

Abstract
RIG‐I is our first line of defense against RNA viruses, serving as a pattern recognition receptor that identifies molecular features common among dsRNA and ssRNA viral pathogens. RIG‐I is maintained in an inactive conformation as it samples the cellular space for pathogenic RNAs. Upon encounter with the triphosphorylated terminus of blunt‐ended viral RNA duplexes, the receptor changes conformation and releases a pair of signaling domains (CARDs) that are selectively modified and interact with an adapter protein (MAVS), thereby triggering a signaling cascade that stimulates transcription of interferons. Here, we describe the structural determinants for specific RIG‐I activation by viral RNA, and we describe the strategies by which RIG‐I remains inactivated in the presence of host RNAs. From the initial RNA triggering event to the final stages of interferon expression, we describe the experimental evidence underpinning our working knowledge of RIG‐I signaling. We draw parallels with behavior of related proteins MDA5 and LGP2, describing evolutionary implications of their collective surveillance of the cell. We conclude by describing the cell biology and immunological investigations that will be needed to accurately describe the role of RIG‐I in innate immunity and to provide the necessary foundation for pharmacological manipulation of this important receptor.
Keywords: anti‐viral response, innate immunity, interferon, pattern recognition receptor, RLR
1. INTRODUCTION
One of the most important cellular defenses against RNA viruses is a large, multidomain protein known as RIG‐I (Retinoic Acid Inducible Gene I), which functions as a pattern recognition receptor (PRR) that triggers early innate immune responses in vertebrate cells. 1 , 2 , 3 RIG‐I is a member of a conserved family of double‐stranded RNA (dsRNA) binding proteins that includes additional innate immune surveillance proteins MDA5 and LGP2. 3 , 4 , 5 By recognizing and responding to different types of viral RNA motifs, this family (known as the RIG‐I‐like receptors or RLRs) provides broad protection against viral infections. These proteins are, in turn, closely related to members of the broader Dicer family, such as DRH3 (Dicer related helicase 3), as all of these proteins share a distinctive set of dsRNA binding domains, and an ATPase core that is catalytically activated only upon binding of dsRNA. 6 , 7 This link between RLRs and Dicer‐like proteins involved in miRNA processing suggests a shared evolutionary heritage and the possibility of cross‐talk between these two systems. 5 The helicase core of the RLR/Dicer family proteins (previously termed DRAs) is distinct, 7 but identifiable as a member of Helicase Superfamily 2 (SF2), and unrooted phylogenetic trees show that the closest SF2 relatives are the DEAD‐box proteins, which are non‐translocative RNA‐dependent ATPases that function as RNA chaperones in the cell. 8
Each of the RLRs was discovered separately, and their shared function in detecting viral infections was only slowly revealed. All three members RLR family were first identified in large screens for gene sequences responsive to diverse stimuli, particularly pathways involved in cancer. 9 , 10 , 11 , 12 A gene sequence named RIG‐I was initially identified in response to transretinoic acid stimulation in 2000, but there was no further characterization of its gene products. 13 One year later, LGP2 was identified from the mouse Stat3/5 locus and determined to share homology with the DEAD‐box proteins, but the function of the protein was not determined. 14 The first RLR gene that was fully cloned and characterized was MDA5 in 2002, in which the helicase core and CARD domains were identified and found to be responsive to dsRNA. 15 Subsequently, RIG‐I was cloned from a pool of interferon‐stimulated genes and found to produce type I interferons in response to dsRNA treatment. 16 Finally, a comprehensive delineation of the RLR family's shared function in antiviral response was conducted in 2005, showing that the RLRs shared a role in restricting viral infections. 17
Since the discovery of RIG‐I and its role as an innate immune receptor, there has been a wealth of studies dissecting the pathway by which viral RNA stimulates activation of RIG‐I and causes the downstream induction of potent interferon responses. 2 , 10 , 16 There has been intense investigation of the molecular determinants by which RIG‐I recognizes viral RNAs and differentiates these pathogenic targets from abundant host RNAs. 18 , 19 , 20 , 21 , 22 , 23 , 24 In addition, the molecular basis for RIG‐I activation and subsequent propagation of the resulting signal has been the subject of numerous biochemical and cell‐based studies. 1 , 2 , 10 , 25 , 26 , 27 Throughout the course of these investigations, many models have been put forth to explain the mechanism of RIG‐I activation, while at the same time parallel breakthroughs in structural biology and immunological tools have led to the continuous refinement of these models and to new ideas for RIG‐I function. With this review, we synthesize the latest structural and biochemical information on RIG‐I, joining it with results from functional analyses and imaging conducted in cells and more recently in whole animals, to provide a comprehensive overview of the molecular mechanism for RIG‐I activation and signaling. In addition, we describe the molecular basis for host‐pathogen discrimination by RIG‐I, and the mechanisms by which this powerful receptor is selectively triggered only upon infection.
2. RIG‐I ON PATROL: LIFE OF THE RECEPTOR IN THE UNINFECTED CELL
RIG‐I is expressed in almost all the nucleated cells without showing tissue‐specific patterns of expression, suggesting a universal role in the surveillance of viral infections. 28 , 29 In the absence of infection, RIG‐I is inactive, adopting an autoinhibited conformation as it patrols the cell. While the receptor is localized primarily in the cytoplasm, a significant subpopulation is also detected within nucleus 30 , 31 , 32 , 33 , 34 and it has been reported in specific subcellular compartments, including mitochondria, microsomes, and mitochondria‐associated membranes. 35 , 36 While patrolling these compartments, RIG‐I continuously samples RNAs that it encounters, dynamically binding and releasing them as it searches for viral RNA targets. As a positively charged SF2 protein, RIG‐I has relatively high affinity for many RNAs, 37 and it has therefore developed active strategies to become selectively stimulated only upon binding viral RNAs that contain specific molecular determinants (vide infra).
The distinctive architecture of RIG‐I includes a pair of amino‐terminal caspase activation and recruitment domains (the signaling domains, or CARDs), an atypical RNA‐dependent ATPase motor domain (which is commonly, if inaccurately, called the Helicase domain) that is comprised of two RecA domains (typically called Hel1 and Hel2) and an alpha‐helical insertion domain (Hel2i) connected via a V‐shaped Pincer motif, and a carboxy‐terminal domain (CTD) 20 , 22 , 24 , 38 (Figure 1). While on patrol in the inactivated state, RIG‐I clasps the CARDs against the surface of Hel2i, locking RIG‐I in an autorepressed conformation that has been visualized in crystal structures of the apo‐RIG‐I, along with HDX‐MS and SAXS studies. 24 , 39 , 40 , 41 When RIG‐I binds to host RNAs, including capped mRNAs, 5′‐monophosphorylated miRNAs, internal stem structures, and other cellular RNAs, association is weak and transient. 18 , 42 , 43 , 44 , 45 , 46 , 47 These brief encounters fail to dislodge the inhibitory loop motif (the Hel2 loop) that blocks the viral RNA recognition pocket that is located within the CTD. 18 , 21 Therefore, despite frequent RIG‐I encounters with host RNAs, the resulting weak complexes fail to stimulate sustained CARD release and trigger downstream activation. 18 , 19 , 43 , 49 , 50 This dynamic sampling process is further facilitated by the binding and hydrolysis of ATP, as described below.
Figure 1.
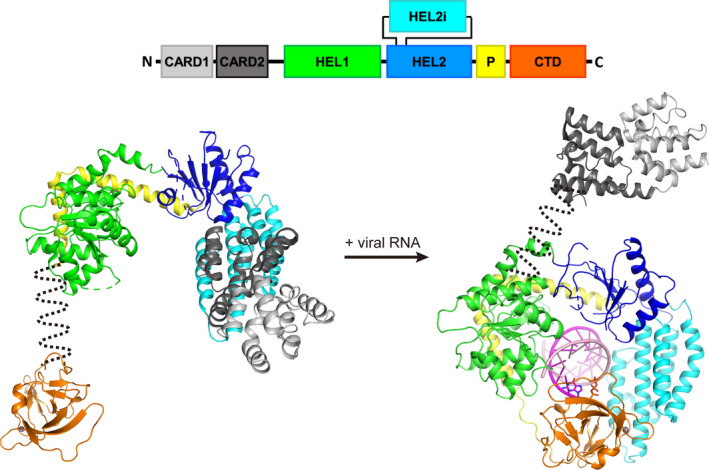
Structure‐based model of RIG‐I activation. RIG‐I contains a pair of amino‐terminal caspase activation and recruitment domains (the signaling domains or CARDs (grey)), an atypical RNA‐dependent ATPase motor domain (which is commonly called the Helicase domain) that is comprised of two RecA domains (typically called Hel1 (green) and Hel2 (blue)) and an alpha‐helical insertion domain (Hel2i (cyan)) connected via a V‐shaped Pincer motif (yellow), and a C‐terminal domain (CTD (orange)). While on patrol in the inactivated state, RIG‐I clasps the CARDs against the surface of Hel2i, locking RIG‐I in an autorepressed conformation. Once engaged with viral RNA, these pathogen RNAs trigger massive conformational changes that anchor the RNA tightly inside the RIG‐I receptor and trigger release of the CARDs, thereby completing the first step of RIG‐I signaling
3. IDENTIFYING THE ENEMY: STRATEGIES FOR RIG‐I RECOGNITION OF PATHOGEN RNA
While RIG‐I patrols its cellular environment, binding and releasing rapidly from host RNAs, it will eventually encounter viral RNA molecules upon infection, resulting in the formation of a stable, signal‐active conformation. Once engaged, pathogen RNAs trigger massive conformational changes that anchor the RNA tightly inside the RIG‐I receptor and trigger release of the CARDs, thereby completing the first step of RIG‐I signaling. 24 , 38 , 48 , 49 This specific, high‐affinity complex requires two structural features on an RNA target, or PAMP (Pathogen Associated Molecular Pattern). 4 , 51 One feature is a blunt, double‐stranded RNA that is at least one helical turn in length. 19 , 52 This is grasped by residues on the inner surface of Hel1, Hel2, and Hel2i, which form a network of polar contacts with ribose 2′‐OH groups and non‐bridging phosphoryl oxygens along the RNA backbone. 22 , 24 , 38 Intriguingly, the majority of contacts involve the ribose sugar rather than the phosphates of the backbone, thereby ensuring that RIG‐I is selectively activated by dsRNA and not DNA. 20 , 22 , 24 , 38 , 53 The dsRNA binding footprint of RIG‐I is between 8 and 10 dsRNA base pairs, and indeed, blunt RNA duplexes as short as 10 base pairs trigger a rapid signaling response and potent IFN induction in cells and whole animals. 19 , 20 , 43 , 54 , 55 , 56 , 57 , 58 In the absence of a closed loop to stabilize the RNA duplex, 20 , 54 the minimal two‐stranded RNA agonist for stimulating RIG‐I in cells has been reported be ~19 bp, 19 , 59 , 60 which may be attributable to the relative instability of shorter RNA duplexes, particularly as they pass through cell membranes or bind to exonucleases.
