

Abstract
The current retrospective study involving a total of 1607 patients was designed to identify clinical and molecular variables that were predictive of inferior myelofibrosis‐free survival (MFS) in WHO‐defined essential thrombocythemia (ET), utilizing three independent patient cohorts: University of Florence, Italy (n = 718); Mayo Clinic, USA (n = 479) and Policlinico Gemelli, Catholic University, Rome, Italy (n = 410). The Florence patient cohort was first examined to identify independent risk factors for MFS, which included age > 60 years (HR 2.5, 95% CI 1.3–4.9), male sex (2.1, 1.2–3.9), palpable splenomegaly (2.1, 1.2–3.9), CALR 1/1‐like or MPL mutation (3.4, 1.9–6.1) and JAK2V617F variant allele frequency > 35% (4.2, 1.6–10.8). Subsequently, an operational molecular risk category was developed and validated in the other two cohorts from Mayo Clinic and Rome: “high molecular risk” category included patients with JAK2V617F VAF >35%, CALR type 1/1‐like or MPL mutations; all other driver mutation profiles were assigned to “low molecular risk” category. The former, compared to the latter molecular risk category, displayed significantly higher risk of fibrotic transformation: Florence cohort with respective fibrotic transformation risk rates of 8% vs. 1.2% at 10 years and 33% vs. 8% at 20 years (p < 0.001; HR 6.1; 95% CI 3.2–11.7); Mayo Cohort, 16% vs. 7% at 10 years and 44% vs. 25% at 20 years (p < 0.001; HR 2.5; 95% CI 1.6–4.1); and Rome cohort 7.8% vs. 4.6% at 10 years and 31.2% vs. 7.1% at 20 years (p = 0.007, HR 2.7; 95% CI 1.3–5.8). The present study provides practically useful risk signals for fibrotic transformation in ET and facilitates identification of patients who require close monitoring and appropriate counseling.
1. INTRODUCTION
Essential thrombocythemia (ET) is one of Ph‐negative myeloproliferative neoplasms (MPN) along with polycythemia vera (PV), overt and pre‐fibrotic myelofibrosis (MF), as defined by the latest World Health Organization (WHO) classification. 1 Essential thrombocythemia is characterized by clonal thrombocytosis associated with an increased risk of thrombo‐hemorrhagic complications, progression to MF and, to a lesser extent, transformation to acute myeloid leukemia. 2 Approximately 90% of ET patients harbor one of three MPN‐specific clonal markers, including JAK2V617F, CALR (type 1/1‐like and type 2/2‐like) and MPL mutations that induce constitutive activation of JAK‐STAT signaling pathway; the remaining 10% are referred to as triple‐negative (TN). 3 Current risk stratification in ET targets likelihood of thrombosis, with important implications on therapy, and relies on International prognostic score in essential thrombocythemia (IPSET) for thrombosis 4 that includes age > 60 years, thrombosis history, cardiovascular risk factors and JAK2V617F mutation, as risk variables. The score was re‐analyzed and led to a refined four‐tiered version, 5 which excluded cardiovascular risk factors evaluation; subsequently this score was validated in a large independent cohort. 6 Classically, survival prediction in ET was based on clinically‐derived risk variables, including age, thrombosis history and leukocyte count. 7 More recently, collaborative studies between the Mayo Clinic, Rochester, USA and University of Florence, Italy, established an integrated clinical and genetic survival risk model for ET (MIPSS‐ET) identifying the presence of SF3B1 and SRSF2 mutations (occurring in ∼10% of patients), age > 60 years, male sex, and leukocytosis (≥ 11 × 109/L) as independent risk factors for reduced overall survival 8 ; this collaborative effort also identified SF3B1 and U2AF1 mutations as risk factors for fibrotic progression and highlighted the protective role on arterial thrombosis of ASXL1, RUNX1 and EZH2 mutations. 9 Myelofibrotic transformation was reported to be significantly higher among CALR‐mutated ET patients than JAK2V617F patients in one initial report. 10 Follow‐up studies were inconsistent, some showing no increased risk of transformation 11 , 12 but one large‐scale study again found a higher risk of transformation to MF. 13 Interestingly, the increase in the incidence of MF transformation among CALR mutated ET patients appears to be restricted to type 1/1‐like mutations while patients with type 2/2‐like mutations displayed no differences when compared to JAK2V617F patients. 14 This is in agreement with a recent study on post‐ET myelofibrosis showing a time to MF progression significantly longer in type 2/2‐like than in type 1/1‐like CALR and JAK2 V617F mutations. 15 An increased risk for MF progression was also reported for patients with MPL mutations in two studies. 12 , 16 Moreover, although an impact of JAK2V617F variant allele frequency (VAF) on risk of thrombosis and MF progression was suggested in PV, 17 , 18 no information is available in large cohorts of ET patients.The aim of the current study was to retrospectively identify clinical and molecular variables that were predictive of inferior myelofibrosis‐free survival (MFS) in WHO‐defined ET, utilizing three independent patient cohorts: University of Florence, Italy (n = 718); Mayo Clinic, USA (n = 479) and Policlinico Gemelli, Catholic University, Rome, Italy (n = 410).
