

Abbreviations
- ALT
alanine aminotransferase
- BLV
bulevirtide
- CHD
chronic hepatitis D
- IFNα
interferon alfa
- LNF
lonafarnib
- NTCP
sodium taurocholate cotransporting polypeptide
- pegIFNα
pegylated interferon alfa
- pegIFNλ
pegylated interferon lambda
- SVR
sustained virological response
- TDF
tenofovir disoproxil fumarate
Efficacious, simple, and well‐tolerated therapies are available to control hepatitis B and cure hepatitis C, but such therapies are not yet available for hepatitis D. The time‐honored interferon alfa (IFNα) remains the only therapy for chronic hepatitis D (CHD).( 1 ) Although HDV induces liver disease only in the setting of a dual infection with HBV, attempts to control CHD by inhibiting the replication of HBV with nucleos(t)ide analogues were of no avail.
Targets for New HDV Therapies
The reason why HDV is refractory to conventional antiviral therapies lies in its unique structure and biology.( 2 ) The HDV genome (1.7 kb) is too small to code for the proteins required for its own replication and instead relies on the replicative machinery of the hepatocyte, requiring from HBV only the HBsAg to enter liver cells and disseminate infection. Thus, the HDV replication cycle is not affected by conventional medications that directly target common viral replication processes, such as the viral polymerase inhibitors used in HBV and HCV infections.
New strategies target interactions of HDV with HBsAg or the infected host, in order to deprive the virus of extrinsic functions critical to its replication cycle.( 1 , 3 ) In this review, we will discuss endpoints of CHD treatment and review the efficacy of new therapies in clinical trials.
Endpoints for CHD Treatment
Similar to chronic hepatitis C, IFNα trials for CHD considered undetectable HDV RNA 6 months after the end of therapy, as evidence of sustained virological response (SVR) and successful therapy.( 1 ) Reported SVR rates are generally low (i.e., ~25%‐30%), and late relapses are common. In a 10‐year follow‐up of the HDTI trial of pegylated IFNα (pegIFNα) with or without adefovir dipivoxil, 8 of 14 (57%) CHD patients who had achieved SVR experienced a virological relapse.( 4 ) The high rate of HDV relapse is not surprising, considering the limited sensitivity of current diagnostic assays for serum HDV RNA with lower detection limit ~15 copies/mL, corresponding to ~930 IU/mL.( 5 , 6 ) Thus, HDV is likely still present in the liver despite undetectable serum HDV RNA, and viral relapse can occur as long as HBV is also present; integration of HBV in the host genome may also contribute to production of the HBsAg required for HDV morphogenesis.( 2 ) Indeed, the term SVR is never used in patients with chronic hepatitis B receiving nucleos(t)ide analogues, given that viral relapse is near universal when treatment is discontinued even after many years of undetectable serum HBV DNA.
Given the low rate of SVR and high rates of late virological relapse with IFNα therapy, alternative endpoints need to be considered in assessing the efficacy of new CHD therapies. These endpoints need to be shown to be associated with clinical benefit, such as biochemical or histological improvement or decreased risk of cirrhosis, decompensation, or HCC. From one small study showing an association of HDV decline with survival benefit,( 7 ) a ≥2‐log reduction in serum HDV RNA from baseline was proposed by an expert panel( 8 ) as initial treatment efficacy in clinical trials for CHD. However, subsequent studies have used this criterion as an off‐treatment endpoint of therapeutic efficacy. This is problematic given that it does not consider the baseline HDV‐RNA level; thus, HDV‐RNA levels below detection after treatment and levels of 10E05 IU/mL (decreased from >10E07 IU/mL) would both be considered to have met the therapeutic endpoint. Furthermore, it assumes that serum HDV‐RNA levels at the end of treatment can be sustained after discontinuation of treatment or that treatment can be continued in the long term in order to maintain the potential for clinical benefit. However, the practicality of long‐term therapy will depend on safety, ease of administration, and costs of the medications used. An alternative endpoint may be a decrease in serum HDV RNA below a certain level, similar to inactive carriers in chronic hepatitis B,( 9 ) but the threshold level of HDV RNA for clinical benefit has not been defined.
