

Abstract
Filgotinib, an oral Janus kinase‐1 preferential inhibitor, is approved in Europe and Japan for adults with rheumatoid arthritis. Patients with rheumatoid arthritis are at higher risk of cardiovascular morbidity/mortality; thus, it is important to understand potential drug‐drug interactions of filgotinib with lipid‐lowering agents. This open‐label, randomized, 2‐way crossover study evaluated the pharmacokinetics of atorvastatin, pravastatin, and rosuvastatin with and without filgotinib coadministration. Healthy participants (N = 27) received single doses of atorvastatin (40 mg) and of a pravastatin (40 mg)/rosuvastatin (10 mg) cocktail—alone or with filgotinib (200 mg once daily for 11 days)—on 2 different occasions with washout in between. Serial pharmacokinetic blood samples were collected, and safety was assessed. Pharmacokinetic parameters were evaluated using 90% confidence intervals (CI) of the geometric least‐squares mean (GLSM) ratio of the test treatment (statin coadministration with filgotinib) vs statin alone, with prespecified lack‐of‐interaction bounds of 0.70 to 1.43. Coadministration of filgotinib did not affect atorvastatin area under the plasma concentration–time curve extrapolated to infinity (AUCinf; [GLSM ratios (90% CI): 0.91 (0.84‐0.99)]), but maximum concentration [Cmax] was slightly lower [0.82 (0.69‐0.99)]. The exposure of 2‐hydroxy‐atorvastatin was unaffected (GLSM ratios [90% CI], 0.98 [0.81‐1.19] for Cmax; 1.11 [1.02‐1.22] for AUCinf). Pravastatin AUCinf was also unaffected (GLSM ratios, 1.22 [1.05‐1.41], but Cmax was slightly higher 1.25 [1.01‐1.54]). Rosuvastatin exposure was moderately higher with filgotinib coadministration—GLSM ratios (90% CI), 1.68 (1.43‐1.97) for Cmax; 1.42 (1.30‐1.57) for AUCinf—but this was not considered clinically relevant. These results indicate that filgotinib has no clinically meaningful effect on exposure of atorvastatin, pravastatin, or rosuvastatin.
Keywords: clinical pharmacology, drug‐drug interactions, pharmacokinetics and drug metabolism, pharmacology, rheumatology
Patients with rheumatoid arthritis (RA) are at an elevated risk of cardiovascular morbidity and mortality (relative risk, 1.4 in men and 1.5 in women). 1 Statin use is recommended for patients with risk‐enhancing factors including RA. 2 A recent position paper suggests that many patients with RA may benefit from intensified lipid‐modifying therapy to reduce cardiovascular risk. 3 Drug‐drug interactions that significantly increase the exposure of statins are unfavorable because high statin exposure has been associated with statin‐induced myopathy. 4 As such, it is important to understand potential interactions of therapies for RA with lipid‐modifying agents.
Filgotinib (brand name Jyseleca®), a once‐daily, oral, Janus kinase (JAK) 1 preferential inhibitor, is approved for use in Europe and Japan in adult patients with moderately to severely active RA who have had an inadequate response to conventional therapies. 5 Filgotinib is ≈5‐ to 30‐fold more selective for inhibition of JAK1 than JAK2. 6 , 7 Filgotinib undergoes rapid absorption following oral dosing and is primarily metabolized by carboxylesterase isoform 2, resulting in the loss of the cyclopropyl carboxylic acid group to form its major circulating metabolite, GS‐829845 (see Namour et al 8 for the structures of filgotinib and its major metabolite). GS‐829845 has similar selectivity for JAK1 as filgotinib but with ≈10‐fold reduced potency based on human whole blood assay and higher (≈16‐ to 20‐fold) systemic exposure. 8
In vitro studies demonstrated that filgotinib and GS‐829845 do not interact with cytochrome P450 enzymes or uridine 5’‐diphospho‐glucuronosyltransferases, and they do not inhibit key drug transporters at clinically relevant concentrations. 9 Filgotinib and GS‐829845 were shown to not affect QT interval. 10 Systemic exposure to filgotinib is not affected by food intake or by coadministration of acid‐reducing agents. 11 Coadministration of filgotinib and oral contraceptives did not affect the pharmacokinetics of the representative oral contraceptive studied (Microgynon [ethinyl estradiol]/levonorgestrel). 12 Age or mild to moderate renal impairment were shown to have only limited effects on the steady‐state exposure of filgotinib. 13
Filgotinib and GS‐829845 are in vitro inhibitors of organic anion transporting peptide (OATP)‐1B1/1B3 isoforms at concentrations that are higher than those that are clinically relevant with half‐maximal inhibitory concentrations (IC50) of 98 μM and >285 μM, respectively, for filgotinib and 260 μM and >473 μM, respectively, for GS‐829845. Steady‐state maximum observed plasma concentration (Cmax) values of filgotinib and GS‐829845 are 5.1 and 12 μM, respectively, following 200 mg once‐daily dosing in RA patients. OATP1B1 and OATP1B3 are the major isoforms of OATP expressed on hepatocytes and are responsible for transporting substrate drugs, including statins, into hepatocytes for further metabolism. 14 , 15
The objective of this study was to clinically evaluate the potential for drug‐drug interactions (DDIs) between filgotinib and 3 statins that are OATP substrates: atorvastatin (cytochrome P450 (CYP) 3A4 and OATP substrate), pravastatin (OATP substrate), and rosuvastatin (breast cancer resistance protein and OATP substrate).
Methods
Ethics Statement
The study protocol was reviewed and approved by the IntegReview institutional review board (Austin, Texas). The study was carried out in accordance with the Declaration of Helsinki and the International Council for Harmonisation Good Clinical Practice guidelines. All participants provided written informed consent before study participation. The study was conducted at Prism Research (St Paul, Minnesota).
