

Abstract
Multiple osteochondromas (MO) is a rare disorder, characterized by benign osteocartilaginous tumors (osteochondromas), arising from the perichondrium of bones. The osteochondromas increase during growth, frequently causing deformities and limitations. Our study aims to analyze the data captured by the Registry of Multiple Osteochondromas, to refine Istituto Ortopedico Rizzoli (IOR) Classification, providing a representative picture of the phenotypic manifestations throughout the lifespan. We conducted a single‐institution cross‐sectional study. Patients were categorized according to IOR Classification, which identifies three patients' classes on the presence/absence of deformities and/or limitations. The present dataset was compared with our previously published data, to refine the classification. Nine hundred sixty‐eight patients were included: 243 children (<10 years), 136 adolescents (10–15 years), and 589 adults. Of the entire population, half patients presented at least one deformity, and one quarter reported at least one limitation. Compared with our previous study, the amount of children was more than doubled and the percentage of mild/moderate cases was notably increased, giving a better disease overview throughout the lifespan and suggesting a different cut‐off for dividing Class II in subclasses. We confirmed that MO is characterized by phenotypic heterogeneity, suggesting that an early classification of the disease may offer a useful tool to follow disease pattern and evolution, to support clinical practice, and to propose timely interventions.
Keywords: classification, functional limitation, multiple osteochondromas, rare disease, skeletal deformity, skeletal dysplasia
1. INTRODUCTION
Multiple osteochondromas (MO, MIM #133700, #133701) is a rare genetic condition with autosomal‐dominant transmission that affects the skeleton (Bovée, 2008). Pathogenic variants on EXT1 and EXT2 genes (OMIM #608177, #608210) lead to MO in 70%–94% of the patients (Ahn et al., 1995; Jennes et al., 2009; Jurik, 2020; Pedrini et al., 2011; Piombo et al., 2018; Stickens et al., 1996; Wuyts et al., 1993); for the remaining 6%–30% of MO patients, the disease cause is still unclear.
The diagnosis is primarily based on clinical and radiological findings. The signature feature of the disease is the presence of benign osteocartilaginous tumors, named osteochondromas or exostoses (OCs), arising from the perichondrium of bones, especially of long bones. The lesions increase in number and dimension during growth, gradually ossifying until skeletal maturity, after which no new OC develops in most cases (Wuyts et al., 1993). These slow‐growing masses depending on the localization can lead to bony deformities, functional limitations, and joint malalignment. Frequent additional clinical manifestations are short stature, impingement of tendons, vascular or neurological compromise, and pain (Bovée, 2008; Wuyts et al., 1993). The most feared complication of MO is the progression of an existing OC to secondary peripheral chondrosarcoma (SPC), malignant cartilage‐producing tumors with an incidence ranging from 0.5% to 20% (Hameetman et al., 2004; Jurik, 2020; Wuyts et al., 1993).
The MO phenotype is heterogeneous—in terms of number of OCs, clinical appearance, and functional limitation—with a notable interindividual and interfamilial variability, ranging from almost inappreciable signs to severe impairment (Stieber & Dormans, 2005). To date, mild MO is treated by palliative management of symptoms, while surgery remains the treatment of choice to prevent or ameliorate deformities, pain, and other MO‐related clinical manifestations (Bovée, 2008; Darilek et al., 2005). A clear picture of factors affecting the disease severity and progression in MO is still lacking. In this context, the identification of classification systems for defining homogeneous cohorts of patients is essential, especially for comparing the outcomes of specific interventions in different types of patients among several centers. In addition, this classification should be easily assessable, minimally invasive, and clinically relevant, to be captured in routine care of MO patients.
To date, many classification systems for staging MO patients have been proposed. Nevertheless, most of them are incomplete, focusing only on a particular aspect of the disease, or require ad hoc investigations. For instance, some authors limited their classification only to the deformities of a limb segment (forearm, ankle) (Jo et al., 2017; Masada et al., 1989; Taniguchi, 1995), while other authors based their classification on extensive radiographic investigations (Alvarez et al., 2006; Fan et al., 2014; Shapiro et al., 1979). Other classifications were tested only in relatively small series and not formally validated (Francannet et al., 2001; Jäger et al., 2007; Porter et al., 2004; Schmale et al., 1994).
In fact, none of these classifications is widely used to describe the MO phenotypes in a clinical setting (Francannet et al., 2001; Jäger et al., 2007; Porter et al., 2004; Schmale et al., 1994); this limits the possibility to understand and monitor the behavior of the disease in different cohorts of patients, thus complicating the management of this condition. In 2013, we proposed the “IOR Classification,” a novel clinical evaluation system for assessing MO patients based on their phenotypic presentation, categorizing patients in three major classes, each divided into two subclasses (Mordenti et al., 2013). The IOR classes were validated formally through an innovative machine learning model on a cohort of 289 MO patients, while subclasses were never tested due to the limited number of patients. This Classification, requiring few clinical information, easily obtained during a routine physical examination, is helpful in monitoring the disease progression. In 2013, was also established and implemented as a routine activity of our Institution, IRCCS Istituto Ortopedico Rizzoli, the Registry of Multiple Osteochondromas (REM).
The present study aims to review and analyze the data collected with the support of the REM during a period of 18 years, in order to investigate the IOR classes and subclasses and to provide a more representative picture of the phenotypic manifestations throughout the lifespan, improving the understanding and management of this rare condition.
2. METHODS
2.1. Participants and setting
This is a cross‐sectional study, investigating a cohort of patients with MO, admitted at a single‐institution, from 2003 to 2020. Our hospital is a national referral center for rare skeletal conditions and the coordinator of the European Reference Network on Rare Bone Disorders since 2017.
Data captured from hospital electronic health records, patients' narrative reports, and medical documentation were stored into a GDPR‐compliant IT platform, on which the REM (ClinicalTrial.gov: NCT04133285) relies; this platform is arranged to manage and update retrospective and prospective data.
