

Abstract
The high mobility group A (HMGA) protein family is composed of three non‐histone chromatin remodeling proteins that act as architectural transcriptional factors. Indeed, although HMGA proteins lack transcriptional activity per se, they bind the minor groove of DNA at AT‐rich sequences, and, interacting with the transcription machinery, are able to modify chromatin modeling, thus regulating the expression of several genes. HMGA proteins have been deeply involved in embryogenesis process, and a large volume of studies has pointed out their key role in human cancer. Here, we review the studies on the role of the HMGA proteins in human hematological malignancies: they are overexpressed in most of the cases and their expression correlates with a reduced survival. In some cases, such as in acute lymphoblastic leukemia and acute myelogenous leukemia, HMGA2 gene rearrangements have been also described. Finally, recent studies evidence a synergism between HMGA and EZH2 in diffuse B‐cell lymphomas, suggesting an innovative therapy for this disease based on the inhibition of the function of both these proteins.
Keywords: EZH2, hematological malignancies, HMGA1, HMGA1 pseudogenes, HMGA2
1. INTRODUCTION
1.1. The high mobility group A proteins in benign and malignant neoplasias
The high mobility group A (HMGA) protein group is composed of three proteins: HMGA1a and HMGA1b that are generated from the same gene via alternative splicing, and HMGA2 encoded by the homonym gene. The HMGA1 gene is located on the chromosome band 6p21, whereas HMGA2 lies on the chromosome band 12q13‐15. HMGA are non‐histone nuclear proteins able to bind AT‐rich regions in the minor groove of DNA through their three basic AT‐hook domains. Although HMGA proteins do not have an “intrinsic” transcriptional activity, they can regulate the expression of several genes, acting as architectural proteins. 1 , 2 , 3 These proteins exert their physiologic role mainly during the embryogenic development, where they are strongly expressed, whereas their expression is low or absent in normal adult tissues. However, HMGA are expressed at high levels in experimental and human malignancies. 3 , 4 Indeed, their overexpression is mainly associated with a highly malignant phenotype, also representing a poor prognostic index since HMGA overexpression often correlates with the presence of metastases, and with a reduced survival in several neoplasias, such as colon, breast, thyroid, esophageal, and larynx carcinomas. 3 , 5 , 6 , 7
Many studies indicate that high levels of HMGA expression have a causal relation to the development of a malignant phenotype. In fact, the suppression of HMGA expression through the transfection of HMGA antisense cDNA constructs in thyroid cells prevents cell malignant transformation after infection of the same cells with murine transforming retroviruses. Consistently, HMGA1 silencing induces apoptotic cell death in human carcinoma cell lines, but not in normal ones. 3 , 8 Noteworthy, HMGA are involved, even though with different mechanisms, in benign tumors. Indeed, HMGA2 gene rearrangements, following chromosomal translocations of the region 12q13–15, where the HMGA2 gene is located, have been often associated with benign tumors of mesenchymal origin, such as uterine leiomyomas, lipomas, and lung hamartomas. 9 , 10 In the vast majority of the human benign tumors, chromosomal breaks are found in the third intron of this gene, giving rise to chimeric transcripts embracing the first three HMGA2 exons (encoding the AT‐hook domains) and ectopic sequences from other genes. 9 , 10 Consistently, the truncated form of HMGA2 is able to transform NIH3T3 cells, 11 and transgenic mice overexpressing both truncated or wild type HMGA2 are characterized by the development of benign tumors. 12 , 13
Chromosomal alterations involving the 6p21.3 region, where the HMGA1 gene is located, have been also reported in benign neoplasias. 14 , 15
The HMGA overexpression contributes to cancer development through several mechanisms that include (a) regulation of genomic stability 16 ; (b) modulation of autophagy 17 ; (c) induction of AP‐1 activity 18 ; (d) stemness regulation 19 ; (e) regulation of p53 expression and function 20 ; (f) cell cycle regulation, 21 (g) induction of chemoresistance 22 (Figure 1).
FIGURE 1.