The ideal endpoint of CHD treatment would be HBsAg loss, similar to the proposed definition for functional HBV cure. This is rarely achieved with IFNα monotherapy; whether a combination of new therapies in development will increase the rate of HBsAg loss remains to be determined.
New Targets for HDV Treatment
Several new targets have been identified for drug development against HDV. Of these, three have been tested in clinical trials (Fig. 1).
HBsAg secretion inhibitor: REP‐2139 is a nucleic acid polymer that interacts with a host chaperone, blocking the assembly/release of subviral HBsAg particles. Given that subviral particles account for >99.99% of HBsAg in the circulation, REP‐2139 might reduce available HBsAg to support HDV particle assembly. Modeling estimates also suggest an accelerated loss of HDV‐infected cells by hitherto unknown mechanisms.( 10 )
Farnesyl‐transferase inhibitor: Lonafarnib (LNF) interferes with the assembly of HDV virion, which requires farnesylation by the host of the large HDAg isoform of the virus.( 11 )
Entry inhibitor: Bulevirtide (BLV; formerly Myrcludex B), a small myristoylated synthetic lipopeptide corresponding to the HBV preS1 sequence, blocks the binding of HBsAg‐enveloped particles to sodium taurocholate cotransporting polypeptide (NTCP), the entry receptor for both HBV and HDV, preventing entry of HDV into hepatocytes.( 12 )
FIG. 1.
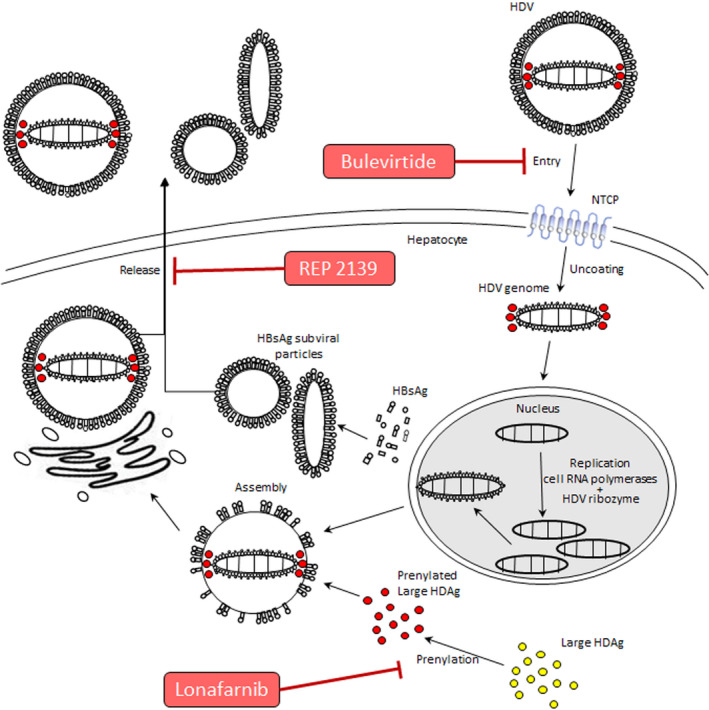
HDV life cycle and sites of action of different antivirals. HDV enters into hepatocytes through NTCP, which is a functional membrane receptor for HDV. Bulevirtide docks to the NTCP, blocking entry of HDV into hepatocytes. Within the hepatocyte, HDV discards the HBsAg coat and migrates to the nucleus. In the nucleus, viral RNA is replicated by the RNA polymerases of the host, with participation of the HDV ribozyme. The HDV ribonucleoprotein migrates to the cytoplasm, where it is coated with the HBsAg to assemble into the virion. For HDV RNA to combine with the HBsAg, it is necessary that the large HDAg of the HDV is farnesylated by a cytoplasmic farnesyl‐transferase. The farnesyl‐transferase inhibitor, LNF, interferes with the farnesylation process, preventing assembly of the virion. Mature HDV virions are released from the hepatocyte by the trans‐Golgi network; the HBsAg secretion inhibitor nucleic acid polymer, REP‐2139, blocks the assembly/release of subviral HBsAg particles, reducing available HBsAg for HDV morphogenesis and export (adapted from Caviglia and Rizzetto( 28 )).