The second structural feature critical for RIG‐I recognition is the presence of multiple phosphates on the 5′ end of the blunt RNA duplex terminus. DsRNA with three phosphates on the 5′ end (p3dsRNA) is most commonly generated during viral genome replication by the 5′‐nucleotide that primes initiation. 61 Crystal structures show that the primary point of contacts between RIG‐I and p3dsRNA are stacking interactions with the terminal base pair, and a network of interactions with the alpha and beta phosphate, which interact with a specialized phosphate recognition pocket in the CTD. Intriguingly, the gamma phosphate is not consistently recognized by the receptor. 21 , 23 , 62 The importance of the alpha and beta phosphate was functionally validated by studies showing that a 5′‐dsRNA diphosphate (p2dsRNA) is the primary RNA requirement for activation of RIG‐I 23 , 63 and subsequent work in cells and in vivo has shown that p2dsRNAs are even more potent than p3dsRNAs as triggers for RIG‐I. 54 , 63 These diphosphorylated RNA PAMPs have been found in reovirus RNAs and other nonhost targets. 63
Unlike p2dsRNA or p3dsRNA duplexes, RNA duplexes terminated by a 5′‐monophosphate (p1dsRNA) are potent antagonists of RIG‐I, as their binding is obstructed by the Hel2 loop insertion within the CTD and they fail to interact productively with RIG‐I. 18 Surprisingly, monophosphate dsRNAs are even more inhibitory than blunt RNA duplexes lacking any phosphates at all (OHdsRNA). 18 Given the presence of pre‐miRNAs in the cytosol, 64 along with other host p1dsRNAs, strongly rejecting these host RNAs is essential to prevent aberrant RIG‐I activation. 18 Given that p1dsRNAs differ from the most potent RIG‐I agonists (the p2dsRNA) by only a single phosphate group, it is clear that RIG‐I has evolved an exceptionally selective strategy for its target PAMP, and that it actively selects against host RNAs. Similarly, blunt dsRNAs lacking any phosphates (OHdsRNA) also fail to stimulate RIG‐I signaling. 18 , 19 Structural studies have shown that OHdsRNAs form an autoinhibited complex with RIG‐I, in which a loop projecting from Hel2 plugs into the same CTD amino acids that are required for high‐affinity interactions with the terminal di‐ and triphosphate groups of the most potent RIG‐I PAMPS. 42 Blocked from interacting with the CTD, OHdsRNAs bind more weakly and may be less able to trigger the steric clash that stimulates release of the CARDs from Hel2i. RIG‐I is also capable of discriminating various types of 5′ terminal cap structures dsRNAs: RIG‐I has been observed to bind tightly and signal from dsRNAs containing a methyl guanosine cap (Cap‐0‐dsRNA), but it fails to bind capped dsRNAs if the first 5′ ribose is 2′OH methylated (Cap‐1‐dsRNA), or if both the first and second 5′ ribose are methylated (Cap‐2‐dsRNA), indicating that RIG‐I is selectively inhibited by 2′‐O‐methyl groups on the terminal and penultimate nucleotide of modified cap structures, rather than by the modified guanosine itself. 21
Prior to the availability of structural data on RIG‐I/dsRNA complexes, and before quantitative functional studies were carried out to elucidate the minimal recognition determinants for RIG‐I ligands in cells and in animals, older models for RIG‐I signaling suggested a requirement for RIG‐I multimerization and even filament formation on long dsRNAs. These models built upon biochemical studies in which extremely high concentrations of recombinant RIG‐I were biochemically combined with RNA duplexes such as synthetic transcribed RNAs and abiological polymers like polyIC. There is a long history of biochemical studies showing that positively charged nucleic acid‐binding proteins, when presented at high concentrations, will form nonspecific, artifactual filaments on DNA and RNA, and this should be cause for caution whenever invoking a filament model for behavior of a nucleic acid‐binding protein. 65 , 66 , 67 In addition, the subsequent abundance of direct evidence showing that fully activating RIG‐I ligands can be short 3pdsRNAs, which are completely functional in vivo, makes it now clear that RIG‐I does not need to oligomerize on RNA as part of its activation mechanism. Indeed, a whole new generation of RIG‐I‐specific immunomodulatory drugs builds on this premise, resulting in small 3pdsRNAs that are now in development as antivirals, vaccine adjuvants, and immunostimulatory anticancer agents. 9 , 12 , 55 , 68 , 69 , 70 Triphosphorylated stem‐loop RNAs (SLRs) as short as 10 to 14 base pairs, which can only bind to one RIG‐I molecule, stimulate RIG‐I‐mediated IFN response in mouse and induce robust antitumor responses. 54 , 55 , 68 , 69
Taken together, crystal structures of RIG‐I:RNA complexes, together with functional analyses in cells and in animals, demonstrate that the CTD forms an extensive network of specific interactions with the 5′ terminus of the RNA duplex, and that RIG‐I caps the blunt terminus of dsRNAs as a monomeric end‐binder. 20 , 24 , 38 , 53 This 1:1 RNA‐RIG‐I complex, in which a single RIG‐I binds to the terminus of 5′ppp‐dsRNA, is necessary and sufficient for RIG‐I signaling. 20 , 54 , 71 While it is possible that additional RIG‐I proteins, or other proteins can assist this end‐binding mode in certain contexts, biochemical studies have clearly demonstrated that RIG‐I does not display cooperative binding to RNA, which is consistent with crystallographic and functional analyses. 6 , 20 This contrasts with MDA5, which exhibits pronounced cooperative binding and filament formation, 72 , 73 or DRH3, which displays clear ATP‐dependent dimerization on RNA. 6 , 74
Despite the highly specific nature of its PAMP, RIG‐I has been reported to respond to a broad array of pathogenic threats. RIG‐I is a first line of defense against a variety of viral families, including paramyxoviruses, coronaviruses, othomyxoviruses, flaviviruses, rotaviruses, filoviruses, reoviruses, hepeviruses, alphaviruses, and arenaviruses. 34 , 63 , 75 , 76 , 77 , 78 , 79 , 80 , 81 , 82 , 83 , 84 Given the high specificity of RIG‐I for p3dsRNA, it may seem surprising that it is able to respond to such a diversity of apparent targets. However, the key structural features recognized by RIG‐I are all brought together during a key stage in the lifecycle of all RNA viruses: replication. The viral genome itself can serve as a RIG‐I agonist, as in the case of dsRNA viral genomes and in panhandle structures formed by binding together the 5′ and 3′ termini of ssRNA genomes during replication. 19 , 63 , 77 , 85 , 86 , 87 , 88 , 89 Additionally, RIG‐I also recognizes genome replication intermediates, such as defective interfering particles (DI) and mini viral RNA (mvRNA), which all contain triphosphorylated panhandle structures at the 5′ terminus. 81 , 90 , 91 , 92 RIG‐I has also been reported to play a small role in the recognition of some DNA viral infections, including both herpesviruses and hepadnaviruses, by recognizing and binding to uncapped transcripts produced by RNA polymerase III (Pol III). 93 , 94 In addition to p3dsRNAs directly generated by viral sources, RIG‐I can also bind and signal from host p3dsRNAs that are generated by RNA polymerase III (Pol III) and released into the cytosol during DNA virus infection. 93 , 94 However, phosphatases such as RNA triphosphatase dual‐specificity phosphatase 11 (DUSP11) may reduce aberrant RIG‐I activation on host RNAs by limiting their prevalence. 95 Finally, RIG‐I signaling has been implicated in responding to intracellular bacterial infections by sensing the RNA products of agents such as Listeria, Salmonella enterica, and Mycobacterium tuberculosis 96 , 97 , 98
4. SOUNDING THE ALARM: PATHOGEN‐TRIGGERED CONFORMATIONAL CHANGES RELEASE THE RIG‐I SIGNALING DOMAINS
The single most critical step in RIG‐I activation, and subsequently the most vital for signaling, is conformational reorganization of the protein, which results in release of the CARDs. In the absence of stimulatory RNA, the CARDs are sequestered by the Hel2i domain, thereby preventing any downstream signaling 24 (Figure 1). Mutants in which Hel2i cannot bind the CARDs constitutively activate IFN production, such as F539A (designed to disrupt Hel2i‐CARDs interactions) and Q517H (found in patients with autoimmune disease called Singleton‐Merten syndrome). 24 , 99 Similarly, isolated CARDs alone are sufficient to drive antiviral signaling, 16 providing further evidence that full solvent accessibility of the CARDs is necessary for RIG‐I signaling.
Considerable evidence has accumulated showing that the ability of specific RNA PAMPs to bind RIG‐I and trigger CARD release correlates directly with the ability to stimulate RIG‐I signaling in cells and in vivo. FRET studies demonstrated that p3dsRNA, but not unstructured RNA, is required to release the CARDs and that removal of RNA with nucleases is sufficient to return RIG‐I to its autoinhibited state. 49 HDX‐MS experiments showed that p3dsRNA significantly increases the solvent accessibility of the CARDs, implying that they are ejected. 39 Similarly, SEC‐SAXS and limited tryptic digestion data suggested that upon the addition of p3dsRNA, RIG‐I becomes extended and conformationally flexible, consistent with released CARDs. 40 FRET, HDX‐MS, SEC‐SAXS, and limited tryptic digest studies all show that p3dsRNA at least 10bp in length induces conformational changes in the RIG‐I protein and robustly activates RIG‐I in cell culture and in vivo. 39 , 40 , 49 , 54 This correlates directly with the ability of 10bp, but not 8bp, 5′ppp‐dsRNA to stimulate RIG‐I signaling in cells. 20 Thus, the ability of an RNA PAMP to cause the conformational switch that results in the “CARDs out” active conformation of RIG‐I correlates directly with its ability to signal in cells and animals.
The ability of RIG‐I to productively eject the CARDs depends on tight, structurally specific RNA binding that may compete for occupancy of the CARD binding site on the surface of the helicase domains (Figure 1). CTD stacking on the base‐paired surface of the 5′ blunt end, along with strong contacts to the alpha and beta phosphates, anchors the RNA in place and stimulates the conformational changes needed to destabilize interactions between the CARDs and Hel2i. 38 , 100 When tight RNA binding by the CTD is disrupted, either from mutations at the CTD interface, or due to structural features of host RNAs, such as the 5′‐cap structures, RNA is only weakly held by the receptor and its off‐rate is fast. 42 , 53 , 62 , 101 Even if the CARDs‐out conformation is sampled, weakly held RNA is rapidly ejected and RIG‐I returns to the autorepressed state. 21 , 100 Similarly, if the RNA is shorter than 10 bp, the RNA fails to form sufficient interactions with the Hel2 interface, and the RNA is ejected in favor of a strong CARDs interface along Hel2i. 20 , 38
5. AVOIDING COLLATERAL DAMAGE: RIG‐I PROOFREADING OF HOST RNA AND THE ROLE OF ATP
RIG‐I is an exquisitely selective, RNA‐triggered switch for initiating the antiviral signaling pathway. Given this fact, investigators have long wondered why RIG‐I contains a well‐conserved active site for ATP binding and hydrolysis, which is very similar to that found in the DEAD‐box family of SuperFamily 2 Helicases (SF2 proteins). Before structural and genetic work on RIG‐I had been completed, it was presumed that ATP binding and hydrolysis played a central role in the molecular mechanism of RIG‐I signaling, and that helicase or translocase activity was essential for the molecular mechanism of RIG‐I signaling. Such functions seemed reasonable at the time because the RNA recognition requirements for RIG‐I binding had not been defined, and it was presumed that RIG‐I was stimulated by long, complex pieces of viral RNA, which might necessitate a variety of translocative mechanical processes, but now it is clear that CARD release, and the very first structural events involved in initiating the signaling pathway, depend only on binding of simple p3dsRNAs, and that ATP hydrolysis is not required for this process. 42 , 43 , 49 , 102 , 103 There is evidence that ATP binding may cause compaction of the RIG‐I/p3dsRNA complex, 20 , 40 and that this may indeed facilitate release of the CARDs. However, measures of RIG‐I CARD release via FRET and SAXS indicate that, with a high‐affinity p3dsRNA ligand, CARD release occurs even without ATP present, 40 , 49 suggesting that ATP binding and hydrolysis are not required for signaling. So if ATP hydrolysis is not essential for the mechanism of signaling, why is this function so highly conserved and important for the protein?