2. METHODS
Study patients were selected from institutional databases and the study was approved by the respective local ethics committees of the University of Florence, Mayo Clinic and Policlinico Gemelli, Catholic University, and was conducted in accordance with the Declaration of Helsinki. Diagnostic criteria for ET and leukemic transformation were according to the 2016 WHO classification. 1 Diagnostic criteria for post‐ET MF were according to the international working group for MPN research and treatment (IWG‐MRT). 19 Treatment approaches were in accordance with what was considered the standard of care at the time of initial diagnosis. The JAK2V617F mutated patients were annotated for VAF in samples collected within 5 years of diagnosis, according to published methods. 12 In order to minimize the inadvertent inclusion of patients with masked PV, JAK2 mutated cases with hemoglobin (Hb) level > 16 g/dl in women and 16.5 g/dl in men were excluded; similarly, cases with anemia defined by sex adjusted Hb level of < 11 g/dl in women and 12.5 g/dl in men were excluded, in order to minimize the chance of inadvertent inclusion of patients with pre‐fibrotic MF. All statistical analyses considered clinical and laboratory parameters obtained at time of initial diagnosis. Differences in the distribution of continuous variables between categories were analyzed by either Mann–Whitney (for comparison of two groups) or Kruskal–Wallis (comparison of three or more groups) test. Patient groups with nominal variables were compared by chi‐square test. Survival analysis was considered from the date of diagnosis to date of death or last contact. Leukemia‐free and myelofibrosis‐free survival calculations considered the transformation event as the uncensored variable. Survival curves were prepared by the Kaplan–Meier method and compared by the log‐rank test. Cox proportional hazard regression model was used for multivariable analysis. A receiver operating characteristic curve (ROC) was used to determine the best thresholds of VAF using area under curve (AUC) estimate. The p values less than 0.05 were considered significant. All the analyses were performed with SPSS software, version 27 (IBM‐Corp) and JMP Pro 14.1.0 software from SAS Institute (Cary, NC).
3. RESULTS
The primary study population included 718 patients from the University of Florence and the observations in this cohort were subsequently validated in two independent cohorts of 479 and 410 patients from the Mayo Clinic, Rochester, MN, USA and Policlinico Gemelli, Catholic University, Rome, Italy, respectively.Main clinical and laboratory characteristics of the University of Florence cohort are highlighted in Table 1. Median age at diagnosis was 57.9 years (range, 12.9–92.9), women 64.8%. Driver mutation distribution was 65.6% JAK2, 12.8% CALR type1/1like, 7.1% CALR type2/2like, 3.6% MPL and 10.9% TN. The median follow‐up was 106.4 months (6.1–421.6), during which 53 patients (7.4%) progressed to MF (23 JAK2V617F, 20 CALR 1/1‐like, four CALR 2/2‐like, five MPL, one TN, p < 0.001) after a median of 146.2 months (8.6–362.9) and 106 patients (14.8%) died. Leukocytosis (≥ 11 × 109/L) was documented in 16.4% of patients and extreme thrombocytosis (≥1000 × 109/L) in 17.5%. Palpable splenomegaly was present in 97 (13.5%) patients; 43 (6%) and 235 (33.1%) patients had constitutional and microcirculatory symptoms. Differences in overall, thrombosis‐free, myelofibrosis‐free (MFS), and leukemia‐free survival among patients stratified by their mutational status are reported in Figure S1. Univariate analysis for MFS identified age > 60 years (p = 0.02; HR 1.9; 95%CI 1.1–3.9), male sex (p = 0.001; HR 2.4; 95%CI 1.4–4.2), palpable splenomegaly (p = 0.002; HR 2.5; 95%CI 1.4–4.5), constitutional symptoms (p = 0.01; HR 2.6; 95%CI 1.2–5.3) along with CALR mutation (p = 0.01; HR 2.1; 95%CI 1.2–3.6) and MPL mutation (p = 0.008; HR 3.2; 95%CI 1.4–4.7) as risk factors for fibrotic progression. Type 1/1‐like CALR patients were more prone to fibrotic transformation compared with type 2/2‐like (p = 0.04; HR 3.2; 95% CI 1.1–9.3), while the risk was lower in JAK2V617F mutated patients (p = 0.01; HR 0.5; 95% CI 0.3–0.9). Multivariable analysis confirmed male sex, splenomegaly, constitutional symptoms, and mutational status (CALR type 1/1‐like and MPL) as independent prognostic factors for MF progression (Table 2). We also found JAK2V617F VAF, as a continuous variable, to be correlated with MF progression (p = 0.002; HR 1; 95% CI 1–1.1).
TABLE 1.
