

Abstract
Introduction
A massive increase of soluble thrombomodulin (sTM) due to variants in the thrombomodulin gene (THBD) has recently been identified as a novel bleeding disorder.
Aim
To investigate sTM levels and underlying genetic variants as a cause for haemostatic impairment and bleeding in a large number of patients with a mild to moderate bleeding disorder (MBD), including patients with bleeding of unknown cause (BUC).
Patients and methods
In 507 MBD patients, sTM levels, thrombin generation and plasma clot formation were measured and compared to 90 age‐ and sex‐matched healthy controls. In patients, genetic analysis of the THBD gene was performed.
Results
No difference in sTM levels between patients and controls was found overall (median ([IQR] 5.0 [3.8‐6.3] vs. 5.1 [3.7‐6.4] ng/ml, p = .762), and according to specific diagnoses of MBD or BUC, and high sTM levels (≥95th percentile of healthy controls) were not overrepresented in patients. Soluble TM levels had no impact on bleeding severity or global tests of haemostasis, including thrombin generation or plasma clot formation. In the THBD gene, no known pathogenic or novel disease‐causing variants affecting sTM plasma levels were identified in our patient cohort.
Conclusion
TM‐associated coagulopathy appears to be rare, as it was not identified in our large cohort of patients with MBD. Soluble TM did not arise as a risk factor for bleeding or altered haemostasis in these patients.
Keywords: anticoagulants, blood coagulation, blood coagulation disorders, haemorrhage, thrombomodulin
1. INTRODUCTION
Patients with mild to moderate bleeding tendency (mild bleeding disorders, MBD) suffer from a life‐long history of diverse bleeding symptoms such as epistaxis, easy bruising, menorrhagia or bleeding after tooth extractions. 1 , 2 , 3 Severe spontaneous bleedings are rare, which is why patients are often not diagnosed until adulthood. 3 Still, under certain haemostatic challenges, such as surgery or childbirth, bleeding can be even life‐threatening. A vast majority of patients with MBD remains without a definite diagnosis, despite thorough haemostatic investigations of plasmatic coagulation and platelet function. 1 Patients with bleeding of unknown cause (BUC) do not differ phenotypically from patients with an established diagnosis, 1 , 4 and were found to have impaired thrombin generation and plasma clot formation. 5 This underlines the urgent need to identify underlying mechanisms of bleeding in BUC patients, 6 leading to better diagnostic and therapeutic approaches. 7
Recently, several groups described a thrombomodulin (TM) ‐associated coagulopathy with a 100‐fold enhancement of soluble TM (sTM). 8 , 9 , 10 , 11 The routinely performed coagulation tests, like the activated partial thromboplastin time (aPTT) or prothrombin time (PT), platelet function, and clotting factor levels were all within the normal range, whereas thrombin generation was impaired. 8
Thrombomodulin is a transmembrane glycoprotein primarily located on endothelial cells of capillary vessels, which is not only involved in coagulation but also in immune processes, inflammation and cell‐proliferation. 12 By forming a complex with thrombin, TM becomes an essential co‐factor for both the natural anticoagulant protein C and also for the thrombin‐mediated activation of the thrombin activatable fibrinolysis inhibitor (TAFI). 12 , 13 Activated protein C (APC) with its co‐factor protein S is a key enzyme for the natural anticoagulation of activated coagulation factors (F) V and VIII. 14 On the other hand, thrombin‐TM complexes also have been shown to delay fibrinolysis by activation of TAFI, independently of APC. 12 , 15 , 16
The encoding gene for TM is THBD, which consists of only one exon on chromosome 20. For the TM‐associated coagulopathy, two variants in the THBD gene have been identified in different families that code for a premature stop codon: c.1611C > A; p.Cys537Stop and c.1487delCM; p.Pro496Argfs*10. These nonsense variants lead to a loss of the C‐terminal portion of the transmembrane helix and the cytoplasmatic domain of TM. 8 The resulting introduction of a negatively charged C‐terminus of TM and the loss of interaction with the cytoplasmatic domain result in the shedding of TM from the endothelium into the plasma. 9 , 17 The high sTM levels induce persistent APC activation, which then further inactivates FVa and FVIIIa. 9
Few genetic variants in the coding region of THBD are observed within the population database (gnomAD_v3). The most prevalent being a non‐synonymous variant rs1042579, NM_000361.3:c.1418C > T p.Ala473Val (GRCh38.p12 chr 20:23048087G > A).
Based on the recent findings in patients with TM‐associated coagulopathy, we systematically investigated levels of sTM in a well‐characterized cohort of patients with MBD, including a large number of BUC patients. Furthermore, we assessed how increased sTM levels affected global coagulation tests, particularly the thrombin generation assay and plasma clot formation, as well as the bleeding phenotype in our patients with MBD. To identify known or novel genetic variants within the THBD gene in MBD, genetic data were analysed.
2. METHODS
2.1. Study design and patients
Recruitment of patients with MBD in the Vienna Bleeding Biobank (VIBB), a prospective single‐centre cohort study, started in October 2009 and is on‐going. Patients aged ≥ 16 years without a previously diagnosed bleeding disorder were included. Detailed inclusion‐ and exclusion criteria were published recently 1 and are summarized in Supplementary table 1. The bleeding phenotype and severity were assessed by two standardized scores, the Vicenza bleeding score (BS) and the ISTH BAT. In the current analysis, 507 patients who were included in the study until March 2017 were investigated.