Inclusion criteria were males and females with a confirmed clinical and/or radiographic diagnosis of MO (Bovée, 2008) and a detailed visit from an orthopedic surgeon. The visit should include at least the following parameters: age, sex, height, weight, number of skeletal sites and bones with OC(s), assessment of upper and lower limbs alignment, assessment of range of motion at the big joints of the upper and lower limbs. When available, inheritance details and genetic background were included. All inclusion criteria had to be satisfied for enrolment in the present study. Patients with missing data that precluded to define the disease class were excluded. For each patient, data were collected at a single time point.
The present study was approved by an independent review committee in September 2020 (12854/2020).
2.2. Data collection and outcome assessment
Clinical data were collected according to a standardized protocol that included patient demographics, inheritance, height, weight, body mass index (BMI), number of OCs, presence and number of skeletal deformities and functional limitations. The number of sites with OCs, the presence of skeletal deformities, and joint limitations were established by both physical examination and imaging data (radiographs, magnetic resonance imaging, computed tomography, and ultrasound), when available. Patients were divided in four subgroups according to the number of sites involved by OCs: <5, 6–10, 11–20, >20. Skeletal deformities, considered as alterations of the normal bone alignment, and functional limitations, considered as a restriction of range of motion, are listed in Table S1.
Based on these data, patients were classified according to the IOR Classification. This system (Table 1) identifies three classes of MO patients characterized by the presence/absence of deformities and/or functional limitations. In addition, each clinical class is subdivided into two subclasses according to the number of affected sites (A and B).
TABLE 1.
IOR Classification: disease severity
| Criteria | Class | Subclass | |
|---|---|---|---|
| No deformities—no functional limitations | I | IA | ≤5 sites with OC |
| IB | >5 sites with OC | ||
| Deformities—no functional limitations | II | IIA | ≤5 sites with deformities |
| IIB | >5 sites with deformities | ||
| Deformities—functional limitations | III | IIIA | 1 site with functional limitation |
| IIIB | >1 site with functional limitation | ||
Abbreviations: IOR, Istituto Ortopedico Rizzoli; OC, osteochondroma.
In case of availability of biologic material (blood, saliva, DNA), genetic analyses were performed to identify pathogenic variant on EXT1 or EXT2 gene.
2.3. Statistical analyses
Patients' characteristics were summarized stratifying by age in three groups (<10, 10–15, ≥16 years) in order to compare the results from the present study with our previously published data. Results were expressed as medians (along with interquartile range, IQR) and percentages for continuous and categorical variables, respectively. Anthropometric measures (height, weight, BMI) were reported both as absolute values and as z‐scores, calculated using Italian reference charts (Cacciari et al., 2006).
Prevalence of demographic and clinical characteristics was compared between groups with χ 2 test or Wilcoxon‐type trend test, where appropriate. The significance level was adjusted with Bonferroni correction for multiplicity when post hoc χ 2 tests were performed to compare the distribution of each category.
All analyses were performed with Stata 11.2 (StataCorp, College Station, TX).
3. RESULTS
Overall, 1065 MO patients were screened for eligibility. Nine hundred sixty‐eight patients (representing 91% of the entire cohort), who fulfilled the inclusion criteria, were analyzed (Figure 1).
FIGURE 1.
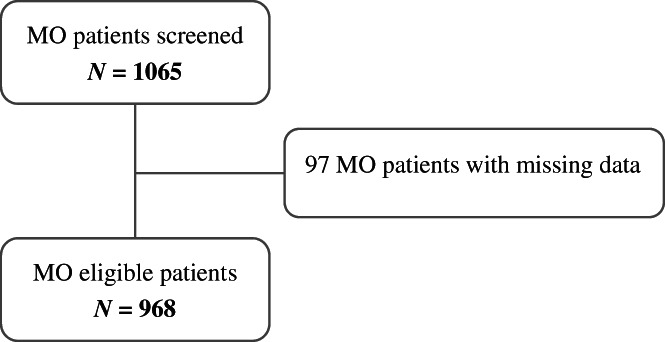
Flow chart of patients included in the study
Patients' demographics and disease characteristics are summarized in Table 2.
TABLE 2.
Patients' demographics and disease characteristics at evaluation
| Population group | ||||||||
|---|---|---|---|---|---|---|---|---|
| <10 years (N = 243) | 10–15 years (N = 136) | ≥16 years (N = 589) | Overall (N = 968) | |||||
| Male (N = 147) | Female (N = 96) | Male (N = 80) | Female (N = 56) | Male (N = 298) | Female (N = 291) | |||
| Age, years | Mean (SD) | 5.68 (2.16) | 5.38 (2.11) | 12.82 (1.65) | 12.50 (1.51) | 38.46 (13.69) | 36.63 (11.86) | 26.00 (17.69) |
| Median (IQR) | 5.38 (3.92, 7.36) | 4.94 (3.76, 6.90) | 12.90 (11.81, 13.89) | 12.12 (11.57, 13.55) | 38.50 (27.00, 47.00) | 36.00 (27.00, 45.00) | 24.00 (9.92, 40.00) | |
| Height, cm | Mean (SD) | 112.10 (13.34) | 109.38 (14.26) | 151.90 (14.49) | 150.80 (9.35) | 168.49 (8.51) | 156.78 (7.31) | 145.86 (25.48) |
