

Summary
Axicabtagene ciloleucel (axi‐cel) is an autologous anti‐CD19 chimeric antigen receptor (CAR) T‐cell therapy approved for relapsed or refractory large B‐cell lymphoma (R/R LBCL). To reduce axi‐cel–related toxicity, several exploratory safety management cohorts were added to ZUMA‐1 (NCT02348216), the pivotal phase 1/2 study of axi‐cel in refractory LBCL. Cohort 4 evaluated the rates and severity of cytokine release syndrome (CRS) and neurologic events (NEs) with earlier corticosteroid and tocilizumab use. Primary endpoints were incidence and severity of CRS and NEs. Patients received 2 × 106 anti‐CD19 CAR T cells/kg after conditioning chemotherapy. Forty‐one patients received axi‐cel. Incidences of any‐grade CRS and NEs were 93% and 61%, respectively (grade ≥ 3, 2% and 17%). There was no grade 4 or 5 CRS or NE. Despite earlier dosing, the cumulative cortisone‐equivalent corticosteroid dose in patients requiring corticosteroid therapy was lower than that reported in the pivotal ZUMA‐1 cohorts. With a median follow‐up of 14·8 months, objective and complete response rates were 73% and 51%, respectively, and 51% of treated patients were in ongoing response. Earlier and measured use of corticosteroids and/or tocilizumab has the potential to reduce the incidence of grade ≥ 3 CRS and NEs in patients with R/R LBCL receiving axi‐cel.
Keywords: large B‐cell lymphoma, axi‐cel, CAR T, corticosteroids, toxicity
Introduction
Axicabtagene ciloleucel (axi‐cel), an autologous anti‐CD19 chimeric antigen receptor (CAR) T‐cell therapy, is approved for treatment of relapsed/refractory large B‐cell lymphoma (R/R LBCL) after ≥ 2 previous systemic therapies. 1 , 2 Regulatory approvals were based on results from cohorts 1 + 2 (N = 101) of ZUMA‐1 (NCT02348216), which evaluated the efficacy and safety of axi‐cel in patients with refractory LBCL. 3 At a median follow‐up of 27·1 months (N = 101), axi‐cel demonstrated objective response, complete response (CR), and ongoing response rates of 83%, 58%, and 39%, respectively. 4 After a median of 51·1 months, median overall survival (OS) was 25·8 months, and the Kaplan–Meier (KM) estimate of the four‐year OS rate was 44%. 5
Cytokine release syndrome (CRS) and neurologic events (NEs) are common in patients receiving anti‐CD19 CAR T‐cell therapies and may be severe or life‐threatening. 6 , 7 , 8 At the two‐year follow‐up of the combined phase 1 + 2 ZUMA‐1 data (N = 108; data cutoff, August 11, 2018), grade ≥ 3 CRS was reported in 11% and grade ≥ 3 NEs were reported in 32% of patients. 4 Most CRS cases and NEs were manageable and reversible. 4 CRS and NEs are thought to initiate by T‐cell activation after CAR engagement of cognate antigen on target cells, leading to CAR T‐cell activation, proliferation, and cytokine release. Subsequent activation of a broad range of ‘bystander’ immune cells, including non‐CAR T cells and myeloid cells, may contribute to efficacy but also amplify these adverse events (AEs). 8 , 9 The interleukin (IL)‐6/IL‐6 receptor pathway has been directly implicated in pathogenesis of severe CRS, 3 , 10 and tocilizumab—a monoclonal antibody against IL‐6 receptor—is indicated for treatment of severe CAR T‐cell–induced CRS. 11
The etiology of NEs is incompletely elucidated and appears to be mediated by excess activation and mobilisation of T and myeloid cells, initiated by strong CAR‐triggered signaling in T cells. 12 , 13 Proposed mechanisms include peripheral cytokine release followed by cytokine diffusion across a breached blood‐brain‐barrier and/or translocation of activated anti‐CD19 CAR T cells and other immune cells—most notably myeloid cells—across the blood‐brain‐barrier, aided by vascular‐occlusive inflammatory injury and leading to a local inflammatory effect. 14 Recent evidence suggests that CD19 expression on brain mural cells, which surround the endothelium and are important for blood‐brain‐barrier integrity, may play a role in NEs associated with CAR T‐cell therapy. 15 Although unclear whether this occurs in all patients, increased blood‐brain‐barrier permeability has been reported in CAR T‐cell–treated patients with severe NEs or oedema. 16 In ZUMA‐1 cohorts 1 + 2, higher levels of CAR T‐cell expansion and serum proinflammatory cytokines (IL‐15, IL‐2, granulocyte‐macrophage colony‐stimulating factor [GM‐CSF]), were more tightly associated with severe NEs versus CRS. Other cytokines—such as interferon (IFN)‐γ, IL‐6, CXCL10, and CCL2—were significantly associated with both severe NEs and CRS. 3 Higher tumour burden (TB) was positively associated with higher rates of NEs and negatively associated with ongoing response rates at one year, 17 and with OS and progression‐free survival (PFS). 18
Exploratory safety management cohorts were added to phase 2 of ZUMA‐1 to evaluate toxicity management strategies in axi‐cel–treated patients. Cohort 3, which evaluated prophylactic tocilizumab on day 2 and the anticonvulsant levetiracetam starting on day 0, appeared to decrease rates of grade ≥ 3 CRS but not grade ≥ 3 NEs. 12 In addition to indicating differences in the pathogenesis of CRS and NEs, these data suggest that down‐modulating additional immune pathways may be required to reduce the rates of severe CRS and NEs. The impact of levetiracetam prophylaxis and earlier corticosteroid and tocilizumab intervention on the incidence and severity of CRS and NEs was assessed in cohort 4, reported herein.
Methods
ZUMA‐1 is a single‐arm, multicentre, registrational study of axi‐cel in R/R LBCL being conducted in the United States, Europe, Canada, and Israel. Cohort 4 procedures were similar to those described for cohorts 1 + 2 3 but differed in the use of levetiracetam prophylaxis and earlier corticosteroid and tocilizumab intervention for managing CRS and NEs (Fig 1).
Fig 1.