Clinical and laboratory variables of 718 patients with essential thrombocythemia from University of Florence cohort, stratified by their driver mutational status
| Clinical and laboratory variables | All patients (n = 718) | JAK2 (n = 471; 65.6%) | CALR type 1/1 like (n = 92; 12.8%) | CALR type 2/2 like (n = 51; 7.1%) | MPL (n = 26; 3.6%) | TN (n = 78; 10.9%) | p valuesa |
|---|---|---|---|---|---|---|---|
| Age in year; median (range) | 57.9 (12.9–92.9) | 62.0 (14.9–92.9) | 52.6 (12.9–85.1) | 48.9 (20.5–84.4) | 54.8 (37.3–89.2) | 49.2 (15.2–81.3) | <0.001 |
| Age ≥ 60 years; n (%) | 333 (46.4) | 253 (53.7) | 30 (32.6) | 13 (25.5) | 11 (42.3) | 26 (33.3) | <0.001 |
| Sex, females; n (%) | 465 (64.8) | 318 (67.5) | 39 (42.6) | 25 (49.0) | 20 (76.9) | 63 (80.8) | <0.001 |
|
White blood cells × 109/L; median (range) N evaluable = 695 |
8.3 (3.7–18.2) | 8.7 (3.7–18.2) | 8.2 (3.9–18.2) | 7.2 (3.8–14.7) | 6.8 (4.0–16.7) | 7.6 (6.4–13.3) | <0.001 |
|
White blood cells ≥11 × 109/L; n (%) N evaluable = 695 |
114 (16.4) | 90 (19.6) | 11 (12.8) | 2 (4.2) | 5 (20.0) | 6 (7.7) | 0.01 |
| Hemoglobin, g/dl; median (range) | 14.1 (11.5–16.5) | 14.4 (11.8–16.5) | 13.8 (11.8–15.9) | 13.7 (11.9–16.3) | 13.6 (11.9–16.1) | 13.5 (11.5–16.4) | <0.001 |
| Hematocrit, %; median (range) | 42.9 (35.1–51.0) | 43.9 (36.0–50.0) | 41.6 (36.7–47.9) | 41,2 (35,6 – 49,4) | 41.3 (38.0–47.0) | 40.9 (35.1–51.0) | <0.001 |
| Platelets, ×109/L; median (range) | 739 (455–2348) | 698 (455–1881) | 823 (504–2000) | 886 (495–2348) | 851.5 (467–1742) | 794 (460–1700) | <0.001 |
| Platelets ≥ 1000 ×109/L; n (%) | 126 (17.5) | 61 (12.9) | 24 (26.1) | 16 (31.4) | 7 (26.9) | 18 (23.1) | 0.001 |
| Palpable splenomegaly; n (%) | 97 (13.5) | 67 (14.2) | 15 (16.3) | 6 (11.8) | 4 (15.4) | 5 (6.4) | 0.3 |
| Constitutional symptoms; n (%) | 43 (6.0) | 27 (5.7) | 5 (5.4) | 3 (5.9) | 2 (7.7) | 6 (7.7) | 0.9 |
|
Major thrombosis before diagnosis; n (%) N evaluable = 633 |
39 (6.2) | 35 (8.6) | 1 (1,2) | 0 (0) | 0 (0) | 3 (3.8) | 0.01 |
|
Major thrombosis at diagnosis; n (%) N evaluable = 631 |
42 (6.7) | 31 (7.8) | 2 (2,4) | 1(2) | 4 (16.0) | 4 (5.1) | 0.07 |
|
Overall thrombosis at follow‐up; n (%) N evaluable = 715 |
99 (13.8) | 69 (14.7) | 14 (15.4) | 7 (14) | 7 (26.9) | 2 (2.6) | 0.01 |
|
Overall major bleeding; n (%) N evaluable = 713 |
59 (8.3) | 41 (8.7) | 9 (10.0) | 2 (3,9) | 3 (11,5) | 4 (5.1) | 0.5 |
|
Overall minor bleeding; n (%) N evaluable = 709 |
90 (12.7) | 54 (11.6) | 13 (14.4) | 7 (14.3) | 7 (26.9) | 9 (11.5) | 0.2 |
|
Cardiovascular risk factors; n (%) N evaluable = 618 |
327 (52.9) | 235 (59.5) | 34 (44.7) | 15 (34.1) | 12 (48.0) | 31 (39.7) | <0.001 |
|
Microcirculatory symptoms; n (%) N evaluable = 709 |
235 (33.1) | 149 (32.0) | 25 (28.1) | 18 (36) | 16 (61.5) | 27 (34.6) | 0.03 |
| Median follow up; (months), range | 106.4 (6.1–421.6) | 105.9 (6.1–421.6) | 104.9 (7.3–417.9) | 101.1 (12.1–403.5) | 140.3 (17.4–275.1) | 106.6 (8.6–390.9) |
0.7 |
| MF progression; n (%) | 53 (7.4) | 23 (4.9) | 20 (21.7) | 4 (7.9) | 5 (19.2) | 1 (1.3) | <0.001 |
| AML progression; n (%) | 12 (1.7) | 9 (1.9) | 1 (0.7) | 0 (0.0) | 2 (7.7) | 0 (0.0) | 0.08 |
| Death; n (%) |
106 (14.8) |
77 (16.3) | 15 (16.3) | 3 (5.9) | 6 (23.1) | 5 (6.4) | 0.04 |
Abbreviations: TN, triple negative.
Significant p values are highlighted in bold.
TABLE 2.