| Median (IQR) | 111.00 (102.00, 122.00) | 107.75 (99.00, 119.00) | 153.50 (141.00, 162.25) | 152.25 (144.75, 157.00) | 168.00 (163.00, 174.00) | 157.00 (152.00, 161.00) | 155.00 (126.00, 165.00) | |
| Height z‐score | Mean (SD) | −0.39 (0.96) | −0.39 (1.00) | −0.58 (1.46) | −0.30 (1.01) | −1.27 (1.37) | −0.99 (1.25) | −0.81 (1.27) |
| Median (IQR) | −0.34 (−0.87, 0.16) | −0.49 (−0.95, 0.39) | −0.36 (−1.51, 0.46) | −0.21 (−0.97, 0.19) | −1.33 (−2.18, −0.40) | −0.96 (−1.81, −0.27) | −0.78 (−1.59, –0.07) | |
| Weight, kg | Mean (SD) | 20.68 (6.71) | 19.52 (7.21) | 46.35 (14.33) | 44.74 (10.11) | 72.73 (14.20) | 55.59 (8.96) | 47.49 (23.20) |
| Median (IQR) | 19.00 (16.00, 24.00) | 18.00 (14.60, 22.00) | 45.60 (36.00, 54.50) | 46.00 (38.00, 52.00) | 70.00 (63.00, 80.00) | 54.00 (49.00, 61.00) | 50.00 (25.00, 64.00) | |
| Weight z‐score | Mean (SD) | −0.48 (1.04) | −0.56 (1.04) | −0.49 (1.20) | −0.35 (0.92) | 0.12 (1.26) | −0.17 (1.05) | −0.24 (1.14) |
| Median (IQR) | −0.39 (−1.25, 0.18) | −0.54 (−1.18, 0.24) | −0.46 (−1.32, 0.29) | −0.40 (−1.06, 0.24) | 0.10 (−0.69, 0.96) | −0.24 (−0.94, 0.58) | −0.24 (−0.99, –0.52) | |
| BMI | Mean (SD) | 16.11 (2.25) | 15.86 (2.21) | 19.44 (3.66) | 19.53 (3.17) | 25.70 (4.50) | 22.82 (4.09) | 20.90 (5.30) |
| Median (IQR) | 15.86 (14.65, 16.96) | 15.51 (14.40, 16.62) | 18.69 (16.88, 21.23) | 19.35 (16.94, 21.56) | 25.02 (22.84, 28.08) | 21.99 (20.17, 24.56) | 20.31 (16.62, 24.22) | |
| BMI z‐score | Mean (SD) | −0.36 (1.20) | −0.36 (1.04) | −0.36 (1.02) | −0.27 (1.01) | 0.85 (1.12) | 0.34 (1.10) | 0.15 (1.22) |
| Median (IQR) | −0.24 (−1.10, 0.46) | −0.24 (−1.03, 0.36) | −0.50 (−1.04, 0.31) | −0.29 (−1.04, 0.46) | 0.91 (0.23, 1.62) | 0.32 (−0.37, 1.07) | 0.22 (−0.69, –0.96) | |
| Inheritance, n (%) | De novo | 65 (44.2) | 43 (44.8) | 27 (34) | 20 (36) | 83 (27.9) | 80 (27.5) | 318 (32.9) |
| Maternal | 31 (21.1) | 23 (24) | 26 (33) | 22 (39) | 89 (29.9) | 79 (27.1) | 270 (27.9) | |
| Paternal | 48 (32.7) | 25 (26) | 23 (29) | 12 (21) | 88 (29.5) | 98 (33.7) | 294 (30.4) | |
| Not available | 3 (2) | 5 (5.2) | 4 (5) | 2 (4) | 38 (12.8) | 34 (11.7) | 86 (8.9) | |
| Malignant Degeneration, n (%) | Presence of SPC | 0 (0) | 1 (1) | 0 (0) | 0 (0) | 38 (12.8) | 17 (5.8) | 56 (5.8) |
| Absence of SPC | 147 (100) | 95 (99) | 80 (100) | 56 (100) | 260 (87.2) | 274 (94.2) | 912 (94.2) | |
Abbreviations: BMI, body mass index; IQR, interquartile range, 25°–75°; SD, standard deviation; SPC, secondary peripheral chondrosarcoma.
The dataset was composed of 243 children <10 years (25%), 136 adolescents aged between 10 and 15 years (14.1%), and 589 adults (60.9%). No significant difference was observed in gender distribution, even if among children the percentage of males (60%) exceeded the percentage of females (40%). Demographics information is reported in Table 2.
The median age at evaluation was 5 years in the youngest group, 12 years in the intermediate group, and 37 years in adults. Average height and weight were lower across all groups (especially among adults) compared with the general Italian population.
Inheritance data were available for 882 patients (91.1%), with 58% of patients reporting parental transmission, almost equally distributed between maternal and paternal transmission (47.9% and 52.1%, respectively). Genetic assessment was performed in 931 cases, 839 of them (90.1%) had an EXT1 or EXT2 variant, while the remaining 9.9% was negative (Table 3). In line with previous studies (Pedrini et al., 2011; Wuyts et al., 1993), pathogenic variants of EXT1 are associated with a more severe phenotype, in comparison with EXT2 and patients with no pathogenic variant are frequently characterized by milder manifestations (Table 3).
TABLE 3.
Distribution of genetic variants and secondary peripheral chondrosarcoma among IOR classes and overall
| Genetic background | |||||||
|---|---|---|---|---|---|---|---|
| Class I (N = 433) | Class II (N = 274) | Class III (N = 224) | Overall (N = 931) | ||||
| Male (N = 222) | Female (N = 211) | Male (N = 140) | Female (N = 134) | Male (N = 142) | Female (N = 82) | ||
| EXT1, n (%) | 85 (19.6) | 117 (27.0) | 86 (31.4) | 98 (35.7) | 114 (50.9) | 61 (27.2) | 561 (60.2) |
| EXT2, n (%) | 100 (23.1) | 68 (15.7) | 43 (15.7) | 27 (9.9) | 22 (9.8) | 18 (8.0) | 278 (29.9) |
| Negative, n (%) | 37 (8.6) | 26 (6.0) | 11 (4.0) | 9 (3.3) | 6 (2.7) | 3 (1.4) | 92 (9.9) |
| Malignant degeneration | |||||||
|---|---|---|---|---|---|---|---|
| Class I (N = 452) | Class II (N = 287) | Class III (N = 229) | Overall (N = 968) | ||||
| Male (N = 232) | Female (N = 220) | Male (N = 147) | Female (N = 140) | Male (N = 146) | Female (N = 83) | ||
| Presence of SPC, n (%) | 14 (3.1) | 6 (1.3) | 7 (2.4) | 4 (1.4) | 17 (7.4) | 8 (3.5) | 56 (5.8) |
| Absence of SPC, n (%) | 218 (48.2) | 214 (47.4) | 140 (48.8) | 136 (47.4) | 129 (56.3) | 75 (32.8) | 912 (94.2) |
Note: Percentages are calculated per each class and overall.