Ninety age‐and sex‐matched healthy controls were recruited by trained healthcare personnel for comparison.
The study has approval by the Ethics Committee of the Medical University Vienna (EC No 603/2009) according to the Declaration of Helsinki of 1975.
2.2. Blood sampling, laboratory analyses
After study inclusion, samples were timely processed to routine laboratory assessments and to storage at the biobank facility of the Medical University of Vienna (www.biobank.at, Supplementary paragraph 1). 18 Routinely performed analyses included the assessment of plasmatic coagulation and platelet function (Supplementary table 2). Assessment of thrombin generation and plasma clot formation is described in Supplementary paragraph 2 and 3.
2.3. Established diagnoses
The diagnostic criteria applied in the study to classify patients are reported in Supplementary table 3. Due to the low number of only 2 patients, patients with dys‐/hypofibrinogenemia were not analysed separately in this study.
2.4. Determination of sTM levels
The determination of sTM was performed using a commercially available ELISA (ab46508 – Thrombomodulin (CD141) Human ELISA Kit – Abcam, Cambridge, United Kingdom) from samples stored at the biobank.
2.5. Statistical and bioinformatic analysis
Statistical analysis was performed with the Statistical Package for Social Sciences (SPSS IBM Version 26.0) and the free open‐source software GNU R version 3.5.3. 19
Variables are described by mean (and standard deviation), or median (and interquartile range), in case of non‐normal distribution. Group comparison was performed using the Student's t‐test or the Wilcoxon‐rank‐sum‐test in case of non‐normal distribution for unadjusted groups and the chi‐square or Fisher's exact test were used for comparing categorical variables. Comparisons between diagnoses as well as genotypes and sTM levels were performed using a Kruskal‐Wallis test.
To evaluate the adjusted differences for sTM levels between patients and healthy controls multiple linear regression analysis (considering sex and age as confounding variables) was applied. The odds ratio (confidence interval 95%) for having sTM values above the pre‐defined cut‐off were calculated with logistic regression (considering sex, age and blood group O as confounding variables).
To account for the number of multiple comparisons performed within the individual secondary research questions, the Bonferroni‐Holm correction was accordingly applied. All p‐values are results of two‐sided tests, and p‐values < .05 were considered as statistically significant.
Genetic data come from genetic high‐throughput sequencing within the ThromboGenomics platform. 20 Bioinformatic analysis is described in detail in Supplementary paragraph 4.
3. RESULTS
3.1. Patients’ characteristics
Five hundred and seven patients with MBD were included in this study, of whom 358 (70.6%) had BUC. Eighty‐four patients (16.5%) had a possible or definite platelet function defect (PFD), and VWD (VWF≤30 IU/dL) or low von‐Willebrand factor (VWF 31–50 IU/dL) was diagnosed in 43 patients (8.5%). Further diagnoses were coagulation factor deficiencies (CFD, n = 21, 4.1%), and hypo‐/dysfibrinogenemia (n = 2, .4%).
Patients’ characteristics and laboratory results are summarized in Table 1. Blood group O was overrepresented in the patient cohort. Patients had a prolonged aPTT and PT and lower levels of von VWF antigen (VWF:Ag) and activity (VWF:RCo) than healthy controls, albeit the values were within the normal range.
TABLE 1.
Demographic and laboratory data of all patients with MBD and healthy controls
| All patients with MBD (n = 507) | Controls (n = 90) | |
|---|---|---|
| Female, n (%) | 412 (81.3) | 73 (81.1) |
| Blood group O, n (%) | 254 (50.1) | 21 (23.3) 1 |
| Positive family history, n (%) | 184 (36.3) | na |
| Caucasian, n (%) | 505 (99.6) | 89 (98.9) |
| mean (SD) | mean (SD) | |
|---|---|---|
| Age, years | 41.6 (15.8) | 42.6 (15.2) |
| BMI, kg/m2 | 24.2 (4.5) | 23.9 (7.1) |
| Haemoglobin, g/dL | 13.6 (1.3) | 14.4 (4.2) |
| Platelet count, x109/L | 250.9 (66.6) | 260.9 (48.6) |
| Fibrinogen, mg/dL | 316.6 (72.6) | 292.8 (63.1) |
| median (IQR) | median (IQR) | |
|---|---|---|
| aPTT‐STA, seconds | 35.8 (33.6‐38.7) | 34.7 (32.9‐36.1) |
| Prothrombin time, % | 95.0 (88.0‐102.0) | 101.0 (91.5‐109.0) |
| FVIII, % | 121.0 (96.0‐155.0) | 135.5 (106.3‐158.8) |
| FIX, % | 105.0 (90.0‐121.0) | 102.5 (89.0‐117.0) |
| VWF:Ag, IU/dL | 95.0 (74.0‐121.0) | 104.0 (89.25‐134.0) |
| VWF:RCo, IU/dL | 79.0 (63.3‐122.0) | 100.0 (77.0‐137.0) |
| mean [SD] | mean [SD] | |
|---|---|---|
| Vicenza bleeding score | 5.7 (3.0) | 0 (0) |
| ISTH BAT 2 | 6.5 (3.5) | na |
Blood group available of 73 healthy controls.