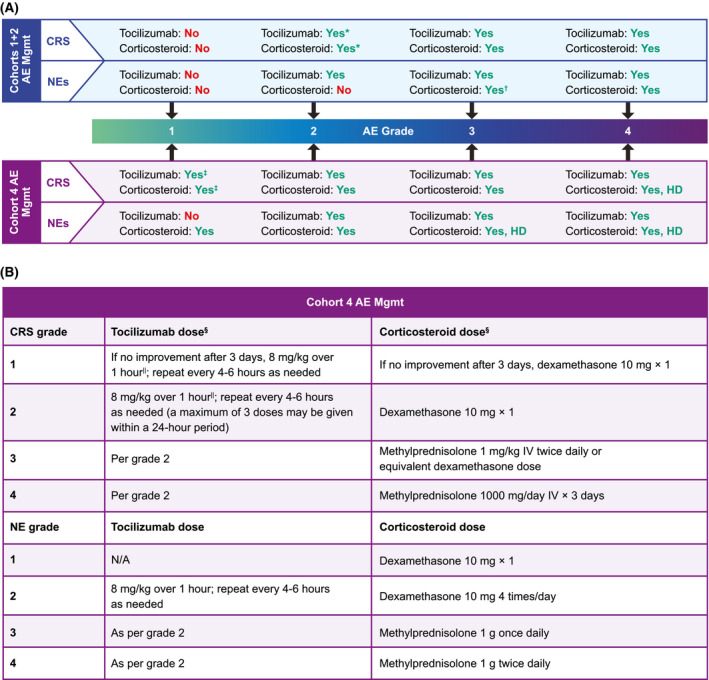
Protocol‐specified AE management in cohorts 1 + 2 and cohort 4 of ZUMA‐1. (A) Comparison of AE management in cohorts 1 + 2 and cohort 4 of ZUMA‐1. ‘Yes’ or ‘No’ indicates whether tocilizumab or corticosteroid was or was not administered, respectively. (B) Tocilizumab and corticosteroid guidelines for AE management in cohort 4 of ZUMA‐1. *Only in case of comorbidities or older age. †Only if no improvement with tocilizumab; use standard dose. ‡If no improvement after three days. §Therapy to be tapered upon improvement of symptoms at investigator’s discretion. ||Not to exceed 800 mg. AE, adverse event; CRS, cytokine release syndrome; HD, high dose; IV, intravenously; N/A, not applicable; NE, neurologic event; Mgmt, management.
Patients
Eligible patients in cohort 4 had R/R LBCL after ≥ 2 systemic lines of therapy or were refractory to first‐line therapy (i.e., best response of progressive disease (PD) or stable disease (SD) to ≥ 4 cycles of first‐line therapy with SD duration ≤ 6 months). See Data S1 for additional eligibility criteria. The study was approved by the institutional review board or ethics commission at each site and was conducted in accordance with the Good Clinical Practice guidelines of the International Conference on Harmonisation. All patients provided written informed consent.
Treatment
Cohort 4 patients received a conditioning regimen of cyclophosphamide (500 mg/m2/day) and fludarabine (30 mg/m2/day) on days −5 to −3, and one dose of axi‐cel (target dose, 2 × 106 CAR T cells/kg; maximum flat dose for patients > 100 kg, 2 × 108 CAR T cells) on day 0. Bridging therapy prior to initiation of conditioning chemotherapy (Table SI) was allowed per investigator’s discretion (e.g., bulky disease or rapidly progressing disease at screening or baseline). Patients received levetiracetam (750 mg orally or intravenously twice daily) starting on day 0 and at the onset of grade ≥ 2 neurologic toxicities if NEs occurred after the discontinuation of prophylactic levetiracetam. If a patient did not experience any grade ≥ 2 neurologic toxicities, levetiracetam was tapered and discontinued as clinically indicated. Corticosteroid therapy was initiated to manage all grade 1 CRS if there was no improvement after three days and for all grade ≥ 1 NEs (Fig 1). Tocilizumab was initiated at grade 1 CRS if there was no improvement after three days, at grade ≥ 2 CRS, and at grade ≥ 2 NE (Fig 1).
Endpoints and analysis
No formal hypothesis was tested; all endpoints were analysed descriptively. The primary endpoint was the incidence and severity of CRS and NEs. CRS was graded per modified Lee et al. criteria 19 and NEs were graded per Common Terminology Criteria for Adverse Events version 4.03. 20 Key secondary endpoints, and clinical pharmacology and biomarker analyses, are described in Data S1.
The modified intent‐to‐treat population included patients enrolled and treated with an axi‐cel dose of ≥ 1 × 106 anti‐CD19 CAR T cells/kg. This analysis set was used for all objective response analyses and endpoints based on objective response. The safety analysis set included all patients treated with any dose of axi‐cel. For patients who received bridging therapy (excluding corticosteroids only), TB was measured after bridging but before conditioning chemotherapy. The cumulative corticosteroid dose was calculated by conversion to a systemic cortisone‐equivalent dose during the initial hospitalisation period.
Propensity score matching (PSM) analysis
Exploratory PSM analysis 21 , 22 was performed to allow a descriptive comparison of results for patients in cohort 4 versus cohorts 1 + 2 (median follow‐up, 15·4 months 3 ) after balancing for the following baseline characteristics: age, Eastern Cooperative Oncology Group (ECOG) performance status, TB, International Prognostic Index score, number of prior lines of chemotherapy, prior platinum use, disease stage, and lactate dehydrogenase (LDH) level (Data S1). Standardised mean difference 23 , 24 within ± 0·2 between cohort 4 and matched cohorts 1 + 2 was used as a criterion to assess covariate balance after PSM.
Results
Patient disposition, baseline and product characteristics
Cohort 4 enrollment commenced in February 2018. Forty‐six patients were enrolled and leukapheresed; 41 patients received the minimum target dose of axi‐cel and comprised both the modified intent‐to‐treat and safety analysis sets (Figure S1). Sixty‐eight percent of patients (n = 28/41) received bridging therapy before axi‐cel with a median reduction in TB among the 17 evaluable patients of 10%. As of the 6 November 2019 data cutoff, the median follow‐up was 14·8 months (range, 8·9–19·9 months).