Abbreviations: EXT, exostosin genes; IOR, Istituto Ortopedico Rizzoli; SPC, secondary peripheral chondrosarcoma.
As previously highlighted by many studies (Hameetman et al., 2004; Jurik, 2020; Pedrini et al., 2011; Wuyts et al., 1993), our results showed that the incidence of malignant transformation of an OC into a SPC is a rare event, affecting 56 patients (5.8%) of the analyzed population, ranging from around 4% in Classes I and II to 10.9% in Class III (p = 0.002). In addition, this feared complication has a higher frequency in males compared with females (12.8% vs. 5.8%) across all classes.
Patients' clinical features and classification of disease, stratified by age group, are reported in Table 4. The class of disease increased significantly with age (Wilcoxon‐type trend test, p < 0.001). Most patients were in Class I (46.7%), almost uniformly across all age groups (61.3%, 45.6%, and 40.9%, respectively). Class II was mainly represented in the two youngest groups (28.8% and 40.4% for <10 years and 10–15 years, respectively), while Class III was most frequent among adults (31.6% vs. 11.3%; p < 0.0001).
TABLE 4.
Patients' clinical features by age groups at evaluation
| Age groups, year | |||||
|---|---|---|---|---|---|
| <10 | 10–15 | ≥16 | Overall | ||
| (N = 243) | (N = 136) | (N = 589) | (N = 968) | ||
| Disease stage, n (%) | Class I | 149 (61.3) | 62 (45.6) | 241 (40.9) | 452 (46.7) |
| Class II | 70 (28.8) | 55 (40.4) | 162 (27.5) | 287 (29.6) | |
| Class III | 24 (9.9) | 19 (14.0) | 186 (31.6) | 229 (23.7) | |
| Number of affected skeletal sites, n (%) | 1–5 | 95 (39.1) | 47 (34.6) | 127 (21.6) | 269 (27.8) |
| 6–10 | 92 (37.9) | 34 (25.0) | 139 (23.6) | 265 (27.4) | |
| 11–20 | 56 (23.0) | 55 (40.4) | 196 (33.3) | 307 (31.7) | |
| >20 | 0 (0) | 0 (0) | 85 (14.4) | 85 (8.8) | |
| Missing data | 0 (0) | 0 (0) | 42 (7.1) | 42 (4.3) | |
| Number of bones affected by OCs | Mean (SD) | 7 (4.06) | 8 (4.37) | 10 (5.49) | 9 (5.15) |
| Median (IQR) | 6 (4, 9) | 7 (4, 11) | 9 (5, 13) | 8 (5, 12) | |
| Number of deformities, n (%) | Without deformities | 155 (63.8) | 69 (50.7) | 277 (47.0) | 501 (51.8) |
| 1 | 29 (11.9) | 21 (15.4) | 84 (14.3) | 134 (13.8) | |
| 2 | 38 (15.6) | 17 (12.5) | 88 (14.9) | 143 (14.8) | |
| 3 | 16 (6.6) | 5 (3.7) | 44 (7.5) | 65 (6.7) | |
| 4 | 2 (0.8) | 12 (8.8) | 44 (7.5) | 58 (6.0) | |
| 5 | 1 (0.4) | 6 (4.4) | 26 (4.4) | 33 (3.4) | |
| ≥6 | 2 (0.8) | 6 (4.4) | 18 (3.1) | 26 (2.7) | |
| Missing data | 0 (0) | 0 (0) | 8 (1.4) | 8 (0.8) | |
| Deformity localizations, n (%) | Without deformity localizations | 155 (63.8) | 69 (50.7) | 277 (47.0) | 501 (51.8) |
| Lower limbs | 36 (14.8) | 29 (21.3) | 110 (18.7) | 175 (18.1) | |
| Upper limbs | 32 (13.2) | 13 (9.6) | 75 (12.7) | 120 (12.4) | |
| Both lower and upper limbs | 20 (8.2) | 25 (18.4) | 115 (19.5) | 160 (16.5) | |
| Missing data | 0 (0) | 0 (0) | 12 (2.0) | 12 (1.2) | |
| Number of functional limitations, n (%) | Without functional limitations | 219 (90.1) | 117 (86) | 398 (67.6) | 734 (75.8) |
| 1 | 18 (7.4) | 10 (7.4) | 76 (12.9) | 104 (10.7) | |
| 2 | 5 (2.1) | 7 (5.1) | 60 (10.2) | 72 (7.4) | |
| ≥3 | 1 (0.4) | 2 (1.5) | 47 (8.0) | 50 (5.2) | |
| Missing data | 0 (0) | 0 (0) | 8 (1.4) | 8 (0.8) | |
| Functional limitation localizations, n (%) | Without functional limitation localizations | 219 (90.1) | 117 (86.0) | 398 (67.6) | 734 (75.8) |
| Lower limbs | 4 (1.6) | 4 (2.9) | 89 (15.1) | 97 (10.0) | |
| Upper limbs | 19 (7.8) | 10 (7.4) | 57 (9.7) | 86 (8.9) | |
| Both lower and upper limbs | 1 (0.4) | 5 (3.7) | 37 (6.3) | 43 (4.4) | |
| Missing data | 0 (0) | 0 (0) | 8 (1.4) | 8 (0.8) | |
Abbreviations: IQR, interquartile range, 25°–75°; OC, osteochondroma(s).
On average, nine bony sites per patient were involved. Notably, only adults exhibited OCs in more than 20 skeletal sites (8.8%).