Univariate and multivariable analysis of clinical and molecular risk factors for myelofibrosis‐free survival (MFS) among 718 patients with essential thrombocythemia from University of Florence cohort, Italy
| Clinical and laboratory variables | Univariate analysis p a (HR; 95% CI) | Multivariable analysis b p a (HR; 95% CI) | Multivariable analysis c p a (HR; 95% CI) | Multivariable analysis d p a (HR; 95% CI) |
|---|---|---|---|---|
| White blood cells (×109/L) (continuous variable) | 0.05 (0.9; 0.8–1) | |||
|
White blood cells ≥11 × 109/L |
0.07 (0.4; 0.1–1.1) | |||
| Hb (g/dl) (continuous variable) | 0.4 (0.9; 0.7–1.1) | |||
| Hct levels (%) (continuous variable) | 0.5 (0.9; 0.8–1.1) | |||
| Platelet count (×109/L) (continuous variable) | 0.05 (0.9; 0.9–1) | |||
|
Platelet count ≥1000 × 109/L |
0.05 (0.5; 0.2–1) | |||
|
Age >60 years |
0.02 (1.9; 1.1–3.9) | 0.005 (2.5; 1.3–4.9) | 0.02 (3.8; 1.3–11.4) | 0.05 (1.9; 0.9–3.5) |
| Age (continuous variable) | 0.03 (1; 1–1.1) | |||
| Male sex | 0.001 (2.4; 1.4–4.2) | 0.02 (2.1; 1.2–3.9) | 0.3 (1.7; 0.7–4.2) | 0.01 (2; 1.2–3.5) |
| Constitutional symptoms | 0.01 (2.6; 1.2–5.3) | 0.06 2.1 (0.9; 4.7) | 0.3 (2.6; 0.5–12.1) | 0.04 (2.2; 1–4.8) |
| Palpable splenomegaly | 0.002 (2.5; 1.4–4.5) | 0.01 (2.1; 1.2–3.9) | 0.003 (4.3; 1.7–11) | 0.02 (2.1; 1.1–3.8) |
| Microcirculatory symptoms | 0.5 (1.2; 0.7–2.2) | |||
| JAK2 mut | 0.01 (0.5; 0.3–0.9) | |||
| CALR 1/1‐like and 2/2‐like | 0.01 (2.1; 1.2–3.6) | |||
| MPL mut | 0.008 3.2 (1.4–7.7) | |||
| TN | 0.09 0.2 (0.02–1.3) | |||
| CALR 1/1‐like vs. 2/2‐like | 0.04 (3.2; 1.1–9.3) | |||
| CALR 1/1‐like/MPL | <0.001 (3.8; 2.2–6.6) | <0.001 (3.4; 1.9–6.1) | ||
| JAK2V617F VAF >35% vs. ≤35% | <0.001 (5.9; 2.4–14.4) | 0.003 (4.2; 1.6–10.8) | ||
| JAK2V617F > 35% vs. all the others | 0.009 (2.2; 1.2–4) | |||
| CALR type 1/1‐like + MPL + JAK2V617F > 35% vs. all the others | <0.001 (6.1; 3.2–11.7) | <0.0001 (5.2; 2.7–10) |
Significant p values are highlighted in bold.
Considering all ET cohort (n = 718) with CALR type 1/1‐like/MPL as molecular risk variable.
Considering only JAK2 patients (n = 471) with VAF >35% as molecular risk variable.
Considering all ET cohort (n = 718) with CALR type 1/1‐like/MPL/ JAK2 VAF >35% as molecular risk variable.
A receiver operating characteristic (ROC) curve, used to determine the best cut‐off level for JAK2V617F VAF predicting fibrotic evolution, showed an area under curve (AUC) of 0.76 (Figure S2) and best VAF value was 35%. Accordingly, we divided JAK2V617F mutated patients in those with VAF ≤ 35% (364; 77.3%) and >35% (107; 22.7%) (Table 3); the latter showed a significantly higher risk for MF progression (p < 0.001; HR 5.9; 95% CI 2.4–14.4) (Figure 1(A)), that remained significant (p = 0.003; HR 4.2; 95% CI 1.6–10.8) in multivariable analysis including age, sex, splenomegaly, and constitutional symptoms. Among the latter covariates, only age > 60 years (p = 0.02; HR 3.8; 95% CI 1.3–11.4) and splenomegaly (p = 0.003; HR 4.3; 95% CI 1.7–11) remained significant (Table 2). Patients with JAK2V617F VAF > 35% were older (p = 0.02), more likely to be male (p = 0.008), and displayed higher hemoglobin (p = 0.02), platelet counts (p = 0.03), palpable splenomegaly (p < 0.001) and constitutional symptoms (p = 0.009). Moreover, patients with VAF > 35% showed a trend for reduced overall survival (p = 0.08; HR 1.5; 95% CI 0.9–2.4) without differences in thrombosis risk (p = 0.9; HR 0.9; 95% CI 0.5–1.6) and leukemic progression (p = 0.3; HR 2; 95% CI 0.5–7.7) (Figure S3). Multivariable analysis, including age, sex, palpable splenomegaly, and constitutional symptoms, confirmed the independent contribution of JAK2V617F VAF > 35% (p < 0.001; HR 4.6; 95% CI 2.1–9.8) and presence of CALR type 1/1‐like and MPL mutations (p < 0.001; HR 6; 95% CI 2.9–12.2) in predicting MF progression.The above noted observations were corroborated by analysis of two separate cohorts of patients with 2016 WHO‐defined ET, according to the above reported criteria to avoid including incidentally cases of masked PV and pre‐fibrotic MF: Mayo Clinic, USA, and Policlinico Gemelli, Catholic University, Rome, Italy. In the Mayo Clinic ET database, 604 patients (median age 58 years, range 18–90; females 63%) fully annotated for driver mutations were identified; information on JAK2V617F VAF was available in 206 of 347 JAK2V617F mutated patients, including 34 (17%) with VAF >35%. Accordingly, the total number of patients that were fully annotated for driver mutations and JAK2V617F VAF were 479: 199 (42%) JAK2V617F, 101 (21%) CALR type 1/1‐like, 87 (18%) CALR type 2/2‐like, 19 (4%) MPL and 73 (15%) TN. Median time of follow‐up was 110 months (4–588). Multivariable analysis confirmed the independent contribution of JAK2V617F VAF >35% (p < 0.01; HR 4.0; 95% CI 1.9–8.2) and presence of CALR type 1/1‐like and MPL mutations (p = 0.003; HR 2.2; 95% CI 1.3–3.8) in predicting MF progression; these risk factors remained significant when analysis was adjusted for age, sex, or palpable splenomegaly; among the latter, only splenomegaly displayed additional borderline significance (p = 0.07). The Policlinico Gemelli, Catholic University database, contained 454 patients (median age 54 years, range 16–92 years; females 67%) who were fully annotated for driver mutations and in 227 JAK2V617F patients VAF was available, including 62 (27%) with VAF >35%. Accordingly, the total number of patients that were fully annotated for driver mutations and JAK2V617F VAF were 410: 227 (55.4%) JAK2V617F, 45 (11%) CALR type 1/1‐like, 33 (8%) CALR type2/2‐like, 19 (4.6%) MPL and 86 (21%) TN. Median time of follow‐up was 75.6 months (4–362.4). Multivariable analysis confirmed the independent contribution of JAK2V617F VAF >35% in predicting transformation to myelofibrosis (p = 0.01; HR, 2.9, 95%CI 1.2–7.0), that remained significant after adjusting for age, sex, and palpable splenomegaly. The latter covariate displayed a borderline significance (p = 0.07); conversely, CALR type 1/1‐like and MPL mutations were not statistically significant.