Among treated patients, the median age was 61 years (range, 19–77; Table I). The most common disease subtype was diffuse LBCL (63%). Most patients (71%) had disease stage III or IV, 63% had had ≥ 3 previous therapies, and 37% had a best response of PD to their most recent chemotherapy. Product characteristics were largely comparable with those previously reported in ZUMA‐1 (Table SII). 3
TABLE 1.
Baseline characteristics.
| Characteristic | Cohorts 1 + 2 (N = 101) 3 | Cohort 4 (N = 41) |
|---|---|---|
| Disease type, n (%) | ||
| DLBCL | 77 (76) | 26 (63) |
| PMBCL | 8 (8) | 2 (5) |
| TFL | 16 (16) | 10 (24) |
| HGBCL | NA* | 3 (7) |
| Age | ||
| Median (range), years | 58·0 (23–76) | 61·0 (19–77) |
| ≥65 years, n (%) | 24 (24) | 13 (32) |
| Male sex, n (%) | 68 (67) | 28 (68) |
| ECOG performance status score of 1, n (%) | 59 (58) | 20 (49) |
| Disease stage, n (%) | ||
| I or II | 15 (15) | 11 (27) |
| III or IV | 86 (85) | 29 (71) |
| IPI score, n (%) | ||
| 0–2 | 55 (54) | 21 (51) |
| 3–4 | 46 (46) | 20 (49) |
| CD19 positivity, n/N (%) † | ||
| Yes | 74 (73) | 22/24 (92) |
| No | 8 (8) | 2/24 (8) |
| Number of previous lines of chemotherapy, n (%) | ||
| 1 | 3 (3) | 0 |
| 2 | 28 (28) | 15 (37) |
| 3 | 29 (29) | 15 (37) |
| 4 | 29 (29) | 8 (20) |
| ≥ 5 | 12 (12) | 3 (7) |
| Previous SCT, n (%) | 25 (25) | 14 (34) |
| PD as best response to most recent chemotherapy, n (%) ‡ | 67 (66) | 15 (37) |
| Median (range) tumour burden by SPD, § mm2 | 3721 (171–23 297) | 2100 (204–24 758) |
| Median (range) LDH, U/l | 356 (116–7802) | 263 (145–4735) |
| Median (range) ferritin, ng/ml | 786 (0·78–10 576) | 393 (23–3457) |
| Refractory subgroup, n (%) | ||
| Primary refractory | 3 (3) | 0 (0) |
| Refractory ≥ 2nd‐line therapy | 77 (76) | 28 (68) |
| Relapsed ≥ 2nd‐line therapy | 0 (0) | 5 (12) |
| Relapsed post‐ASCT | 21 (21) | 8 (20) |
ASCT, autologous stem cell transplant; DLBCL, diffuse large B‐cell lymphoma; ECOG, Eastern Cooperative Oncology Group; HGBCL, high‐grade B‐cell lymphoma; IPI, International Prognostic Index; LDH, lactate dehydrogenase; NA, not applicable; PD, progressive disease; PMBCL, primary mediastinal B‐cell lymphoma; SCT, stem cell transplant; SPD, sum of the products of diameters; TFL, transformed follicular lymphoma.
As ZUMA‐1 was initiated under the 2008 World Health Organisation classifications of B‐cell lymphomas, high‐grade subtypes were not considered separate entities in standard of care practice and therefore not identified in cohorts 1 + 2; rather, such patients were considered to have DLBCL not otherwise specified or DLBCL which was unclassifiable when using the 2008 criteria.
For cohort 4, archival and on‐study pretreatment tumour biopsy ascertainment rate was 59% (24/41) by central confirmation of diagnosis. Two additional patients had missing confirmatory diagnosis due to absence of tumour tissue within the biopsy specimen sent for central assessment.
For patients who had not relapsed post‐ASCT.
For cohort 4, at the last observation before conditioning chemotherapy; may have been measured before or after bridging in patients who received bridging.
Safety
All patients experienced AEs, with 98% experiencing at least one grade ≥ 3 event—most frequently neutropenia (39%), decreased neutrophil count (29%), anaemia (24%), and pyrexia (24%; Table II). Any‐grade infection was reported in 25 (61%) patients, with worst grade 3, 4, and 5 occurring in eight (20%), one (2%), and one (2%) patient, respectively. There were two deaths due to AEs and both were reported as related to conditioning chemotherapy (day 13 pneumonia) or previous chemotherapy (day 354 acute myeloid leukaemia; shown by retrospective analysis to have transformed from underlying myelodysplastic syndrome already present at leucapheresis). Grade ≥ 3 cytopenias present on or after day 30 were reported in 39% of patients (Table SIII).
TABLE 2.
Incidence and severity of TEAEs.*
| Cohort 4 (N = 41) | |||
|---|---|---|---|
| Any grade | Worst grade 3 | Worst grade 4 | |
| Any, n (%) | 41 (100) | 12 (29) | 22 (54) |
| Pyrexia | 39 (95) | 10 (24) | 0 (0) |
| Diarrhoea | 25 (61) | 4 (10) | 0 (0) |
| Hypotension | 25 (61) | 4 (10) | 0 (0) |
| Anaemia | 19 (46) | 10 (24) | 0 (0) |
| Fatigue | 19 (46) | 3 (7) | 0 (0) |
| Headache | 16 (39) | 1 (2) | 0 (0) |
| Neutropenia | 16 (39) | 4 (10) | 12 (29) |
| Nausea | 12 (29) | 0 (0) | 0 (0) |
| Neutrophil count decreased | 12 (29) | 1 (2) | 11 (27) |
| Chills | 11 (27) | 0 (0) | 0 (0) |
| Cough | 10 (24) | 0 (0) | 0 (0) |
| Platelet count decreased | 10 (24) | 2 (5) | 2 (5) |
| Somnolence | 8 (20) | 3 (7) | 0 (0) |
| Dizziness | 7 (17) | 0 (0) | 0 (0) |
| Encephalopathy | 7 (17) | 2 (5) | 0 (0) |
| Leucopenia | 7 (17) | 1 (2) | 5 (12) |
| Tachycardia | 7 (17) | 1 (2) | 0 (0) |
| Thrombocytopenia | 7 (17) | 4 (10) | 1 (2) |
| Back pain | 6 (15) | 0 (0) | 0 (0) |
| Constipation | 6 (15) | 0 (0) | 0 (0) |
| Hypocalemia | 6 (15) | 1 (2) | 0 (0) |
| Hypophosphataemia | 6 (15) | 4 (10) | 0 (0) |
| Hypoxia | 6 (15) | 3 (7) | 0 (0) |
| Tremor | 6 (15) | 0 (0) | 0 (0) |
| Vomiting | 6 (15) | 1 (2) | 0 (0) |
| White blood cell count decreased | 6 (15) | 1 (2) | 5 (12) |
TEAE, treatment‐emergent adverse event.