Of the entire population, 459 patients (47.4%) presented at least one skeletal deformity, with almost all of them having less than 5 (433 out of 459; 94.3%). In addition, the skeletal deformities were almost equally distributed between lower and upper limbs (18.1% and 12.4%, respectively).
Seven hundred thirty‐four patients (75.8%) did not present any functional limitation. Of the 226 patients with limitations, 122 (54%) presented at least two joints involved.
Patients with joint limitations were mostly adults (183 out of 226; 80.9%). The limitations, as well as the deformities, were almost equally distributed between lower and upper limbs (42.9% and 38.1%, respectively).
The present dataset was compared with the data from our previous study (Mordenti et al., 2013), both in terms of patients' distribution across age groups (Figure 2a) and across IOR classes (Figure 2b).
FIGURE 2.
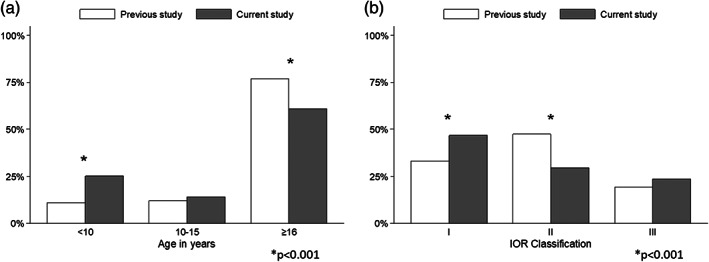
Comparison between current dataset and our previously published data. (a) Patients distribution by age. The bar chart shows the comparison of patients' distribution across age groups. (b) Patients distribution by Istituto Ortopedico Rizzoli (IOR) Classification. The bar chart represents the patients' distribution across the IOR classes
The mean age of the entire cohort was significantly lower in the current dataset, because of a substantial increase in number of children included (Figure 2a). In detail, the distribution of patients across age groups was 25%, 14%, 61% (<10, 10–15, ≥16 years, respectively), and the percentage of children was more than doubled compared with the previous study (from 11% to 25%; post‐hoc χ 2, p < 0.001). Concerning the patients' distribution among the IOR classes, we observed a significant increase in Classes I and III and a reduction of Class II (post‐hoc χ 2, p < 0.001; Figure 2b).
The analysis of distribution between subclasses revealed that, while Subclasses A and B were well balanced both in Classes I and III (60%/40% and 45%/55%, respectively), Class II showed an uneven distribution between Subclasses A and B (94% vs. 6%). Therefore, in order to balance the distribution of patients across the IOR Classification, we applied a different cut‐off for Subclass II, moving the value from 5 to 2. The updated distribution between IIA and IIB subclasses changed as follows: 59% in IIA and 41% in IIB for adults, 60% in IIA and 40% in IIB for the intermediate age group, and 79% in IIA and 21% in IIB for the youngest population. Table 5 summarizes the grouping of MO patients among the classes and subclasses with the original cut‐off and the proposed revision (χ 2 , p < 0.001). Revised IOR Classification is shown in Table 6 and Figure 3.
TABLE 5.
Comparison between IOR Classification and revised IOR Classification patient distributions
| IOR Class | N (%) | IOR Subclassification | N (%) | Revised IOR Subclassification | N (%) | |
|---|---|---|---|---|---|---|
| <10 years | I | 149 (61.3) | IA (≤5 sites with OC) | 71 (29.2) | IA (≤5 sites with OC) | 71 (29.2) |
| IB (>5 sites with OC) | 78 (32.1) | IB (>5 sites with OC) | 78 (32.1) | |||
| II | 70 (28.8) | IIA (≤5 sites with deformities) | 69 (28.4) | IIA (≤2 sites with deformities) | 55 (22.6) | |
| IIB (>5 sites with deformities) | 1 (0.4) | IIB (>2 sites with deformities) | 15 (6.2) | |||
| III | 24 (9.9) | IIIA (1 site with functional limitation) | 18 (7.4) | IIIA (1 site with functional limitation) | 18 (7.4) | |
| IIIB (>1 site with functional limitation) | 6 (2.5) | IIIB (>1 site with functional limitation) | 6 (2.5) | |||
| 10–15 years | I | 62 (45.6) | IA (≤5 sites with OC) | 31 (22.8) | IA (≤5 sites with OC) | 31 (22.8) |
| IB (>5 sites with OC) | 31 (22.8) | IB (>5 sites with OC) | 31 (22.8) | |||
| II | 55 (40.4) | IIA (≤5 sites with deformities) | 50 (36.8) | IIA (≤2 sites with deformities) | 33 (24.3) | |
| IIB (>5 sites with deformities) | 5 (3.7) | IIB (>2 sites with deformities) | 22 (16.2) | |||
| III | 19 (14.0) | IIIA (1 site with functional limitation) | 10 (7.4) | IIIA (1 site with functional limitation) | 10 (7.4) | |
| IIIB (>1 site with functional limitation) | 9 (6.6) | IIIB (>1 site with functional limitation) | 9 (6.6) | |||
| ≥16 years | I | 241 (40.9) | IA (≤5 sites with OC) | 77 (13.1) | IA (≤5 sites with OC) | 77 (13.1) |
| IB (>5 sites with OC) | 164 (27.8) | IB (>5 sites with OC) | 164 (27.8) | |||
| II | 162 (27.5) | IIA (≤5 sites with deformities) | 152 (25.8) | IIA (≤2 sites with deformities) | 95 (16.1) | |
| IIB (>5 sites with deformities) | 10 (1.7) | IIB (>2 sites with deformities) | 67 (11.4) | |||
| III | 186 (31.6) | IIIA (1 site with functional limitation) | 76 (12.9) | IIIA (1 site with functional limitation) | 76 (12.9) | |
| IIIB (>1 site with functional limitation) | 110 (18.7) | IIIB (>1 site with functional limitation) | 110 (18.7) | |||
| OVERALL | I | 452 (46.7) | IA (≤5 sites with OC) | 179 (18.5) | IA (≤5 sites with OC) | 179 (18.5) |
| IB (>5 sites with OC) | 273 (28.2) | IB (>5 sites with OC) | 273 (28.2) | |||
| II | 287 (29.6) | IIA (≤5 sites with deformities) | 271 (28.0) | IIA (≤2 sites with deformities) | 183 (18.9) | |
| IIB (>5 sites with deformities) | 16 (1.7) | IIB (>2 sites with deformities) | 104 (10.7) | |||
| III | 229 (23.7) | IIIA (1 site with functional limitation) | 104 (10.7) | IIIA (1 site with functional limitation) | 104 (10.7) | |
| IIIB (>1 site with functional limitation) | 125 (12.9) | IIIB (>1 site with functional limitation) | 125 (12.9) |
Abbreviations: IOR, Istituto Ortopedico Rizzoli; OC, osteochondroma(s).