ISTH bleeding score available of 250 patients (49.3%).
Abbreviations: na, not available; BMI, body mass index; aPTT‐STA, activated partial thromboplastin time, according to the STA coagulation analyzer; FVIII, factor VIII activity; FIX, factor IX activity, VWF: Ag, Von Willebrand Antigen; VWF:RCo, Von Willebrand factor ristocetin cofactor activity.
3.2. Levels of soluble thrombomodulin
Soluble TM was not altered in patients when comparing to 90 sex‐ and age‐matched healthy controls (median [IQR] 5.0 [3.8‐6.3] vs. 5.1 [3.7‐6.4] ng/ml, p = .762). In the patient group, men had higher sTM levels than women (5.8 [4.4‐7.2] vs. 4.8 [3.7‐6.1] ng/ml, p < .001), which could not be seen in healthy controls (men: 5.9 [4.8‐6.3] vs. women: 5.0 [3.5‐6.4] ng/ml, p = .116). No strong correlations with age (r = .133, p = .003) or BMI (r = .077, p = .085) were identified in patients.
In multivariate analysis when adjusting for sex and age, there was no significant difference in sTM levels between patients and healthy controls (Table 2). When analysing sTM values according to established diagnoses of bleeding disorders or BUC, no differences in sTM levels were found in one of the subgroups in comparison to healthy controls (Figure 1, Table 2). There was no overall difference between subgroups of diagnoses either (Kruskal‐Wallis, p = .091).
TABLE 2.
Soluble TM values and odds ratio for high sTM values (≥ 14.7 ng/ml) in comparison to healthy controls in all patients and according to established diagnoses
| sTM, ng/ml | sTM ≥ 95th percentile( ≥ 14.7 ng/ml) | |||||
|---|---|---|---|---|---|---|
| n (%) | median [IQR] | p | p 1 | n (%) | OR 1 [95% CI] | |
| Healthy controls | 90 (100) | 5.1 [3.7‐6.4] | na | na | 4 (5.54) | na |
| All patients with MBD | 507 (100) | 5.0 [3.8‐6.3] | .762 | .306 | 10 (2.0) | 1.8 [.5‐6.9] |
| BUC | 358 (70.6) | 5.0 [3.8‐6.3] | .789 | .661 | 6 (1.7) | 2.4 [.6‐10.0] |
| PFD 2 | 84 (16.6) | 5.1 [3.7‐7.1] | .759 | .724 | 4 (4.4) | .8 [.1‐4.4] |
| VWF≤50 IU/dL 1 | 43 (8.5) | 4.5 [3.2‐5.9] | .202 | .482 | 0 (0) | na |
| CFD | 21 (4.1) | 4.9 [4.4‐6.0] | .913 | .900 | 1 (4.8) | .6 [.4‐9.2] |
Abbreviations: MBD, mild bleeding disorder; sTM, soluble thrombomodulin; OR, odds ratio; IQR, interquartile range; BUC, bleeding of unknown cause; PFD, platelet function defect; VWF, von Willebrand factor; CFD, coagulation factor deficiency.
adjusted for sex, age and blood group O in multivariate analysis.
one patient had both a PFD and VWF ≤50 IU/dL, two patients also had mild FXI deficiency.
FIGURE 1.
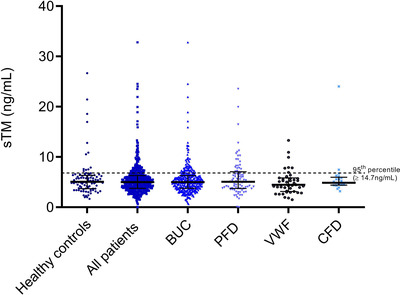
Scatter plot (including mean and SD) of soluble TM levels in patients and healthy controls. Abbreviations: sTM, soluble thrombomodulin; BUC, bleeding of unknown cause; PFD, platelet function defects; VWF, von Willebrand factor ≤ 50 IU/dL; CFD, clotting factor deficiency
To identify outliers of sTM levels in our patients, a cut‐off according to the 95th percentile of sTM levels in healthy controls (≥ 14.7 ng/ml) was defined. No increased number of patients above the predefined cut‐off was identified (Table 2). In line, also in separate analysis of patients with established diagnoses or BUC, no increased number of patients with very high sTM levels above the cut‐off was found.
3.3. Correlation of sTM with the bleeding phenotype
When analysing the influence of sTM on the bleeding phenotype, no clear correlation was found with the number of bleeding manifestations (r = ‐.088, p = .048), the Vicenza BS (r = ‐.055, p = .214) or the ISTH BAT (r = ‐.067, p = .292). No moderate or strong correlations between the bleeding scores and sTM levels were observed in the separate analysis of each diagnosis and BUC, also after adjustment for sex (Table 3). Mild positive correlations between sTM levels and the number of bleeding symptoms and the Vicenza BS were found in patients with VWF ≤50 IU/ml after adjustment for sex in linear regression analysis (Table 3).
TABLE 3.