Study Design and Population
This was a phase 1, randomized, 2‐way crossover, open‐label, single‐ and multiple‐dose, single‐center study in healthy adult participants (Figure 1). Eligible participants included surgically sterile men and nonpregnant, nonlactating women between 18 and 55 years of age, with a body mass index ≥19 and ≤30 kg/m2, normal 12‐lead electrocardiogram, normal renal function, and no significant medical history, who were in general good health at the time of screening (≤28 days before the first dose) as determined by the investigator. Exclusion criteria included receipt of any investigational drug or device (30 days before the first dose), positive coronavirus disease 2019 real‐time reverse transcriptase polymerase chain reaction testing on screening and admission, use of any prescription or over‐the‐counter medications (except vitamins, acetaminophen, ibuprofen, and/or hormonal contraceptive medications) or herbal products within 28 days of commencing study drug dosing, and no current alcohol or substance use.
Figure 1.
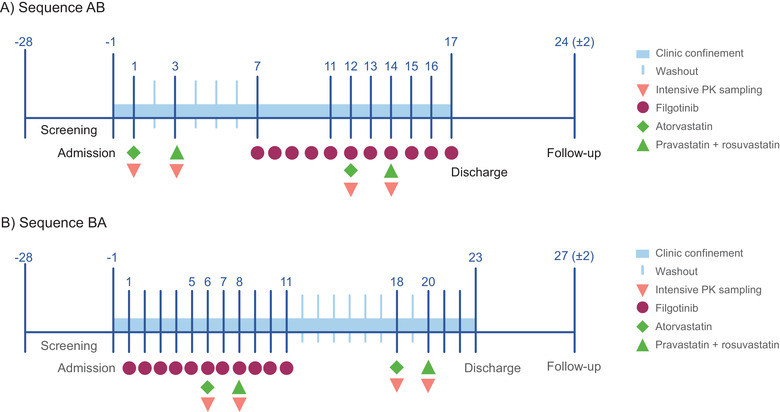
Study schematic for (A) sequence AB and (B) sequence BA. Samples collected during intensive PK sampling. PK, pharmacokinetics.
Following screening and day −1 assessments, participants were randomized 1:1 to 1 of 2 treatment sequences (AB or BA). Treatment A (reference treatment) was a single dose of atorvastatin 40 mg, followed by a washout period of 1 day and a single dose of a combination of pravastatin 40 mg (Bristol‐Myers Squibb, Princeton, New Jersey)/rosuvastatin 10 mg (AstraZeneca Pharmaceuticals, Wilmington, Delaware). Treatment B (test treatment) was filgotinib 200 mg administered once daily for 11 days, with a single dose of atorvastatin 40 mg (Pfizer, Dublin, Ireland) administered on day 6 followed by a single dose of a combination of pravastatin 40 mg/rosuvastatin 10 mg administered on day 8.
All doses were administered by qualified study center staff; mouth checks were performed to ensure that the medication was taken, and time of dose was recorded. All study treatments were administered with 240 mL water in the morning at approximately the same time following an overnight fast. Participants fasted for 2 hours after dosing on days without blood sampling; on days when pharmacokinetic samples were collected, participants fasted until 4 hours after dosing and were then provided with a standardized meal.
Pharmacokinetic Sampling
Plasma concentrations of atorvastatin, 2‐hydroxy‐atorvastatin (active metabolite of atorvastatin), pravastatin, and rosuvastatin were determined, and pharmacokinetic parameters were evaluated. Participants were assigned to 1 of 2 sequences, described above, either statins alone first or in combination with filgotinib first (Figure 1). Intensive pharmacokinetic sampling occurred relative to dosing of study drug on the following schedules: blood samples were collected from participants at 5 minutes before dosing and 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 16, 24, and 48 hours after dosing. Samples were also collected at 5, 10, and 36 hours after dosing on days when atorvastatin was administered and 72 hours after dosing when pravastatin/rosuvastatin was administered. All samples were collected on ice and stored at −70˚C until processing.
Safety Assessments
Safety was monitored throughout the study, at screening, and at discharge for all participants. All participants were contacted via telephone 7 ± 2 days after the last administration of the study drug as a follow‐up. On days of drug administration, vital signs were assessed before drug dosing (except for days 2, 4, and 10). Symptom‐driven physical examination was conducted during clinic confinement. Assessments of adverse events (AEs) were performed throughout the study with AEs recorded as per the Medical Dictionary for Regulatory Activities and severity recorded by Common Terminology Criteria for Adverse Events version 5.0. AEs associated with laboratory abnormalities were graded on the basis of clinical severity of the underlying condition.
Bioanalytical Procedures
Plasma samples were collected and subsequently analyzed at QPS, LLC (Newark, Delaware) by commercially available bioanalytical methods for each of the statins studied; the methods were validated at QPS according to criteria established in US Food and Drug Administration guidance. Each method used liquid‐liquid extraction to isolate the analytes and their added corresponding isotopically labeled analytes as internal standards from the sample. Additionally, each method used high‐performance liquid chromatography‐tandem mass spectrometry for quantitation. Details of the methods are presented in Table 1. Linear calibration curves were generated from drug‐to‐internal standards peak area ratios of calibration standards using least‐squares regression analysis with 1/(nominal concentration) 2 weighting. All study samples were analyzed within the frozen storage stability durations established for each method.
Table 1.