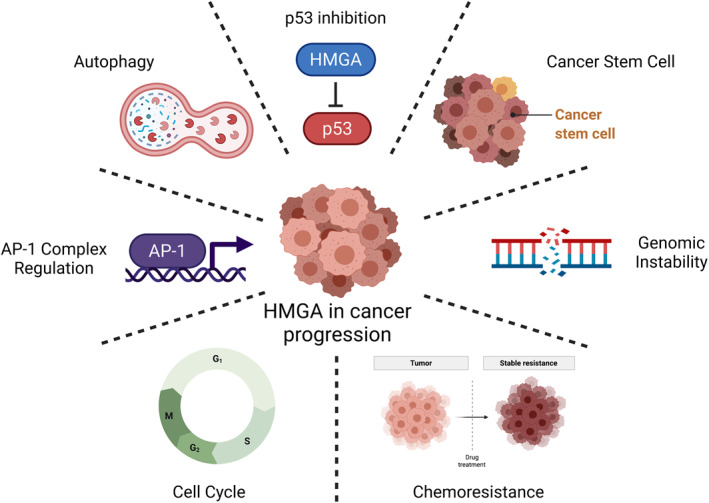
Main molecular mechanisms by which high mobility group A proteins are involved in human cancer. Created with BioRender.com
1.2. Modulation of HMGA expression: role of microRNAs and HMGA1 pseudogenes
Although several studies have assessed that HMGA overexpression is a feature of human cancer, the molecular mechanisms that control HMGA levels remain largely unexplored. In the last decade, a novel class of molecules, the microRNA (miRNAs), emerges as key regulator of gene expression at post‐transcriptional level. MiRNAs are small RNA fragments (19‐25nt) that are able to bind the 3′ untranslated region (UTR) of several transcripts, leading to mRNA degradation or inhibition of protein translation. 23 , 24 Interestingly, several studies reported that the HMGA expression levels are strongly regulated by miRNAs in human cancer, such as pituitary adenomas (mir‐15, mir‐16, miR‐34b, mir‐214, and mir‐761), 25 , 26 thyroid carcinomas 27 and seminomas (Let‐7), 28 and breast cancer. 29 Furthermore, chromosomal rearrangements that lead to the loss of HMGA2 3′UTR have been reported in mesenchymal benign tumors, thus abrogating the repression exerted by several miRNAs on this gene, and then inducing its overexpression. 30 , 31
It has been recently found that HMGA1 expression levels are post‐transcriptionally regulated by its pseudogenes. Indeed, eight HMGA1 pseudogenes have been identified in human genome. 32 , 33 The most characterized HMGA1‐pseudogenes are HMGA1P6 and HMGA1P7. They differ from HMGA1 just for few mismatches located in its coding and non‐coding sequences (5′ and 3′UTRs of the HMGA1 gene). Consequently, they share target regions for miRNAs that have been already validated to target HMGA1 and other cancer‐related genes, such as HMGA2, EZH2, and VEGF, then enhancing their expression through a competitive endogenous RNA (ceRNA) mechanism. 32 , 34 , 35 Consistently, HMGA1P6 and HMGA1P7 overexpression increases cell migration, invasiveness, and proliferation. Interestingly, their expression has been found drastically upregulated in several human cancers types. 7 , 32 , 36 , 37
The HMGA1P1, HMGA1P2, and HMGA1P3, although classified as pseudogenes, may represent a sort of competitor proteins for HMGA1 wild‐type with different post‐translational modifications, since bioinformatic analyses revealed that no point mutations affect the translation capability. 33 Moreover, another HMGA1‐pseudogene, HMGA1‐p, is able to compete with HMGA1 3′ UTR for a critical RNA stability factor, the αCP1. 38
1.3. HMGA genes in hematopoiesis
Several studies evidence that HMGA proteins play a substantial role in lymphohematopoietic differentiation. Indeed, the disruption of one or both alleles of the Hmga1 gene in mouse embryonic stem cells (mESC) is able to alter lymphohematopoietic differentiation. 39 A reduced number of T‐cell precursors and an increased number of B‐cell ones was observed in Hmga1−/− ES cells suggesting that HMGA1 would induce the differentiation of lymphoid precursors to T rather than to B lymphocytes, probably by regulating the expression of several cytokines that are able to control B‐ and T‐cell proliferation and differentiation. 39 Moreover, HMGA1 impairs megakaryocyte and erythroid differentiation processes by negatively regulating GATA‐1 expression, a critical factor for both megakaryocyte growth regulation 40 and erythroid differentiation. 41
It has been reported that also HMGA2 has a critical role in the induction of self‐renewal of mouse 42 and human hematopoietic stem cells (HSCs). Consistently, a strong induction of HMGA2 expression levels has been found in CD34+ cells 43 taking part in colony‐forming potential of cord blood. 44 Moreover, recent studies have revealed that HMGA2 is crucial for a physiological development of erythroid lineage, whereas its silencing is able to reduce myeloid progenitor cells without affecting their differentiation abilities. 45
1.4. HMGA in hematological malignancies
1.4.1. HMGA genes in myelogenous leukemias
Acute myelogenous leukemia (AML) and chronic myelogenous leukemia (CML) represent the main types of myelogenous leukemia. AML is the most frequent leukemia in adults, with about 30% of new diagnoses. Myeloid leukemia derives from the block of the differentiation and the accumulation of the granulocytic and monocytic blasts. 46 The main treatment is based on Cytarabine and anthracyclines, but, unfortunately, the vast majority of patients relapses and long‐term survival is very low. CML is a myeloproliferative disease that consists in the clonal expansion of hematological progenitor cells. 47 CML occurrence is deeply associated with a peculiar chromosomal translocation t(9; 22)–(q34; q11), the so‐called Philadelphia chromosome, where a chimeric tyrosine‐kinase (BCR‐ABL protein) is generated, leading to the deregulation of cell growth pathways. Thus, the improved knowledge of CML molecular biology brought the development of the first targeted therapy for a human neoplasia, which specifically blocks the altered kinase activity of the chimeric BCR‐ABL protein. 48
Several studies have underlined the role of HMGA2 in myeloid malignancies. Indeed, Odero and colleagues reported several cytogenetic alterations of 12q13‐15 chromosomal region in six myeloid neoplasia cases. In all the analyzed cases, the cytogenetic abnormalities induced HMGA2 overexpression, suggesting the important function played by HMGA2 in myeloid neoplasms. 49