To understand the role of ATP, it is important to realize that RIG‐I has two jobs: A. It needs to initiate signaling upon binding to the “right RNA” (from a pathogen). B. It needs to prevent signaling upon binding to the “wrong RNA” (host RNAs). Both of these jobs are essential for the overall function and evolutionary tuning of the RIG‐I sensor. Juggling these tasks is incredibly difficult, however, because the “right RNA” is rare and dilute, while the “wrong RNA” continually surrounds RIG‐I as a dense, concentrated soup. Therefore, staying inactivated is arguably the most important and challenging task of the RIG‐I receptor, as constitutive activation results in pathological forms of inflammation (vide infra). As discussed in the section on ligand recognition, one of the ways that RIG‐I distinguishes host from pathogen RNA is through structural selectivity: Host RNAs (lacking 3pdsRNA termini) are blocked from entering the high‐affinity CTD binding site by an autoinhibitory loop that projects from Hel2, thereby reducing RNA affinity and CARD release. 18 , 21 But no autoinhibitory mechanism is perfect, so to reduce the impact of CARD release and signaling from host dsRNA, RIG‐I uses a backup proofreading strategy that requires ATP. 42 , 43 , 50 , 104 , 105 Like the closely related DEAD‐box proteins, 8 the major role of ATP in RLRs is not in directional molecular motion, but in enhancing the rate by which RIG‐I binds, samples and releases potential RNA targets (kinetic proofreading). Kinetic and functional studies have shown that RIG‐I affinity for dsRNA is modulated by the binding and hydrolysis of ATP. 42 , 43 , 102 , 104 , 105 , 106 Much like the related DEAD‐box proteins, 8 , 107 cycles of ATP binding and hydrolysis accelerate the process of RNA interrogation and dissociation. 42 , 43 , 102 , 104 , 105 , 106 As a result, only the highest‐affinity p3dsRNA ligands remain bound for sufficient amounts of time to initiate signaling. Other RNAs fall off without maintaining the RIG‐I conformation that culminates in sustained CARD release. 18 , 19 , 20 , 21 , 22 , 23 , 24
Some of the best evidence that ATP plays a key role in proofreading comes from studies on RIG‐I mutations that are implicated in human disease. 2 , 42 , 106 , 108 , 109 Under certain pathological conditions, RIG‐I can become inappropriately activated by host RNAs, causing massive collateral inflammatory damage. This can be due to ATPase site mutants in RIG‐I, 42 , 43 , 50 or dysregulation in the abundance or type of host RNA molecules in the cytoplasm. 110 , 111 , 112 Defects in enzymes involved in RNA decay or degradation pathways flood the cytoplasm with a high concentration of low‐affinity ligands that overwhelm the ability of RIG‐I and other RLRs to successfully discriminate host from pathogen RNA. 110 , 111 , 112 , 113 , 114
Specifically, RIG‐I has been implicated in certain autoinflammatory diseases and interferonopathies. Clinical studies have identified several single‐nucleotide polymorphisms (SNPs) in RIG‐I, including ATPase‐active site mutants E373A and C268F. These result in an atypical form of Singleton‐Merten syndrome (SMS) in patients who express excessive amounts of IFN. Biochemical and cell‐based studies have shown that variant E373A slows down RNA‐dependent ATP hydrolysis by RIG‐I, which in turns leads to constitutive activation by endogenous dsRNA. 50 , 104 Other ATPase‐active site mutants, such as C268F, also contribute to inappropriate activation by host RNA. 102 In both of these cases, ATPase activity of RIG‐I is damaged, either through defects in ATP binding or hydrolysis. This leads to inappropriate stimulation of RIG‐I by host ligands, such as double‐stranded RNAs that lack a triphosphate. 42 , 43 , 50 Similarly, patients with defects in RNA decay pathways (either in exosomal proteins or endonucleases such as RNAse L) accumulate high concentrations of RNA in the cytoplasm, triggering dysregulated RIG‐I response. 110 , 111 , 112 , 113 , 114 Taken together, these clinical and mechanistic data underscore the critical role of proofreading in the function of RIG‐I, and the importance of ATP in this process. 2 , 42 , 106 , 108 , 109
Although ATP binding and hydrolysis are not strictly required for signaling on high affinity, optimized ligands such as short, synthetic 3pdsRNA hairpins, it would be a mistake to assume that ATP cannot play a role in the mechanical function of RIG‐I on more complex RNA targets. Single‐molecule 115 and kinetic 42 studies have demonstrated that RIG‐I is capable of undergoing slow, directional translocation on dsRNA molecules and that this requires ATP. In the cell, few RNAs present perfect target structures for recognition, and so it remains possible that RIG‐I actively translocates along candidate targets, searching for RNA regions that could function as stimulatory ligands. Furthermore, RNAs are often coated by proteins, and it has been well documented that SF2 proteins can undergo ATP‐stimulated conformational changes on RNA that “push” away proteins in their path (RNPase activity). 116 , 117 , 118 Although such behavior has never been demonstrated for an RLR, it remains possible that RIG‐I uses ATP‐powered translocative processes to strip proteins away from candidate RNA targets, thereby exposing the blunt 3pdsRNA terminus for specific interaction.
6. KEEPING THE SIGNAL ON: THE CRITICAL ROLE OF POST‐TRANSLATIONAL MODIFICATIONS
Once RIG‐I has tightly bound its RNA PAMP, the liberated CARDs are capable of transmitting the active RIG‐I signal to the downstream transmembrane adaptor mitochondrial antiviral signaling protein (MAVS) (Figure 2). However, numerous protein cofactors intervene between RNA binding and MAVS transmission, either by enhancing or repressing the RIG‐I signal. The most essential cofactors are those that modify active RIG‐I via post‐translational modification, including ubiquitination and dephosphorylation, to enhance signaling. 119 , 120 , 121 , 122 The selective interaction of these cofactors with RIG‐I after CARD release is likely to be essential for stabilizing the activated form of RIG‐I and may serve as a checkpoint prior to signal transmission.
Figure 2.
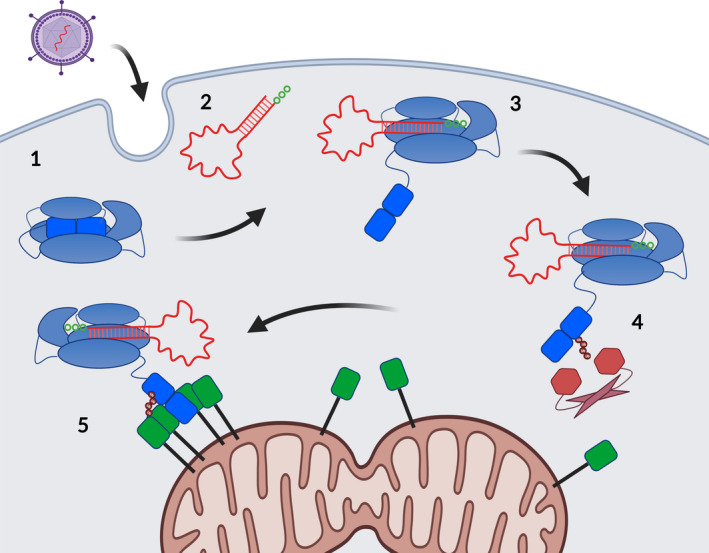
The RIG‐I signaling pathway. 1. In the absence of viral RNA, RIG‐I patrols the cell with the CARDs (bright blue) packed tightly against the other protein domains. 2. Upon viral entry and generation of a blunt dsRNA with two or three phophates (p3dsRNA, red RNA with green phosphate circles), 3. RIG‐I binds tightly to the p3dsRNA terminus, and the tethered CARDs are displaced into the cytosol. 4. Liberated CARDs are then modified by cofactors (red) that append ubiquitin (small red circles), which prevents return to autorepressed state. 5. Finally, active RIG‐I moves to the mitochondria and nucleates MAVS oligomerization
Ubiquitination of RIG‐I CARDs plays a key role in RIG‐I signaling, but the mechanism for how ubiquitination enhances RIG‐I, and the identity of the E3 ligase responsible for ubiquitination remain the subject of lively debate. 120 , 123 , 124 RIG‐I K63‐linked ubiquitination was first correlated with active signaling in 2007, and mutations blocking ubiquitination at lysine 172 dramatically reduce signaling. 120 The E3 ligase identified as responsible for this K63‐linked ubiquitination is TRIM25, which has been shown to coimmunoprecipitate with isolated RIG‐I CARD domains. 120 Knockdown of TRIM25 was reported to inhibit ubiquitination of CARDs. 120 Similarly, full reconstitution of the RIG‐I signaling pathway in vitro was found to require cytosolic extracts, and K63‐linked polyubiquitin chains in particular, for RIG‐I signaling to be fully active. 125 Finally, structural studies of ubiquitinated CARDs, which were expressed as recombinant, isolated domains (ie, lacking RNA binding, helicase, or CTD regions), supported a “lock‐washer” model of RIG‐I activation, where TRIM25 selectively recognized a tetramer of active RIG‐I CARDs, and bound them together via K63‐linked ubiquitin chains. 126 According to this model, multiple active RIG‐I molecules would be required to form this tetramer, thereby enabling it to serve as a checkpoint for errant activation. However, all of the biochemical and structural studies implicating TRIM25 ubiquitination were founded on experiments that employed free CARD domains in isolation, rather than full‐length RIG‐I, or RIG‐I in a cellular context. 120 Furthermore, TRIM25 has been implicated in multiple antiviral pathways, so its specific role in RIG‐I activation is still being elucidated. 127 , 128 , 129 A second E3 ligase, Riplet, has also been proposed as the cofactor responsible for ubiquitination of RIG‐I CARDs in the context of full‐length RIG‐I. 130 , 131 Riplet depletion directly impairs RIG‐I signaling and type‐I interferon production both in cells 132 and in vivo, 121 giving greater weight to Riplet as the E3 ligase responsible for K63‐linked ubiquitination. The current Riplet‐based model by which ubiquitination enhances RIG‐I signaling suggests that it acts as a bridging cofactor, linking together RIG‐I complexes. 130
Both TRIM25 and Riplet models for ubiquitination‐enhanced signaling propose a role for ubiquitination in the formation of large RIG‐I oligomers and filaments. However, attempts to identify RIG‐I oligomerization in cellulo with a split‐luciferase protein complementation assay failed to identify significant oligomerization upon stimulation with RNA. 71 A significantly simpler explanation consistent with all the available data is that K63‐ubiquitination of CARDs creates a large, covalent steric obstruction on the signaling domain. This would effectively lock RIG‐I in the “CARDs out” active conformation by sterically preventing the CARDS from rebinding Hel2i and readopting the autoinhibited state. 24 Regardless of the mechanism by which it ultimately enhances RIG‐I signaling, ubiquitination plays a central role in the process, and this is underscored by the observation that knockdown of cellular deubiquitinases (DUBs), including USP3, USP21, and CYLD greatly enhances RIG‐I signaling. 133 , 134 , 135
Ubiquitination is not the only PTM that modifies RIG‐I signaling. In the autoinhibited state, RIG‐I CARDs are phosphorylated at S8 and T170, and this has been postulated to prevent E3 ligase binding and ubiquitination. 136 , 137 Upon RNA binding and CARD release, these sites undergo dephosphorylation by phosphatases PP1α and PP1γ, potentially facilitating greater association with either an E3 ligase or MAVS. 122 Consistent with this, PP1α and PP1γ‐depleted cells show elevated CARDs phosphorylation, and IFN‐β production is correspondingly impaired. 138 Thus, dephophorylation may also be a required step in RIG‐I signaling, which is otherwise unidentified in cell‐free biochemical assays using purified, unphosphorylated RIG‐I.