Details of the Bioanalytical Methods for the Evaluated Statins
| Method: | Atorvastatin/2‐OH‐atorvastatin | Pravastatin | Rosuvastatin | |
|---|---|---|---|---|
| Analyte | Atorvastatin | 2‐OH‐Atorvastatin | Pravastatin | Rosuvastatin |
| Internal standard | Atorvastatin‐d 5 | 2‐OH‐Atorvastatin‐d 5 | Pravastatin‐d 3 | Rosuvastatin‐d 6 |
| Extraction method | Liquid‐liquid | Liquid‐liquid | Liquid‐liquid | |
| HPLC column | Waters Acquity UPLC HSS T3, 1.8 μm, 2.1 x 50 mm | Waters Acquity UPLC BEH C18, 2.1 x 50 mm, 1.7 μm | Phenomenex Luna C18(2), 2 x 30 mm, 5 μm | |
|
Mobile phase (Gradient components A and B) |
A: Water:acetic acid at 100:0.1 (v:v) B: Acetonitrile: acetic acid at 100:0.1 (v:v) |
A: Water:formic acid at 100:0.1 (v:v) B: Acetonitrile:water: formic acid at 95:5:0.1 (v:v:v) |
A: Water:formic acid at 100:0.1 (v:v) B: Methanol:formic acid at 100:0.1 (v:v) |
|
| Retention time | 1.33 min | 1.22 min | 1.16 min | 2.6 min |
| Mass spectrometer settings | ||||
| Ionization | Positive Turbo Ion Spray® | Negative Turbo Ion Spray® | Positive Turbo Ion Spray® | |
| Mode | MRM | MRM | MRM | |
| m/z monitored | ||||
| Analyte | 559.3 → 440.3 | 575.3 → 440.3 | 423.2 → 321.2 | 482.2 → 258.2 |
| Internal Standard | 564.3 → 445.3 | 580.3 → 445.3 | 426.2 → 321.2 | 488.2 → 264.2 |
| LLOQ | 0.1 ng/mL | 0.1 ng/mL | 0.1 ng/mL | 0.05 ng/mL |
| Precision (%CV) | ||||
| Within‐day | ≤4.7 | ≤5.9 | ≤4.4 | ≤6.0 |
| Between‐day | ≤3.7 | ≤5.0 | ≤3.8 | ≤5.7 |
| Accuracy (%RE) | ||||
| Within‐day | −2.7 to 3.3 | −0.7 to 5.0 | −2.3 to 4.0 | −1.6 to 10.0 |
| Between‐day | −2.0 to 2.5 | 0.0 to 3.8 | −1.9 to 2.7 | 1.2 to 6.0 |
%CV, coefficient of variation, expressed as a percentage; %RE, relative error, expressed as a percentage; LLOQ, lower limit of quantitation; MRM, multiple reaction monitoring.
Pharmacokinetic Analysis
Pharmacokinetic parameters of atorvastatin, 2‐hydroxy‐atorvastatin, pravastatin, and rosuvastatin were calculated by noncompartmental methods using Phoenix WinNonlin 8.2 (Certara, Princeton, New Jersey). Calculated parameters included area under the plasma concentration–time curve (AUC) from time 0 to the last quantifiable concentration, AUC extrapolated to infinity (AUCinf), Cmax, time to maximal concentration, and the terminal phase elimination half‐life. The 2‐hydroxy‐atorvastatin to atorvastatin metabolite‐to‐parent molar ratios of AUCinf and Cmax were calculated. Postdose samples that were below the lower limit of quantitation were treated as missing data in the noncompartmental analyses. For summary purposes, values that were below the lower limit of quantitation were treated as 0 at predose and postdose time points.
Statistical Methods
Plasma concentrations and pharmacokinetic parameters were summarized using descriptive statistics by treatment. An analysis of variance using a mixed‐effects model with treatment, period, and sequence as fixed effects and participant as a random effect was fitted to the natural logarithmic transformation of the primary pharmacokinetic parameters (AUC from time 0 to the last quantifiable concentration, AUCinf, Cmax) for each analyte. Two‐sided 90% confidence intervals (CIs) were calculated for the ratios of geometric least‐squares mean (GLSM) of primary pharmacokinetic parameters for treatment B versus treatment A. A lack of pharmacokinetic interaction was concluded if the 90% CIs for the GLSM ratios were contained within a range of 0.70 and 1.43 for atorvastatin, 2‐hydroxy‐atorvastatin, pravastatin, and rosuvastatin for AUC and Cmax. This 0.70 to 1.43 boundary was selected on the basis of the dose/exposure relationship for the statins studied, which indicates that a 30% change in exposure is not clinically relevant, and these no‐effect boundaries have been previously used in other DDI studies with statins. 16 , 17 , 18 With 25 participants enrolled, the estimated 2‐sided 90% CI of the percent GLSM ratio for treatment B versus treatment A was within 0.70 to 1.43, with >80% probability if the true GLSM ratio were 1.0 and a standard deviation of no more than 0.569 on a natural logarithmic scale, based on prior pharmacokinetic studies.
Results
A total of 27 participants enrolled in the study, and 25 completed all study treatments. Two participants were enrolled but discontinued study treatments: 1 participant—the only participant of Asian descent in the study—who was administered filgotinib plus the statins, discontinued before the period of administration of the statins alone due to a grade 3 increase in creatine phosphokinase, and a second participant discontinued due to difficulty with blood draws. The majority of participants were White women with a mean age of 30 years. Baseline demographics were balanced between treatment sequences (Table 2).
Table 2.
Baseline Demographics
| Treatment Sequence AB n = 14 | Treatment Sequence BA n = 13 | Total N = 27 | |
|---|---|---|---|
| Age, y | |||
| Median (range) | 32 (20‐43) | 26 (19‐43) | 30 (19‐43) |
| Mean (SD) | 31 (7.2) | 29 (7.6) | 30 (7.3) |
| Sex, n (%) | |||
| Female | 14 (100) | 12 (92.3) | 26 (96.3) |
| Male | 0 | 1 (7.7) | 1 (3.7) |
| Race, n (%) | |||
| Asian | 0 | 1 (7.7) | 1 (3.7) |
| Black or African American | 3 (21.4) | 2 (15.4) | 5 (18.5) |
| White | 10 (71.4) | 9 (69.2) | 19 (70.4) |
| Other | 1 (7.1) | 1 (7.7) | 2 (7.4) |
| Ethnicity, n (%) | |||
| Hispanic or Latino | 0 | 1 (7.7) | 1 (3.7) |
| Not Hispanic or Latino | 14 (100) | 12 (92.3) | 26 (96.3) |
| Body weight, kg | |||
| Median (range) | 65.6 (54.6‐90.9) | 72.2 (55.6‐84.5) | 67.8 (54.6‐90.9) |
| Mean (SD) | 68.1 (9.70) | 70.7 (9.39) | 69.4 (9.46) |
| Body mass index, kg/m2 | |||
| Median (range) | 24.7 (19.3‐29.8) | 26.1 (19.2‐29.1) | 24.8 (19.2‐29.8) |
| Mean (SD) | 25.1 (3.07) | 25.4 (3.01) | 25.3 (2.99) |
SD, standard deviation.