Subsequently, the relation between HMGA2 and homeobox A9 (HOXA9) genes in the regulation of myeloid leukemia cell differentiation has been investigated. Indeed, HOXA9 has been found upregulated in about 70% of AML patients and its role in the differentiation blockage of immature leukemic blasts has been also reported. 50 , 51 Conversely, HMGA2 silencing experiments led to monocytic–granulocytic differentiation of myeloid leukemia cell lines and also of primary cells, where the HMGA2 knockdown increases CD11b positive population. Interestingly, it has been shown that HMGA2 silencing decreases HOXA9 expression levels, thus suggesting its positive regulation by HMGA2. 52
Intriguingly, it has been demonstrated that genomic mutations of the transcription factor Runt‐related transcription factor 1 (RUNX1) increase self‐renewal capability, halting granulocytic differentiation in human leukemia models by deregulating HMGA2 expression. 53 Moreover, mice knocked‐out for the RUNX1 gene showed an accumulation of both myeloid and granulocyte‐macrophage progenitors, and that this phenotype was reverted through the inactivation of Hmga2 gene, underlining that HMGA2 is a transcriptional target of RUNX1 and critical regulator of differentiation and expansion of myeloid lineage. 54
Consistently, HMGA2 has been found deeply involved in several molecular pathways that drive leukemogenesis process and resistance to therapeutic approaches. Indeed, HMGA2 was overexpressed in subsets of human AML samples mainly due to chromosomal rearrangements. Moreover, the HMGA2 overexpression was especially high in patients without remission, and, noteworthy, the low HMGA2‐ and high HMGA2‐expression levels significantly correlated with the remission rates after therapy. In agreement with these results, the silencing of HMGA2 in several AML‐derived cell lines led to increased cell viability and growth inhibition. Interestingly, Tan et al. reported that HMGA2 silencing was also able to inhibit PI3K/Akt signaling pathway and cell proliferation by decreasing mTOR expression levels. 55 Furthermore, it has been recently reported that HMGA2 is able to modulate daunorubicin (DNR) sensitivity, an anthracycline‐based chemotherapy drug used in the vast majority of first‐line standard therapeutic protocols, in AML cells. 55 , 56 Therefore, HMGA2 silencing was able to increase the DNR effects on AML cells, whereas HMGA2 overexpressing experiments gave opposite results. These findings are in agreement with previous studies reporting that the modulation of HMGA2 is able to regulate chemoresistance in several cancer types. 57 , 58
The role of HMGA protein overexpression in leukemia progression was also supported by the analysis of survival data of 430 AML patients correlated with HMGA2 expression, indicating HMGA2 overexpression as a powerful prognostic marker in AML. Indeed, increased HMGA2 expression levels correlated with a lower rate of complete remission, a worse 3‐year overall survival and relapse‐free survival. 59 Moreover, high HMGA2 expression levels were significantly correlated with the probability of the treatment failure of the primary anthracycline and cytarabine therapies. Notably, a HMGA2 test based on qRT‐PCR analysis has been developed showing high reproducibility, specificity, and may represent also a potential cost reduction for AML diagnosis/prognosis processes. 60 Therefore, the high expression of HMGA2 may represent an important novel prognostic marker, complementing the current AML clinical and genetic prognostic factors.
1.4.2. HMGA genes in lymphocytic leukemias
Lymphocytic leukemias may be divided in two main groups: acute lymphoblastic leukemia (ALL) and chronic lymphocytic leukemia (CLL).
ALL derives from the lymphoid line of blood cells and is characterized by the hyperproduction of immature lymphocytes. ALL represents the most frequent form of childhood cancer, being responsible for 25% of all pediatric cancer cases. 61 The most recurrent form of ALL is the B‐lineage cell subtype (B‐ALL), where a clonal expansion of precursor of B‐cell lymphoblasts is found in bone marrow, blood or other tissues. Commonly, ALL is characterized by a rapid progression and a high mortality rate if not adequately treated. 61
CLL is the clonal expansion of CD5+ B cells that appear as small mature lymphocytes accumulating in blood, bone marrow and lymphoid tissues. CLL progression is characterized by a wide range of outcomes: from the patients that need a rapid treatment to live‐long untreated cases. Unfortunately, in few patients, CLL may evolve rapidly in a more aggressive form of large cell lymphoma, thus representing the Richter's syndrome (RS). 61
Several evidence indicate a role of altered expression of the HMGA genes in these malignancies. Indeed, two independent studies reported high HMGA1 expression levels in all the analyzed ALL (6 and 28, respectively), whereas its expression was undetectable in normal cells from peripheral blood of healthy volunteers. 62 , 63 Moreover, HMGA1 expression correlates with relapse in pediatric ALL, since gene expression profile of leukemic blasts from relapsed B‐ALL children showed a significant increase of HMGA1 expression levels in leukemic blasts from patients with early and late relapses. Intriguingly, using the “Oncomine” database, HMGA1 was among the top 10% of the most overexpressed genes, with a fold change higher than 3 compared to normal bone marrow 64 (Figure 2).
FIGURE 2.