7. FINDING A PARTNER IN DEFENSE: A MODEL FOR IFN INDUCTION INVOLVING RIG‐I‐MAVS INTERACTION
MAVS is absolutely essential for detecting, amplifying, and relaying the active RIG‐I signal to produce an interferon response. First identified by four different research teams in 2005, MAVS contains an N‐terminal CARD, a proline‐rich region, and a C‐terminal transmembrane domain. 139 , 140 , 141 , 142 Genetic deletion of MAVS completely blocks the RIG‐I‐mediated interferon response, signifying that MAVS is a primary mediator of RIG‐I signaling. 143 MAVS has been found to associate with several membranous organelles, including the mitochondrial network, mito‐associated membrane (MAM) of the ER, and peroxisomes, which have been found to produce distinct downstream signaling responses. 144 , 145 Specifically, the peroxisomal compartment has been implicated in interferon‐independent transcription of antiviral genes to stimulate a rapid host response via IRF1. 144 Regardless of which MAVS subpopulation is stimulated by RIG‐I, MAVS must be membrane‐bound to propagate RIG‐I signaling. 144 This could be due to a high concentration of essential signaling components within the transmembrane region, or more likely, it suggests that MAVS requires a specific two‐dimensional orientation of membrane components for downstream signaling.
MAVS mediates and amplifies the RIG‐I active signal by polymerization of a large oligomeric complex (Figure 2). The first study to explore MAVS oligomerization in cellulo involved purifying large, full‐length MAVS aggregates from mitochondrial extracts in virus‐infected cells, and then showing that these extracts were capable of dimerizing IRF3 (which is a hallmark of interferon induction, see next section). 146 The amino‐terminal CARD is essential for both oligomerization and downstream signaling: not only does expression of ΔCARD‐MAVS fail to signal, 141 but expression of a truncated MAVS mRNA transcript from an internal start codon, which similarly lacks the CARD, represses downstream interferon signaling. 147 The MAVS CARD is believed to form two interactions that are essential for propagating this signal: first by binding to the liberated CARDs of activated RIG‐I and second by forming homotypic interactions with additional MAVS monomers, thereby leading to a form of activated oligomerization. The primary experimental method for characterizing both these interactions has been the formation of large, in vitro filaments composed of unanchored MAVS CARD. For example, in biochemical and structural studies, suggesting that the second CARD of RIG‐I contacts the MAVS CARD, mutants of the putative CARD‐CARD interaction were found to disrupt in vitro filament formation and prevent RIG‐I signaling in cells. 126 Similarly, mutations within MAVS CARD that disrupt prion‐like filament formation of MAVS CARD alone, including E26 and R64, fail to stimulate interferon when overexpressed in cells. 148 , 149 Subsequent studies exploring the nucleation of MAVS have utilized similar purified filament formation assays as a proxy for function, 146 , 150 relying upon the prior correlation to claim an effect on downstream signaling. 126 The only direct evidence in cellulo of CARD‐CARD interactions between RIG‐I and MAVS and for higher‐order oligomerization of MAVS comes from FRET studies in which isolated RIG‐I CARD domains were shown to slightly increase the FRET signal of overexpressed YFP‐MAVS and CFP‐MAVS fusions. 151 Collectively, these studies indicate that MAVS is likely to form CARD‐CARD interactions with other MAVS monomers to form a larger complex that propagates the signal further. However, super‐resolution imaging of labeled MAVS found no evidence of MAVS filaments larger than 80 nm, which is the limit of resolution, raising questions about the actual size of functional MAVS oligomers in vivo. 152 Thus, while the MAVS CARD interactions with both RIG‐I CARDs and MAVS CARD are clearly important for downstream signaling, the size and composition of the active MAVS signaling complex remain unclear. 119 , 153
8. MAVS ACTIVATION LEADS TO NUCLEAR TRANSLOCATION OF KEY TRANSCRIPTION FACTORS
Once MAVS has been activated by RIG‐I and an activated complex of MAVS has formed, this assembly functions as a signaling hub for a cascade of phosphorylation and ubiquitination, leading ultimately to the activation of IRF3 and NF‐κB transcription factors. 139 , 140 The key players in this signaling hub include the TNF‐α‐associated recruitment factors (TRAFs), Tank‐binding kinase 1 (TBK1), and the IκB kinase complex (IKK). 140 , 148 , 154
The TRAFs are a family of 7 proteins that play a role in many immune signaling pathways, and their function relies primarily on two domains: a scaffolding domain and an E3 ligase domain. 155 Several TRAFs, including TRAF2, TRAF3, TRAF5, and TRAF6, are recruited to MAVS via three TRAF‐binding motifs. 151 The association of TRAF factors to MAVS is required for downstream signaling, as mutations to all three motifs block the interferon response. 156 Though the mechanism by which TRAFs distinguish active oligomeric MAVS from inactive MAVS is unclear, they are essential for the recruitment of both TBK1 and the IKK complex. 148 The E3 ligase domain of the TRAFs plays a key role in recruitment of these downstream factors, 157 suggesting that the polyubiquitin chains made by the MAVS‐bound TRAFs are recognized by the IKK complex and TBK1. 158 However, the TBK1 complex can also be recruited by TRAFs lacking the E3 ligase domain, suggesting that there are non‐ubiquitin mechanisms for downstream activation. 158
TBK1 and the IKK complex are, as their names suggest, kinases which set off a phosphorylation cascade leading to the activation of transcription factors. 159 The recruitment of IKK complexes leads to IKKb phosphorylation via trans‐activation. 160 Once active, the IKK(αβγ) complex is able to phosphorylate IkB, leading to its degradation. Phosphorylation of IkB leads to its ubiquitination and degradation by the proteasome, 161 freeing NF‐κB to translocate to the nucleus and promote transcription of type I interferons. The IKK complex also phosphorylates TBK1 in trans, enhancing TBK1 activity. 162 Once phosphorylated and active, TBK1 is key to IRF3 activation, as TBK1 kinase activity was found to phosphorylate serine and threonine residues on MAVS, which may enable IRF recruitment. 156 TBK1 also binds and phosphorylates IRF3 directly. 163 Phosphorylated IRF3 (pIRF3) dimerizes and migrates to the nucleus, and promotes transcription of type I interferon. 164
9. RIG‐I SIGNALING IS CONSERVED AMONG DIFFERENT SPECIES
Given the essential role RIG‐I plays in countering viral threats, it is perhaps unsurprising that it is well‐conserved among our close relatives. In mice, for instance, total deletion of RIG‐I leads to death within 3 weeks after birth, with massive liver degeneration, suggesting additional important functions of the RIG‐I protein. 165 While closely related RIG‐I othologues are found in all mammals, in other vertebrates, conservation of RIG‐I is more limited. 166 , 167 , 168 , 169 , 170 , 171 , 172 Protein sequence alignment of RIG‐I among mammals, birds, and fish reveals that the domain organization of RIG‐I is similar in these classes, and that the key residues involved in RNA recognition, ATP binding, and hydrolysis are all conserved (Figure 3). Furthermore, crystal structures of truncated human, mouse, and duck RIG‐I constructs reveal a highly conserved architecture of RIG‐I, particularly the key residues in the RNA binding pocket of the CTD (H847, K858, K861, and K888) and in the catalytic core of the ATPase domain (K270). 24 , 38 , 173 Moreover, RIG‐I orthologues in birds and fish have been shown to play a key role in producing an IFN response to viral infections, indicating that the function of RIG‐I is also conserved across species. 166 , 167 , 168 , 169 , 170 , 171 , 172 Altogether, structural and cellular studies of RIG‐I orthologues reveal conserved RIG‐I structure and function across many classes of vertebrates, suggesting that in species where the RIG‐I gene is retained, its essential function in responding to viral infections is also conserved.
Figure 3.
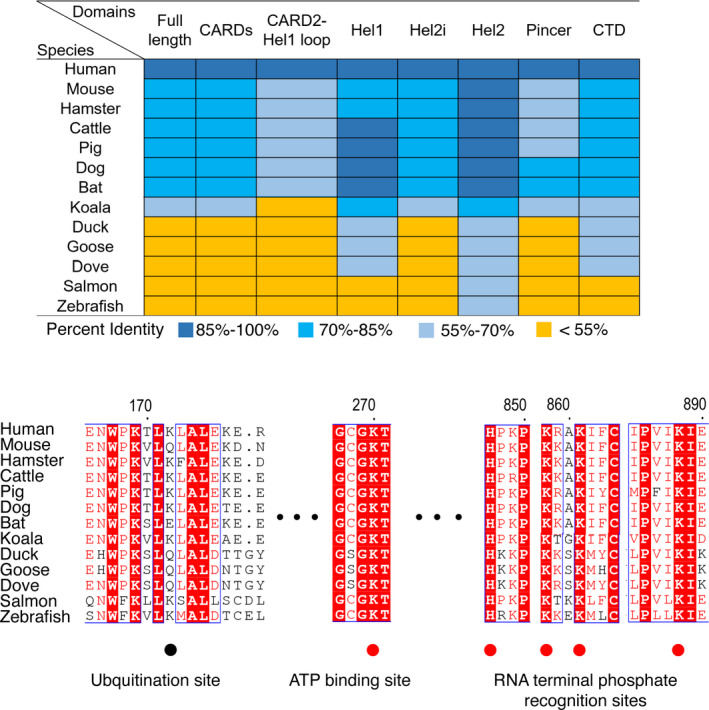
The conservation of RIG‐I among different species. RIG‐I protein sequences from selected species are aligned to human RIG‐I using local alignment using EMBOSS (https://www.bioinformatics.nl/cgi‐bin/emboss/matcher). The identity scores of these alignments are recorded in the table, colored based on the identity scores (100%–86% in dark blue, 85%–70% in blue, 70%–55% in light blue, and <55% in orange). The conserved residues involved in ATP binding, ATP hydrolysis, and RNA terminal phosphate recognition are labeled with red dots, while the K63‐linked ubiquitination site in human RIG‐I CARDs is labeled with a black dot
While the structure and function of core RIG‐I domains are conserved in most vertebrates, loss of the RIG‐I gene has been observed in some species, such as Chinese tree shrew and chicken, 174 , 175 although more studies on RIG‐I gene identification and RIG‐I signaling in other vertebrates will be necessary to comprehensively understand its distribution. Furthermore, certain regions of RIG‐I display significant sequence variation, even within the same species, 176 suggesting potential variation in either RIG‐I sensitivity or the dispensibility of various cofactors. Regions that display significant variation include the CARD2‐Hel1 linker, the length of which varies significantly. The variation in loop length, and subsequent changes in RIG‐I sensitivity, has been proposed as evidence of co‐evolution of RIG‐I and viruses. For example, a comparative study of 19 different mammals reveals sequence variations in CARD2‐Hel1 loop and the Pincer‐CTD loop. 177 Variation in RIG‐I sequences between rabbit species that are sensitive or resistant to myxomatosis suggests that RIG‐I structural changes can influence viral susceptibility. 178 Similarly, the ubiquitination sites of the CARDs found in mammals are not conserved in birds (Figure 3), 24 , 179 suggesting that ubiquitination of the CARDs might occur in a different location, or function differently, in avian species. When viewed comprehensively, RIG‐I recognition of viral RNA and promotion of an antiviral interferon response is conserved within vertebrates, although differences in RIG‐I sensitivity and regulated mechanism remain possible.