TABLE 3.
Clinical characteristics of JAK2V617F positive ET patients from University of Florence cohort stratified by their variant allele frequency (VAF, ≤ 35% vs. > 35%)
| Clinical and laboratory variables | All JAK2 patients (n = 471) | JAK2 V617F VAF ≤35% (n = 364; 77.3%) | JAK2 V617F VAF > 35% (n = 107; 22.7%) | p valuesa |
|---|---|---|---|---|
| Age in year; median (range) | 62.0 (14.9–92.9) | 60.1 (14.9–92.9) | 66.5 (17.5–85.3) | 0.02 |
| Age ≥ 60 years; n (%) | 253 (53.7) | 183 (50.3) | 70 (65.4) | 0.01 |
| Sex females; n (%) | 318 (67.5) | 258 (70.9) | 60 (56.1) | 0.008 |
|
White blood cells × 109/L; median (range) N evaluable = 458 |
8.7 (3.7–18.2) | 8.6 (4.0–18.2) | 9.2 (3.7–18.2) | 0.1 |
|
White blood cells ≥11 × 109/L; n (%) N evaluable = 458 |
90 (19.6) | 67 (18.9) | 23 (22.3) | 0.3 |
| Hemoglobin, g/dl; median (range) | 14.2 (11.8–16.5) | 14.2 (11.8–16.5) | 14.6 (11.8–16.5) | 0.02 |
|
Hematocrit, % median (range) |
43.8 (36.0–50.0) | 43.4 (36.0–50.0) | 45 (37.0–50.0) | 0.003 |
| Platelets × 109/L; median (range) | 698 (455–1881) | 694 (455–1867) | 728 (455–1881) | 0.03 |
| Platelets ≥1000 × 109/L; n (%) | 61 (12.9) | 43 (11.8) | 18 (16.8) | 0.3 |
| Palpable splenomegaly; n (%) | 67 (14.2) | 40 (11.0) | 27 (25.2) | 0.0003 |
| Constitutional symptoms; n (%) | 27 (5.7) | 15 (4.1) | 12 (11.2) | 0.009 |
|
Major thrombosis before diagnosis; n (%) N evaluable = 405 |
35 (8.6) | 25 (8.0) | 10 (11.0) |
0.4 |
|
Major thrombosis at diagnosis; n (%) N evaluable = 397 |
31 (7.8) | 21 (6.8) | 10 (11.2) | 0.09 |
|
Overall thrombosis at follow‐up; n (%) N evaluable = 470 |
69 (14.7) | 52 (14.3) | 17 (15.9) | 0.7 |
|
Overall major bleeding; n (%) N evaluable = 470 |
41 (8.7) | 34 (9.4) | 7 (6.5) | 0.4 |
|
Overall minor bleeding; n (%) N evaluable = 467 |
54 (11.6) | 40 (11.1) | 14 (13.1) | 0.6 |
|
Cardiovascular risk factors; n (%) N evaluable = 395 |
235 (59.5) | 186 (60.0) | 49 (57.6) | 0.7 |
|
Microcirculatory symptoms; n (%) N evaluable = 466 |
149 (32.0) | 113 (31.4) | 36 (34.0) | 0.6 |
| MF progression; n (%) | 23 (4.9) | 7 (1.9) | 16 (14.9) | <0.001 |
| AML progression: n (%) | 9 (1.9) | 5 (1.4) | 4 (3.7) | 0.1 |
| Death; n (%) | 77 (16.3) | 51 (13.9) | 28 (25.9) | 0.003 |
Abbreviations: VAF, variant allele frequency.
Significant p values are highlighted in bold.
FIGURE 1.