Percentages based on total number of participants in the safety analysis set. Treatment A was a single dose of atorvastatin 40 mg, followed by a 1‐day washout and a single dose of pravastatin 40 mg/rosuvastatin 10 mg; treatment B was filgotinib 200 mg once daily for 11 days, atorvastatin 40 mg on day 6, followed by pravastatin 40 mg/rosuvastatin 10 mg on day 8.
Pharmacokinetics
Atorvastatin
Mean plasma concentration–time profiles of atorvastatin and 2‐hydroxy‐atorvastatin with and without filgotinib administration are shown in Figure 2A and 2B, respectively. The corresponding pharmacokinetic parameters and statistical comparisons for atorvastatin and 2‐hydroxy‐atorvastatin are presented in Table 3. The GLSM ratio and 90% CIs were within the lack‐of‐interaction boundary for atorvastatin AUC and 2‐hydroxy‐atorvastatin AUC and Cmax when atorvastatin was administered with filgotinib relative to when atorvastatin was administered alone. Atorvastatin Cmax appeared to be 18% lower when administered with filgotinib relative to when administered alone. Atorvastatin metabolite‐to‐parent molar ratio (2‐hydroxy‐atorvastatin to atorvastatin) for Cmax and AUC were within the prespecified lack‐of‐interaction criteria when coadministered with filgotinib.
Figure 2.
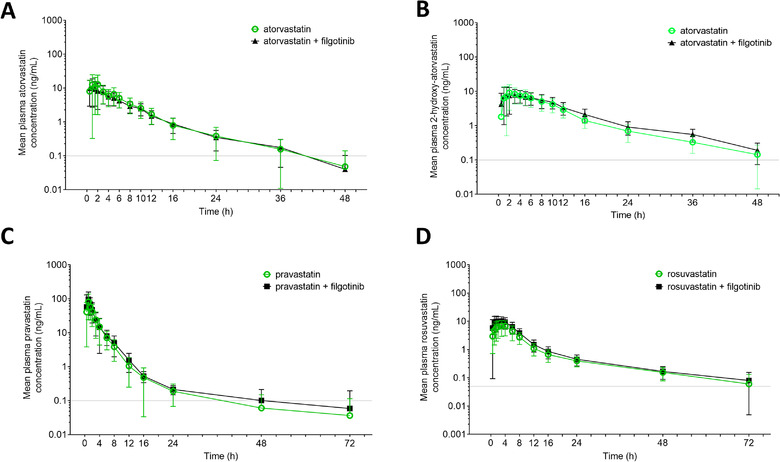
Mean (± SD) plasma concentrations of (A) atorvastatin, (B) 2‐hydroxy‐atorvastatin, (C) pravastatin, and (D) rosuvastatin with or without coadministration with filgotinib. 2‐hydroxy‐atorvastatin is an active metabolite of atorvastatin. Gray horizontal lines indicate lower limit of quantitation. H, hours; SD, standard deviation.
Table 3.
Atorvastatin, 2‐Hydroxy‐Atorvastatin, Pravastatin, and Rosuvastatin Pharmacokinetic Parameters With and Without Filgotinib 200 mg
| Effect of Filgotinib on Atorvastatin PK | |||
|---|---|---|---|
| PK Parameter | Atorvastatin Alone (n = 25) | Atorvastatin + Filgotinib (n = 26) | GLSM Ratio (90% CI) |
| AUCinf, ng · h/mL | 80.8 (49.2) | 71.8 (38.0) | 0.91 (0.84‐0.99) |
| AUClast, ng · h/mL | 78.8 (49.6) | 70.2 (38.6) | 0.91 (0.84‐0.99) |
| Cmax, ng/mL | 19.7 (68.6) | 15.0 (52.1) | 0.82 (0.69‐0.99) |
| tmax, h a | 1.5 (1.0‐2.0) | 1.0 (0.5‐3.0) | … |
| t1/2, h a | 6.9 (4.1‐9.0) | 7.4 (6.4‐9.2) | … |
| t1/2, h b | 7.16 (2.97) | 7.40 (2.39) | … |
| Effect of Filgotinib on 2‐Hydroxy‐Atorvastatin PK | |||
|---|---|---|---|
| PK Parameter | Atorvastatin Alone (n = 25) | Atorvastatin + Filgotinib (n = 26) | GLSM Ratio (90% CI) |
| AUCinf, ng · h/mL | 96.2 (38.4) | 109 (36.4) | 1.11 (1.02‐1.22) |
| AUClast, ng · h/mL | 93.0 (39.8) | 106 (36.8) | 1.12 (1.02‐1.23) |
| Cmax, ng/mL | 12.4 (53.3) | 11.7 (43.9) | 0.98 (0.81‐1.19) |
| tmax, h a | 2.0 (1.5‐4.1) | 2.5 (1.5‐4.0) | … |
| t1/2, h a | 9.1 (7.1‐10.9) | 9.4 (8.4‐10.1) | … |
| t1/2, h b | 9.91 (4.32) | 9.30 (1.49) | … |
| AUCinf (molar ratio of 2‐hydroxy‐atorvastatin to atorvastatin) | 1.3 (35.1) | 1.6 (26.3) | 1.22 (1.15‐1.29) |
| Cmax (molar ratio of 2‐hydroxy‐atorvastatin to atorvastatin) | 0.7 (43.9) | 0.8 (29.3) | 1.19 (1.06‐1.33) |
| Effect of Filgotinib on Pravastatin PK | |||
|---|---|---|---|
| PK Parameter | Pravastatin Alone c (n = 25) | Pravastatin + Filgotinib (n = 26) | GLSM Ratio (90% CI) |
| AUCinf, ng · h/mL | 201 (57.8) | 235 (58.5) | 1.22 (1.05‐1.41) |
| AUClast, ng · /mL | 200 (58.0) | 233 (59.2) | 1.22 (1.05‐1.41) |
| Cmax, ng/mL | 84.2 (67.3) | 99.2 (66.2) | 1.25 (1.01‐1.54) |
| tmax, h a | 1.0 (1.0‐1.0) | 1.0 (1.0‐1.0) | … |
| t1/2, h a | 4.9 (3.5‐8.6) | 5.6 (3.3‐8.5) | … |
| t1/2, h b | 6.48 (4.97) | 6.57 (4.39) | … |
| Effect of Filgotinib on Rosuvastatin PK | |||
|---|---|---|---|
| PK Parameter | Rosuvastatin Alone c (n = 25) | Rosuvastatin + Filgotinib (n = 26) | GLSM Ratio (90% CI) |
| AUCinf, ng · h/mL | 66.0 (44.8) | 92.3 (37.9) | 1.42 (1.30‐1.57) |
| AUClast, ng · h/mL | 62.6 (46.4) | 89.3 (38.8) | 1.46 (1.32‐1.62) |
| Cmax, ng/mL | 7.5 (55.3) | 12.3 (48.8) | 1.68 (1.43‐1.97) |