10. THE RLRS AS A DIVERSIFIED DEFENSE SYSTEM: MDA5 AND LGP2 COMPLEMENT RIG‐I BY RECOGNIZING A DIFFERENT SPECTRUM OF RNA MOTIF STRUCTURES
While RIG‐I signaling has been the focus of this review, it is not alone in responding to RNA viruses. In addition to the arsenal of TLR receptors, 180 the closely related PRRs MDA5 and LGP2 perform complementary roles in surveillance against viral pathogens. 3 MDA5 (melanoma differentiation‐associated protein 5), is also a double‐stranded RNA‐dependent ATPase, with a domain architecture similar to RIG‐I in that it contains both a caspase recruitment domain (CARDs) and an almost identical set of RNA helicase and RNA binding motifs. 15 , 38 , 181 MDA5 localizes in the cytoplasm, as demonstrated by confocal fluorescence microscopy using a GFP‐MDA5 fusion protein, where it functions to sense cytoplasmic viral RNAs and induce antiviral responses via MAVS. 15 , 17 , 182 , 183 , 184 , 185 SAXS data for full‐length MDA5 indicate that the CARDs have an open and flexible conformation and that they are not sequestered in the autoinhibited state like the CARDs of RIG‐I. 24 , 182 , 186 , 187 Additionally, the CTD has a different function in MDA5, as it has no RNA binding affinity and instead is required for cooperative filament assembly. 72 , 188 , 189 , 190 Similar to RIG‐I, RNA binding causes the MDA CARDs to interact with MAVS, ultimately leading to the transcription of the genes encoding IFNs. 146 , 150 , 191 Crystal structures have shown that MDA5 can bind short dsRNA as a monomer, with a domain organization that resembles those previously shown for RIG‐I/RNA complexes. 38 , 192 Whereas MDA5 forms a C‐ring structure that binds to the internal duplex structure of dsRNA, RIG‐I has been shown to form an O‐ring structure that caps the end of dsRNA. 38 , 186 , 192 , 193 These differences in RNA binding properties are reflected in the substrate specificity and mode of signaling. MDA5 senses longer dsRNA species and secondary structures and shows no requirement for terminal di‐or triphosphates. 61 , 63 , 72 , 73 , 77 , 192 , 194 Cooperative binding of MDA5 in a head‐to‐tail arrangement of dsRNA induces assembly of MDA5 into helical filaments, which nucleate the assembly of MAVS into an active polymeric form. 72 , 186 , 192 , 195 The N‐terminal tandem CARDs of MDA5, which are essential for signaling, cannot be visualized in structures of MDA5 filaments. 188 ATP hydrolysis efficiently promotes MDA5/RNA filament disassembly, but on long dsRNA, due to stabilization by the filament architecture, not every ATP hydrolysis event triggers MDA5 dissociation from RNA. 72 , 195 Protease protection assays show that ATP hydrolysis regulates the conformation of the CARDs of MDA5 and the increased protection of CARDs in long dsRNA is consistent with its oligomerization within the filament. 192 In the current proposed model for MDA5 in IFN induction, the tandem CARDs form patches along MDA5 filaments, inducing oligomerization into elongated structures that activate MAVS. 72 , 125 , 192 , 196
Less is known about the third RLR family member, LGP2 (the laboratory of genetics and physiology 2), which is structurally homologous to RIG‐I and MDA5, but lacks CARDs. Despite its lack of signaling domains, LGP2 is consistently observed to play an important role in modulating signaling by MDA5 and RIG‐I. 14 , 17 , 153 , 197 , 198 , 199 , 200 Crystal structures of chicken LGP2 with dsRNA show that the helicase domain of LGP2 resembles that of MDA5, although the CTD of LGP2 caps the ends of dsRNA in a fashion similar to that of RIG‐I. 38 , 192 , 193 LGP2 displays high RNA binding affinity and the isolated CTD of LGP2 has a comparatively higher RNA binding affinity than the CTD of RIG‐I. 189 , 193 , 201 Unique among the RLRs, LGP2 can recognize various types of RNAs, irrespective of length or 5′ phosphate ends, which gives LGP2 more versatility when binding to viral RNAs. 189 , 193 , 201 , 202 Structural analysis by limited protease digestion assay suggests that LGP2 induces significant conformational changes in MDA5 in the presence of RNA, promoting presentation of its CARDs. 195 In addition, LGP2 mutants that are defective in ATP hydrolysis fail to enhance MDA5‐mediated signaling, 203 indicating that ATPase activity of LGP2 is important when responding to viral infection. Although the exact mechanism by which LGP2 enhances MDA5‐mediated antiviral signaling is still unclear, accumulating evidence in the form of ATP hydrolysis assays, electrophoretic mobility shift assays, and electron microscopy suggest that LGP2 and MDA5 work together to detect viral RNA and generate a stronger antiviral response. 103 , 195 , 204 LGP2 may facilitate MDA5/RNA interactions or regulate MDA5 filament assembly, presumably through heterocomplex formation, potentially resulting in alternative MDA5 filaments that are somehow active for signaling. 195 , 197 , 200 , 203 Taken together, the functional diversity of RLRs may be linked to their inherent differences in RNA recognition, binding, and signaling mechanism. RIG‐I, MDA5, and LGP2 appear to play non‐redundant roles by recognizing complementary groups and distinct features of RNAs, thereby providing a complete surveillance system that detects a broad spectrum of pathogenic RNAs.
11. CONCLUDING REMARKS
The carefully coordinated mechanical properties of RIG‐I enable it to serve as our front‐line response against the most deadly viral pathogens while maintaining a selectivity that prevents it from turning on its host. RIG‐I is an exquisitely sensitive molecular switch that adopts different functional conformations in response to stimuli. Viral p3dsRNA is the trigger for CARDs presentation and the initiation of IFN signaling, while ATP is the trigger for rapid release of host RNAs and active proofreading. A wealth of new functional data from cell biology, imaging, and whole animal experiments, combined with ever more sophisticated structural biology and biochemical methods, has made it possible to refine the working models for RIG‐I activation and selectivity. These reveal that the RLRs as a group recognize and respond to RNAs quite differently, thereby providing broad protection against a diversity of viral threats. But despite these advances, central questions remain unanswered, and concrete physical evidence for physical models of RLR function is still lacking. For example, there is no direct, functional information on the interplay between RIG‐I CARDs and those of MAVS and we have never visualized the subcellular structures that actually stimulate signaling by RLRs. Information on the specific functional role for RIG‐I post‐translational modifications and on the participation of accessory cofactors (particularly those that may be cell‐type or tissue‐specific) remain unclear and largely speculative. But these missing pieces only serve to underscore the many ways that RLR signaling remains a vibrant and biomedically critical area of research. Indeed, study of RLRs will reveal much about the fundamental biology of innate immunity, and their pharmacological manipulation promises to bring new therapies for cancer, infection, and autoimmunity.
CONFLICT OF INTEREST
Yale University has submitted patent applications for Stem‐loop RNAs. AMP has founded a company (RIGImmune) to develop SLRs as therapeutic agents.
AUTHOR CONTRIBUTIONS
DT and WW designed the figures. DT, WW, and AMP designed the review organization. All authors contributed to the writing, revision, and editing of the manuscript.
ACKNOWLEDGEMENTS
We would like to thank Akiko Iwasaki for offering valuable comments on the manuscript. The Pyle laboratory is supported by NIH grants, and by Howard Hughes Medical Institute. DT is supported by a Pre‐doctoral Fellowship in Virology (T32AI055403), and by the Gruber Foundation. is a AMP Howard Hughes Medical Institute Investigator. Figure 2 was assembled using BioRender software.
Thoresen D, Wang W, Galls D, Guo R, Xu L, Pyle AM. The molecular mechanism of RIG‐I activation and signaling. Immunol Rev. 2021;304:154–168. 10.1111/imr.13022
This article is part of a series of reviews covering RNA Regulation in Immunity appearing in Volume 304 of Immunological Reviews.
REFERENCES
- 1. Chow KT, Gale M Jr, Loo YM. RIG‐I and other RNA sensors in antiviral immunity. Annu Rev Immunol. 2018;36:667‐694. [DOI] [PubMed] [Google Scholar]
- 2. Rehwinkel J, Gack MU. RIG‐I‐like receptors: their regulation and roles in RNA sensing. Nat Rev Immunol. 2020;20:537‐551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brisse M, Ly H. Comparative structure and function analysis of the RIG‐I‐like receptors: RIG‐I and MDA5. Front Immunol. 2019;10:1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fitzgerald ME, Rawling DC, Vela A, Pyle AM. An evolving arsenal: viral RNA detection by RIG‐I‐like receptors. Curr Opin Microbiol. 2014;20:76‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Barral PM, Sarkar D, Su Z‐Z, et al. Functions of the cytoplasmic RNA sensors RIG‐I and MDA‐5: key regulators of innate immunity. Pharmacol Ther. 2009;124:219‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fitzgerald ME, Vela A, Pyle AM. Dicer‐related helicase 3 forms an obligate dimer for recognizing 22G‐RNA. Nucleic Acids Res. 2014;42:3919‐3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Luo D, Kohlway A, Pyle AM. Duplex RNA activated ATPases (DRAs): platforms for RNA sensing, signaling and processing. RNA Biol. 2013;10:111‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jankowsky E. RNA helicases at work: binding and rearranging. Trends Biochem Sci. 2011;36:19‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Elion DL, Cook RS. Harnessing RIG‐I and intrinsic immunity in the tumor microenvironment for therapeutic cancer treatment. Oncotarget. 2018;9:29007‐29017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Loo YM, Gale M Jr. Immune signaling by RIG‐I‐like receptors. Immunity. 2011;34:680‐692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Duan Y, Li Z, Cheng S,et al. Nasopharyngeal carcinoma progression is mediated by EBER‐triggered inflammation via the RIG‐I pathway. Cancer Lett. 2015;361:67‐74. [DOI] [PubMed] [Google Scholar]
- 12. Wu Y, Wu X, Wu L, Wang X, Liu Z. The anticancer functions of RIG‐I‐like receptors, RIG‐I and MDA5, and their applications in cancer therapy. Transl Res. 2017;190:51‐60. [DOI] [PubMed] [Google Scholar]
- 13. Liu T‐X, Zhang J‐W, Tao J, et al. Gene expression networks underlying retinoic acid–induced differentiation of acute promyelocytic leukemia cells. Blood. 2000;96:1496‐1504. [PubMed] [Google Scholar]
- 14. Cui Y, Li M, Walton KD, et al. The Stat3/5 locus encodes novel endoplasmic reticulum and helicase‐like proteins that are preferentially expressed in normal and neoplastic mammary tissue. Genomics. 2001;78:129‐134. [DOI] [PubMed] [Google Scholar]
- 15. Kang D‐C, Gopalkrishnan RV, Wu Q, et al. mda‐5: An interferon‐inducible putative RNA helicase with double‐stranded RNA‐dependent ATPase activity and melanoma growth‐suppressive properties. Proc Natl Acad Sci USA. 2002;99:637‐642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yoneyama M, Kikuchi M, Natsukawa T, et al. The RNA helicase RIG‐I has an essential function in double‐stranded RNA‐induced innate antiviral responses. Nat Immunol. 2004;5:730‐737. [DOI] [PubMed] [Google Scholar]
- 17. Yoneyama M, Kikuchi M, Matsumoto K, et al. Shared and unique functions of the DExD/H‐box helicases RIG‐I, MDA5, and LGP2 in antiviral innate immunity. J Immunol. 2005;175:2851‐2858. [DOI] [PubMed] [Google Scholar]
- 18. Ren X, Linehan MM, Iwasaki A, Pyle AM. RIG‐I selectively discriminates against 5'‐Monophosphate RNA. Cell Rep 2019;26:2019‐2027 e2014. [DOI] [PubMed] [Google Scholar]