TEAEs that occurred in ≥ 15% of patients and includes all grade ≥ 3 events that occurred in > 10% of patients.
The overall incidence of CRS was 93%, grade 3 CRS occurred in only 2% of patients (Table III), and there were no grade 4 CRS events or deaths in the setting of CRS. The most common grade 3 symptoms of CRS were pyrexia (24%), hypotension (8%), and hypoxia (5%). The median time to onset of CRS was two days, with a median duration of 6·5 days, and all CRS events resolved by the data cutoff. NEs occurred in 61% of patients, with an incidence of grade ≥ 3 NEs of 17% (Table III). The most common grade ≥ 3 NEs in cohort 4 were somnolence (7%), confusional state (7%), and encephalopathy (5%). There were no grade 4 or 5 NEs. Notably, grade ≥ 3 NEs were limited to patients who received bridging therapy. The median time to onset of NEs was six days, with a median duration of eight days. Three patients had ongoing NEs as of the data cutoff (Table SIV). Bridging therapy did not contribute to a reduction in the incidence of grade ≥ 3 CRS (bridging, 1/28 [4%]; no bridging, 0/13 [0%]) or NEs (bridging, 7/28 [25%]; no bridging, 0/13 [0%]). A total of 73% of patients received corticosteroids. Among those who received corticosteroids, the cumulative cortisone‐equivalent corticosteroid dose was 939 mg, and 43% received ≥ 5 doses (Table SV). Tocilizumab was administered to 76% of patients.
TABLE 3.
Incidence, severity, onset, and duration of CRS and NEs.
| TEAE | Cohort 4 (N = 41) |
|---|---|
| CRS | |
| Any, n (%) | 38 (93) |
| Worst grade 1, n (%) | 13 (32) |
| Worst grade 2, n (%) | 24 (59) |
| Worst grade 3, n (%) | 1 (2) |
| Worst grade 4, n (%) | 0 |
| Worst grade 5, n (%) | 0 |
| Median (range) time to onset of any grade CRS, days | 2·0 (1·0–8·0) |
| Median (range) duration, days | 6·5 (2·0–16·0) |
| NEs | |
| Any, n (%) | 25 (61) |
| Worst grade 1, n (%) | 14 (34) |
| Worst grade 2, n (%) | 4 (10) |
| Worst grade 3, n (%) | 7 (17) |
| Worst grade 4, n (%) | 0 |
| Worst grade 5, n (%) | 0 |
| Median (range) time to onset of any grade NE, days | 6·0 (1·0–93·0) |
| Median (range) duration, days | 8·0 (1·0–144·0) |
CRS, cytokine release syndrome; NE, neurologic event; TEAE, treatment‐emergent adverse event.
Efficacy
The investigator‐assessed objective response rate (ORR) in cohort 4 was 73%, with a CR rate of 51% (Fig 2). While the study was not designed to evaluate the effect of bridging therapy, comparable ORRs were observed in patients who did and did not receive bridging therapy (71% vs. 77%, respectively), although the CR rate was numerically lower in patients who received bridging therapy (46% vs. 62%). The KM estimate of the 12‐month duration of response rate was 71%, and 51% of treated patients remained in response as of the data cutoff date. Response did not appear to be affected by corticosteroid use (Figure S2). Median PFS was not reached (95% CI, 3·0 months—not estimable [NE]; Figure S3A), and the KM estimate of the 12‐month PFS rate was 57%. Median PFS in patients who achieved CR, partial response, or no response was not reached (95% CI, NE–NE), 6·1 months (95% CI, 1·3 months—NE), and 1·4 months (95% CI, 0·2–1·9 months), respectively (Figure S3B). Among all patients, median OS was not reached (95% CI, 15·8 months—NE), and the KM estimate of the 12‐month OS rate was 68%.
Fig 2.
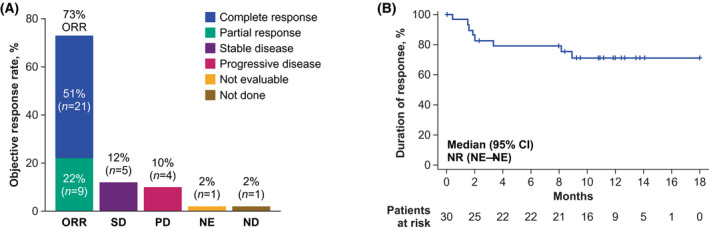
ORR and duration of response. (A) ORR of patients in cohort 4 and rates of SD and PD. Response could not be evaluated in two patients: one patient died of pneumonia before the first assessment, and one patient had a positive result from positron emission tomography with suspected inflammation. (B) Kaplan–Meier curve of duration of response. CR, complete response; NE, not estimable; NR, not reached; ORR, objective response rate; PD, progressive disease; PR, partial response; SD, stable disease.
Biomarker analyses
Median peak CAR T‐cell expansion for cohort 4 was 52·9 cells/μl blood and was observed within 14 days after axi‐cel infusion (Fig 3A). Post‐treatment median levels of key inflammatory serum biomarkers associated with CRS and/or NEs—including IFN‐γ, IL‐2, IL‐6, IL‐8, IL‐15, MCP‐1, GM‐CSF, C‐reactive protein (CRP), and ferritin 3 —peaked during the first week after axi‐cel infusion (Fig 3B; Table SVI). Cohort 4 patients with evaluable samples and grade ≥ 3 NEs had numerically greater post‐infusion (day 5) cerebrospinal fluid levels of IFN‐γ, IL‐15, IL‐2Rα, IL‐6, IL‐8, CRP, and ferritin versus those with grade 0–1 NEs, despite low and comparable baseline levels (Figure S4). A similar pattern was observed for serum biomarkers (Figure S5).