Correlation of sTM values with the number of bleeding manifestations and bleeding scores in all patients and according to each diagnosis
| Number of bleeding manifestations | Vicenza BS | ISTH BAT | ||||
|---|---|---|---|---|---|---|
| r | β | r | β | r | β | |
| All patients with MBD | −.088* | .023 | .002 | .010 | −.067 | −.012 |
| BUC | −.126* | .006 | −.084 | −.020 | −.059 | .006 |
| PFD | −.073 | −.103 | .018 | −.008 | −.054 | −.049 |
| VWF≤50 IU/dL | .097 | .311* | .125 | .313* | −.129 | −.061 |
| CFD | .254 | −.159 | .030 | −.285 | .110 | −.703 |
Abbreviations: BS, bleeding score; BAT, bleeding assessment tool; BUC, bleeding of unknown cause; PFD, platelet function defect; VWF, von Willebrand factor; CFD, coagulation factor deficiency.
r, correlation coefficient (Spearman‐rho).
β, regression coefficient (linear regression with adjustment for age, sex and blood group O).
Weak (.2‐.4), moderate (.4‐.6), strong (.6‐.8) and very strong (.8‐1.0) correlations.
Significance: *p < .05; **p < .01; ***p < .001; ns: not significant.
When comparing the bleeding phenotype between patients with sTM levels < 95th percentile of healthy controls and those with values ≥ 95th percentile, there was no significant difference in the number of bleeding manifestations or bleeding severity (Supplementary table 4). Patients with BUC and very high sTM values ≥ 14.7 ng/ml had a slightly higher number of bleeding manifestations and ISTH BAT values, but lacking significant difference. There was no difference in the prevalence of specific bleeding symptoms between patients with sTM values above or below 14.7 ng/ml (Supplementary table 5).
3.4. Correlation of sTM levels with global coagulation tests, thrombin generation and fibrin clot formation
No moderate or strong correlations were identified between sTM values and aPTT, PT, parameters of thrombin generation or plasma clot formation (Table 4). The PT showed a weak inverse correlation with sTM levels in PFD patients only.
TABLE 4.
Correlation of sTM with global coagulation tests and parameters of thrombin generation and plasma clot formation in all patients with MBD and according to diagnoses
| Controls | All patients with MBD | BUC | PFD | CFD | |
|---|---|---|---|---|---|
| n = 90 | n = 507 | n = 358 | n = 84 | n = 21 | |
| aPTT, seconds | −.168 ns | −.015 ns | −.008 ns | −.027 ns | .286 ns |
| PT, % | .109 ns | −.096* | −.074 ns | ‐.329 *** | .029 ns |
| Thrombin generation assay | n = 46 | n = 507 | n = 358 | n = 84 | n = 21 |
| Lag time, min | −.180 ns | .097* | .106 * | .072 ns | .412 ns |
| Velocity index, nmol/L/min | .222 ns | −.038 ns | −.083 ns | .103 ns | −.133 ns |
| Peak thrombin, nmol/L | .211 ns | −.048 ns | −.093 ns | .070 ns | −.052 ns |
| TTP, min | −.169 ns | .056 ns | .081 ns | −.028 ns | .285 ns |
| AUC, nmol/L x min | .082 ns | −.079 ns | −.107 * | −.038 ns | −.009 ns |
| Plasma clot assay | n = 46 | n = 507 | n = 358 | n = 84 | n = 21 |
| Lag time, min | −.150 | .062 ns | .110 * | −.110 ns | .178 ns |
| ΔAbs, OD 405 nm | .168 ns | .002 ns | −.032 ns | .146 ns | .196 ns |
| TTP, min | −.049 | .045 ns | .070 ns | −.036 ns | .213 ns |
| Vmax, OD/min | .263 ns | −.057 ns | −.110 * | .048 ns | −.021 ns |
| CLT, min | .365* | −.040 ns | −.053 ns | .023 ns | −.390 ns |
sTM, soluble thrombomodulin; BUC, bleeding of unknown cause; PFD, platelet function defects; aPTT, activated partial thromboplastin time; PT, prothrombin time; AUC, area under the curve; CLT, clot lysis time; OD, optical density; TTP, time to peak.
Weak (r = .2‐.4), moderate (r = .4‐.6), strong (r = .6‐.8) and very strong (r = .8‐1.0) correlations (Spearman).
Significance: *p < .05; **p < .01; ***p < .001; ns: not significant.
There were no differences in global coagulation tests, parameters of thrombin generation and plasma clot formation between patients with high sTM levels above and below the 95th percentile of healthy controls (Table 5). Also, in the separate analysis of each diagnosis and BUC there was no difference in global coagulation tests, parameters of thrombin generation and plasma clot formation between patients with high sTM levels above and below the 95th percentile of healthy controls (data not shown).
TABLE 5.