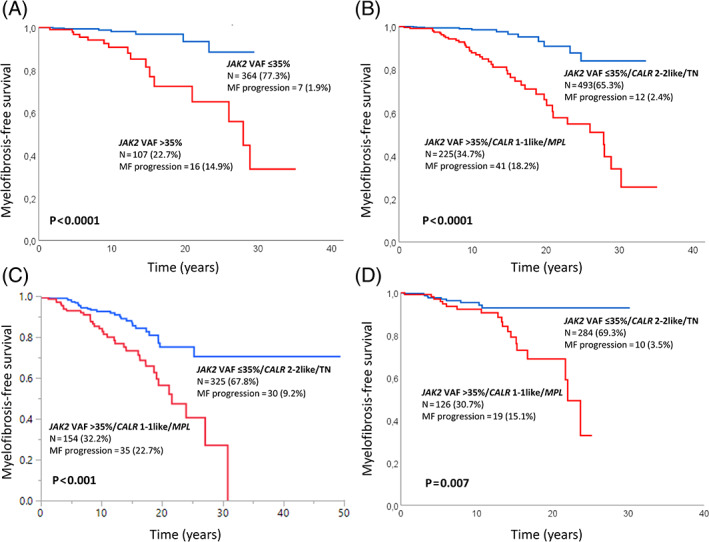
(A) Myelofibrosis‐free survival (MFS) among 471 JAK2V617F mutated 2016‐WHO essential thrombocythemia (ET) patients from University of Florence cohort, stratified by their VAF, ≤ 35% vs. >35%; (B) MFS among 7 182 016‐WHO ET patients from University of Florence cohort, fully annotated for driver mutation type and JAK2V617F VAF, stratified by high‐risk (JAK2V617F VAF >35%, CALR type 1/1‐like and MPL) and low‐risk (JAK2V617F VAF ≤35%, CALR type 2/2‐like and TN) for MF progression. (C) External validation of the molecular model in 479 ET patients from Mayo Clinic, USA and (D) 410 from Policlinico Gemelli, Catholic University, Italy [Color figure can be viewed at wileyonlinelibrary.com]
Subsequently, using the University of Florence patient cohort, we developed a two‐tiered, driver mutation‐based, model for risk of MF progression in ET identifying a high molecular risk (JAK2V617F VAF > 35%, CALR type 1/1‐like and MPL) and low molecular risk (JAK2V617F VAF ≤ 35%, CALR type 2/2‐like and TN) category. Kaplan‐Meier analysis documented a significantly different MFS (p < 0.001; HR 6.1; 95% CI 3.2–11.7) with respective risk rates of MF evolution of 1.2% (population at risk, n = 195) and 8% (n = 100) at 10 years and 8% (n = 40) and 33% (n = 26) at 20 years, respectively for the low and high‐risk category (Figure 1(B)). The model remained statistically significant (p < 0.001; HR 5.2; 95%CI 2.7–10) in multivariable analysis including age, sex, splenomegaly, and constitutional symptoms; among the latter covariates, male sex (p = 0.01; HR 2; 95% CI 1.2–3.5), splenomegaly (p = 0.02; HR 2.1; 95% CI 1.1–3.8) and constitutional symptoms (p = 0.04; HR 2.2; 95% CI 1–4.8) remained independently significant whereas age > 60 years displayed a borderline significance (p = 0.05), (Table 2).
This operational molecular model developed in University of Florence cohort was validated in the other two independent cohorts. In the Mayo Clinic cohort, the rate of progression to MF was 9% (events = 30) and 23% (events = 35) in the low and high‐risk category, with respective risk rates of 7% (population at risk; n = 124) and 16% (n = 65) at 10 years, and 25% (n = 30) and 44% (n = 14) at 20 years. Figure 1(C) illustrates application of the Florence molecular model for MF progression in ET, in the Mayo Clinic validation cohort (p < 0.001; HR 2.5; 95% CI 1.6–4.1). In the Catholic University, Policlinico Gemelli cohort, the rate of progression to MF was 3.5% (events n = 10) and 15% (events n = 19) in the low and high‐risk category, with respective risk rates of 4.6% (population at risk; n = 80) and 7.8% (n = 59) at 10 years, and 7.1% (n = 6) and 31.2% (n = 9) at 20 years. Application of the molecular model, using the Policlinico Gemelli cohort, is shown in Figure 1(D) (p = 0.007, HR 2.7; 95% CI 1.3–5.8).
4. DISCUSSION
To date, while many studies have investigated the presence of risk factors impacting on survival and an increased risk of thrombosis in ET, few data are available on the risk of progression to MF. With limitations imposed by the retrospective nature of analysis, the current study included a remarkable series of 1607 patients from three independent institutions, identified a category of patients at high risk for MF progression among 2016 WHO‐defined ET patients. Firstly, Florence patient cohort was examined to identify independent risk factors for MFS, which included age > 60 years (HR 2.5, 95% CI 1.3–4.9), male sex (2.1, 1.2–3.9), palpable splenomegaly (2.1, 1.2–3.9), CALR 1/1‐like or MPL mutation (3.4, 1.9–6.1) and JAK2V617F variant allele frequency > 35% (4.2, 1.6–10.8). Subsequently, an operational molecular risk category was developed and validated in the other two independent cohorts from Mayo Clinic and Rome. “High molecular risk” category included patients with JAK2V617F VAF > 35%, CALR type 1/1‐like or MPL mutations whereas all other driver mutation profiles were assigned to “low molecular risk” category. All the clinical and molecular variables analyzed in the present study are routinely collected at diagnosis, including the VAF of JAK2V617F that is provided by the majority of assays employed in diagnostic laboratories. From a practical standpoint, the findings provided by the current study may be useful for patients' counseling and, prospectively, stratification in clinical trials. Most recently, targeted next generation sequencing (NGS) has revealed the presence of other mutations in many ET patients, with the most frequent being TET2 (16%) and ASXL1 (11%). In the same study, SH2B3, SF3B1, U2AF1, TP53, IDH2, and EZH2 mutations were associated with inferior overall, leukemia‐free or myelofibrosis‐free survival. 20 Consequently, these notions were incorporated in a molecular‐clinical integrated prognostic model for survival (MIPSS‐ET). 8 Future collaborative efforts will investigate possible correlations between the VAF of JAK2V617F and the presence/absence of additional mutations. Moreover, we have found a significantly higher risk of fibrotic progression in patients carrying type 1/1‐like CALR mutations than in those with type 2/2‐like. This result was highlighted in a previous study including a large cohort of ET patients, but without a multivariable model. 14 Interestingly, our observations are in according with a recent experimental paper, 21 in which the authors generated heterozygous and homozygous conditional inducible knock‐in (KI) mice expressing a chimeric murine CALR del‐52 (type 1) or ins‐5 (type 2) with the human mutated C‐terminal tail to investigate their pathogenic effects on hematopoiesis. Thus, Del52 induces greater phenotypic changes than ins5 including thrombocytosis, leucocytosis, splenomegaly, bone marrow hypocellularity, megakaryocytic lineage amplification. Moreover, homozygous del52 KI mice displayed features of a penetrant myelofibrosis‐like disorder with extramedullary hematopoiesis linked to splenomegaly, megakaryocyte hyperplasia and the presence of reticulin fibers. In this regard, it would be interesting to evaluate in future studies the in vivo impact, if any, of VAF both for CALR type 1/1 like and type 2/2‐like in large cohorts of ET patients.