- 19. Schlee M, Roth A, Hornung V, et al. Recognition of 5' triphosphate by RIG‐I helicase requires short blunt double‐stranded RNA as contained in panhandle of negative‐strand virus. Immunity. 2009;31:25‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kohlway A, Luo D, Rawling DC, Ding SC, Pyle AM. Defining the functional determinants for RNA surveillance by RIG‐I. EMBO Rep. 2013;14:772‐779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Devarkar SC, Wang C, Miller MT, et al. Structural basis for m7G recognition and 2'‐O‐methyl discrimination in capped RNAs by the innate immune receptor RIG‐I. Proc Natl Acad Sci USA. 2016;113:596‐601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jiang F, Ramanathan A, Miller MT, et al. Structural basis of RNA recognition and activation by innate immune receptor RIG‐I. Nature. 2011;479:423‐427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Luo D, Kohlway A, Vela A, Pyle AM. Visualizing the determinants of viral RNA recognition by innate immune sensor RIG‐I. Structure. 2012;20:1983‐1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kowalinski E, Lunardi T, McCarthy AA, et al. Structural basis for the activation of innate immune pattern‐recognition receptor RIG‐I by viral RNA. Cell. 2011;147:423‐435. [DOI] [PubMed] [Google Scholar]
- 25. Reikine S, Nguyen JB, Modis Y. Pattern recognition and signaling mechanisms of RIG‐I and MDA5. Front Immunol. 2014;5:342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chan YK, Gack MU. RIG‐I‐like receptor regulation in virus infection and immunity. Curr Opin Virol. 2015;12:7‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maelfait J, Beyaert R. Emerging role of ubiquitination in antiviral RIG‐I signaling. Microbiol Mol Biol Rev. 2012;76:33‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Uhlen M, Fagerberg L, Hallstrom BM, et al. Proteomics. Tissue‐based map of the human proteome. Science 2015;347:1260419. [DOI] [PubMed] [Google Scholar]
- 29. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805‐820. [DOI] [PubMed] [Google Scholar]
- 30. Goubau D, Deddouche S, Reis e Sousa C. Cytosolic sensing of viruses. Immunity. 2013;38:855‐869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hartmann G. Nucleic acid immunity. Adv Immunol. 2017;133:121‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li W, Chen H, Sutton T, Obadan A, Perez DR. Interactions between the influenza A virus RNA polymerase components and retinoic acid‐inducible gene I. J Virol. 2014;88:10432‐10447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu G, Lu Y, Thulasi Raman SN, et al. Nuclear‐resident RIG‐I senses viral replication inducing antiviral immunity. Nat Commun. 2018;9:3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mateer EJ, Paessler S, Huang C. Visualization of double‐stranded RNA colocalizing with pattern recognition receptors in arenavirus infected cells. Front Cell Infect Microbiol. 2018;8:251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Esser‐Nobis K, Hatfield LD, Gale M Jr. Spatiotemporal dynamics of innate immune signaling via RIG‐I‐like receptors. Proc Natl Acad Sci USA. 2020;117:15778‐15788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Thul PJ, Åkesson L, Wiking M, et al. A subcellular map of the human proteome. Science. 2017;356:eaal3321. [DOI] [PubMed] [Google Scholar]
- 37. Vela A, Fedorova O, Ding SC, Pyle AM. The thermodynamic basis for viral RNA detection by the RIG‐I innate immune sensor. J Biol Chem. 2012;287:42564‐42573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Luo D, Ding SC, Vela A, et al. Structural insights into RNA recognition by RIG‐I. Cell. 2011;147:409‐422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zheng J, Yong HY, Panutdaporn N, et al. High‐resolution HDX‐MS reveals distinct mechanisms of RNA recognition and activation by RIG‐I and MDA5. Nucleic Acids Res. 2015;43:1216‐1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shah N, Beckham SA, Wilce JA, Wilce MCJ. Combined roles of ATP and small hairpin RNA in the activation of RIG‐I revealed by solution‐based analysis. Nucleic Acids Res. 2018;46:3169‐3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zheng J, Wang C, Chang MR, et al. HDX‐MS reveals dysregulated checkpoints that compromise discrimination against self RNA during RIG‐I mediated autoimmunity. Nat Commun. 2018;9:5366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Devarkar SC, Schweibenz B, Wang C, Marcotrigiano J, Patel SS. RIG‐I uses an ATPase‐powered translocation‐throttling mechanism for kinetic proofreading of RNAs and oligomerization. Mol Cell. 2018;72:355‐368.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rawling DC, Fitzgerald ME, Pyle AM. Establishing the role of ATP for the function of the RIG‐I innate immune sensor. Elife 2015;4:e09391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ren X, Linehan MM, Iwasaki A, Pyle AM. RIG‐I recognition of RNA targets: the influence of terminal base pair sequence and overhangs on affinity and signaling. Cell Rep. 2019;29:3807‐3815.e3. [DOI] [PubMed] [Google Scholar]
- 45. Jiang M, Zhang S, Yang Z, et al. Self‐recognition of an inducible host lncRNA by RIG‐I feedback restricts innate immune response. Cell. 2018;173:906‐919.e13. [DOI] [PubMed] [Google Scholar]
- 46. Chen YG, Kim MV, Chen X, et al. Sensing self and foreign circular RNAs by intron identity. Mol Cell. 2017;67:228‐238.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen YG, Chen R, Ahmad S, et al. N6‐methyladenosine modification controls circular RNA immunity. Mol Cell. 2019;76(1):96‐109.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rawling DC, Pyle AM. Parts, assembly and operation of the RIG‐I family of motors. Curr Opin Struct Biol. 2014;25:25‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dickey TH, Song B, Pyle AM. RNA binding activates RIG‐I by releasing an autorepressed signaling domain. Sci Adv 2019;5:eaax3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Louber J, Brunel J, Uchikawa E, Cusack S, Gerlier D. Kinetic discrimination of self/non‐self RNA by the ATPase activity of RIG‐I and MDA5. BMC Biol. 2015;13:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kawai T, Akira S. Toll‐like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637‐650. [DOI] [PubMed] [Google Scholar]
- 52. Schmidt A, Schwerd T, Hamm W, et al. 5'‐triphosphate RNA requires base‐paired structures to activate antiviral signaling via RIG‐I. Proc Natl Acad Sci USA. 2009;106:12067‐12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kolakofsky D, Kowalinski E, Cusack S. A structure‐based model of RIG‐I activation. RNA. 2012;18:2118‐2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Linehan MM, Dickey TH, Molinari ES, et al. A minimal RNA ligand for potent RIG‐I activation in living mice. Sci Adv. 2018;4:e1701854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Elion DL, Jacobson ME, Hicks DJ, et al. Therapeutically active RIG‐I agonist induces immunogenic tumor cell killing in breast cancers. Cancer Res. 2018;78:6183‐6195. [DOI] [PubMed] [Google Scholar]
- 56. Yong HY, et al. Structure‐guided design of immunomodulatory RNAs specifically targeting the cytoplasmic viral RNA sensor RIG‐I. FEBS Lett. 2019;593:3003‐3014. [DOI] [PubMed] [Google Scholar]
- 57. Ho V, Yong HY, Chevrier M, et al. RIG‐I activation by a designer short RNA ligand protects human immune cells against Dengue virus infection without causing cytotoxicity. J Virol. 2019;93:e00102–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Luke JM, Simon GG, Soderholm J, et al. Coexpressed RIG‐I agonist enhances humoral immune response to influenza virus DNA vaccine. J Virol. 2011;85:1370‐1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ranjith‐Kumar CT, Murali A, Dong W, et al. Agonist and antagonist recognition by RIG‐I, a cytoplasmic innate immunity receptor. J Biol Chem. 2009;284:1155‐1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Marq JB, Hausmann S, Veillard N, Kolakofsky D, Garcin D. Short double‐stranded RNAs with an overhanging 5' ppp‐nucleotide, as found in arenavirus genomes, act as RIG‐I decoys. J Biol Chem. 2011;286:6108‐6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hornung V, Ellegast J, Kim S, et al. 5'‐Triphosphate RNA is the ligand for RIG‐I. Science. 2006;314:994‐997. [DOI] [PubMed] [Google Scholar]
- 62. Wang Y, Ludwig J, Schuberth C, et al. Structural and functional insights into 5'‐ppp RNA pattern recognition by the innate immune receptor RIG‐I. Nat Struct Mol Biol. 2010;17:781‐787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Goubau D, Schlee M, Deddouche S, et al. Antiviral immunity via RIG‐I‐mediated recognition of RNA bearing 5'‐diphosphates. Nature. 2014;514:372‐375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Li N, You X, Chen T, et al. Global profiling of miRNAs and the hairpin precursors: insights into miRNA processing and novel miRNA discovery. Nucleic Acids Res. 2013;41:3619‐3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wingfield PT, Stahl SJ, Payton MA, et al. HIV‐1 Rev expressed in recombinant Escherichia coli: purification, polymerization, and conformational properties. Biochemistry. 1991;30:7527‐7534. [DOI] [PubMed] [Google Scholar]
- 66. Jensen DE, von Hippel PH. DNA "melting" proteins. I. Effects of bovine pancreatic ribonuclease binding on the conformation and stability of DNA. J Biol Chem. 1976;251:7198‐7214. [PubMed] [Google Scholar]
- 67. Swisher JFA, Rand E, Cedar H, Pyle AM. Analysis of putative RNase sensitivity and protease insensitivity of demethylation activity in extracts from rat myoblasts. Nucleic Acids Res. 1998;26:5573‐5580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jiang X, Muthusamy V, Fedorova O, et al. Intratumoral delivery of RIG‐I agonist SLR14 induces robust antitumor responses. J Exp Med. 2019;216:2854‐2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Heidegger S, Wintges A, Stritzke F, et al. RIG‐I activation is critical for responsiveness to checkpoint blockade. Sci Immunol. 2019;4:eaau8943. [DOI] [PubMed] [Google Scholar]
- 70. Cheng YS, Xu F. Anticancer function of polyinosinic‐polycytidylic acid. Cancer Biol Ther. 2010;10:1219‐1223. [DOI] [PubMed] [Google Scholar]
- 71. Louber J, Kowalinski E, Bloyet L‐M, et al. RIG‐I self‐oligomerization is either dispensable or very transient for signal transduction. PLoS One. 2014;9:e108770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Peisley A, Lin C, Wu B, et al. Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition. Proc Natl Acad Sci USA. 2011;108:21010‐21015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Peisley A, Jo MH, Lin C, et al. Kinetic mechanism for viral dsRNA length discrimination by MDA5 filaments. Proc Natl Acad Sci USA. 2012;109:E3340‐3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Matranga C, Pyle AM. Double‐stranded RNA‐dependent ATPase DRH‐3: insight into its role in RNAsilencing in Caenorhabditis elegans . J Biol Chem. 2010;285:25363‐25371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Samanta M, Iwakiri D, Kanda T, Imaizumi T, Takada K. EB virus‐encoded RNAs are recognized by RIG‐I and activate signaling to induce type I IFN. EMBO J. 2006;25:4207‐4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Chiu YH, Macmillan JB, Chen ZJ. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG‐I pathway. Cell. 2009;138:576‐591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Rehwinkel J, Tan CP, Goubau D, et al. RIG‐I detects viral genomic RNA during negative‐strand RNA virus infection. Cell. 2010;140:397‐408. [DOI] [PubMed] [Google Scholar]
- 78. Schnell G, Loo YM, Marcotrigiano J, Gale M Jr. Uridine composition of the poly‐U/UC tract of HCV RNA defines non‐self recognition by RIG‐I. PLoS Pathog. 2012;8:e1002839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Uzri D, Greenberg HB. Characterization of rotavirus RNAs that activate innate immune signaling through the RIG‐I‐like receptors. PLoS One. 2013;8:e69825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Sato S, Li K, Kameyama T, et al. The RNA sensor RIG‐I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity. 2015;42:123‐132. [DOI] [PubMed] [Google Scholar]