Fig 3.
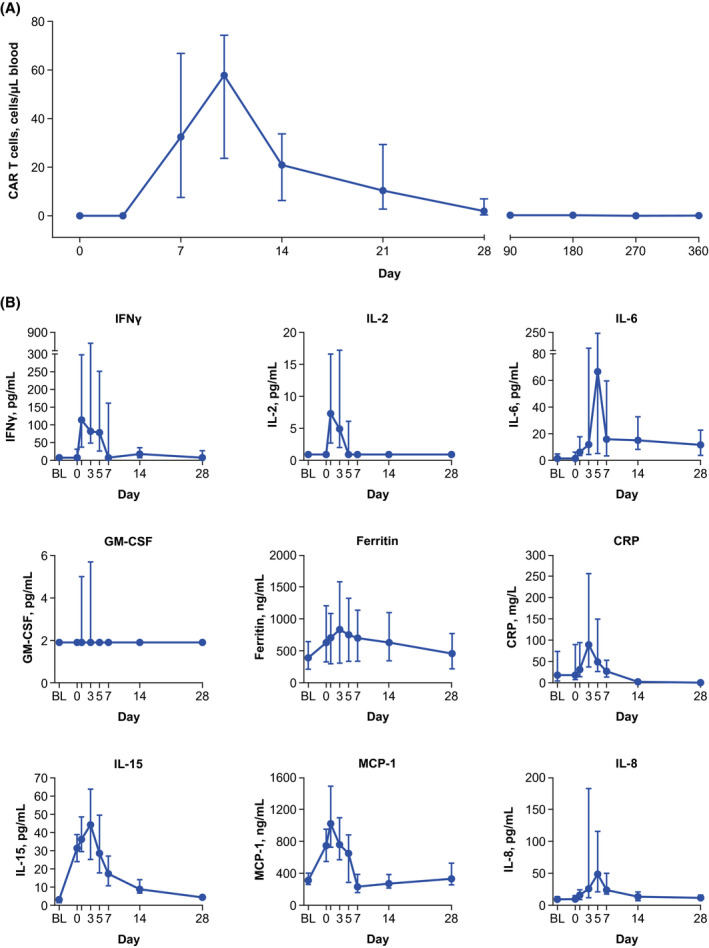
CAR T‐cell expansion and key soluble serum biomarker levels over time. (A) Median (Q1, Q3) blood levels of CAR T cells over time. (B) Median (Q1, Q3) levels of key soluble serum inflammatory biomarkers plotted against time. BL, baseline; CAR, chimeric antigen receptor; CRP, C‐reactive protein; GM‐CSF, granulocyte‐macrophage colony–stimulating factor; IFN, interferon; IL, interleukin; MCP‐1, monocyte chemoattractant protein‐1.
PSM analysis
The incidence of grade ≥ 3 CRS and grade ≥ 3 NEs observed in cohort 4 (2% and 17%, respectively) was numerically lower than in cohorts 1 + 2 (12% and 29%, respectively). 3 Because cohort 4 was not designed for statistical comparison with cohorts 1 + 2, an exploratory PSM analysis 21 , 22 was used to match these cohorts with respect to key baseline characteristics. Following PSM, baseline disease and product characteristics were generally similar between patients in cohort 4 and cohorts 1 + 2, although fewer cohort 4 patients had baseline ECOG performance status of 1 (49% vs. 68%; Table SVII). Notably, the differences in grade ≥ 3 CRS and NEs observed between patients in cohorts 1 + 2 and cohort 4 before PSM were maintained after matching. Although CR rates after PSM were numerically lower in cohort 4 versus cohorts 1 + 2, ongoing response rates remained comparable. Clinical outcomes were corroborated by lower levels of key inflammatory soluble biomarkers associated with CAR‐related inflammatory events (e.g., IFN‐γ, IL‐2, IL‐8, C‐reactive protein, ferritin, GM‐CSF), 3 , 10 and by generally comparable peak CAR T‐cell levels in cohort 4 versus cohorts 1 + 2 before and after PSM. The median cumulative cortisone‐equivalent corticosteroid dose required to manage CRS or NEs remained lower in cohort 4 (939 mg) than in matched cohorts 1 + 2 (6886 mg; Table SVIII).
Discussion
AE management in CAR T‐cell therapy is an evolving field with ongoing efforts to improve the safety profile of this treatment without compromising the durable clinical benefit. To this end, ZUMA‐1 cohort 4 patients received corticosteroid and/or tocilizumab intervention earlier than the pivotal cohorts 1 + 2. 3 , 4 Numerically lower rates of grade ≥ 3 CRS and NEs were observed in cohort 4 (2% and 17%, respectively) than in cohorts 1 + 2 (12% and 29%), suggesting that earlier intervention with corticosteroids and/or tocilizumab has the potential to improve the safety profile of axi‐cel in patients with R/R LBCL. In patients treated with corticosteroids, the median cumulative cortisone‐equivalent dose was 939 mg in cohort 4 versus 6388 mg in cohorts 1 + 2, suggesting that earlier corticosteroid use does not increase cumulative corticosteroid dose. Furthermore, this revised safety management regimen did not appear to negatively affect the ongoing response rate at one year (cohort 4: 51%; cohorts 1 + 2: 42%), although additional follow‐up is needed to assess the long‐term durability of responses.
Differences in baseline characteristics and cohort sizes should be considered when comparing cohort 4 with cohorts 1 + 2. Cohort 4 patients had lower levels of inflammatory serum biomarkers (e.g., ferritin or LDH) at baseline, and a lower proportion of patients had PD in response to the most recent line of therapy. 4 , 25 Cohort 4 also had lower TB, which has previously been associated with lower rates of NEs and increased efficacy. 17 , 18 To overcome these limitations and reduce bias in the absence of a randomised trial, PSM 21 , 22 was applied to cohorts 1 + 2 and cohort 4. This statistical method adjusts for potential imbalances in baseline disease characteristics between cohorts, thereby providing a more balanced and robust comparison. 23 , 26 It should be noted that although PSM controls for known imbalances between cohorts, other sources of bias may exist due to unmeasured confounding variables. Although minor differences in pretreatment characteristics remained after matching, the aforementioned differences in toxicity outcomes observed between patients in cohort 4 and cohorts 1 + 2 before PSM were maintained after matching, supporting the benefit of earlier corticosteroid and/or tocilizumab. PSM also had little effect on peak CAR T‐cell levels, and ongoing response rates at one year remained comparable, suggesting no negative impact of earlier corticosteroid and/or tocilizumab use on long‐term outcomes.