TABLE 6.
Revised IOR Classification: disease severity
| Criteria | Class | Subclass | |
|---|---|---|---|
| No deformities—no functional limitations | I | IA | ≤5 sites with OC |
| IB | >5 sites with OC | ||
| Deformities—no functional limitations | II | IIA | ≤2 sites with deformities |
| IIB | >2 sites with deformities | ||
| Deformities—functional limitations | III | IIIA | 1 site with functional limitation |
| IIIB | >1 site with functional limitation | ||
Abbreviations: IOR, Istituto Ortopedico Rizzoli; OC, osteochondroma(s).
FIGURE 3.
Disease worsening. A graphical visualization of three classes according to multiple osteochondromas (MO) severity
4. DISCUSSION
We described the largest European cohort of MO patients, adding further evidence on how this disorder affects individuals throughout their lifetime. Consistently with the available literature, we confirmed a slightly higher prevalence of MO in males and a shorter stature of MO patients in comparison with the Italian population (Beltrami et al., 2016; Bovée, 2008; Mordenti et al., 2020; Pedrini et al., 2011; Schmale et al., 1994). Similarly, familial inheritance was present in more than half of patients. In addition, the vast majority of patients had an EXT pathogenic variant, and the distribution of pathogenic variants is remarkably higher in EXT1 gene (Bovée, 2008; Francannet et al., 2001; Jennes et al., 2009; Pedrini et al., 2011), showing a most severe phenotype. Nonetheless, in our population, the rate of malignant degeneration of an OC is in line with literature data (Hameetman et al., 2004; Jurik, 2020; Pedrini et al., 2011; Wuyts et al., 1993). The incidence of this event seems to increase in Class III; however, this evidence requires a larger cohort to allow for a more adequate control of confounding factors.
Interestingly, we found that more than half of patients would not develop skeletal deformities, and only less than one‐fourth would experience one or more functional limitations, during adulthood, thus confirming the overall benign natural course of the disease.
The distribution among classes changed due to a significant increase of mild cases. This was related to the proportion of children aged less than 10 years, that in the present study was more than doubled in comparison with the previous Classification study (Mordenti et al., 2013). In addition, the presence of a larger series allowed us to refine the IOR Classification, evaluating also subclass distribution. In particular, the Subclasses A and B of Class II resulted as not representative of moderate MO manifestations. Our data showed that the presence of >5 deformities is a rare event, resulting in an inappropriate cut‐off for clinical severity description. Accordingly, we modified the criteria for moving from Subclasses IIA to IIB (>2 deformities, instead of >5 deformities). This redefinition leads to a more balanced patient distribution and a more useful tool for patients' classification. The centralization of patients with rare bone disorders in a specialized institution, the establishment of the REM and the related dissemination activities may allow early screening, anticipated management, and timely interventions in children. All these aspects may ultimately influence the disease progression and the potential long‐term disability, reducing early skeletal deterioration and improving daily activities (Speerin et al., 2014).
We acknowledge that our study has few limitations. The classification was assessed and validated in a previous study (Mordenti et al., 2013) and was mainly based on the surgeon's physical examination. Nonetheless, some concerns remain due to the absence of a radiographic assessment of the whole body that could result in an underestimation of the OCs number. Moreover, the lack of detailed surgical history in some patients could partially lead to misclassification. In addition, despite the large number of MO patients enrolled, the IOR Classification may need multicentric studies as our cohort was collected at a single‐institution. Further research incorporating additional clinical data and surgical information, is needed to assess the prognostic value of the revised Classification.
5. CONCLUSION
We described the largest European cohort of MO patients, stratified according to a classification system based on clinical examination. We confirmed that MO presents with heterogeneous phenotypes and severity, often changing during patients' skeletal growth. The refined IOR Classification is capable to describe and monitor the disease manifestations, thus supporting natural history study. Moreover, this system may represent a useful tool to follow disease pattern and evolution, to predict the outcomes, and to propose appropriate and timely interventions.
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
AUTHOR CONTRIBUTIONS
Marina Mordenti: conceptualization (lead), methodology (equal), project administration (lead), supervision (lead), validation (lead), formal analysis (equal), investigation (equal), data curation (equal), visualization (lead), writing—original draft (lead), writing—review (lead) and editing (equal). Maria Gnoli: conceptualization (equal), investigation (equal), data curation (equal), methodology (equal), validation (equal), formal analysis (equal), visualization (lead), writing—original draft (lead), writing—review (lead) and editing (lead). Manila Boarini: conceptualization (equal), methodology (equal), validation (equal), formal analysis (equal), data curation (equal), visualization (equal), writing—original draft (equal), writing—review and editing (equal). Giovanni Trisolino: conceptualization (equal), methodology (equal), validation (equal), formal analysis (equal), data curation (equal), visualization (equal), writing—original draft (equal), writing—review and editing (equal). Andrea Evangelista: conceptualization (equal), methodology (lead), formal analysis (lead), data curation (equal), visualization (equal), writing—original draft (equal), writing—review (equal) and editing (equal). Elena Pedrini: validation (equal), investigation (equal), data curation (equal), writing—original draft (equal), writing—review and editing (equal). Serena Corsini: validation (equal), investigation (equal), data curation (equal), writing—original draft (equal), writing—review and editing (equal). Morena Tremosini: validation (equal), investigation (equal), data curation (equal), writing—original draft (equal), writing—review and editing (equal). Eric Staals: validation (equal), investigation (equal), data curation (equal), writing—original draft (equal), writing—review and editing (equal). Diego Antonioli: validation (equal), investigation (equal), data curation (equal), writing—original draft (equal), writing—review and editing (equal). Stefano Stilli: conceptualization (equal), investigation (equal), resources (equal), supervision (equal), writing—review (equal) and editing (equal). Davide M. Donati: conceptualization (equal), investigation (equal), resources (equal), supervision (equal), writing—review (equal) and editing (equal). Luca Sangiorgi: conceptualization (lead), investigation (equal), methodology (equal), resources (equal), supervision (equal), writing‐original draft (equal), writing‐review, editing (equal).