- 81. Sanchez David RY, Combredet C, Sismeiro O, et al. Comparative analysis of viral RNA signatures on different RIG‐I‐like receptors. Elife. 2016;5:e11275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chazal M, Beauclair G, Gracias S, et al. RIG‐I recognizes the 5' region of Dengue and Zika Virus genomes. Cell Rep. 2018;24:320‐328. [DOI] [PubMed] [Google Scholar]
- 83. Zhang Y, Dittmer DP, Mieczkowski PA, et al. RIG‐I Detects Kaposi's sarcoma‐associated herpesvirus transcripts in a RNA polymerase III‐independent manner. MBio 2018;9:e00823–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Sooryanarain H, Heffron CL, Meng XJ. The U‐Rich untranslated region of the hepatitis E virus induces differential type I and type III interferon responses in a host cell‐dependent manner. MBio 2020;11.e03103–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Weber M, Gawanbacht A, Habjan M, et al. Incoming RNA virus nucleocapsids containing a 5'‐triphosphorylated genome activate RIG‐I and antiviral signaling. Cell Host Microbe. 2013;13:336‐346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Liu G, Park HS, Pyo HM, Liu Q, Zhou Y. Influenza A virus panhandle structure is directly involved in RIG‐I activation and interferon induction. J Virol. 2015;89:6067‐6079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Chiang C, Beljanski V, Yin K, et al. Sequence‐specific modifications enhance the broad‐spectrum antiviral response activated by RIG‐I agonists. J Virol. 2015;89:8011‐8025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Goulet M‐L, Olagnier D, Xu Z, et al. Systems analysis of a RIG‐I agonist inducing broad spectrum inhibition of virus infectivity. PLoS Pathog. 2013;9:e1003298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hoffmann E, Stech J, Guan Y, Webster RG, Perez DR. Universal primer set for the full‐length amplification of all influenza A viruses. Archiv Virol. 2001;146:2275‐2289. [DOI] [PubMed] [Google Scholar]
- 90. Xu J, Mercado‐López X, Grier JT, et al. Identification of a natural viral RNA motif that optimizes sensing of viral RNA by RIG‐I. MBio 2015;6:e01265‐01215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Velthuis AJW, Long JC, Bauer DLV, et al. Mini viral RNAs act as innate immune agonists during influenza virus infection. Nat Microbiol. 2018;3:1234‐1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Baum A, Sachidanandam R, Garcia‐Sastre A. Preference of RIG‐I for short viral RNA molecules in infected cells revealed by next‐generation sequencing. Proc Natl Acad Sci USA. 2010;107:16303‐16308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Zhao Y, Ye X, Dunker W, Song Y, Karijolich J. RIG‐I like receptor sensing of host RNAs facilitates the cell‐intrinsic immune response to KSHV infection. Nat Commun. 2018;9:4841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Chiang JJ, Sparrer KMJ, van Gent M, et al. Viral unmasking of cellular 5S rRNA pseudogene transcripts induces RIG‐I‐mediated immunity. Nat Immunol. 2018;19:53‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Choi JH, Burke JM, Szymanik KH, et al. DUSP11‐mediated control of 5′‐triphosphate RNA regulates RIG‐I sensitivity. Genes Dev. 2020;34:1697‐1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Cheng Y, Schorey JS. Extracellular vesicles deliver Mycobacterium RNA to promote host immunity and bacterial killing. EMBO Rep. 2019;20:e46613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Schmolke M, Patel JR, de Castro E, et al. RIG‐I detects mRNA of intracellular Salmonella enterica serovar Typhimurium during bacterial infection. MBio. 2014;5:e01006‐01014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Hagmann CA, Herzner AM, Abdullah Z, et al. RIG‐I detects triphosphorylated RNA of Listeria monocytogenes during infection in non‐immune cells. PLoS One. 2013;8:e62872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Ferreira CR, Crow YJ, Gahl WA, et al. DDX58 and classic singleton‐merten syndrome. J Clin Immunol. 2019;39:75‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Rawling DC, Kohlway AS, Luo D, Ding SC, Pyle AM. The RIG‐I ATPase core has evolved a functional requirement for allosteric stabilization by the Pincer domain. Nucleic Acids Res. 2014;42:11601‐11611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Lu C, Xu H, Ranjith‐Kumar CT, et al. The structural basis of 5' triphosphate double‐stranded RNA recognition by RIG‐I C‐terminal domain. Structure. 2010;18:1032‐1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Lässig C, Lammens K, Gorenflos López JL, et al. Unified mechanisms for self‐RNA recognition by RIG‐I Singleton‐Merten syndrome variants. Elife. 2018;7:e38958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Jang MA, Kim EK, Now H, et al. Mutations in DDX58, which encodes RIG‐I, cause atypical Singleton‐Merten syndrome. Am J Hum Genet. 2015;96:266‐274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Lässig C, Matheisl S, Sparrer KMJ, et al. ATP hydrolysis by the viral RNA sensor RIG‐I prevents unintentional recognition of self‐RNA. Elife. 2015;4:e10859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Ramanathan Anand, Devarkar Swapnil C., Jiang Fuguo, et al. The autoinhibitory CARD2‐Hel2i Interface of RIG‐I governs RNA selection. Nucleic Acids Research. 2016;44(2):896–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Fitzgerald ME, Rawling DC, Potapova O, et al. Selective RNA targeting and regulated signaling by RIG‐I is controlled by coordination of RNA and ATP binding. Nucleic Acids Res. 2017;45:1442‐1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Cao W, Coman MM, Ding S, et al. Mechanism of Mss116 ATPase reveals functional diversity of DEAD‐Box proteins. J Mol Biol. 2011;409:399‐414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Lu C, MacDougall M. RIG‐I‐like receptor signaling in singleton‐merten syndrome. Front Genet. 2017;8:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Shigemoto T, Kageyama M, Hirai R, et al. Identification of loss of function mutations in human genes encoding RIG‐I and MDA5: implications for resistance to type I diabetes. J Biol Chem. 2009;284:13348‐13354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Yang S, Deng P, Zhu Z, et al. Adenosine deaminase acting on RNA 1 limits RIG‐I RNA detection and suppresses IFN production responding to viral and endogenous RNAs. J Immunol. 2014;193:3436‐3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Lamers MM, van den Hoogen BG, Haagmans BL. ADAR1: "Editor‐in‐Chief" of cytoplasmic innate immunity. Front Immunol. 2019;10:1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Liddicoat BJ, Chalk AM, Walkley CR. ADAR1, inosine and the immune sensing system: distinguishing self from non‐self. Wiley Interdiscip Rev RNA. 2016;7:157‐172. [DOI] [PubMed] [Google Scholar]
- 113. Malathi K, Dong B, Gale M Jr, Silverman RH. Small self‐RNA generated by RNase L amplifies antiviral innate immunity. Nature. 2007;448:816‐819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Manivannan P, Siddiqui MA, Malathi K. RNase L amplifies interferon signaling by inducing protein kinase R‐mediated antiviral stress granules. J Virol. 2020;94:e00205–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Myong S, Cui S, Cornish PV, et al. Cytosolic viral sensor RIG‐I is a 5'‐triphosphate‐dependent translocase on double‐stranded RNA. Science. 2009;323:1070‐1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Jankowsky E, Gross CH, Shuman S, Pyle AM. Active disruption of an RNA‐protein interaction by a DExH/D RNA helicase. Science. 2001;291:121‐125. [DOI] [PubMed] [Google Scholar]
- 117. Pyle AM. Translocation and unwinding mechanisms of RNA and DNA helicases. Annu Rev Biophys. 2008;37:317‐336. [DOI] [PubMed] [Google Scholar]
- 118. Pyle AM. RNA helicases and remodeling proteins. Curr Opin Chem Biol. 2011;15:636‐642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Chiang C, Gack MU. Post‐translational control of intracellular pathogen sensing pathways. Trends Immunol. 2017;38:39‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Gack MU, Shin YC, Joo C‐H, et al. TRIM25 RING‐finger E3 ubiquitin ligase is essential for RIG‐I‐mediated antiviral activity. Nature. 2007;446:916. [DOI] [PubMed] [Google Scholar]
- 121. Oshiumi H, Miyashita M, Inoue N, et al. The ubiquitin ligase riplet is essential for RIG‐I‐dependent innate immune responses to RNA virus infection. Cell Host Microbe. 2010;8:496‐509. [DOI] [PubMed] [Google Scholar]
- 122. Wies E, Wang MK, Maharaj NP, et al. Dephosphorylation of the RNA sensors RIG‐I and MDA5 by the phosphatase PP1 is essential for innate immune signaling. Immunity. 2013;38:437‐449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Arimoto K‐I, Takahashi H, Hishiki T, et al. Negative regulation of the RIG‐I signaling by the ubiquitin ligase RNF125. Proc Natl Acad Sci USA. 2007;104:7500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Oshiumi H, Matsumoto M, Hatakeyama S, Seya T. Riplet/RNF135, a RING finger protein, ubiquitinates RIG‐I to promote interferon‐β induction during the early phase of viral infection*. J Biol Chem. 2009;284:807‐817. [DOI] [PubMed] [Google Scholar]
- 125. Zeng W, Sun L, Jiang X, et al. Reconstitution of the RIG‐I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell. 2010;141:315‐330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Peisley A, Wu B, Xu H, Chen ZJ, Hur S. Structural basis for ubiquitin‐mediated antiviral signal activation by RIG‐I. Nature. 2014;509:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Choudhury NR, Nowak JS, Zuo J, et al. Trim25 is an RNA‐specific activator of Lin28a/TuT4‐mediated uridylation. Cell Rep. 2014;9:1265‐1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Manokaran G, Finol E, Wang C, et al. Dengue subgenomic RNA binds TRIM25 to inhibit interferon expression for epidemiological fitness. Science. 2015;350:217‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Meyerson NR, Zhou L, Guo YR, et al. Nuclear TRIM25 specifically targets influenza virus ribonucleoproteins to block the onset of RNA chain elongation. Cell Host Microbe 2017;22:627‐638.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Cadena C, Ahmad S, Xavier A, et al. Ubiquitin‐dependent and ‐independent roles of E3 ligase RIPLET in innate immunity. Cell. 2019;177:1187‐1200.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Hayman TJ, Hsu AC, Kolesnik TB, et al. RIPLET, and not TRIM25, is required for endogenous RIG‐I‐dependent antiviral responses. Immunol Cell Biol. 2019;97:840‐852. [DOI] [PubMed] [Google Scholar]
- 132. Shi Y, Yuan B, Zhu W, et al. Ube2D3 and Ube2N are essential for RIG‐I‐mediated MAVS aggregation in antiviral innate immunity. Nat Commun. 2017;8:15138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Cui J, Song Y, Li Y, et al. USP3 inhibits type I interferon signaling by deubiquitinating RIG‐I‐like receptors. Cell Res. 2014;24:400‐416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Fan Y, Mao R, Yu Y, et al. USP21 negatively regulates antiviral response by acting as a RIG‐I deubiquitinase. J Exp Med. 2014;211:313‐328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Friedman CS, O’Donnell MA, Legarda‐Addison D, et al. The tumour suppressor CYLD is a negative regulator of RIG‐I‐mediated antiviral response. EMBO Rep. 2008;9:930‐936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Gack MU, Nistal‐Villan E, Inn KS, Garcia‐Sastre A, Jung JU. Phosphorylation‐mediated negative regulation of RIG‐I antiviral activity. J Virol. 2010;84:3220‐3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Maharaj NP, Wies E, Stoll A, Gack MU. Conventional protein kinase C‐alpha (PKC‐alpha) and PKC‐beta negatively regulate RIG‐I antiviral signal transduction. J Virol. 2012;86:1358‐1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Sun Z, Ren H, Liu Y, Teeling JL, Gu J. Phosphorylation of RIG‐I by casein kinase II inhibits its antiviral response. J Virol. 2011;85:1036‐1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Kawai T, Takahashi K, Sato S, et al. IPS‐1, an adaptor triggering RIG‐I‐ and Mda5‐mediated type I interferon induction. Nat Immunol. 2005;6:981‐988. [DOI] [PubMed] [Google Scholar]