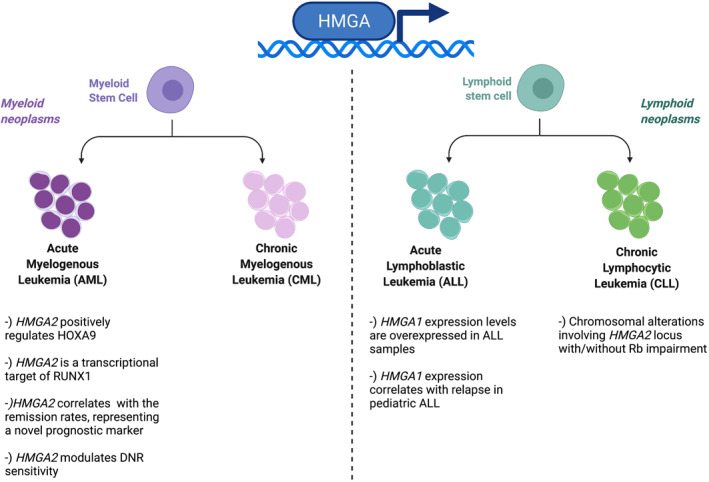
High mobility group A protein roles in myeloid and lymphoid neoplasms. Created with BioRender.com
To better understand the involvement of HMGA1 overexpression in ALL development, transgenic mice bearing Hmga1 gene under the control of the murine H‐2K promoter and immunoglobulin μ enhancer were generated. Interestingly, these mice developed lymphoid tumors in 100% of cases with a mean age of death of 4.8 months. Pathological lymphoblasts were detected also in other body compartments such as spleen, lymph nodes, bone marrow, and peripheral blood, underlining the ongoing leukemia pathology. Moreover, the fluorescence‐activated cell‐sorting (FACS) analysis pointed out CD3+, CD4−, CD8+, and alfa beta TCR + T‐cell markers, whereas cells were negative for natural killer 1.1, thus suggesting a mature T‐cell phenotype and T‐ALL diagnosis. 63
Since the abrogation of the CDKN2A tumor suppressor locus is a feature of almost all T‐ALL cases, inducing the leukemic transformation, a mouse strain transgenic for Hmga1 and null for Cdkn2a (HMGA1a/Cdkn2a −/− ) was generated providing an important model of human T‐ALL, a neoplasia of thymocytes characterized by the rapid accumulation of the T lymphocytes precursors. Indeed HMGA1a overexpression enhanced the T‐ALL malignant phenotype in these mice. 65
Interestingly, HMGA proteins seem to be involved also in CLL pathology. Indeed, Santulli and colleagues reported a case of Richter transformation with a 12q13 translocation t(12; 14) (q13; q32h), the chromosomic region where HMGA2 locus is located. Fluorescence in situ hybridization (FISH) analysis confirmed that HMGA2 gene was involved in the 12q chromosomal translocation, revealing also that the chromosomal breakpoint occurred in HMGA2 intron 3, which was transferred to a derivative chromosome 6. Furthermore, through immunohistochemistry (IHC) experiments performed on bone marrow smear, HMGA2 has been found specifically overexpressed in the blasts. Interestingly, further analyses revealed that HMGA2 translocation has been associated with chromosomal abnormalities of 13q14 region, where Rb1 gene is located, suggesting that the cooperation between inappropriate HMGA2 expression with impairment of Rb functions may have a role in the progression of this disease 66 (Figure 2). Consistently, this hypothesis would be in line with reported data showing a synergistic effect of HMGA overexpression and cell cycle alterations in the development of pituitary adenoma 21 , 67 and T‐cell lymphoma. 65
Notably, the most recurrent chromosomal anomaly in atypical CLL is represented by the trisomy of the chromosome 12, which is associated with the amplification of 12q13–15 region and this chromosomal alteration is a feature of most aggressive CLL, indicating its association with cancer progression. 68 Moreover, in a case of ALL, a t(9; 12) (p22; q14) chromosomal translocation, involving the HMGA2 locus has been reported. Interestingly, by using specific probes, FISH analyses revealed the absence of the most common HMGA2 breakpoints described so far and occurring in third intron. Further experiments pointed out that HMGA2 transcript overexpressed in blast cells do not have the carboxy‐terminal tail, corresponding to exons 3 and 4. 69 Consistently, two cases of AML with a chromosome 12 anomaly where new HMGA2 splice variants have been found. Intriguingly, these variants did not have the linker region and the acidic carboxy‐terminal domain. 70
In order to better understand the HMGA2 role in ALL and CLL, an engineered mouse strain bearing the human HMGA2 gene under control of the VH promoter/Eμ enhancer was created. About 90% of Eμ‐HMGA2 transgenic mice showed abnormal lymph nodes and spleens, and immunophenotype analyses reported the hyperproliferation of CD5+CD4+, CD5+CD8+, or CD5+CD8+CD4+ T‐cell populations in these organs. These altered populations may be comparable to those that characterize the human T‐ALL. 71
The 12‐q14.3 locus was also analyzed by microsatellite (MST) markers showing submicroscopic deletions in 20 of the 78 ALL cases (26%), with the highest frequency in Ph‐negative B‐cell ALL (13 of 27, 48%). Interestingly, the analysis of deletion frequencies of MST markers along this locus underlines that the targeted gene of deletion is likely located within a 170‐kb region starting about 65 kb upstream of HMGA2 and ending in the intron 3 of HMGA2. 72 These data strongly suggest that the submicroscopic deletions may deregulate HMGA2 expression, likely having a role in ALL pathogenesis, particularly in Ph‐negative B‐cell ALL.