Supporting information
Table S1 Diagnostic criteria for skeletal deformities and functional limitations
ACKNOWLEDGMENTS
We would acknowledge patients and their family for their participation in this study. We are grateful to the Registry of Multiple Osteochondromas (REM) and ACAR Onlus—that has been supporting it since 2013—that allow us to perform the study. Furthermore, we express our thanks to the regional project “GE.D.I.—Genotype‐phenotype Data Integration platform” (PG/2015/795506) and NSI s.r.l. for IT platform that hosts the REM Registry. All the authors of this publication are members of the European Reference Network on Rare Bone Disorders—Project ID No. 739543.
Mordenti, M. , Gnoli, M. , Boarini, M. , Trisolino, G. , Evangelista, A. , Pedrini, E. , Corsini, S. , Tremosini, M. , Staals, E. L. , Antonioli, D. , Stilli, S. , Donati, D. M. , & Sangiorgi, L. (2021). The Rizzoli Multiple Osteochondromas Classification revised: describing the phenotype to improve clinical practice. American Journal of Medical Genetics Part A, 185A:3466–3475. 10.1002/ajmg.a.62470
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available upon reasonable request to the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- Ahn, J. , Lüdecke, H. J. , Lindow, S. , Horton, W. A. , Lee, B. , Wagner, M. J. , & Wells, D. E. (1995). Cloning of the putative tumour suppressor gene for hereditary multiple exostoses (EXT1). Nature Genetics, 11(2), 137–143. 10.1038/ng1095-137 [DOI] [PubMed] [Google Scholar]
- Alvarez, C. , Tredwell, S. , De Vera, M. , & Hayden, M. (2006). The genotype‐phenotype correlation of hereditary multiple exostoses. Clinical Genetics, 70(2), 122–130. 10.1111/j.1399-0004.2006.00653.x [DOI] [PubMed] [Google Scholar]
- Beltrami, G. , Ristori, G. , Scoccianti, G. , Tamburini, A. , & Capanna, R. (2016). Hereditary multiple Exostoses: A review of clinical appearance and metabolic pattern. Clinical cases in mineral and bone metabolism: the official journal of the Italian Society of Osteoporosis, Mineral Metabolism, and Skeletal Diseases, 13(2), 110–118. 10.11138/ccmbm/2016.13.2.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bovée, J. V. (2008). Multiple osteochondromas. Orphanet journal of rare diseases, 3, 3. 10.1186/1750-1172-3-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacciari, E. , Milani, S. , Balsamo, A. , Spada, E. , Bona, G. , Cavallo, L. , Cerutti, F. , Gargantini, L. , Greggio, N. , Tonini, G. , & Cicognani, A. (2006). Italian cross‐sectional growth charts for height, weight and BMI (2 to 20 yr). Journal of Endocrinological Investigation, 29(7), 581–593. 10.1007/BF03344156 [DOI] [PubMed] [Google Scholar]
- Darilek, S. , Wicklund, C. , Novy, D. , Scott, A. , Gambello, M. , Johnston, D. , & Hecht, J. (2005). Hereditary multiple exostosis and pain. Journal of pediatric orthopedics, 25(3), 369–376. 10.1097/01.bpo.0000150813.18673.ad [DOI] [PubMed] [Google Scholar]
- Fan, X.‐L. , Han, Z.‐J. , Gong, X.‐Y. , Xiang, J.‐J. , Zhu, L.‐L. , & Chen, W.‐H. (2014). Morphological classification for prediction of malignant transformation in multiple exostoses. European Review for Medical and Pharmacological Sciences, 18(6), 840–845. [PubMed] [Google Scholar]
- Francannet, C. , Cohen‐Tanugi, A. , Le Merrer, M. , Munnich, A. , Bonaventure, J. , & Legeai‐Mallet, L. (2001). Genotype‐phenotype correlation in hereditary multiple exostoses. Journal of medical genetics, 38(7), 430–434. 10.1136/jmg.38.7.430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hameetman, L. , Bovée, J. V. , Taminiau, A. H. , Kroon, H. M. , & Hogendoorn, P. C. (2004). Multiple osteochondromas: clinicopathological and genetic spectrum and suggestions for clinical management. Hereditary cancer in clinical practice, 2(4), 161–173. 10.1186/1897-4287-2-4-161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäger, M. , Westhoff, B. , Portier, S. , Leube, B. , Hardt, K. , Royer‐Pokora, B. , Goßheger, G. , & Krauspe, R. (2007). Clinical outcome and genotype in patients with hereditary multiple exostoses. Journal of Orthopaedic Research: Official Publication of the Orthopaedic Research Society, 25(12), 1541–1551. 10.1002/jor.20479 [DOI] [PubMed] [Google Scholar]
- Jennes, I. , Pedrini, E. , Zuntini, M. , Mordenti, M. , Balkassmi, S. , Asteggiano, C. G. , Casey, B. , Bakker, B. , Sangiorgi, L. , & Wuyts, W. (2009). Multiple osteochondromas: Mutation update and description of the multiple osteochondromas mutation database (MOdb). Human Mutation, 30(12), 1620–1627. 10.1002/humu.21123 [DOI] [PubMed] [Google Scholar]