We acknowledge potential limitations in the interpretation of findings reported herein. First, the retrospective nature of the study that may harbor intrinsic selection biases, and second, the absence of a centralized histopathological review of BM biopsies. In this respect, it is reasonable to entertain the possibility that some of the cases in the current study population might have represented occult cases of early/prefibrotic MF or masked PV. Moreover, as recently reported, 22 , 23 we recognize that a definite diagnosis of early cases of PV may require red cell mass (RCM) determination by isotopic methods. However, this is really far from a routine clinical scenario, therefore we made our best to adhere to the latest WHO criteria that uses BM histopathology along with subnormal erythropoietin levels as a potential surrogates. Finally, to minimize the inadvertent inclusion of cases with masked‐PV we excluded those cases that exceeded the Hb/Hct threshold recommended by WHO, whereas cases with anemia were excluded as potential misdiagnosis of pre‐fibrotic MF. On the other hand, it should be emphasized that histopathology analysis was performed, and clinical and hematological information were collected and evaluated, in centers of huge experience in MPNs, that may add to the quality and cleanness of the series reported in this manuscript.
Finally, the possibility that the current model merely selects morphologically bypassed pre‐fibrotic MF cases or, in other words, the fact that VAF of JAK2V617F may help, together with clinical and histopathological picture, in discriminating true ET from pre‐MF cases is an intriguing issue that might be studied in large cohorts of patients with a centralized histopathological review by a panel of experts. 24 However, from a practical standpoint, this possibility by itself does not undermine the main objective of the current study, which is to identify patients who require close monitoring and appropriate counseling.
CONFLICT OF INTEREST
V.D.S. received personal fees for advisory board and/or lectures from AbbVie, AOP Orphan Pharmaceutical, and Novartis, and research grants from AbbVie and Novartis. P.G. received personal fees for advisory board and/or lectures from Novartis. A.M.V. received personal fees for advisory board and/or lectures from Novartis, AbbVie, AOP Pharmaceuticals, B.M.S. and Incyte. All other authors have no conflict to report.
AUTHOR CONTRIBUTIONS
Giuseppe G. Loscocco, Paola Guglielmelli, Valerio De Stefano, Ayalew Tefferi and Alessandro M. Vannucchi designed research, analyzed data, and wrote the manuscript. All other authors contributed clinical and/or molecular data and revised and approved the final version of the manuscript.
Supporting information
Figure S1. Overall (A), myelofibrosis‐free (B), leukemia‐free (C) and thrombosis‐free (D) survival curves in 718 ET patients from University of Florence stratified by their mutational status.
Figure S2. ROC curve for JAK2V617F VAF as a risk variable for MF progression.
Figure S3. Kaplan Meyer curves for overall (A), thrombosis‐free (B) and leukemia‐free (C) survival in JAK2 ET patients stratified by their variant allele frequency (Blue curve VAF ≤35% vs. red curve >35%).
ACKNOWLEDGMENTS
This work has received financial support from Associazione Italiana per la Ricerca sul Cancro (AIRC) 5 × 1000 call “Metastatic disease: the key unmet need in oncology” to MYNERVA (MYeloid NEoplasms Research Venture AIRC), project #21267; Bando Ricerca Finalizzata Ministero della Salute RF‐2016‐02362930. Open Access Funding provided by Universita degli Studi di Firenze within the CRUI‐CARE Agreement.
Loscocco GG, Guglielmelli P, Gangat N, et al. Clinical and molecular predictors of fibrotic progression in essential thrombocythemia: A multicenter study involving 1607 patients. Am J Hematol. 2021;96(11):1472-1480. doi: 10.1002/ajh.26332
Giuseppe G. Loscocco, Paola Guglielmelli, Valerio De Stefano, Ayalew Tefferi, and Alessandro M. Vannucchi contributed equally to this work.