The therapeutic potential of all three strategies has been shown in phase 2 trials,( 13 , 14 , 15 ) and LNF and BLV are moving into phase 3 trials.
Efficacy in Clinical Trials
HBsAg Secretion Inhibitor
A small study of REP 2139‐Ca given for 15 weeks as monotherapy, followed by add‐on pegIFNα for 15 weeks and then pegIFNα monotherapy for another 33 weeks in 12 CHD patients, found that at the end of therapy 7 patients had undetectable HDV RNA and 5 had cleared HBsAg.( 13 ) After 3.5 years of follow‐up, 7 of 11 patients had undetectable HDV DNA and 4 had HBsAg loss.( 16 ) Further studies are needed to confirm these impressive response rates.
Farnesyl‐Transferase Inhibitor
A pilot trial showed that LNF monotherapy, given orally, decreased serum HDV‐RNA levels, but all patients experienced gastrointestinal side effects (Table 1). Subsequent studies assessed split doses as well as combination with the cytochrome P450 3A4 inhibitor, ritonavir, to allow for lower doses of LNF while preserving its antiviral activity. The best results with this dual combination were reported in the LOWR‐2 study,( 17 ) where HDV RNA fell below the detection limit in 5 of 13 patients assigned to receive LNF 50 mg b.i.d. with ritonavir 100 mg b.i.d. for 24 weeks. Varying doses of LNF from 50 to 100 mg plus ritonavir 100 mg q.d. were studied in LOWR‐3,( 18 ) and gradually escalating doses of LNF from 50 to 100 mg b.i.d. plus ritonavir 100 mg b.i.d. were studied in LOWR‐4.( 19 ) Higher doses of LNF were associated with more adverse effects. For the LIFT‐HDV study, LNF 50 mg b.i.d. and ritonavir 100 mg b.i.d. were combined with pegylated interferon lambda (pegIFNλ) at weekly doses of 180 µg for 24 weeks.( 20 ) Serum HDV RNA became undetectable at the end of treatment in 11 of 26 patients, with 5 maintaining their response 24 weeks after the end of therapy, but none lost HBsAg. In contrast to IFNα, IFNλ has less side effects. PegIFNλ monotherapy has also been shown to be efficacious in the LIMT study,( 21 ) where 5 of 14 patients had undetectable HDV RNA at the end of 48‐week treatment and at 24‐week follow‐up. In the ongoing phase 3 D‐LIVR study, LNF plus ritonavir are combined with pegIFNα for 48 weeks. In view of the need for long‐term therapies, it is likely that the side effects of LNF, though mitigated by ritonavir, may remain a concern, particularly when added to those of pegIFNα.
TABLE 1.