| tmax, h a | 3.0 (2.0‐4.0) | 2.5 (1.5‐3.0) | … |
| t1/2, h a | 18.3 (13.5‐22.0) | 15.4 (12.9‐20.8) | … |
| t1/2, h b | 18.40 (6.89) | 16.53 (4.50) | … |
AUCinf, area under the concentration‐time curve from 0 to infinity; AUClast, area under the concentration‐time curve from 0 to last PK observation; CI, confidence interval; Cmax, maximum observed plasma concentration; %CV, coefficient of variation; GLSM, geometric least‐squares mean; PK, pharmacokinetic; t1/2, terminal elimination half‐life; tmax, time to maximum concentration.
All data presented as mean (%CV) with exceptions shown in the footnote.
Presented as median (Q1‐Q3).
Presented as mean (standard deviation).
Administered as part of a pravastatin/rosuvastatincocktail.
Pravastatin
Mean plasma concentration–time profiles of pravastatin with and without filgotinib administration are shown in Figure 2C. The corresponding pravastatin pharmacokinetic parameters and statistical comparisons are presented in Table 3. There was no effect of filgotinib coadministration on pravastatin AUC, and the GLSM ratio and 90% CIs were within the lack‐of‐interaction boundaries. The Cmax of pravastatin appeared to be increased by 25% with filgotinib coadministration relative to pravastatin alone.
Rosuvastatin
Mean plasma concentration–time profiles of rosuvastatin with and without filgotinib administration are shown in Figure 2D. The corresponding rosuvastatin pharmacokinetic parameters and statistical comparisons are presented in Table 3. Relative to rosuvastatin alone, filgotinib coadministration increased rosuvastatin AUCinf and Cmax by 42% and 68%, respectively. Consistent with the observed pharmacokinetic changes, the GLSM ratio and 90% CIs were above the lack‐of‐interaction boundary for rosuvastatin AUC and Cmax.
A summary of changes in pharmacokinetic parameters observed for atorvastatin, 2‐hydroxy‐atorvastatin, pravastatin, and rosuvastatin with filgotinib coadministration including the prespecified boundaries are shown in Figure 3.
Figure 3.
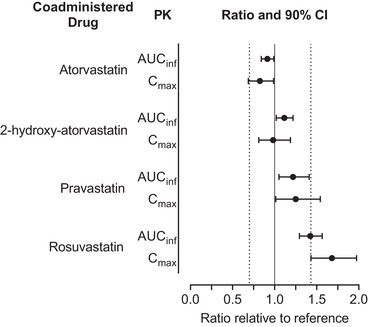
Effect of filgotinib on exposures of atorvastatin, 2‐hydroxy‐atorvastatin, pravastatin, and rosuvastatin. Dashed vertical lines indicate prespecified boundaries (0.70‐1.43). AUCinf, area under the curve from time 0 extrapolated to infinity; CI, confidence interval; Cmax, maximum observed concentration; PK, pharmacokinetics.
Safety
Eleven (40.7%) participants had a treatment‐emergent AE related to study drug; 3 (11.5%) events occurred during atorvastatin treatment, 1 (4.0%) in the pravastatin plus rosuvastatin arm, 8 (30.8%) in the filgotinib arm, 1 (3.8%) in the filgotinib plus atorvastatin arm, and 2 (7.7%) in the filgotinib plus pravastatin plus rosuvastatin arm. The majority of treatment‐emergent AEs were mild or moderate (n = 5 each). One participant (3.7%), a 24‐year‐old Asian female who received filgotinib, experienced an asymptomatic grade 3 AE of blood creatine phosphokinase increase. The investigator assessed the event as AE related to all study drugs (statins plus filgotinib), and the event resulted in premature discontinuation of all study drugs and study discontinuation. The most common AEs were headache (n = 5 [19.2%] filgotinib, n = 2 [7.7%] atorvastatin, and n = 1 [4.0%] pravastatin plus rosuvastatin) and nausea (n = 3 [11.5%] filgotinib and n = 1 [3.8%] filgotinib plus atorvastatin); all were mild or moderate.
Discussion
This phase 1 study was conducted to assess the potential effect of filgotinib and its major metabolite, GS‐829845, on the pharmacokinetics of atorvastatin, pravastatin, and rosuvastatin. There is an elevated cardiovascular risk for patients with RA, and statins are recommended for use in patients with risk‐enhancing factors, such as RA. 2 , 3 Studies have reported dyslipidemia can affect up to one‐third of patients with RA. 19 Therefore, it is necessary to understand potential interactions of RA therapies with statins.