Despite the theoretical concern that immunosuppressive agents may abrogate CAR T‐cell expansion and anti‐tumour response, 27 earlier and measured introduction of corticosteroid in cohort 4 did not substantially affect either. Cohort 4 showed generally comparable peak CAR T‐cell levels versus cohorts 1 + 2 but lower levels of key inflammatory serum biomarkers associated with CAR‐related inflammatory events (e.g., IFN‐γ, IL‐2, IL‐8, and GM‐CSF). 3 , 10 These findings corroborated clinical outcomes, suggesting that early use of corticosteroids may have a greater impact on immune cell cytokine production than on CAR T‐cell expansion and anti‐tumour activity. Furthermore, corticosteroids are known to modulate T‐cell and myeloid cell activity, thereby decreasing proinflammatory cytokine levels with potential beneficial impact on blood‐brain‐barrier permeability and local inflammatory processes. 28
The results presented here are consistent with the primary analysis of ZUMA‐1 (cohorts 1 + 2), which suggested no substantial effect of corticosteroid use on ORR (corticosteroid, 78% [58–91%]; no corticosteroid, 84% [73–91%]). 3 Retrospective analyses of real‐world data have delivered conflicting results regarding the impact of corticosteroid use on clinical outcomes after axi‐cel in R/R LBCL. 29 , 30 However, in the larger of these two studies (N = 298), multivariate analysis demonstrated no significant difference in PFS, CR rates, or OS in patients treated with corticosteroids versus treated without. 30 It is important to note that the clinical applicability of these studies is unclear given their retrospective nature and potential imbalances in baseline characteristics (e.g., TB) 17 , 18 , 31 , 32 in patients requiring corticosteroids versus those not requiring corticosteroids. Although studies of other CAR T‐cell products in B‐cell acute lymphoblastic leukaemia have also not been designed to assess the impact of corticosteroid use, published analyses have shown no substantial effect of corticosteroid use on CAR T‐cell expansion or anti‐tumour response. 33 , 34
Finally, we cannot fully rule out the contribution of additional differences in intervention beyond corticosteroid and/or tocilizumab use. However, it is unlikely that prophylactic levetiracetam resulted in fewer grade ≥ 3 NEs in cohort 4 versus cohorts 1 + 2, given that prophylactic levetiracetam was used in ZUMA‐1 safety management cohort 3, which actually demonstrated a numerical increase in grade ≥ 3 NEs. 12 While cohort 4 patients could receive bridging therapy, bridging was not associated with a reduction in the incidence of grade ≥ 3 CRS or NEs in cohort 4. It should also be noted that, at time of enrollment, none of the 40 cohort 4 sites were considered a CAR T‐experienced centre, and 90% had never before treated a patient with CAR T cells. Thus, the experience of the investigators is unlikely to have contributed to the improved safety of axi‐cel in cohort 4.
In conclusion, earlier and measured use of corticosteroids and/or tocilizumab has the potential to reduce the incidence of grade ≥ 3 CRS and NEs through down‐modulating key proinflammatory soluble serum biomarkers, including cytokines, without notably affecting CAR T‐cell expansion and ongoing response rates in patients with R/R LBCL receiving axi‐cel. This approach offers important additive information to further inform patient care.
Data sharing statement
Gilead is committed to sharing clinical trial data with external medical experts and scientific researchers in the interest of advancing public health. As such, Gilead shares anonymised individual patient data (IPD) upon request or as required by law and/or regulation. Qualified external researchers may request IPD for studies of Gilead compounds approved in the United States and the European Union with a marketing authorisation date on or after 1 January 2014, and are publicly listed on clinicaltrials.gov or the European Union‐Clinical Trials Register (EU CTR). For studies of newly approved compounds or indication, the IPD will be available for request six months after approval from the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA). Such requests are at Gilead’s discretion and are dependent on the nature of the request, the merit of the research proposed, availability of the data, and the intended use of the data. If Gilead agrees to the release of clinical data for research purposes, the requestor will be required to sign a data‐sharing agreement, to ensure protection of patient confidentiality before the release of any data.
Funding
The study was supported by Kite, a Gilead Company.
Author contributions
MST, AB, AK, and MJK conceived and designed the study; all authors provided study material or patients, collected or assembled data, participated in drafting and revising the manuscript, approved the final version of the manuscript, and are accountable for all aspects of this work.