Virological Responses to Ritonavir‐Boosted Lonafarnib Treatment With or Without pegIFNα or pegIFNλ
| Study | Treatment Arms | Rx Duration (Weeks) | N | EOT HDV RNA ≥2‐log Decline/BLQ* | 24 Weeks Post‐Rx HDV RNA BLQ*/Undetectable | HBsAg Loss |
|---|---|---|---|---|---|---|
| LOWR‐2( 17 ) | 24 | 6 | 1/0 | NR | NR | |
| LNF 25 mg b.i.d. | ||||||
| + RTV 100 mg b.i.d. | ||||||
| LNF 50 mg b.i.d. | 24 | 13 | 06‐May | |||
| + RTV 100 mg b.i.d. | ||||||
| LNF 25 mg b.i.d. | 24 | 5 | 03‐Mar | |||
| + RTV 100 mg b.i.d. | ||||||
| + pegIFNα2a 180 μg q.w. | ||||||
| LNF 50 mg b.i.d. | 24 | 4 | 04‐Feb | |||
| + RTV 100 mg b.i.d. | ||||||
| + pegIFNα2a 180 μg q.w. | ||||||
| LOWR‐3( 18 ) | LNF 50 or 75 or 100 mg q.d. | 12 or 24 | 21 | 6/4 † | NR | NR |
| + RTV 100 mg q.d. | ||||||
| LOWR‐4( 19 ) | Starting dose LNF 50 mg b.i.d. + RTV 100 mg b.i.d., escalating every 2‐4 weeks to LNF 75 mg b.i.d. + RTV 100 mg b.i.d. followed by LNF 100 mg b.i.d. + RTV 100 mg b.i.d. | 24 | 15 | 04‐Jan | 3/0 | NR |
| LIFT HDV( 20 ) | LNF 50 mg b.i.d. | 24 | 26 | 25‐Nov | NR/5 | 0 |
| + RTV 100 mg b.i.d. | ||||||
| + pegIFNλ 180 μg q.w. | ||||||
| LIMT HDV( 21 ) | pegIFNλ 120 μg q.w. | 48 | 19 | 04‐Mar | 02‐Mar | NR |
| pegIFNλ 180 μg q.w. | 14 | 07‐May | 05‐May |
The quantitative HDV‐RNA assays used in the above trials had a lower limit of quantification of 40 IU/mL (LIFT HDV) or 14 IU/mL (LOWR‐2, LOWR‐3, LOWR‐4, and LIMT HDV).
Aggregate data from all six treatment arms.
Abbreviations: BLQ, below the limit of quantification; pegIFNα2a, pegylated interferon alfa‐2a, RTV, ritonavir; NR, not reported; Rx, treatment; EOT, end of treatment.
Entry Inhibitor
BLV has been studied as monotherapy and in combination with pegIFNα and/or tenofovir disoproxil fumarate (TDF; Table 2). It is administered daily by the s.c. route and is generally well tolerated despite a dose‐dependent bile acid increase.
TABLE 2.
Virological Responses to Bulevirtide Treatment With or Without pegIFNα2a and TDF
| Study | Treatment Arms | N | EOT HDV RNA* | Posttreatment HDV RNA* | HBsAg Response |
|---|---|---|---|---|---|
| MYR 202( 22 ) | ≥2‐log decrease or undetectable after 24 weeks of BLV | ≥2‐log decrease or undetectable 24 weeks after end of BLV | HBsAg loss 24 weeks after end of BLV | ||
| TDF 12 weeks then BLV 2 mg + TDF 24 weeks then TDF 24 weeks | 28 | 13 | 2 | 1 | |
| TDF 12 weeks then BLV 5 mg + TDF 24 weeks then TDF 24 weeks | 32 | 15 | 3 | 2 | |
| TDF 12 weeks then BLV 10 mg + TDF 24 weeks then TDF 24 weeks | 30 | 23 | 3 | 0 | |
| TDF 60 weeks | 30 | 1 | 0 | 0 | |
| MYR 203( 23 , 24 ) | 48 weeks of treatment in all arms | Undetectable at EOT | ≥2‐log decrease/undetectable at 24 weeks post‐EOT | ≥1‐log decrease/negative at 24 weeks post‐EOT | |
| pegIFNα2a 180 μg | 15 | 2 | 0/0 | 0/0 | |
| BLV 2 mg + pegIFNα2a 180 μg | 15 | 9 | 03‐Aug | 06‐Apr | |
| BLV 5 mg + pegIFNα2a 180 μg | 15 | 6 | 03‐Apr | 2/0 | |
| BLV 10 mg + pegIFNα2a 180 µg | 15 | 13 | 04‐Jan | 01‐Jan | |
| BLV 2 mg | 15 | 2 | 04‐Jan | 0/0 | |
| TDF + BLV 5 mg b.i.d. | 15 | 6 | 02‐May | 0/0 |
For all trials, the lower limit of quantification of the HDV‐RNA assay was 14 IU/mL.
Abbreviations: pegIFNα2a, pegylated interferon alfa‐2a; EOT, end of treatment.