Based on in vitro studies, filgotinib inhibited OATP1B1 and OATP1B3 at exposures that are significantly greater than the clinically relevant range. Filgotinib inhibited OATP1B1 and OATP1B3 with an IC50 of 98 μM and >285 μM, respectively, which is ≈19‐ and 56‐fold higher than the steady‐state Cmax of filgotinib in patients with RA following a 200‐mg once‐daily dose. GS‐829845, filgotinib's active metabolite, inhibited OATP1B1 and OATP1B3 with an IC50 of 260 μM and >473 μM, respectively, which is at least 22‐ and 39‐fold higher than the steady state Cmax of GS‐829845 in patients with RA following a 200‐mg once‐daily dose of filgotinib. Therefore, filgotinib was not expected to significantly increase exposure of statins via inhibition of the OATP transporters at the clinically relevant dose.
The recommended dose of filgotinib for the treatment of moderately to severely active RA is 200 mg once daily. 5 Filgotinib does not accumulate in plasma following multiple once‐daily dosing, and steady‐state filgotinib plasma concentrations are achieved in approximately 2 to 3 days. GS‐829845, the major active metabolite, demonstrates ≈2‐fold accumulation following multiple once‐daily dosing of filgotinib, with steady‐state plasma concentrations achieved in ≈4 days. Therefore, in the current study, filgotinib was administered as 200‐mg once‐daily doses for 5 days before being coadministered with atorvastatin, pravastatin, or rosuvastatin to ensure the maximal effect of filgotinib and GS‐829845 was characterized for potential OATP inhibition. Furthermore, filgotinib was administered alone for an additional 1 day and 3 days after being coadministered with atorvastatin and pravastatin/rosuvastatin, respectively, to ensure any potential effect of filgotinib and GS‐829845 on OATP drug transporter was sustained during washout of the statin.
The atorvastatin dose evaluated in the study (40 mg) was selected because it is representative of the middle of the recommended dose range of 10 to 80 mg used in clinical practice to treat hyperlipidemia or prevent cardiovascular disease. 20 In addition, drug interactions resulting in significant changes in atorvastatin exposure generally involve inhibition of CYP3A4; thus, large increases in atorvastatin exposure were not expected to occur when atorvastatin was administered with filgotinib, since filgotinib does not inhibit CYP3A. The 40‐mg dose of pravastatin was selected because it is representative of a dose used for treatment of hyperlipidemia and for cardiovascular disease prevention, and it has been established as an in vivo probe for assessing OATP at this dose. Rosuvastatin is administered at doses ranging from 5 to 40 mg once daily; a 10‐mg dose was selected as a clinically relevant dose representative of the range used in clinical practice, and it has been established as an in vivo probe substrate for OATP at this dose. 21 Rosuvastatin and pravastatin were administered as a cocktail of drug transporter probe substrates. The use of this cocktail as a measure of OATP and breast cancer resistance protein activity was confirmed in a prior study, in which these probe drugs were administered simultaneously with no interactions with each other. 22 , 23
With multiple filgotinib 200 mg once‐daily administrations, there was no increase in the exposure of atorvastatin (CYP3A4 and OATP substrate). The apparent small decrease (18%) in atorvastatin Cmax when coadministered with filgotinib is not considered clinically relevant based on the exposure‐efficacy relationship for atorvastatin, which indicates that a reduction in low‐density lipoprotein cholesterol is more likely to be correlated with total daily exposure (AUC) rather than Cmax. 24 , 25 , 26 There was no increase in the systemic exposure (AUC) of pravastatin, but a small increase in pravastatin Cmax (25%) was observed. This small increase in pravastatin Cmax is not considered clinically relevant on the basis of the dose/exposure‐safety data for pravastatin. 27
Of the 3 evaluated statins, only rosuvastatin showed a small but consistent increase in AUC and Cmax. However, the magnitude of observed change (42% and 68% increase in AUCinf and Cmax, respectively) is not considered clinically relevant based on the known dose/exposure‐response relationship of rosuvastatin. 28 Across the approved dose range for rosuvastatin, the dose‐response relationship for safety is comparable to other widely used statins. 29 , 30 Results from clinical trials and postmarketing surveillance in a broad patient population revealed no cases of rhabdomyolysis in patients receiving 10 to 40 mg of rosuvastatin, and myopathy related to rosuvastatin occurred in ≤0.03% of patients receiving rosuvastatin. 28 Further, across the approved dose range, no significant increases in creatinine kinase or alanine aminotransferase levels have been observed with increases in dose. Above the approved dose range, a dose‐dependent increase was observed. Development of severe myopathy or rhabdomyolysis occurred at an increased incidence only at the 80‐mg dose. Consistently, guidance from the American Heart Association on statin interactions describes the magnitude of drug interaction as minor if the AUC increase is >1.25 to <2. 31 Interaction of rosuvastatin with drugs that result in a <2‐fold increase in exposure of rosuvastatin (such as rosuvastatin interactions with eltrombopag 75 mg once daily, darunavir 600 mg/ritonavir 100 mg twice daily, tipranavir/ritonavir combination 500 mg/200 mg, dronedarone 400 mg twice daily, and itraconazole 200 mg once daily) were not noted in the US prescribing information as being clinically relevant or necessitating rosuvastatin dose adjustment. 32
Collectively, these data indicate a lack of clinically relevant interaction between filgotinib and atorvastatin, pravastatin, or rosuvastatin. Given that atorvastatin, pravastatin, and rosuvastatin are sensitive OATP substrates and the highest impact of filgotinib on the evaluated statin exposure is limited (<50% increase in rosuvastatin AUC), filgotinib is not anticipated to have clinically meaningful interactions with other sensitive substrates of the OATP transporter (eg, pitavastatin, simvastatin, and valsartan).