Moreover, it is remarkable to note that 12q13 chromosomal translocations have been found in a subset of acute non‐lymphoblastic leukemia with a poor prognosis. Indeed, Brynes and colleagues reported that the totality of these patients presented blasts immaturity, suggesting a deregulation of early hematopoiesis. 72 , 73
1.5. HMGA1 overexpression in human B‐cell lymphomas: correlation with Enhancer of Zeste Homolog 2 (EZH2) expression
Recently, an increased expression of HMGA1 has been detected in a panel of hematological neoplasias including a selection of human follicular lymphomas, mantle cell lymphomas, and diffuse large B‐cell lymphomas. Intriguingly, also EZH2 was overexpressed in the same tissue samples with a significant correlation with HMGA1 levels. These data were also confirmed by the analysis of other DLBCL cases reported in the Cancer Genome Atlas (TCGA) database. EZH2 is an E2F‐regulated gene necessary for cell cycle progression since it regulates G2/M transition. 74 , 75 Several human malignancies such as lymphoma, melanoma, prostate, breast, colon, bladder, and liver cancer are characterized by EZH2 protein overexpression. 76 , 77 , 78 In these tumors, EZH2 expression was related with tumor cell proliferation, an aggressive clinical behavior, and poor outcome. Many studies report that EZH2 upregulation has a significant role in the progress of some hematological malignancies. 79 , 80 , 81
Consistently with the correlated HMGA1 and EZH2 expression, HMGA1 is able to bind to EZH2 promoter regions enhancing its transcription. As expected, inhibition of HMGA1 expression leads to the reduction of the EZH2 levels resulting in an inhibition of human lymphoma cell lines proliferation and migration rate. Finally, these studies recognize HMGA1 as an EZH2 activator, revealing a new molecular mechanism sustaining EZH2 overexpression in human cancers and a synergy of these two proteins in tumor progression (Figure 3). Intriguingly, by analyzing the data available on TCGA database, it has been pointed out a strong correlation between the HMGA1P1 and EZH2 levels, suggesting a role also for this HMGA1‐pseudogene in the lymphomagenesis process by upregulating EZH2 expression. 82
FIGURE 3.

HMGA1 is able to bind Enhancer of Zeste Homolog 2 promoter region and upregulate its expression levels in human lymphomas. Created with BioRender.com
Therefore, these studies propose a novel approach for the therapy of human lymphoma based on the combined impairment of EZH2 and HMGA functions. 82
1.6. HMGA overexpressing mice develop lymphomas
The role of HMGA protein overexpression in lymphomas is supported by animal models overexpressing the HMGA genes. Indeed, mice carrying the wild‐type or truncated form of Hmga2 gene under the CMV promoter developed lymphomas at 1 year of age. 83 These tumors were classified as natural killer–T/natural killer cell lymphomas. The mechanism underlying their development is based on the ability of HMGA2 to transcriptionally activate interleukin‐2 (IL2), interleukin‐15 (IL15), and their receptors. 83 , 84 Similar results were obtained analyzing mice overexpressing the wild‐type form of Hmga1 gene. 83 Moreover, a form of aggressive lymphoma, compatible with a mature T‐cell phenotype, developed between 1 and 8.5 months of age also in transgenic mice carrying Hmga1a flanked by the H‐2K promoter and immunoglobulin intronic enhancer. 63
Recently, transgenic HMGA1P6‐ and HMGA1P7‐ overexpressing mouse models have been also generated. 32 , 85 , 86 At 12 months of age, about 50% of these mice were affected by splenomegaly and different mouse anatomical districts were invaded by lymphoid cells. Indeed, the pathological spleens revealed disseminate and monotonous lymphoid cells destroying the splenic parenchyma with the loss of the characteristic structures and germinal centers. These neoplastic cells were characterized as B cells, suggesting the diagnosis of B‐cell lymphomas for the HMGA1P7 mouse model. 85
The mRNA profile of the neoplastic spleens, compared with the normal ones, showed that the genes blocked by B‐cell receptor inhibitors in diffuse large B‐cell lymphoma (DLBCL) were notably enhanced in the mouse pathological spleens. Indeed, the decreased genes were enriched of transcripts downregulated in post‐Germinal Center (GC) of BCL 6 dependent B‐cell lymphomas, and expressed in the GC B‐cell‐type DLBCL signature. Therefore, the lymphoproliferative lesions developed in these mice match with DLBCL of the non‐GCB type. 85
2. CONCLUSIONS
Numerous studies have confirmed that the inhibition of HMGA protein expression and/or function has an adverse consequence on proliferation and metastatic ability of tumor cells. Then, the focus of the next years will be to use all this data in order to discover new therapeutic approaches that will help patients affected by HMGA1‐overexpressing tumors. It is worth note that a HMGA‐based therapy could have a wide range of applications representing an excellent tool for hematological malignancies and cancers of different origins overexpressing these proteins. Then, the development of cancer therapies based on the inhibition of HMGA proteins has been the subject of several studies. It has been reported that trabectedin, already used for the therapy of human ovary cancer, exerts its cytotoxic properties on tumor cells through its ability to impair HMGA protein functions. However, the antitumoral effects of trabectedin treatment in HMGA positive cancer patients have to be better evaluated by further studies.
Moreover, several miRNAs able to target the HMGA genes have been identified; then, a miRNA treatment of patients affected by hematological malignancies could also be taken in consideration.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
TRANSPARENT PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1002/hon.2934.
ACKNOWLEDGMENT
This research received no specific grant from any funding agency in the public, commercial, or not‐for‐profit sectors.