In the MYR 202 trial,( 22 ) 90 patients received TDF for 12 weeks followed by BLV (2, 5, or 10 mg) plus TDF for 24 weeks, and then TDF for 24 weeks, whereas 30 patients received TDF monotherapy for 60 weeks. HDV RNA decrease by ≥2 log (or undetectable) was observed in 46%‐77% patients at the end of BLV therapy, with the highest response rate in the group that received BLV 10‐mg doses. However, at the end of follow‐up (i.e., 24 weeks after the end of BLV therapy), 7%‐10% of patients had maintained these HDV responses. Three patients lost HBsAg (1 in the BLV 2‐mg group and 2 in the 5‐mg group, but none in the 10‐mg group) whereas none of the patients in the TDF monotherapy group had HDV‐RNA or HBsAg respo‐nse.
In the MYR 203 study,( 23 , 24 ) 90 patients were enrolled into six groups (15 patients each) of 48‐week treatment. The primary endpoint of undetectable HDV RNA at week 72 was achieved in 8 (53%), 4 (27%), and 1 (7%) patients who received a combination of pegIFNα and 2, 5, and 10 mg of BLV, respectively, compared to 1 (7%) patient who received 2‐mg BLV monotherapy, none who received pegIFNα monotherapy, and 3 (33%) who received 10 mg of BLV and TDF. At week 72, a ≥1‐log decrease in HBsAg was observed only in patients who received a combination of pegIFNα and BLV, with the highest response in those who received BLV 2‐mg doses, but not in those who received BLV or pegIFNα monotherapy or a combination of BLV and TDF. HBsAg became undetectable in 4 of 15 of patients treated with pegIFNα and BLV 2 mg.
Results of the MYR 202 study suggest that BLV 10 mg has better antiviral efficacy than the 2‐ or 5‐mg doses when used in the absence of pegIFNα; good clinical results were also reported in three anecdotal cases of CHD while on therapy with BLV 10 mg.( 25 ) However, the lower dose of 2 mg appeared to be superior in the MYR 203 study where BLV was used in combination with pegIFNα. The reasons why BLV 10 mg was inferior to BLV 2 mg when used in combination with pegIFNα are unclear. Thus, although it has been proposed that s.c. injections of 10‐mg BLV monotherapy daily may be used for long‐term treatment of CHD, further studies are needed to establish the long‐term safety (bile acid increase, in particular in patients with cirrhosis) and acceptability (daily injections) as well as efficacy (maintained suppression/undetectable HDV RNA, normal alanine aminotransferase [ALT], and HBsAg loss) of this approach. Despite the known side effects of pegIFNα, the combination of 2 mg of BLV and pegIFNα had the best response and deserves further studies. In addition, the possibility of combining BLV with pegIFNλ, which has less side effects, should also be evaluated. One finding in support of BLV monotherapy is that many patients normalized ALT during therapy despite persistent HDV viremia. In the MYR 203 trial, 11 patients receiving 2‐mg BLV monotherapy normalized ALT, though 9 still had detectable HDV RNA at the end of therapy, and 3 patients maintained normal ALT at the end of follow‐up.
Ongoing studies might shed more light on the optimal regimen of BLV and its efficacy as monotherapy or in combination with pegIFNα. One study (MYR 204) is evaluating 48‐week treatment of the combination of 2 or 10 mg of BLV with pegIFNα, compared to pegIFNα alone, and 144 weeks of BLV 10‐mg monotherapy. Another study is a phase 3 study (MYR 301) evaluating 2‐ versus 10‐mg BLV monotherapy for 144 weeks versus 10‐mg BLV monotherapy for 96 weeks. The primary outcome of the MYR 301 study is a combined response of undetectable HDV RNA or a ≥2‐log decrease in HDV RNA plus ALT normalization at 48 weeks (ClinicalTrials.gov accession no.: NCT03852719).