This study provides an evaluation of the safety and tolerability of a single dose of statins with filgotinib. The majority of AEs were mild or moderate in nature, and no new safety signals were observed with filgotinib exposure in combination with statins compared to the individual drugs. Given the low potential for OATP‐mediated DDIs at clinical concentrations, statins were an allowed concomitant medication in the phase 2 and phase 3 studies of filgotinib in patients with RA. 33 , 34 , 35 In a post hoc analysis of pooled data from these 5 studies, no increases in statin‐induced AEs, such as muscle or liver toxicities, were observed following coadministration of statins with filgotinib (N = 387 patients with RA on filgotinib and concomitant statin treatment). Results from the current DDI study confirm that filgotinib has no clinically relevant effect on exposure of statins.
This study evaluated the OATP DDI liability of filgotinib in healthy participants rather than a targeted patient population; however, healthy participants are recommended for evaluating potential DDIs. In addition, filgotinib and GS‐829845 exposure is comparable between healthy participants and patients with RA, and the mechanism of OATP DDI is not disease specific. Therefore, it is to be expected that these results can be extrapolated to the intended patient population.
Conclusion
Filgotinib, at its 200‐mg once‐daily dosage, had no clinically relevant effect on the exposure of atorvastatin, pravastatin, and rosuvastatin. Results of this study confirm that filgotinib has a low potential to affect the pharmacokinetics of concomitantly administered drugs that are OATP substrates at the clinically relevant exposures.
Conflicts of Interest
K.A., C.N., Q.G., M.A., T.T., and A.O. are employees and shareholders of Gilead Sciences, Inc. Writing and editorial support was provided by Kathleen Pieper, PhD, and Helen Rodgers, PhD, of AlphaScientia, LLC, and was funded by Gilead Sciences, Inc. This work was supported by Gilead Sciences, Inc.
Data Sharing
Anonymized individual patient data will be shared upon request for research purposes dependent on the nature of the request, the merit of the proposed research, the availability of the data, and its intended use. The full data‐sharing policy for Gilead Sciences, Inc., can be found at https://www.gileadclinicaltrials.com/transparency‐policy/.
Dr. Ahmed A. Othman is a Fellow of the American College of Clinical Pharmacology.
References
- 1. Piepoli MF, Hoes AW, Agewall S, et al. 2016 European Guidelines on cardiovascular disease prevention in clinical practice: The Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts) developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Eur Heart J. 2016;37:2315‐2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: a report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines. Circulation. 2019;139:e1082‐e143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hollan I, Ronda N, Dessein P, et al. Lipid management in rheumatoid arthritis: a position paper of the Working Group on Cardiovascular Pharmacotherapy of the European Society of Cardiology. Eur Heart J Cardiovasc Pharmacother. 2020;6:104‐114. [DOI] [PubMed] [Google Scholar]
- 4. Turner RM, Pirmohamed M. Statin‐related myotoxicity: a comprehensive review of pharmacokinetic, pharmacogenomic and muscle components. J Clin Med. 2019;9(1):22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dhillon S, Keam SJ. Filgotinib: first approval. Drugs. 2020;80:1987‐1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Traves PG, Murray B, Campigotto F, Galien R, Meng A, Di Paolo JA. JAK selectivity and the implications for clinical inhibition of pharmacodynamic cytokine signalling by filgotinib, upadacitinib, tofacitinib and baricitinib. Ann Rheum Dis. 2021;80(7):865‐875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Van Rompaey L, Galien R, van der Aar EM, et al. Preclinical characterization of GLPG0634, a selective inhibitor of JAK1, for the treatment of inflammatory diseases. J Immunol. 2013;191:3568‐3577. [DOI] [PubMed] [Google Scholar]
- 8. Namour F, Diderichsen PM, Cox E, et al. Pharmacokinetics and pharmacokinetic/pharmacodynamic modeling of filgotinib (GLPG0634), a selective JAK1 inhibitor, in support of phase IIB dose selection. Clin Pharmacokinet. 2015;54:859‐874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Namour F, Desrivot J, Van der Aa A, Harrison P, Tasset C, van't Klooster G. Clinical confirmation that the selective JAK1 inhibitor filgotinib (GLPG0634) has a low liability for drug‐drug interactions. Drug Metab Lett. 2016;10:38‐48. [DOI] [PubMed] [Google Scholar]
- 10. Anderson K, Xin Y, Zheng H, et al. Filgotinib, a JAK1 inhibitor, has no effect on QT interval in healthy subjects. Clin Pharmacol Drug Dev. 2020;9:32‐40. [DOI] [PubMed] [Google Scholar]
- 11. Anderson K, Zheng H, Kotecha M, et al. The relative bioavailability and effects of food and acid‐reducing agents on filgotinib tablets in healthy subjects. Clin Pharmacol Drug Dev. 2019;8:585‐594. [DOI] [PubMed] [Google Scholar]
- 12. Begley R, Anderson K, Watkins TR, et al. Lack of drug‐drug interaction between filgotinib, a selective JAK1 inhibitor, and oral hormonal contraceptives levonorgestrel/ethinyl estradiol in healthy volunteers. Clin Pharmacol Drug Dev. 2021;10:376‐383. [DOI] [PubMed] [Google Scholar]
- 13. Namour F, Fagard L, Van der Aa A, Harrison P, Xin Y, Tasset C. Influence of age and renal impairment on the steady state pharmacokinetics of filgotinib, a selective JAK1 inhibitor. Br J Clin Pharmacol. 2018;84:2779‐2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hsiang B, Zhu Y, Wang Z, et al. A novel human hepatic organic anion transporting polypeptide (OATP2). Identification of a liver‐specific human organic anion transporting polypeptide and identification of rat and human hydroxymethylglutaryl‐CoA reductase inhibitor transporters. J Biol Chem. 1999;274:37161‐37168. [DOI] [PubMed] [Google Scholar]