Global coagulation tests, parameters of thrombin generation and plasma clot formation according to sTM values (< 95th vs. ≥ 95th percentile of the healthy controls)
| sTM < 95th percentile (< 14.7 ng/ml) | sTM ≥ 95th percentile (≥ 14.7 ng/ml) | p | BHC | |
|---|---|---|---|---|
| aPTT, seconds, median (IQR) | 35.8 (33.6‐38.7) | 38.1 (35.6‐45.4) | .056 | ns |
| PT, %, mean (SD) | 95.2 (11.4) | 89.3 (12.0) | .106 | ns |
| Thrombin generation assay 1 | ||||
| Lag time, min, mean (SD) | 11.3 (3.1) | 10.9 (2.3) | .656 | ns |
| Velocity index, nmol/l/min, median (IQR) | 29.7 (15.4‐50.1) | 25.1 (13.1‐32.3) | .194 | ns |
| Peak thrombin, nmol/L, mean (SD) | 236.1 (119.8) | 190.2 (95.7) | .282 | ns |
| TTP, min, median (IQR) | 18.6 (15.6‐22.1) | 19.6 (17.9‐21.4) | .525 | ns |
| AUC, nmol/l x min, mean (SD) | 3167.2 (754.8) | 3021.8 (1452.4) | .786 | ns |
| Plasma clot formation assay 2 | ||||
| Lag time, min, median (IQR) | 10.6 (7.7‐14.3) | 9.6 (6.5‐22.3) | .892 | ns |
| ΔAbs, OD 405 nm, mean (SD) | .72 (.18) | .63 (.16) | .114 | ns |
| TTP, min, median (IQR) | 19.5 (14.5‐23.9) | 16.1 (12.1‐34.0) | .646 | ns |
| Vmax, OD/min, mean (SD) | .13 (.10) | .11 (.09) | .186 | ns |
| CLT, min, median (IQR) | 15.6 (13.1‐18.9) | 13.9 (12.2‐21.0) | .715 | ns |
Abbreviations: IQR, interquartile range; BHC, Bonferroni‐Holm correction; TTP, time to peak; AUC, area under the curve; CLT, clot lysis time; OD, optical density
data of thrombin generation assay available of 493 (99.2%) patients sTM < 95th percentile and 8 (80.0%) patients with sTM ≥ 95th percentile.
data of plasma clot assay available of 497 (100%) patients with sTM < 95th percentile and 10 (100%) patients with sTM ≥ 95th percentile.
3.5. Genetic analysis
Sequencing and genotyping data were available in 503 of the 507 patients (99%). The known variations c.1611C > A; p.Cys537Stop and c.1487delCM; p.Pro496Argfs*10, causal for a TM‐associated coagulopathy, were not identified in our patients. The most common variant was found in rs1042579 with 18 homozygous individuals (3.5%), and 151 heterozygous patients (30%). There was no impact of this variant on sTM levels in individuals being homozygous, heterozygous or not carrying the variation rs1042579 (Supplementary figure 1). Coverage analysis did not show any evidence of copy number variations for the THBD gene region in patients with above‐average sTM levels. In addition, nine other variants were found in 23 individuals (Supplementary table 6). All of them were present as heterozygous variants, none of them were predicted by VEP to have a high impact and no significant correlation with sTM levels was found.
4. DISCUSSION
In this study, we did not identify increased levels of sTM in plasma of patients with MBD compared to healthy subjects, also when analysing patients with BUC or established diagnoses of MBD separately. Soluble TM levels were not associated with the bleeding phenotype. Global coagulation tests, and parameters of thrombin generation and plasma clot formation were not affected by increased sTM values. In bioinformatic analysis of the TM encoding gene (THBD), we did not identify variants that influenced sTM levels.
Thrombomodulin is a transmembrane protein expressed on endothelial cells of all vessels, which besides its traditional role in coagulation and fibrinolysis, possesses several other functions. 12 Depending on the structural region of TM, it interferes with the complement system and leukocyte trafficking, therefore playing a major role in inflammation. Under physiological conditions, very low levels of sTM in plasma are detectable. 21 During inflammation, immune cells are also able to induce cleavage of endothelial TM resulting in the shedding of TM into circulating blood. Enhanced levels of sTM can therefore be seen as a biomarker for endothelial damage. The exact shedding mechanism is not fully understood, and translational data suggest that proteinases belonging to the matrix metalloproteinases family to be involved in the shedding process. 22 Interestingly, Jourdy et al. did not find high shear stress to play a role in the shedding of TM. 21 Also, an increase of sTM has been reported in disease like rheumatoid arthritis, systemic lupus erythematosus and systemic sclerosis, where sTM was found to be associated with disease activity. 23 , 24 Patients in our cohort are selected according to precise exclusion criteria covering also acute inflammatory processes. 1
In affected patients with TM‐ associated coagulopathy, levels of sTM are up to 100‐fold higher than the normal reference of around 3–8 ng/ml. 9 , 11 , 25 In our cohort, no such massively increased sTM levels were found. Nevertheless, also a slight increase in sTM was identified as a risk factor for clinical relevant and major bleeding in patients on oral anticoagulants. 26 , 27 These findings emphasize the possible role of sTM as a clinically relevant marker of increased bleeding risk. In our cohort of patients with MBD and BUC, no associations of sTM values with bleeding severity and the bleeding phenotype were identified. Our recent findings and those of others suggest, that bleeding scores have a low sensitivity for diagnosing a bleeding disorder and that most mild to moderate bleeding symptoms are not predictive for diagnosing a MBD. 3 , 4 The lack of correlations of sTM with the bleeding score might also base on a multifactorial aggregation of distinct risk factors that are only partially responsible for the bleeding outcome in BUC patients, as reviewed by Mezzano et al. 7 For example, we recently identified blood group O as a risk factor for increased bleeding severity. 6 This hypothesis of a multifactorial aggregation of risk factors underlying the bleeding tendency in patients with MBD was also addressed in our analysis by including patients with a definite diagnosis of a bleeding disorder. In these patients with a known haemostatic defect, the individual bleeding phenotype often is independent of residual coagulation factor levels. 4 , 28 Amongst other factors, also natural anticoagulants have been shown to impact the individual bleeding tendency in patients with an established bleeding disorder. 29 Nevertheless, in this analysis we could not observe an impact of sTM on the bleeding severity in patients irrespective of the diagnosis of a bleeding disorder.