Although the phase 3 trials are still ongoing, the European Medicines Agency has afforded a conditional marketing authorization to BLV on July 31, 2020 under the trade name Hepcludex.( 26 ) The recommended dose was 2 mg even though this dose of BLV when used as monotherapy was inferior to 10 mg. The optimal treatment duration was stated as unknown and the recommendation was to continue treatment as long as it is associated with clinical benefit, though it did not specify how clinical benefit should be measured and the product information acknowledged the lack of data on long‐term impact (>48 weeks) of bile salt increase induced by BLV. CHD is designated a rare disease by both the U.S. Food and Drug Administration and European Medicines Agency, allowing treatment for CHD to be approved under the Orphan Drug Act; however, this is not equivalent to authorization of a drug while phase 3 trials are ongoing.
Future Treatments
Two therapeutic approaches can be envisioned. Similar to HBV functional cure, one approach is finite therapy with a combination of BLV or LNF (with ritonavir) and pegIFNα or pegIFNλ with the goal of undetectable HDV RNA and HBsAg loss off treatment in a high percentage of patients; the other is simple and safe long‐term maintenance therapy similar to nucleos(t)ide analogues for HBV, based on BLV or LNF (with ritonavir) monotherapy, with the goal of keeping HDV RNA undetectable or suppressed in the presence of the HBsAg. Different levels of virological responses should be separately reported in clinical trials and correlated with biochemical and clinical responses to help identify a virological threshold that correlates with the inactive HDV carrier state. It will also be important to assess therapy efficacy in terms of histological endpoints by performing sequential liver biopsies.
Given the central role of HBsAg in HDV infection, in addition to drugs directly targeting the HDV life cycle, clinical trials evaluating the combination of these direct‐acting antivirals and drugs that specifically inhibit production of HBsAg, notably interfering RNAs and antisense oligonucleotides that have demonstrated safety and efficacy in decreasing HBsAg levels in patients with chronic hepatitis B,( 27 ) should be evaluated.
Author Contributions
A.S.L., F.N. and M.R.: drafting of the article, contribution to conception and design, analysis and interpretation of data. T.A., P.F.: critical interpretation of data, revision for intellectual content, final approval of the version to be published.
Acknowledgment
P.F. is supported by the Intramural Programs of the Division of Intramural Research of the National Institute of Allergy and Infectious Diseases, National Institutes of Health. Open Access Funding provided by Universita degli Studi di Torino within the CRUI‐CARE Agreement. [Correction added on 10 May, after first online publication: Open Access Funding provided by Universita degli Studi di Torino within the CRUI‐CARE Agreement funding statement has been added.]
Potential conflict of interest: Dr. Lok advises for and received grants from TARGET. She advises for Ambys, Bristol‐Myers Squibb, CLEAR‐B, GNI, and Eli Lilly. She received grants from Gilead. Dr. Negro consults for, advises for, and received grants from Gilead. He advises for AbbVie and Merck. Dr. Asselah consults for, advises for, is on the speakers’ bureau for, and received grants from Eiger, Gilead, and Myr. Dr. Rizzetto advises for Gilead.
References
Author names in bold designate shared co‐first authorship.