Conflict of interest
MST has served in a consultancy or advisory role for Amgen, Kite, a Gilead Company, Celgene, Regeneron and Roche, and received research funding from Amgen, Kite, a Gilead Company, Regeneron, Roche and MacroGenics. TvM has received honoraria from Kite, a Gilead Company, and has served in a consultant or advisory role for Janssen. RH has received honoraria from Bristol Myers Squibb, Novartis, Celgene, Janssen, MSD, Kite, a Gilead Company, Roche, and ADC Therapeutics and has served in a consultancy or advisory role for Kite, a Gilead Company. MCM has served in a consultancy or advisory role for Janssen‐Cilag, Gilead, and Alnylam and has received travel support from Celgene. KB has served in a consultancy or advisory role for Kite, a Gilead Company, Roche, Sandoz, and Takeda; received honoraria from Kite, a Gilead Company, Celgene, Roche and Takeda; and received travel support from Roche. PJL has served in a consultancy or advisory role for Takeda, Servier, Roche, Bristol Myers Squibb, Celgene, Sandoz, and Genmab and received research funding from Takeda and Servier. CT has served in a consultancy or advisory role for Novartis, Roche, Celgene, and Janssen; received honoraria from Gilead, Novartis, Roche, Celgene and Janssen; received research funding from Roche; and received travel support from Gilead, Novartis, Roche and Janssen. MW has served in a consultancy or advisory role for Kite, a Gilead Company, Novartis, Bristol Myers Squibb, AstraZeneca, Pfizer, Merck, Genmab and Boehringer Ingelheim; received honoraria from Novartis, Bristol Myers Squibb, AstraZeneca, Pfizer and Merck; and received travel support from Novartis, Bristol Myers Squibb and AstraZeneca. KWS has received honoraria from and has served in a consultancy or advisory role for Kite, a Gilead Company. JK has served in a consultancy or advisory role for AbbVie, Bristol Myers Squibb, Gilead, Karyopharm, Merck, Roche and Seattle Genetics; received honoraria from Amgen, AstraZeneca, Bristol Myers Squibb, Celgene, Gilead, Janssen, Karyopharm, Merck, Novartis, Roche and Seattle Genetics; and received research funding from Roche and Janssen. UD served in a consultancy or advisory role for AbbVie, Amgen, CPT, Gilead, Janssen, Novartis and Takeda; received honoraria from AbbVie, Amgen, Celgene, CPT, Gilead, Janssen, Novartis, Roche and Takeda; received research funding from Amgen, Celgene and Roche; and received travel support from AbbVie and Janssen. YZ, MS, JJK, and AK are employed by Kite, a Gilead Company, and have stock or other ownership in Gilead Sciences. SV is employed by and has received research funding and travel support from Kite, a Gilead Company, and has stock or other ownership in Gilead Sciences. JD is employed by Kite, a Gilead Company; has served in a consultancy or advisory role for GliaCure/Tufts; and has received patents, royalties or other intellectual property from Patent US8598141 (Dec 03, 2013). AB is employed by Kite, a Gilead Company, and has stock or other ownership in, has served in a consultancy or advisory role for, and received travel support from Gilead Sciences. JMR is employed by Kite, a Gilead Company. VP is employed by and has received honoraria and travel support from Kite, a Gilead Company; has stock or other ownership in Gilead Sciences; and has patents, royalties or other intellectual property from Genentech. MJK has served in a consultancy or advisory role for and received honoraria from Kite, a Gilead Company, Novartis and Miltenyi; received research funding from Roche, Takeda and Celgene; and received travel support from Kite, a Gilead Company, Novartis and Miltenyi. The remaining author (IA) declares no competing financial interests.
Supporting information
Data S1. Supplemental methods
Table SI. Bridging therapy regimens.*
Table SII. Summary of product characteristics.
Table SIII. Incidence of worst grade ≥ 3 neutropenia, thrombocytopenia, and anaemia present on or after day 30 following axi‐cel infusion.
Table SIV. Summary of neurologic events unresolved at data cutoff.
Table SV. Cumulative dose and frequency of corticosteroid use.
Table SVI. Summary of serum biomarkers.
Table SVII. Comparison of baseline and product characteristics between patients in cohorts 1 + 2 and cohort 4 before and after propensity score matching.
Table SVIII. Comparison of efficacy and safety outcomes and CAR T‐cell and soluble serum biomarker levels between patients in cohorts 1 + 2 and cohort 4 before and after propensity score matching.
Fig S1. Patient disposition diagram.
Fig S2. Best response by corticosteroid use.
Fig S3. Progression‐free survival in cohort 4. (A) All treated patients. (B) Best overall response subgroups.
Fig S4. Selected CSF analysis at baseline and day 5 and association with neurologic events.
Fig S5. Selected serum analysis at baseline and day 5 and association with neurologic events.
Acknowledgements
We thank all investigators, coordinators, study‐site personnel, and the patients and their families for participating in this study. Kite, a Gilead Company, was involved in the development of the study protocol and in data collection, analysis, and interpretation. We would like to acknowledge Drs. Zahid Bashir, Justin Chou, Aurora Liao, and Yan Zheng for their contributions to data analysis and interpretation. Editorial and writing assistance during the development of this article was provided by Skye Geherin, PhD, and Ashley Skorusa, PhD, of Nexus Global Group Science, a Vaniam Group Company, and funded by Kite, a Gilead Company.
References
- 1.YESCARTA® (axicbatagene ciloleucel) Summary of product characteristics. Kite Pharma EU B.V.; 2021.
- 2. YESCARTA® (axicabtagene ciloleucel) Prescribing information. Kite Pharma, Inc; 2021. [Google Scholar]
- 3. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T‐cell therapy in refractory large B‐cell lymphoma. N Engl J Med. 2017;377(26):2531–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long‐term safety and activity of axicabtagene ciloleucel in refractory large B‐cell lymphoma (ZUMA‐1): a single‐arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019;20(1):31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jacobson C, Locke FL, Ghobadi A, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long‐term survival and gradual recovery of B cells in patients with refractory large B cell lymphoma treated with axicabtagene ciloleucel (axi‐cel). Blood. 2020;136(Suppl 1):40–2. [Google Scholar]
- 6. Maruffi M, Sposto R, Oberley MJ, Kysh L, Orgel E. Therapy for children and adults with mixed phenotype acute leukemia: a systematic review and meta‐analysis. Leukemia. 2018;32(7):1515–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sun W, Malvar J, Sposto R, Verma A, Wilkes JJ, Dennis R, et al. Outcome of children with multiply relapsed B‐cell acute lymphoblastic leukemia: a therapeutic advances in childhood leukemia & lymphoma study. Leukemia. 2018;32(11):2316–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Santomasso B, Bachier C, Westin J, Rezvani K, Shpall EJ. The other side of CAR T‐cell therapy: cytokine release syndrome, neurologic toxicity, and financial burden. Am Soc Clin Oncol Educ Book. 2019;39:433–44. [DOI] [PubMed] [Google Scholar]
