bassez G, Chapoy E, Bastuji‐Garin S, et al. Type 2 myotonic dystrophy can be predicted by the combination of type 2 muscle fiber central nucleation and scattered atrophy. J Neuropathol Exp Neurol. 2008;67:319‐325. [DOI] [PubMed] [Google Scholar]
- 43. Schoser BG, Schneider‐Gold C, Kress W, et al. Muscle pathology in 57 patients with myotonic dystrophy type 2. Muscle Nerve. 2004;29:275‐281. [DOI] [PubMed] [Google Scholar]
- 44. Lexell J. Human aging, muscle mass, and fiber type composition. J Gerontol A Biol Sci Med Sci. 1995;50:11‐16. [DOI] [PubMed] [Google Scholar]
- 45. Hanisch F, Müller T, Dietz A, et al. Phenotype variability and histopathological findings in centronuclear myopathy due to DNM2 mutations. J Neurol. 2011;258:1085‐1090. [DOI] [PubMed] [Google Scholar]
- 46. Böhm J, Yiş U, Ortaç R, et al. Case report of intrafamilial variability in autosomal recessive centronuclear myopathy associated to a novel BIN1 stop mutation. Orphanet J Rare Dis. 2010;5:35‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Demonbreun AR, McNally EM. Dynamin 2 the rescue for centronuclear myopathy. J Clin Invest. 2014;124:976‐978. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supplementary Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.