We found higher sTM values in men than in women in our patients, which is in agreement with earlier studies in healthy controls, 25 nevertheless, our healthy subjects did not reach a significant significance.
In line with reported results in patients with a TM‐ associated coagulopathy, we did not see a strong correlation between sTM levels and PT or aPTT. 9 , 11 Interestingly, there was a weak negative correlation between sTM levels and the PT, suggesting an impaired haemostatic potential which only occurred in patients with PFD. Due to the versatile and probably also unknown properties of sTM, we do not have an obvious explanation for this association, which could also be by chance from a statistical point of view. In detail, the anticoagulant function of sTM bases on protein C activation, which then, together with its cofactor PS, cleaves FVa and FVIIIa and thus inhibits coagulation. 12 , 30 Therefore, the PT is not sensitive for this intrinsic mechanism. Additionally, the aPTT assay is usually not affected by high sTM levels either, as in this assay thrombin is formed very rapidly so that the anticoagulation properties of sTM cannot come into effect. 9 On the other hand, a reduced thrombin generation was observed in patients with a TM‐associated coagulopathy. 9 In our cohort, we did not find an effect of sTM levels on thrombin generation or plasma clot formation in the overall cohort. This suggests that sTM must reach a very high threshold to affect thrombin generation and thus the haemostatic balance.
THBD is a small gene consisting of one exon, and most described variations in this gene were associated with thrombotic events and atypical haemolytic‐uremic syndrome. 31 , 32 , 33 The two mutations in the THBD gene underlying the TM‐ associated coagulopathy were only found in single cases and families, which suggests a rare inheritance trait. In our cohort, we could not identify these mutations.
In total, 10 variations within the THBD gene were identified in our cohort, but none of them had a significant impact on sTM levels. The most common variant in our study was the rs1042579 (GRCh38.p12 chr 20: NC_000020.11:g.23048087G > A/T), which had the same prevalence in our cohort as in the general population (gnomAD, Fischer's exact test for Hardy‐Weinberg equilibrium p = .5271). 34 This variation results in an Alanine to Valine substitution (p.Ala473Val) and is classified as benign based on the clinical significance as reported dbSNP. This variant was also reported by Langdown et al in a heterozygous form in one subject with TM‐associated coagulopathy, whereas it was not present in other family members who had a milder clinical phenotype. 9 On the other hand, homozygous genotypes of rs1042579 have been associated with coronary heart disease in some studies, 35 , 36 , 37 but data are inconclusive. 38 , 39 In our study, this variant did not impact levels of sTM.
Our study has some limitations: we were able to analyse the targeted exonic region of the THBD gene, while the surrounding non‐coding areas not covered by sufficient sequencing reads could not be analysed from our data on whole‐exome sequencing of a high‐throughput sequencing panel of predefined genes within the Thrombogenomics project. 20 In this previous study a molecular diagnostic rate of only 3.2 % was achieved in BUC patients. This urges for more comprehensive and broader genetic investigations to identify relevant disease‐causing variants in patients with MBD. Another limitation is that in our cohort and control group the majority of patients were of European ancestry, as this represents the population of our geographic region and was confirmed by genotyping.
According to our data, TM‐associated coagulopathy is rare and apparently of limited relevance in patients with a mild to moderate bleeding tendency. Nevertheless, TM‐associated coagulopathy should be suspected especially in patients with severe undefined bleeding. In a case of intra‐abdominal bleeding during surgery in a patient, who was later found to suffer from TM‐associated coagulopathy, even a broad therapeutic approach using desmopressin, tranexamic acid and fresh frozen plasma could not stop the bleeding. Bleeding was finally controlled by transfusion of platelets, which contain partially activated and intrinsically APC‐resistant FV. 8 Langdown et al reported TM‐associated coagulopathy in a family with massive bleeding following physical trauma and surgery, as well as spontaneous intraabdominal bleedings in two affected family members. 9 The patients reported by Westbury et al suffered from a lifelong history of muscle and joint bleeding after minor trauma. 11 Published data underline, that in patients with severe bleeding manifestations, including the above mentioned symptoms, or an undefined severe bleeding tendency TM‐associated coagulopathy should be considered as a differential diagnosis.
In summary, we did not find an increase of sTM in patients with MBD and there was no effect of sTM‐levels on the bleeding phenotype. Furthermore, sTM levels did not affect thrombin generation or plasma clot properties. We conclude that TM‐associated coagulopathy appears to be very rare and that the level of sTM within the range that we observed in our patients with a bleeding tendency, was neither associated with bleeding severity nor with any specific bleeding manifestation.
CONFLICT OF INTEREST
The authors have no conflict of interest regarding this manuscript.