Funding information Associazione Italiana per la Ricerca sul Cancro (AIRC) 5×1000 call “Metastatic disease: the key unmet need in oncology” to MYNERVA (MYeloid NEoplasms Research Venture AIRC), Grant/Award Number: project #21267; Bando Ricerca Finalizzata Ministero della Salute, Grant/Award Number: RF‐2016‐02362930
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon request. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- 1. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391‐2405. 10.1182/blood-2016-03-643544 [DOI] [PubMed] [Google Scholar]
- 2. Tefferi A, Pardanani A. Essential thrombocythemia. N Engl J Med. 2019;381(22):2135‐2144. 10.1056/NEJMcp1816082 [DOI] [PubMed] [Google Scholar]
- 3. Loscocco GG, Guglielmelli P, Vannucchi AM. Impact of mutational profile on the management of myeloproliferative neoplasms: a short review of the emerging data. Onco Targets Ther. 2020;13:12367‐12382. 10.2147/OTT.S287944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barbui T, Finazzi G, Carobbio A, et al. Development and validation of an international prognostic score of thrombosis in World Health Organization‐essential thrombocythemia (IPSET‐thrombosis). Blood. 2012;120(26):5128‐5133; quiz 5252. 10.1182/blood-2012-07-444067 [DOI] [PubMed] [Google Scholar]
- 5. Barbui T, Vannucchi AM, Buxhofer‐Ausch V, et al. Practice‐relevant revision of IPSET‐thrombosis based on 1019 patients with WHO‐defined essential thrombocythemia. Blood Cancer J. 2015;5(11):e369. 10.1038/bcj.2015.94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Haider M, Gangat N, Lasho T, et al. Validation of the revised international prognostic score of thrombosis for essential thrombocythemia (IPSET‐thrombosis) in 585 Mayo clinic patients. Am J Hematol. 2016;91(4):390‐394. 10.1002/ajh.24293 [DOI] [PubMed] [Google Scholar]
- 7. Passamonti F, Thiele J, Girodon F, et al. A prognostic model to predict survival in 867 World Health Organization–defined essential thrombocythemia at diagnosis: a study by the international working group on myelofibrosis research and treatment. Blood. 2012;120(6):1197‐1201. 10.1182/blood-2012-01-403279 [DOI] [PubMed] [Google Scholar]
- 8. Tefferi A, Guglielmelli P, Lasho TL, et al. Mutation‐enhanced international prognostic systems for essential thrombocythaemia and polycythaemia vera. Br J Haematol. 2020;189(2):291‐302. 10.1111/bjh.16380 [DOI] [PubMed] [Google Scholar]
- 9. Guglielmelli P, Gangat N, Coltro G, et al. Mutations and thrombosis in essential thrombocythemia. Blood Cancer J. 2021;11(4):77. 10.1038/s41408-021-00470-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391‐2405. 10.1056/NEJMoa1312542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rumi E, Pietra D, Ferretti V, et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood. 2014;123(10):1544‐1551. 10.1182/blood-2013-11-539098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rotunno G, Mannarelli C, Guglielmelli P, et al. Impact of calreticulin mutations on clinical and hematological phenotype and outcome in essential thrombocythemia. Blood. 2014;123(10):1552‐1555. 10.1182/blood-2013-11-538983 [DOI] [PubMed] [Google Scholar]
- 13. Grinfeld J, Nangalia J, Baxter EJ, et al. Classification and personalized prognosis in myeloproliferative neoplasms. N Engl J Med. 2018;379(15):1416‐1430. 10.1056/NEJMoa1716614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pietra D, Rumi E, Ferretti VV, et al. Differential clinical effects of different mutation subtypes in CALR‐mutant myeloproliferative neoplasms. Leukemia. 2016;30(2):431‐438. 10.1038/leu.2015.277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rotunno G, Pacilli A, Artusi V, et al. Epidemiology and clinical relevance of mutations in postpolycythemia vera and postessential thrombocythemia myelofibrosis: a study on 359 patients of the AGIMM group. Am J Hematol. 2016;91(7):681‐686. 10.1002/ajh.24377 [DOI] [PubMed] [Google Scholar]
- 16. Elala YC, Lasho TL, Gangat N, et al. Calreticulin variant stratified driver mutational status and prognosis in essential thrombocythemia. Am J Hematol. 2016;91(5):503‐506. 10.1002/ajh.24338 [DOI] [PubMed] [Google Scholar]
- 17. Vannucchi AM, Antonioli E, Guglielmelli P, et al. Prospective identification of high‐risk polycythemia vera patients based on JAK2(V617F) allele burden. Leukemia. 2007;21(9):1952‐1959. 10.1038/sj.leu.2404854 [DOI] [PubMed] [Google Scholar]
- 18. Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the international working group for myelofibrosis research and treatment. Blood. 2009;113(13):2895‐2901. 10.1182/blood-2008-07-170449 [DOI] [PubMed] [Google Scholar]
- 19. Barosi G, Mesa RA, Thiele J, et al. Proposed criteria for the diagnosis of post‐polycythemia vera and post‐essential thrombocythemia myelofibrosis: a consensus statement from the international working group for myelofibrosis research and treatment. Leukemia. 2008;22(2):437‐438. 10.1038/sj.leu.2404914 [DOI] [PubMed] [Google Scholar]
- 20. Tefferi A, Lasho TL, Guglielmelli P, et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016;1(1):21‐30. 10.1182/bloodadvances.2016000216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Benlabiod C, Cacemiro M d C, Nédélec A, et al. Calreticulin del52 and ins5 knock‐in mice recapitulate different myeloproliferative phenotypes observed in patients with MPN. Nat Commun. 2020;11(1):4886. 10.1038/s41467-020-18691-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Silver RT, Krichevsky S. Distinguishing essential thrombocythemia JAK2 V617F from polycythemia vera: limitations of erythrocyte values. Haematologica. 2019;104(11):2200‐2205. 10.3324/haematol.2018.213108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maslah N, Soret J, Dosquet C, et al. Masked polycythemia vera: analysis of a single center cohort of 2480 red cell masses. Haematologica. 2020;105(3):e95‐e97. 10.3324/haematol.2018.215582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Barbui T, Vannucchi AM, Guglielmelli P, de Stefano V, Rambaldi A. An agenda for future research projects in polycythemia vera and essential thrombocythemia. Haematologica. 2020;105(8):1999‐2003. 10.3324/haematol.2019.246207 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Overall (A), myelofibrosis‐free (B), leukemia‐free (C) and thrombosis‐free (D) survival curves in 718 ET patients from University of Florence stratified by their mutational status.
Figure S2. ROC curve for JAK2V617F VAF as a risk variable for MF progression.
Figure S3. Kaplan Meyer curves for overall (A), thrombosis‐free (B) and leukemia‐free (C) survival in JAK2 ET patients stratified by their variant allele frequency (Blue curve VAF ≤35% vs. red curve >35%).
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon request. The data are not publicly available due to privacy or ethical restrictions.