- 9. Chen P‐H, Lipschitz M, Weirather JL, Jacobson C, Armand P, Wright K, et al. Activation of CAR and non‐CAR T cells within the tumor microenvironment following CAR T cell therapy. JCI Insight. 2020;5(12):e134612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Teachey DT, Lacey SF, Shaw PA, Melenhorst JJ, Maude SL, Frey N, et al. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T cell therapy for acute lymphoblastic leukemia. Cancer Discov. 2016;6(6):664–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. ACTEMRA (tocilizumab) [package insert]. South San Francisco, CA: Genentech, Inc; 2018. [Google Scholar]
- 12. Locke FL, Neelapu SS, Bartlett NL, Lekakis LJ, Jacobson CA, Braunschweig I, et al. Preliminary results of prophylactic tocilizumab after axicabtagene ciloleucel (axi‐cel; KTE‐C19) treatment for patients with refractory, aggressive non‐Hodgkin lymphoma (NHL). Blood. 2017;130(Suppl 1):1547. [Google Scholar]
- 13. Brudno JN, Lam N, Vanasse D, Shen Y‐W, Rose JJ, Rossi J, et al. Safety and feasibility of anti‐CD19 CAR T cells with fully human binding domains in patients with B‐cell lymphoma. Nat Med. 2020;26(2):270–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brudno JN, Kochenderfer JN. Recent advances in CAR T‐cell toxicity: mechanisms, manifestations and management. Blood Rev. 2019;34:45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Parker KR, Migliorini D, Perkey E, Yost KE, Bhaduri A, Bagga P, et al. Single‐cell analyses identify brain mural cells expressing CD19 as potential off‐tumor targets for CAR‐T immunotherapies. Cell. 2020;183(1):126–42.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gust J, Hay KA, Hanafi L‐A, Li D, Myerson D, Gonzalez‐Cuyar LF, et al. Endothelial activation and blood‐brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR‐T cells. Cancer Discov. 2017;7(12):1404–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Locke F, Rossi J, Neelapu S, Jacobson CA, Miklos D, Ghobadi A, et al. Tumor burden, inflammation, and product attributes determine outcomes of axi‐cel in large B‐cell lymphoma. Blood Adv. 2020;4(19):4898–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dean EA, Mhaskar RS, Lu H, Mousa MS, Krivenko GS, Lazaryan A, et al. High metabolic tumor volume is associated with decreased efficacy of axicabtagene ciloleucel in large B‐cell lymphoma. Blood Adv. 2020;4(14):3268–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124(2):188–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. U.S. Department of Health and Human Services. Common Terminology Criteria for Adverse Events (CTCAE) Version 4.03. 2010. National Institutes of Health National Cancer Institute.
- 21. Rosenbaum PR, Rubin DB. The central role of the propensity score in observational studies for causal effects. Biometriks. 1983;70(1):41–55. [Google Scholar]
- 22. Austin PC. An introduction to propensity score methods for reducing the effects of confounding in observational studies. Multivariate Behav Res. 2011;46(3):399–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Austin PC. A critical appraisal of propensity‐score matching in the medical literature between 1996 and 2003. Stat Med. 2008;27(12):2037–49. [DOI] [PubMed] [Google Scholar]
- 24. Imai K, King G, Stuart EA. Misunderstandings between experimentalists and observationalists about causal inference. J R Stat Soc. 2008;171:481–502. [Google Scholar]
- 25. Topp M, Van Meerten T, Houot R, Minnema MC, Milpied N, Lugtenburg PJ, et al. Earlier steroid use with axicabtagene ciloleucel (Axi‐Cel) in patients with relapsed/refractory large B cell lymphoma. Blood. 2019;134(Suppl 1):243. [Google Scholar]
- 26. Zhang Z, Kim HJ, Lonjon G, Zhu Y. Balance diagnostics after propensity score matching. Ann Transl Med. 2019;7(1):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224):224ra25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zielińska KA, Van Moortel L, Opdenakker G, De Bosscher K, Van den Steen PE. Endothelial response to glucocorticoids in inflammatory diseases. Front Immunol. 2016;7:592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Strati P, Ahmed S, Furqan F, Fayad LE, Lee HJ, Iyer SP, et al. Prognostic impact of corticosteroids on efficacy of chimeric antigen receptor T‐cell therapy in large B‐cell lymphoma. Blood. 2021;137(23):3272–6. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nastoupil LJ, Jain MD, Feng L, Spiegel JY, Ghobadi A, Lin YI, et al. Standard‐of‐care axicabtagene ciloleucel for relapsed or refractory large B‐cell lymphoma: results from the US Lymphoma CAR T Consortium. J Clin Oncol. 2020;38(27):3119–28. [online ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gauthier J, Hirayama AV, Hay KA, Li D, Sheih A, Wu V, et al. Factors associated with duration of response after CD19‐specific CAR‐T cell therapy for refractory/relapsed B‐cell non‐Hodgkin lymphoma. J Clin Oncol. 2018;36(Suppl 15):7567. [Google Scholar]
- 32. Jacobson CA, Hunter B, Armand P, Kamihara Y, Ritz J, Rodig SJ, et al. Axicabtagene ciloleucel in the real world: outcomes and predictors of response, resistance and toxicity. Blood. 2018;132(Suppl 1):92. [Google Scholar]
- 33. Gardner RA, Ceppi F, Rivers J, Annesley C, Summers C, Taraseviciute A, et al. Preemptive mitigation of CD19 CAR T‐cell cytokine release syndrome without attenuation of antileukemic efficacy. Blood. 2019;134(24):2149–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu S, Deng B, Yin Z, Pan J, Lin Y, Ling Z, et al. Corticosteroids do not influence the efficacy and kinetics of CAR‐T cells for B‐cell acute lymphoblastic leukemia. Blood Cancer J. 2020;10(2):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplemental methods
Table SI. Bridging therapy regimens.*
Table SII. Summary of product characteristics.
Table SIII. Incidence of worst grade ≥ 3 neutropenia, thrombocytopenia, and anaemia present on or after day 30 following axi‐cel infusion.
Table SIV. Summary of neurologic events unresolved at data cutoff.
Table SV. Cumulative dose and frequency of corticosteroid use.
Table SVI. Summary of serum biomarkers.
Table SVII. Comparison of baseline and product characteristics between patients in cohorts 1 + 2 and cohort 4 before and after propensity score matching.
Table SVIII. Comparison of efficacy and safety outcomes and CAR T‐cell and soluble serum biomarker levels between patients in cohorts 1 + 2 and cohort 4 before and after propensity score matching.
Fig S1. Patient disposition diagram.
Fig S2. Best response by corticosteroid use.
Fig S3. Progression‐free survival in cohort 4. (A) All treated patients. (B) Best overall response subgroups.
Fig S4. Selected CSF analysis at baseline and day 5 and association with neurologic events.
Fig S5. Selected serum analysis at baseline and day 5 and association with neurologic events.