AUTHORSHIP CONTRIBUTIONS
J. Gebhart, I. Pabinger, C. Ay and D. Mehic designed the study; J. Gebhart, I. Pabinger, C. Ay, D. Mehic and S. Hofer recruited patients; H. Haslacher processed and stored the samples; D. Mehic performed statistical analyses; K. Downes provided the genetic data; A. Tolios and M. Haimel analyzed the genetic data; I. Pabinger, J. Gebhart, D. Mehic interpreted the data; D. Mehic, J. Gebhart and A. Tolios. wrote the manuscript, which was reviewed, edited and finally approved by all authors.
Supporting information
Supporting information
ACKNOWLEDGEMENTS
This project was supported by the Anniversary Fund of the Austrian National Bank (project number 18500). The Vienna Bleeding Biobank was supported by an unrestricted grant of CSL Behring. The genetic analysis of the DNA samples of the reported cases within the ThromboGenomics project was supported by the NIHR BioResource, Cambridge, England.
Mehic D, Tolios A, Hofer S, et al. Thrombomodulin in patients with mild to moderate bleeding tendency. Haemophilia. 2021;27:1028–1036. 10.1111/hae.14433
DATA AVAILABILITY STATEMENT
Data available on request from the corresponding author.
REFERENCES
- 1. Gebhart J, Hofer S, Panzer S, et al. High proportion of patients with bleeding of unknown cause in persons with a mild‐to‐moderate bleeding tendency: results from the Vienna Bleeding Biobank (VIBB). Haemophilia. 2018;24(3):405‐413. [DOI] [PubMed] [Google Scholar]
- 2. Quiroga T, Mezzano D. Is my patient a bleeder? A diagnostic framework for mild bleeding disorders. Hematol Am Soc Hematol Educ Progr. 2012;2012:466‐474. [DOI] [PubMed] [Google Scholar]
- 3. Rodeghiero F, Pabinger I, Ragni M, et al. Fundamentals for a Systematic Approach to Mild and Moderate Inherited Bleeding Disorders: an EHA Consensus Report. HemaSphere. 2019;3(5):e286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gebhart J, Hofer S, Kaider A, et al. The discriminatory power of bleeding assessment tools in adult patients with a mild to moderate bleeding tendency. Eur J Intern Med. 2020;78:34–40. [DOI] [PubMed] [Google Scholar]
- 5. Hofer S, Ay C, Rejto J, et al. Thrombin‐generating potential, plasma clot formation, and clot lysis are impaired in patients with bleeding of unknown cause. J Thromb Haemost. 2019.17:1478–1488. 10.1111/jth.14529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mehic D, Hofer S, Jungbauer C, et al. Association of ABO blood group with bleeding severity in patients with bleeding of unknown cause. Blood Adv. 2020;4(20):5157‐5164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mezzano D, Quiroga T. Diagnostic challenges of inherited mild bleeding disorders: a bait for poorly explored clinical and basic research. J Thromb Haemost. 2019;17(2):257‐270. [DOI] [PubMed] [Google Scholar]
- 8. Dargaud Y, Scoazec JY, Wielders SJH, et al. Characterization of an autosomal dominant bleeding disorder caused by a thrombomodulin mutation. Blood. 2015;125(9):1497‐1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Langdown J, Luddington RJ, Huntington JA, et al. A hereditary bleeding disorder resulting from a premature stop codon in thrombomodulin (p.Cys537Stop). Blood. 2014;124(12):1951‐1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Burley K, Whyte CS, Westbury SK, et al. Altered fibrinolysis in autosomal dominant thrombomodulin‐associated coagulopathy. Blood. 2016;128(14):1879‐1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Westbury SK, Whyte CS, Stephens J, et al. A new pedigree with thrombomodulin‐associated coagulopathy in which delayed fibrinolysis is partially attenuated by co‐inherited TAFI deficiency. J Thromb Haemost. 2020;18:2209–2214. 10.1111/jth.14990 [DOI] [PubMed] [Google Scholar]
- 12. Loghmani H, Conway EM. Exploring traditional and nontraditional roles for thrombomodulin. Blood. 2018;132(2):148‐158. [DOI] [PubMed] [Google Scholar]
- 13. Esmon CT, Esmon NL, Harris KW. Complex formation between thrombin and thrombomodulin inhibits both thrombin‐catalyzed fibrin formation and factor V activation. J Biol Chem. 1982;257(14):7944‐7947. [PubMed] [Google Scholar]
- 14. Griffin JH, Zlokovic BV, Mosnier LO. Protein C anticoagulant and cytoprotective pathways. Int J Hematol. 2012;95(4):333‐345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wu C, Kim PY, Swystun LL, et al. Activation of protein C and thrombin activable fibrinolysis inhibitor on cultured human endothelial cells. J Thromb Haemost. 2016;14(2):366‐374. [DOI] [PubMed] [Google Scholar]
- 16. Foley JH, Kim PY, Hendriks D, et al. Evaluation of and recommendation for the nomenclature of the CPB2 gene product (also known as TAFI and proCPU): communication from the SSC of the ISTH. J Thromb Haemost. 2015;13(12):2277‐2278. [DOI] [PubMed] [Google Scholar]