- 15. Khurana V, Minocha M, Pal D, Mitra AK. Role of OATP‐1B1 and/or OATP‐1B3 in hepatic disposition of tyrosine kinase inhibitors. Drug Metabol Drug Interact. 2014;29:179‐190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cooper KJ, Martin PD, Dane AL, Warwick MJ, Schneck DW, Cantarini MV. Effect of itraconazole on the pharmacokinetics of rosuvastatin. Clin Pharmacol Ther. 2003;73:322‐329. [DOI] [PubMed] [Google Scholar]
- 17. Harvey RD, Aransay NR, Isambert N, et al. Effect of multiple‐dose osimertinib on the pharmacokinetics of simvastatin and rosuvastatin. Br J Clin Pharmacol. 2018;84:2877‐2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Otani N, Wakuda H, Imai H, et al. No effect of digoxin on rosuvastatin pharmacokinetics in healthy subjects: utility of oita combination for clinical drug‐drug interaction study. Clin Transl Sci. 2019;12:513‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dougados M, Soubrier M, Antunez A, et al. Prevalence of comorbidities in rheumatoid arthritis and evaluation of their monitoring: results of an international, cross‐sectional study (COMORA). Ann Rheum Dis. 2014;73:62‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Adams SP, Tsang M, Wright JM. Lipid‐lowering efficacy of atorvastatin. Cochrane Database Syst Rev. 2015:CD008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Simonson SG, Raza A, Martin PD, et al. Rosuvastatin pharmacokinetics in heart transplant recipients administered an antirejection regimen including cyclosporine. Clin Pharmacol Ther. 2004;76:167‐77. [DOI] [PubMed] [Google Scholar]
- 22. Huang L, Wang Y, Grimm S. ATP‐dependent transport of rosuvastatin in membrane vesicles expressing breast cancer resistance protein. Drug Metab Dispos. 2006;34:738‐742. [DOI] [PubMed] [Google Scholar]
- 23. Lutz J, Kirby B, Song Q, Massetto B, Kearney B, Mathias A. Simultaneous evaluation of P‐gp, OATP, and BCRP activity using a three‐drug cocktail in healthy volunteers. [Abstract ITC‐020]. Presented at American Society for Clinical Pharmacology & Therapeutics (ASCPT) 2017 Annual Meeting, March 15‐18, 2017. Washington, DC.
- 24. Cilla DD, Jr. , Whitfield LR, Gibson DM, Sedman AJ, Posvar EL. Multiple‐dose pharmacokinetics, pharmacodynamics, and safety of atorvastatin, an inhibitor of HMG‐CoA reductase, in healthy subjects. Clin Pharmacol Ther. 1996;60:687‐695. [DOI] [PubMed] [Google Scholar]
- 25. Stern RH, Yang BB, Hounslow NJ, MacMahon M, Abel RB, Olson SC. Pharmacodynamics and pharmacokinetic‐pharmacodynamic relationships of atorvastatin, an HMG‐CoA reductase inhibitor. J Clin Pharmacol. 2000;40:616‐623. [PubMed] [Google Scholar]
- 26. Vargo R, Adewale A, Behm MO, Mandema J, Kerbusch T. Prediction of clinical irrelevance of PK differences in atorvastatin using PK/PD models derived from literature‐based meta‐analyses. Clin Pharmacol Ther. 2014;96:101‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Iwaki Y, Lee W, Sugiyama Y. Comparative and quantitative assessment on statin efficacy and safety: insights into inter‐statin and inter‐individual variability via dose‐ and exposure‐response relationships. Expert Opin Drug Metab Toxicol. 2019;15:897‐911. [DOI] [PubMed] [Google Scholar]
- 28. Brewer HB, Jr. Benefit‐risk assessment of Rosuvastatin 10 to 40 milligrams. Am J Cardiol. 2003;92:23K‐9K. [DOI] [PubMed] [Google Scholar]
- 29. Brown WV, Bays HE, Hassman DR, et al. Efficacy and safety of rosuvastatin compared with pravastatin and simvastatin in patients with hypercholesterolemia: a randomized, double‐blind, 52‐week trial. Am Heart J. 2002;144:1036‐1043. [DOI] [PubMed] [Google Scholar]
- 30. Jones PH, Davidson MH, Stein EA, et al. Comparison of the efficacy and safety of rosuvastatin versus atorvastatin, simvastatin, and pravastatin across doses (STELLAR* trial). Am J Cardiol. 2003;92:152‐160. [DOI] [PubMed] [Google Scholar]
- 31. Wiggins BS, Saseen JJ, Page RL 2nd, et al. Recommendations for management of clinically significant drug‐drug interactions with statins and select agents used in patients with cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2016;134:e468‐e95. [DOI] [PubMed] [Google Scholar]
- 32. Kellick KA, Bottorff M, Toth PP, The National Lipid Association's Safety Task Force. A clinician's guide to statin drug‐drug interactions. J Clin Lipidol. 2014;8:S30‐S46. [DOI] [PubMed] [Google Scholar]
- 33. Kavanaugh A, Kremer J, Ponce L, et al. Filgotinib (GLPG0634/GS‐6034), an oral selective JAK1 inhibitor, is effective as monotherapy in patients with active rheumatoid arthritis: results from a randomised, dose‐finding study (DARWIN 2). Ann Rheum Dis. 2017;76:1009‐1019. [DOI] [PubMed] [Google Scholar]
- 34. Westhovens R, Rigby WFC, van der Heijde D, et al. Filgotinib in combination with methotrexate or as monotherapy versus methotrexate monotherapy in patients with active rheumatoid arthritis and limited or no prior exposure to methotrexate: the phase 3, randomised controlled FINCH 3 trial. Ann Rheum Dis. 2021;80(6):727‐738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Westhovens R, Taylor PC, Alten R, et al. Filgotinib (GLPG0634/GS‐6034), an oral JAK1 selective inhibitor, is effective in combination with methotrexate (MTX) in patients with active rheumatoid arthritis and insufficient response to MTX: results from a randomised, dose‐finding study (DARWIN 1). Ann Rheum Dis. 2017;76:998‐1008. [DOI] [PubMed] [Google Scholar]