- 1. Rizzetto M, Hamid S, Negro F. The changing context of hepatitis D. J Hepatol 2021;74:1200‐1211. [DOI] [PubMed] [Google Scholar]
- 2. Sureau C, Negro F. The hepatitis delta virus: replication and pathogenesis. J Hepatol 2016;64(1 Suppl.):S102‐S116. [DOI] [PubMed] [Google Scholar]
- 3. Asselah T, Loureiro D, Tout I, Castelnau C, Boyer N, Marcellin P, et al. Future treatments for hepatitis delta virus infection. Liver Int 2020;40(Suppl. 1):54‐60. [DOI] [PubMed] [Google Scholar]
- 4. Wranke A, Hardtke S, Heidrich B, Dalekos G, Yalçin K, Tabak F, et al. Ten‐year follow‐up of a randomized controlled clinical trial in chronic hepatitis delta. J Viral Hepat 2020;27:1359‐1368. [DOI] [PubMed] [Google Scholar]
- 5. Mederacke I, Bremer B, Heidrich B, Kirschner J, Deterding K, Bock T, et al. Establishment of a novel quantitative hepatitis D virus (HDV) RNA assay using the Cobas TaqMan platform to study HDV RNA kinetics. J Clin Microbiol 2010;48:2022‐2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wedemeyer H, Yurdaydin C, Hardtke S, Caruntu FA, Curescu MG, Yalcin K, et al. Peginterferon alfa‐2a plus tenofovir disoproxil fumarate for hepatitis D (HIDIT‐II): a randomised, placebo controlled, phase 2 trial. Lancet Infect Dis 2019;19:275‐286. [DOI] [PubMed] [Google Scholar]
- 7. Farci P, Roskams T, Chessa L, Peddis G, Mazzoleni AP, Scioscia R, et al. Long‐term benefit of interferon alpha therapy of chronic hepatitis D: regression of advanced hepatic fibrosis. Gastroenterology 2004;126:1740‐1749. [DOI] [PubMed] [Google Scholar]
- 8. Yurdaydin C, Abbas Z, Buti M, Cornberg M, Esteban R, Etzion O, et al.; Hepatitis Delta International Network (HDIN) . Treating chronic hepatitis delta: the need for surrogate markers of treatment efficacy. J Hepatol 2019;70:1008‐1015. [DOI] [PubMed] [Google Scholar]
- 9. European Association for the Study of the Liver . EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J Hepatol 2017;2017:370‐398. [DOI] [PubMed] [Google Scholar]
- 10. Shekhtman L, Cotler SJ, Hershkovich L, Uprichard SL, Bazinet M, Pantea V, et al. Modelling hepatitis D virus RNA and HBsAg dynamics during nucleic acid polymer monotherapy suggest rapid turnover of HBsAg. Sci Rep 2020;10:7837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Glenn JS. Prenylation of HDAg and antiviral drug development. Curr Top Microbiol Immunol 2006;307:133‐149. [DOI] [PubMed] [Google Scholar]
- 12. Tu T, Urban S. Virus entry and its inhibition to prevent and treat hepatitis B and hepatitis D virus infections. Curr Opin Virol 2018;30:68‐79. [DOI] [PubMed] [Google Scholar]
- 13. Bazinet M, Pântea V, Cebotarescu V, Cojuhari L, Jimbei P, Albrecht J, et al. Safety and efficacy of REP 2139 and pegylated interferon alfa‐2a for treatment‐naive patients with chronic hepatitis B virus and hepatitis D virus co‐infection (REP 301 and REP 301‐LTF): a non‐randomised, open‐label, phase 2 trial. Lancet Gastroenterol Hepatol 2017;2:877‐889. [DOI] [PubMed] [Google Scholar]
- 14. Koh C, Canini L, Dahari H, Zhao X, Uprichard SL, Haynes‐Williams V, et al. Oral prenylation inhibition with lonafarnib in chronic hepatitis D infection: a proof‐of‐concept randomised, double‐blind, placebo‐controlled phase 2A trial. Lancet Infect Dis 2015;15:1167‐1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bogomolov P, Alexandrov A, Voronkova N, Macievich M, Kokina K, Petrachenkova M, et al. Treatment of chronic hepatitis D with the entry inhibitor myrcludex B: first results of a phase Ib/IIa study. J Hepatol 2016;65:490‐498. [DOI] [PubMed] [Google Scholar]