Open Access Funding provided by Consiglio Nazionale delle Ricerche within the CRUI‐CARE Agreement.
De Martino M, Esposito F, Fusco A. Critical role of the high mobility group A proteins in hematological malignancies. Hematol Oncol. 2022;40(1):3‐11. 10.1002/hon.2934
Contributor Information
Francesco Esposito, Email: francesco.esposito2@unina.it.
Alfredo Fusco, Email: alfusco@unina.it.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCE
- 1. Vignali R, Marracci S. HMGA genes and proteins in development and evolution. Int J Mol Sci. 2020;21(2):654. doi: 10.3390/ijms21020654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sgarra R, Zammitti S, Lo Sardo A, et al. HMGA molecular network: from transcriptional regulation to chromatin remodeling. Biochim Biophys Acta. 2010;1799(1–2):37‐47. doi: 10.1016/j.bbagrm.2009.08.009 [DOI] [PubMed] [Google Scholar]
- 3. Fusco A, Fedele M. Roles of HMGA proteins in cancer. Nat Rev Cancer. 2007;7(12):899‐910. doi: 10.1038/nrc2271 [DOI] [PubMed] [Google Scholar]
- 4. De Martino M, Fusco A, Esposito F. HMGA and cancer: a review on patent literatures. Recent Pat Anti‐Cancer Drug Discov. 2019;14(3):258‐267. doi: 10.2174/1574892814666190919152001 [DOI] [PubMed] [Google Scholar]
- 5. Pallante P, Sepe R, Puca F, Fusco A. High mobility group a proteins as tumor markers. Front Med. 2015;2:15. doi: 10.3389/fmed.2015.00015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Palumbo A, Jr , Da Costa NM, Esposito F, et al. HMGA2 overexpression plays a critical role in the progression of esophageal squamous carcinoma. Oncotarget. 2016;7(18):25872‐25884. doi: 10.18632/oncotarget.8288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Palumbo A, Jr , De Martino M, Esposito F, et al. HMGA2, but not HMGA1, is overexpressed in human larynx carcinomas. Histopathology. 2018;72(7):1102‐1114. doi: 10.1111/his.13456 [DOI] [PubMed] [Google Scholar]
- 8. Berlingieri MT, Pierantoni GM, Giancotti V, Santoro M, Fusco A. Thyroid cell transformation requires the expression of the HMGA1 proteins. Oncogene. 2002;21(19):2971‐2980. doi: 10.1038/sj.onc.1205368 [DOI] [PubMed] [Google Scholar]
- 9. Ashar HR, Fejzo MS, Tkachenko A, et al. Disruption of the architectural factor HMGI‐C: DNA‐binding AT hook motifs fused in lipomas to distinct transcriptional regulatory domains. Cell. 1995;82(1):57‐65. doi: 10.1016/0092-8674(95)90052-7 [DOI] [PubMed] [Google Scholar]
- 10. Schoenmakers EF, Wanschura S, Mols R, Bullerdiek J, Van den Berghe H, Van de Ven WJ. Recurrent rearrangements in the high mobility group protein gene, HMGI‐C, in benign mesenchymal tumours. Nat Genet. 1995;10(4):436‐444. doi: 10.1038/ng0895-436 [DOI] [PubMed] [Google Scholar]
- 11. Fedele M, Berlingieri MT, Scala S, et al. Truncated and chimeric HMGI‐C genes induce neoplastic transformation of NIH3T3 murine fibroblasts. Oncogene. 1998;17(4):413‐418. doi: 10.1038/sj.onc.1201952 [DOI] [PubMed] [Google Scholar]
- 12. Arlotta P, Tai AK, Manfioletti G, Clifford C, Jay G, Ono SJ. Transgenic mice expressing a truncated form of the high mobility group I‐C protein develop adiposity and an abnormally high prevalence of lipomas. J Biol Chem. 2000;275(19):14394‐14400. doi: 10.1074/jbc.m000564200 [DOI] [PubMed] [Google Scholar]
- 13. Fedele M, Battista S, Manfioletti G, Croce CM, Giancotti V, Fusco A. Role of the high mobility group A proteins in human lipomas. Carcinogenesis. 2001;22(10):1583‐1591. doi: 10.1093/carcin/22.10.1583 [DOI] [PubMed] [Google Scholar]
- 14. Tkachenko A, Ashar HR, Meloni AM, Sandberg AA, Chada KK. Misexpression of disrupted HMGI architectural factors activates alternative pathways of tumorigenesis. Cancer Res. 1997;57(11):2276‐2280. [PubMed] [Google Scholar]
- 15. Kazmierczak B, Dal Cin P, Wanschura S, et al. HMGIY is the target of 6p21.3 rearrangements in various benign mesenchymal tumors. Genes Chromosom Cancer. 1998;23(4):279‐285. [PubMed] [Google Scholar]
- 16. Pierantoni GM, Conte A, Rinaldo C, et al. Deregulation of HMGA1 expression induces chromosome instability through regulation of spindle assembly checkpoint genes. Oncotarget. 2015;6(19):17342‐17353. doi: 10.18632/oncotarget.3944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Conte A, Paladino S, Bianco G, et al. High mobility group A1 protein modulates autophagy in cancer cells. Cell Death Differ. 2017;24(11):1948‐1962. doi: 10.1038/cdd.2017.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vallone D, Battista S, Pierantoni GM, et al. Neoplastic transformation of rat thyroid cells requires the junB and fra‐1 gene induction which is dependent on the HMGI‐C gene product. EMBO J. 1997;16(17):5310‐5321. doi: 10.1093/emboj/16.17.5310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Puca F, Colamaio M, Federico A, et al. HMGA1 silencing restores normal stem cell characteristics in colon cancer stem cells by increasing p53 levels. Oncotarget. 2014;5(10):3234‐3245. doi: 10.18632/oncotarget.1914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Frasca F, Rustighi A, Malaguarnera R, et al. HMGA1 inhibits the function of p53 family members in thyroid cancer cells. Cancer Res. 2006;66(6):2980‐2989. doi: 10.1158/0008-5472.CAN-05-2637 [DOI] [PubMed] [Google Scholar]