- 140. Meylan E, Curran J, Hofmann K, et al. Cardif is an adaptor protein in the RIG‐I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167‐1172. [DOI] [PubMed] [Google Scholar]
- 141. Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF‐kappaB and IRF 3. Cell. 2005;122:669‐682. [DOI] [PubMed] [Google Scholar]
- 142. Xu LG, et al. VISA is an adapter protein required for virus‐triggered IFN‐beta signaling. Mol Cell. 2005;19:727‐740. [DOI] [PubMed] [Google Scholar]
- 143. Sun Q, Sun L, Liu H‐H, et al. The specific and essential role of MAVS in antiviral innate immune responses. Immunity. 2006;24:633‐642. [DOI] [PubMed] [Google Scholar]
- 144. Dixit E, Boulant S, Zhang Y, et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell. 2010;141:668‐681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Horner SM, Liu HM, Park HS, Briley J, Gale M Jr. Mitochondrial‐associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc Natl Acad Sci USA. 2011;108:14590‐14595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Hou F, Sun L, Zheng H, et al. MAVS forms functional prion‐like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146:448‐461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Brubaker SW, Gauthier AE, Mills EW, Ingolia NT, Kagan JC. A Bicistronic MAVS transcript highlights a class of truncated variants in antiviral immunity. Cell. 2014;156:800‐811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Liu S, Chen J, Cai X, et al. MAVS recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. Elife. 2013;2:e00785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Cai X, Chen J, Xu H, et al. Prion‐like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell. 2014;156:1207‐1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Wu B, Peisley A, Tetrault D, et al. Molecular imprinting as a signal‐activation mechanism of the viral RNA sensor RIG‐I. Mol Cell. 2014;55:511‐523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Tang ED, Wang CY. MAVS self‐association mediates antiviral innate immune signaling. J Virol. 2009;83:3420‐3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Hwang MS, et al. MAVS polymers smaller than 80 nm induce mitochondrial membrane remodeling and interferon signaling. FEBS J. 2019;286:1543‐1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Quicke KM, Diamond MS, Suthar MS. Negative regulators of the RIG‐I‐like receptor signaling pathway. Eur J Immunol. 2017;47:615‐628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Saha SK, Pietras EM, He JQ, et al. Regulation of antiviral responses by a direct and specific interaction between TRAF3 and Cardif. EMBO J. 2006;25:3257‐3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Park HH. Structure of TRAF family: current understanding of receptor recognition. Front Immunol. 2018;9:1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Liu S, Cai X, Wu J, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science. 2015;347:aaa2630. [DOI] [PubMed] [Google Scholar]
- 157. Deng LWC, Spencer E, Yang L, et al. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin‐conjugating enzyme complex and a unique polyubiquitin chain. 2000. [DOI] [PubMed]
- 158. Fang R, Jiang Q, Zhou X, et al. MAVS activates TBK1 and IKKε through TRAFs in NEMO dependent and independent manner. PLoS Pathog. 2017;13:e1006720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Edwards MR, Slater L, Johnston SL. Signalling pathways mediating type I interferon gene expression. Microbes Infect. 2007;9:1245‐1251. [DOI] [PubMed] [Google Scholar]
- 160. Xu GLY, Li Q, Napolitano G, et al. Crystal structure of inhibitor of κB kinase β. 2011. [DOI] [PMC free article] [PubMed]
- 161. Henkel TMT, Alkalay I, Krönke M, Ben‐Neriah Y, Baeuerle PA. Rapid proteolysis of I kappa B‐alpha is necessary for activation of transcription factor NF‐kappa B. 1993. [DOI] [PubMed]
- 162. Fang R, Jiang Q, Zhou X, et al. MAVS activates TBK1 and IKKε through TRAFs in NEMO dependent and independent manner. PLoS Pathog. 2017;13:e1006720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Fitzgerald KA, Faia KL, Rowe DC, et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. 2003. [DOI] [PubMed]
- 164. Honda KTA, Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. 2006. [DOI] [PubMed]
- 165. Kato H, Sato S, Yoneyama M, et al. Cell type‐specific involvement of RIG‐I in antiviral response. Immunity. 2005;23:19‐28. [DOI] [PubMed] [Google Scholar]
- 166. Gong XY, Zhang QM, Gui JF, Zhang YB. SVCV infection triggers fish IFN response through RLR signaling pathway. Fish Shellfish Immunol. 2019;86:1058‐1063. [DOI] [PubMed] [Google Scholar]
- 167. Miranzo‐Navarro D, Magor KE. Activation of duck RIG‐I by TRIM25 is independent of anchored ubiquitin. PLoS One. 2014;9:e86968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Helin AS, Wille M, Atterby C, et al. Expression of immune genes RIG‐I and Mx in mallard ducks infected with low pathogenic avian influenza (LPAI): A dataset. Data in brief. 2018;18:1562‐1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Huo H, Wang Y, Wang D, et al. Duck RIG‐I restricts duck enteritis virus infection. Vet Microbiol. 2019;230:78‐85. [DOI] [PubMed] [Google Scholar]
- 170. Sun X, Li W, Liu E, et al. In vivo cellular and molecular study on duck spleen infected by duck Tembusu virus. Vet Microbiol. 2019;230:32‐44. [DOI] [PubMed] [Google Scholar]
- 171. Sun Y, et al. Goose MAVS functions in RIG‐I‐mediated IFN‐beta signaling activation. Dev Comp Immunol. 2019;93:58‐65. [DOI] [PubMed] [Google Scholar]
- 172. Sun Y, Ding N, Ding S, et al. Goose RIG‐I functions in innate immunity against Newcastle disease virus infections. Mol Immunol. 2013;53:321‐327. [DOI] [PubMed] [Google Scholar]
- 173. Civril F, Bennett M, Moldt M, et al. The RIG‐I ATPase domain structure reveals insights into ATP‐dependent antiviral signalling. EMBO Rep. 2011;12:1127‐1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Barber MR, Aldridge JR Jr, Webster RG, Magor KE. Association of RIG‐I with innate immunity of ducks to influenza. Proc Natl Acad Sci USA. 2010;107:5913‐5918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Xu L, Yu D, Fan Y, et al. Loss of RIG‐I leads to a functional replacement with MDA5 in the Chinese tree shrew. Proc Natl Acad Sci USA. 2016;113:10950‐10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Vasseur E, Patin E, Laval G, et al. The selective footprints of viral pressures at the human RIG‐I‐like receptor family. Hum Mol Genet. 2011;20:4462‐4474. [DOI] [PubMed] [Google Scholar]
- 177. Cagliani R, Forni D, Tresoldi C, et al. RIG‐I‐like receptors evolved adaptively in mammals, with parallel evolution at LGP2 and RIG‐I. J Mol Biol. 2014;426:1351‐1365. [DOI] [PubMed] [Google Scholar]
- 178. Lemos de Matos A, McFadden G, Esteves PJ. Evolution of viral sensing RIG‐I‐like receptor genes in Leporidae genera Oryctolagus, Sylvilagus, and Lepus. Immunogenetics. 2014;66:43‐52. [DOI] [PubMed] [Google Scholar]
- 179. Kato K, Ahmad S, Zhu Z, et al. Structural analysis of RIG‐I‐like receptors reveals ancient rules of engagement between diverse RNA helicases and TRIM ubiquitin ligases. Mol Cell. 2021;81:599‐613.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Kawai T, Akira S. Antiviral signaling through pattern recognition receptors. J Biochem. 2007;141:137‐145. [DOI] [PubMed] [Google Scholar]
- 181. Dias AG, Sampaio NG, Rehwinkel J. A balancing act: MDA5 in antiviral immunity and autoinflammation. Trends Microbiol. 2019;27:75‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Kato H, Takeuchi O, Sato S, et al. Differential roles of MDA5 and RIG‐I helicases in the recognition of RNA viruses. Nature. 2006;441:101‐105. [DOI] [PubMed] [Google Scholar]
- 183. Wu B, Hur S. How RIG‐I like receptors activate MAVS. Curr Opin Virol. 2015;12:91‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Andrejeva J, et al. The V proteins of paramyxoviruses bind the IFN‐inducible RNA helicase, mda‐5, and inhibit its activation of the IFN‐beta promoter. Proc Natl Acad Sci USA. 2004;101:17264‐17269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Gitlin L, Barchet W, Gilfillan S, et al. Essential role of mda‐5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci USA. 2006;103:8459‐8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Berke IC, Li Y, Modis Y. Structural basis of innate immune recognition of viral RNA. Cell Microbiol. 2013;15:386‐394. [DOI] [PubMed] [Google Scholar]
- 187. Berke IC, Modis Y. MDA5 cooperatively forms dimers and ATP‐sensitive filaments upon binding double‐stranded RNA. EMBO J. 2012;31:1714‐1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Yu Q, Qu K, Modis Y. Cryo‐EM structures of MDA5‐dsRNA filaments at different stages of ATP hydrolysis. Mol Cell 2018;72(6):999‐1012.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Takahasi K, Kumeta H, Tsuduki N, et al. Solution structures of cytosolic RNA sensor MDA5 and LGP2 C‐terminal domains: identification of the RNA recognition loop in RIG‐I‐like receptors. J Biol Chem. 2009;284:17465‐17474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Li X, Lu C, Stewart ,t al. Structural basis of double‐stranded RNA recognition by the RIG‐I like receptor MDA5. Arch Biochem Biophys. 2009;488:23‐33. [DOI] [PubMed] [Google Scholar]
- 191. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14:36‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Wu B, Peisley A, Richards C, et al. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell. 2013;152:276‐289. [DOI] [PubMed] [Google Scholar]
- 193. Uchikawa E, Lethier M, Malet H, et al. Structural analysis of dsRNA binding to anti‐viral pattern recognition receptors LGP2 and MDA5. Mol Cell. 2016;62:586‐602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Pichlmair A, Schulz O, Tan CP, et al. RIG‐I‐mediated antiviral responses to single‐stranded RNA bearing 5'‐phosphates. Science. 2006;314:997‐1001. [DOI] [PubMed] [Google Scholar]
- 195. Duic I, Tadakuma H, Harada Y, et al. Viral RNA recognition by LGP2 and MDA5, and activation of signaling through step‐by‐step conformational changes. Nucleic Acids Res. 2020;48:11664‐11674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Loo Y‐M, Fornek J, Crochet N, et al. Distinct RIG‐I and MDA5 signaling by RNA viruses in innate immunity. J Virol. 2008;82:335‐345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Rodriguez KR, Bruns AM, Horvath CM. MDA5 and LGP2: accomplices and antagonists of antiviral signal transduction. J Virol. 2014;88:8194‐8200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Zhu Z, Zhang X, Wang G, Zheng H. The laboratory of genetics and physiology 2: emerging insights into the controversial functions of this RIG‐I‐like receptor. Biomed Res Int. 2014;2:1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Satoh T, Kato H, Kumagai Y, et al. LGP2 is a positive regulator of RIG‐I‐ and MDA5‐mediated antiviral responses. Proc Natl Acad Sci USA. 2010;107:1512‐1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Bruns AM, Horvath CM. LGP2 synergy with MDA5 in RLR‐mediated RNA recognition and antiviral signaling. Cytokine. 2015;74:198‐206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Bamming D, Horvath CM. Regulation of signal transduction by enzymatically inactive antiviral RNA helicase proteins MDA5, RIG‐I, and LGP2. J Biol Chem. 2009;284:9700‐9712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Pippig DA, Hellmuth JC, Cui S, et al. The regulatory domain of the RIG‐I family ATPase LGP2 senses double‐stranded RNA. Nucleic Acids Res. 2009;37:2014‐2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Bruns AM, Leser GP, Lamb RA, Horvath CM. The innate immune sensor LGP2 activates antiviral signaling by regulating MDA5‐RNA interaction and filament assembly. Mol Cell. 2014;55:771‐781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Sanchez David RY, Combredet C, Najburg V, et al. LGP2 binds to PACT to regulate RIG‐I‐ and MDA5‐mediated antiviral responses. Sci Signal 2019;12:eaar3993. [DOI] [PubMed] [Google Scholar]