- 16. Bazinet M, Pântea V, Cebotarescu V, Cojuhari L, Jimbei P, Anderson M, et al. Persistent control of hepatitis B virus and hepatitis delta virus infection following REP 2139‐Ca and pegylated interferon therapy in chronic hepatitis B virus/hepatitis delta virus coinfection. Hepatol Commun 2020;5:189‐202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yurdaydin C, Idilman R, Keskin O, Kalkan Ç, Karakaya FM, Çaliskan A, et al. A phase 2 dose‐optimization study of lonafarnib with ritonavir for the treatment of chronic delta hepatitis—analysis from the LOWR HDV‐2 study using the Robogene real‐time qPCR HDV RNA assay. J Viral Hepat 2018;25(Suppl. 2):10.28685917 [Google Scholar]
- 18. Koh C, Surana P, Han T, Fryzek N, Kapuria D, Etzion O, et al. A phase 2 study exploring once daily dosing of ritonavir boosted lonafarnib for the treatment of chronic delta hepatitis—end of study results from the LOWR HDV‐3 study. J Hepatol 2017;66:S101‐S102. [Google Scholar]
- 19. Wedemeyer H, Port K, Deterding K, Wranke A, Kirschner J, Martins EB, et al. A phase 2 dose‐escalation study of lonafarnib plus ritonavir in patients with chronic hepatitis D: final results from the lonafarnib with ritonavir in HDV‐4 (LOWR HDV‐4). J Hepatol 2017;66:S24. [Google Scholar]
- 20. Koh C, Hercun J, Rahman F, Huang A, Da B & Surana P et al. A phase 2 study of peginterferon lambda, lonafarnib and ritonavir for 24 weeks: end‐of‐treatment results from the LIFT HDV study. The Liver Meeting, October 30, 2020 (oral late breaker L08). https://assets.website‐files.com/5f3d77cd56d46907a50fb8d9/5f9d9c2057efc43f55b78db7_2020%20TLMdX%20Late‐breaking%20Abstracts‐%20Oct%2030.pdf. Accessed May 7, 2021.
- 21. Etzion O, Hamid SS, Lurie Y, Gane E, Bader N, Yardeni D, et al. End of study results from LIMT HDV study: 36% durable virologic response at 24 weeks post‐treatment with pegylated interferon lambda monotherapy in patients with chronic hepatitis delta virus infection. J Hepatol 2019;70(Suppl.):e32. [Google Scholar]
- 22. Wedemeyer H, Bogomolov P, Blank A, Allweiss L, Dandri‐Petersen M, Bremer B, et al. Final results of a multicenter, open‐label phase 2b clinical trial to assess safety and efficacy of Myrcludex B in combination with tenofovir in patients with chronic HBV/HDV co‐infection. J Hepatol 2018;68(Suppl.):S3. [Google Scholar]
- 23. Wedemeyer H, Schoeneweis K, Bogomolov PO, Voronka V, Chulanov V, Stepanova T, et al. Final results of a multicenter, open‐label phase 2 clinical trial (MYR203) to assess safety and efficacy of myrcludex B in combination with PEG‐interferon alpha 2a in patients with chronic HBV/HDV co‐infection. J Hepatol 2019;70:e81. [Google Scholar]
- 24. Wedemeyer H, Schöneweis K, Bogomolov PO, Chulanov V, Stepanova T, Viacheslav M, et al. 48 weeks of high dose (10 mg) bulevirtide as mono‐therapy or with peginterferon alfa‐2a in patients with chronic HBV/HDV coinfection. J Hepatol 2020;73:S52‐S53. [Google Scholar]
- 25. Loglio A, Ferenci P, Uceda Renteria SC, Tham CYL, van Bömmel F, Borghi M, et al. Excellent safety and effectiveness of high‐dose Myrcludex‐B monotherapy administered for 48 weeks in HDV‐related compensated cirrhosis: a case report of 3 patients. J Hepatol 2019;71:834‐839. [DOI] [PubMed] [Google Scholar]
- 26. European Medicines Agency . Hepcludex. https://www.ema.europa.eu/en/medicines/human/EPAR/hepcludex. Accessed April 15, 2021.
- 27. Yuen MF, Schiefke I, Yoon JH, Ahn SH, Heo J, Kim JH, et al. RNA interference therapy with ARC‐898 520 results in prolonged hepatitis B surface antigen response in patients with chronic hepatitis B 899 infection. Hepatology 2020;72:19‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Caviglia GP, Rizzetto M. Treatment of hepatitis D: an unmet medical need. Clin Microbiol Infect 2020;26:824‐827. [DOI] [PubMed] [Google Scholar]