- Jo, A. R. , Jung, S. T. , Kim, M. S. , Oh, C. S. , & Min, B. J. (2017). An evaluation of forearm deformities in hereditary multiple exostoses: Factors associated with radial head dislocation and comprehensive classification. The Journal of Hand Surgery, 42(4). 10.1016/j.jhsa.2017.01.010, 42, 292.e1, 292.e8 [DOI] [PubMed] [Google Scholar]
- Jurik, A. G. (2020). Multiple hereditary exostoses and enchondromatosis. Best Practice & Research. Clinical Rheumatology, 34(3), 101505. 10.1016/j.berh.2020.101505 [DOI] [PubMed] [Google Scholar]
- Masada, K. , Tsuyuguchi, Y. , Kawai, H. , Kawabata, H. , Noguchi, K. , & Ono, K. (1989). Operations for forearm deformity caused by multiple osteochondromas. The Journal of bone and joint surgery. British volume, 71(1), 24–29. 10.1302/0301-620X.71B1.2914999 [DOI] [PubMed] [Google Scholar]
- Mordenti, M. , Ferrari, E. , Pedrini, E. , Fabbri, N. , Campanacci, L. , Muselli, M. , & Sangiorgi, L. (2013). Validation of a new multiple osteochondromas classification through switching neural networks. American Journal of Medical Genetics. Part A, 161A(3), 556–560. 10.1002/ajmg.a.35819 [DOI] [PubMed] [Google Scholar]
- Mordenti, M. , Shih, F. , Boarini, M. , Pedrini, E. , Gnoli, M. , Antonioli, D. , Tremosini, M. , & Sangiorgi, L. (2020). The natural history of multiple osteochondromas in a large Italian cohort of pediatric patients. Bone, 139, 115499. 10.1016/j.bone.2020.115499 [DOI] [PubMed] [Google Scholar]
- Pedrini, E. , Jennes, I. , Tremosini, M. , Milanesi, A. , Mordenti, M. , Parra, A. , Sgariglia, F. , Zuntini, M. , Campanacci, L. , Fabbri, N. , Pignotti, E. , Wuyts, W. , & Sangiorgi, L. (2011). Genotype‐phenotype correlation study in 529 patients with multiple hereditary exostoses: Identification of “protective” and “risk” factors. The Journal of Bone and Joint Surgery, 93(24), 2294–2302. 10.2106/JBJS.J.00949 [DOI] [PubMed] [Google Scholar]
- Piombo, V. , Jochmann, K. , Hoffmann, D. , Wuelling, M. , & Vortkamp, A. (2018). Signaling systems affecting the severity of multiple osteochondromas. Bone, 111, 71–81. 10.1016/j.bone.2018.03.010 [DOI] [PubMed] [Google Scholar]
- Porter, D. E. , Lonie, L. , Fraser, M. , Dobson‐Stone, C. , Porter, J. R. , Monaco, A. P. , & Simpson, A. H. R. W. (2004). Severity of disease and risk of malignant change in hereditary multiple exostoses. A genotype‐phenotype study. The Journal of bone and joint surgery. British volume, 86(7), 1041–1046. 10.1302/0301-620x.86b7.14815 [DOI] [PubMed] [Google Scholar]
- Schmale, G. A. , Conrad, E. U. , & Raskind, W. H. (1994). The natural history of hereditary multiple exostoses. The Journal of Bone and Joint Surgery. American volume, 76(7), 986–992. 10.2106/00004623-199407000-00005 [DOI] [PubMed] [Google Scholar]
- Shapiro, F. , Simon, S. , & Glimcher, M. J. (1979). Hereditary multiple exostoses. Anthropometric, roentgenographic, and clinical aspects. The Journal of Bone and Joint Surgery, 61(6A), 815–824. [PubMed] [Google Scholar]
- Speerin, R. , Slater, H. , Li, L. , Moore, K. , Chan, M. , Dreinhöfer, K. , Ebeling, P. R. , Willcock, S. , & Briggs, A. M. (2014). Moving from evidence to practice: Models of care for the prevention and management of musculoskeletal conditions. Best Practice & Research . Clinical Rheumatology, 28(3), 479–515. 10.1016/j.berh.2014.07.001 [DOI] [PubMed] [Google Scholar]
- Stickens, D. , Clines, G. , Burbee, D. , Ramos, P. , Thomas, S. , Hogue, D. , Hecht, J. T. , Lovett, M. , & Evans, G. A. (1996). The EXT2 multiple exostoses gene defines a family of putative tumour suppressor genes. Nature Genetics, 14(1), 25–32. 10.1038/ng0996-25 [DOI] [PubMed] [Google Scholar]
- Stieber, J. R. , & Dormans, J. P. (2005). Manifestations of hereditary multiple exostoses. The Journal of the American Academy of Orthopaedic Surgeons, 13(2), 110–120. 10.5435/00124635-200503000-00004 [DOI] [PubMed] [Google Scholar]
- Taniguchi, K. (1995). A practical classification system for multiple cartilaginous exostosis in children. Journal of pediatric orthopedics, 15(5), 585–591. 10.1097/01241398-199509000-00007 [DOI] [PubMed] [Google Scholar]
- Wuyts, W. , Schmale, G. A. , Chansky, H. A. , & Raskind, W. H. (1993). Hereditary multiple osteochondromas. In Adam M. P., Ardinger H. H., Pagon R. A., Wallace S. E., Bean L. J., Mirzaa G., & Amemiya A. (Eds.), GeneReviews®. University of Washington, Seattle. http://www.ncbi.nlm.nih.gov/books/NBK1235/ [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Diagnostic criteria for skeletal deformities and functional limitations
Data Availability Statement
The data that support the findings of this study are available upon reasonable request to the corresponding author. The data are not publicly available due to privacy or ethical restrictions.