- 17. Hsu Y, Shi G, Kuo C, et al. Thrombomodulin is an ezrin‐interacting protein that controls epithelial morphology and promotes collective cell migration. FASEB J. 2012;26(8):3440‐3452. [DOI] [PubMed] [Google Scholar]
- 18. Haslacher H, Gerner M, Hofer P, et al. Usage data and scientific impact of the prospectively established fluid bioresources at the hospital‐based meduni wien biobank. Biopreserv Biobank. 2018;16(6):477‐482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Core Team R. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. 2020. [Google Scholar]
- 20. Downes K, Megy K, Duarte D, et al. Diagnostic high‐throughput sequencing of 2396 patients with bleeding, thrombotic, and platelet disorders. Blood. 2019;134(23):2082‐2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jourdy Y, Enjolras N, Le Quellec S, et al. Why patients with THBD c.1611C>A (p.Cys537X) nonsense mutation have high levels of soluble thrombomodulin?. PLoS One. 2017;12(11):e0188213. 10.1371/journal.pone.0188213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Menschikowski M, Hagelgans A, Eisenhofer G, et al. Reducing agents induce thrombomodulin shedding in human endothelial cells. Thromb Res. 2010;126(2):e88‐93. [DOI] [PubMed] [Google Scholar]
- 23. Boffa MC, Karmochkine M. Thrombomodulin: an overview and potential implications in vascular disorders. Lupus. 1998;7(2):S120‐5. [DOI] [PubMed] [Google Scholar]
- 24. Conway EM. Thrombomodulin and its role in inflammation. Semin Immunopathol. 2012;34(1):107‐125. [DOI] [PubMed] [Google Scholar]
- 25. Ohlin A‐K, Larsson K, Hansson M. Soluble thrombomodulin activity and soluble thrombomodulin antigen in plasma. J Thromb Haemost. 2005;3(5):976‐982. [DOI] [PubMed] [Google Scholar]
- 26. Lind M, Boman K, Johansson L, et al. Thrombomodulin as a marker for bleeding complications during warfarin treatment. Arch Intern Med. 2009;169(13):1210‐1215. [DOI] [PubMed] [Google Scholar]
- 27. Toorop MMA, van Rein N, Cannegieter SC, et al. High soluble thrombomodulin is associated with an increased risk of major bleeding during treatment with oral anticoagulants: a case‐cohort study. Thromb Haemost. 2021;121(1):70‐75. [DOI] [PubMed] [Google Scholar]
- 28. Moenen F, Nelemans PJ, Schols SEM, et al. The diagnostic accuracy of bleeding assessment tools for the identification of patients with mild bleeding disorders: a systematic review. Haemophilia. 2018;24(4):525‐535. [DOI] [PubMed] [Google Scholar]
- 29. Mehic D, Colling M, Pabinger I, et al. Natural anticoagulants: a missing link in mild to moderate bleeding tendencies. Haemophilia. 2021. 27:701–709. 10.1111/hae.14356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Owen WG, Esmon CT. Functional properties of an endothelial cell cofactor for thrombin catalyzed activation of protein C. J Biol Chem. 1981;256(11):5532‐5535. [PubMed] [Google Scholar]
- 31. Kunz G, Ireland HA, Stubbs PJ, et al. Identification and characterization of a thrombomodulin gene mutation coding for an elongated protein with reduced expression in a kindred with myocardial infarction. Blood. 2000;95(2):569‐576. [PubMed] [Google Scholar]
- 32. Kunz G, Öhlin A‐K, Adami A, et al. Naturally occurring mutations in the thrombomodulin gene leading to impaired expression and function. Blood. 2002;99(10):3646‐3653. [DOI] [PubMed] [Google Scholar]
- 33. Delvaeye M, Noris M, De Vriese A, et al. Thrombomodulin mutations in atypical hemolytic–uremic syndrome. N Engl J Med. 2009;361(4):345‐357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434‐443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cole JW, Roberts SC, Gallagher M, et al. Thrombomodulin Ala455Val polymorphism and the risk of cerebral infarction in a biracial population: the stroke prevention in young women study. BMC Neurol. 2004;4(1):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sugiyama S, Hirota H, Kimura R, et al. Haplotype of thrombomodulin gene associated with plasma thrombomodulin level and deep vein thrombosis in the Japanese population. Thromb Res. 2007;119(1):35‐43. [DOI] [PubMed] [Google Scholar]
- 37. Wu KK, Aleksic N, Ahn C, et al. Thrombomodulin Ala455Val polymorphism and risk of coronary heart disease. Circulation. 2001;103(10):1386‐1389. [DOI] [PubMed] [Google Scholar]
- 38. Faioni EM, Franchi F, Castaman G, et al. Mutations in the thrombomodulin gene are rare in patients with severe thrombophilia. Br J Haematol. 2002;118(2):595‐599. [DOI] [PubMed] [Google Scholar]
- 39. Aleksic N, Folsom AR, Cushman M, Heckbert SR, Tsai MY Wu KK. Prospective study of the A455V polymorphism in the thrombomodulin gene, plasma thrombomodulin, and incidence of venous thromboembolism: the LITE Study. J Thromb Haemost. 2003;1(1):88‐94. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Data Availability Statement
Data available on request from the corresponding author.