- 21. Fedele M, Visone R, De Martino I, et al. HMGA2 induces pituitary tumorigenesis by enhancing E2F1 activity. Cancer Cell. 2006;9(6):459‐471. doi: 10.1016/j.ccr.2006.04.024 [DOI] [PubMed] [Google Scholar]
- 22. D'Angelo D, Mussnich P, Rosa R, Bianco R, Tortora G, Fusco A. High mobility group A1 protein expression reduces the sensitivity of colon and thyroid cancer cells to antineoplastic drugs. BMC Cancer. 2014;14:851. doi: 10.1186/1471-2407-14-851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281‐297. doi: 10.1016/s0092-8674(04)00045-5 [DOI] [PubMed] [Google Scholar]
- 24. Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6(11):857‐866. doi: 10.1038/nrc1997 [DOI] [PubMed] [Google Scholar]
- 25. Palmieri D, D'Angelo D, Valentino T, et al. Downregulation of HMGA‐targeting microRNAs has a critical role in human pituitary tumorigenesis. Oncogene. 2012;31(34):3857‐3865. doi: 10.1038/onc.2011.557 [DOI] [PubMed] [Google Scholar]
- 26. D'Angelo D, Borbone E, Palmieri D, et al. The impairment of the High Mobility Group A (HMGA) protein function contributes to the anticancer activity of trabectedin. Eur J Cancer. 2013;49(5):1142‐1151. doi: 10.1016/j.ejca.2012.10.014 [DOI] [PubMed] [Google Scholar]
- 27. Pallante P, Battista S, Pierantoni GM, Fusco A. Deregulation of microRNA expression in thyroid neoplasias. Nat Rev Endocrinol. 2014;10(2):88‐101. doi: 10.1038/nrendo.2013.223 [DOI] [PubMed] [Google Scholar]
- 28. De Martino M, Esposito F, Pellecchia S, et al. HMGA1‐regulating microRNAs let‐7a and miR‐26a are downregulated in human seminomas. Int J Mol Sci. 2020;21(8):3014. doi: 10.3390/ijms21083014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhao XX, Yuan QZ, Mu DP, et al. MicroRNA‐26a inhibits proliferation by targeting high mobility group AT‐hook 1 in breast cancer. Int J Clin Exp Pathol. 2015;8(1):368‐373. [PMC free article] [PubMed] [Google Scholar]
- 30. Hebert C, Norris K, Scheper MA, Nikitakis N, Sauk JJ. High mobility group A2 is a target for miRNA‐98 in head and neck squamous cell carcinoma. Mol Cancer. 2007;6:5. doi: 10.1186/1476-4598-6-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee YS, Dutta A. The tumor suppressor microRNA let‐7 represses the HMGA2 oncogene. Genes Dev. 2007;21(9):1025‐1030. doi: 10.1101/gad.1540407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Esposito F, De Martino M, Petti MG, et al. HMGA1 pseudogenes as candidate proto‐oncogenic competitive endogenous RNAs. Oncotarget. 2014;5(18):8341‐8354. doi: 10.18632/oncotarget.2202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. De Martino M, Forzati F, Arra C, Fusco A, Esposito F. HMGA1‐pseudogenes and cancer. Oncotarget. 2016;7(19):28724‐28735. doi: 10.18632/oncotarget.7427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. De Martino M, Forzati F, Marfella M, et al. HMGA1P7‐pseudogene regulates H19 and Igf2 expression by a competitive endogenous RNA mechanism. Sci Rep. 2016;6:37622. doi: 10.1038/srep37622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. De Martino M, Palma G, Azzariti A, Arra C, Fusco A, Esposito F. The HMGA1 pseudogene 7 induces miR‐483 and miR‐675 upregulation by activating Egr1 through a ceRNA mechanism. Genes (Basel). 2017;8(11):330. doi: 10.3390/genes8110330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Palumbo Junior A, de Sousa VPL, Esposito F, et al. Overexpression of HMGA1 figures as a potential prognostic factor in endometrioid endometrial carcinoma (EEC). Genes (Basel). 2019;10(5):372. doi: 10.3390/genes10050372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Esposito F, De Martino M, D'Angelo D, et al. HMGA1‐pseudogene expression is induced in human pituitary tumors. Cell Cycle. 2015;14(9):1471‐1475. doi: 10.1080/15384101.2015.1021520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chiefari E, Iiritano S, Paonessa F, et al. Pseudogene‐mediated posttranscriptional silencing of HMGA1 can result in insulin resistance and type 2 diabetes. Nat Commun. 2010;1:40. doi: 10.1038/ncomms1040 [DOI] [PubMed] [Google Scholar]
- 39. Battista S, Pentimalli F, Baldassarre G, et al. Loss of Hmga1 gene function affects embryonic stem cell lympho‐hematopoietic differentiation. FASEB J. 2003;17(11):1496‐1527. doi: 10.1096/fj.02-0977fje [DOI] [PubMed] [Google Scholar]
- 40. Shivdasani RA. Molecular and transcriptional regulation of megakaryocyte differentiation. Stem Cell. 2001;19(5):397‐407. doi: 10.1634/stemcells.19-5-397 [DOI] [PubMed] [Google Scholar]
- 41. Blobel GA, Simon MC, Orkin SH. Rescue of GATA‐1‐deficient embryonic stem cells by heterologous GATA‐binding proteins. Mol Cell Biol. 1995;15(2):626‐633. doi: 10.1128/mcb.15.2.626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ikeda K, Mason PJ, Bessler M. 3'UTR‐truncated Hmga2 cDNA causes MPN‐like hematopoiesis by conferring a clonal growth advantage at the level of HSC in mice. Blood. 2011;117(22):5860‐5869. doi: 10.1182/blood-2011-02-334425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rommel B, Rogalla P, Jox A, et al. HMGI‐C, a member of the high mobility group family of proteins, is expressed in hematopoietic stem cells and in leukemic cells. Leuk Lymphoma. 1997;26(5‐6):603‐607. doi: 10.3109/10428199709050896 [DOI] [PubMed] [Google Scholar]
- 44. Cesana M, Guo MH, Cacchiarelli D, et al. A CLK3‐HMGA2 alternative splicing axis impacts human hematopoietic stem cell molecular identity throughout development. Cell Stem Cell. 2018;22(4):575‐588. doi: 10.1016/j.stem.2018.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kumar P, Beck D, Galeev R, et al. HMGA2 promotes long‐term engraftment and myeloerythroid differentiation of human hematopoietic stem and progenitor cells. Blood Adv. 2019;3(4):681‐691. doi: 10.1182/bloodadvances.2018023986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. McCulloch EA. Stem cells in normal and leukemic hemopoiesis (Henry Stratton Lecture, 1982). Blood. 1983;62(1):1‐13. [PubMed] [Google Scholar]
- 47. Faderl S, Talpaz M, Estrov Z, O'Brien S, Kurzrock R, Kantarjian HM. The biology of chronic myeloid leukemia. N Engl J Med. 1999;341(3):164‐172. doi: 10.1056/NEJM199907153410306 [DOI] [PubMed] [Google Scholar]
- 48. Hehlmann R, Hochhaus A, Baccarani M, European L. Chronic myeloid leukaemia. Lancet. 2007;370(9584):342‐350. doi: 10.1016/S0140-6736(07)61165-9 [DOI] [PubMed] [Google Scholar]
- 49. Odero MD, Grand FH, Iqbal S, et al. Disruption and aberrant expression of HMGA2 as a consequence of diverse chromosomal translocations in myeloid malignancies. Leukemia. 2005;19(2):245‐252. doi: 10.1038/sj.leu.2403605 [DOI] [PubMed] [Google Scholar]
- 50. Golub TR, Slonim DK, Tamayo P, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286(5439):531‐537. doi: 10.1126/science.286.5439.531 [DOI] [PubMed] [Google Scholar]
- 51. Thorsteinsdottir U, Kroon E, Jerome L, Blasi F, Sauvageau G. Defining roles for HOX and MEIS1 genes in induction of acute myeloid leukemia. Mol Cell Biol. 2001;21(1):224‐234. doi: 10.1128/MCB.21.1.224-234.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tan L, Xu H, Chen G, et al. Silencing of HMGA2 reverses retardance of cell differentiation in human myeloid leukaemia. Br J Cancer. 2018;118(3):405‐415. doi: 10.1038/bjc.2017.403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gerritsen M, Yi G, Tijchon E, et al. RUNX1 mutations enhance self‐renewal and block granulocytic differentiation in human in vitro models and primary AMLs. Blood Adv. 2019;3(3):320‐332. doi: 10.1182/bloodadvances.2018024422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lam K, Muselman A, Du R, et al. Hmga2 is a direct target gene of RUNX1 and regulates expansion of myeloid progenitors in mice. Blood. 2014;124(14):2203‐2212. doi: 10.1182/blood-2014-02-554543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tan L, Wei X, Zheng L, et al. Amplified HMGA2 promotes cell growth by regulating Akt pathway in AML. J Cancer Res Clin Oncol. 2016;142(2):389‐399. doi: 10.1007/s00432-015-2036-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. O'Donnell MR, Abboud CN, Altman J, et al. NCCN clinical practice guidelines acute myeloid leukemia. J Natl Compr Cancer Netw. 2012;10(8):984‐1021. doi: 10.6004/jnccn.2012.0103 [DOI] [PubMed] [Google Scholar]
- 57. Xu X, Wang Y, Deng H, Liu C, Wu J, Lai M. HMGA2 enhances 5‐fluorouracil chemoresistance in colorectal cancer via the Dvl2/Wnt pathway. Oncotarget. 2018;9(11):9963‐9974. doi: 10.18632/oncotarget.24133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. D'Angelo D, Mussnich P, Arra C, Battista S, Fusco A. Critical role of HMGA proteins in cancer cell chemoresistance. J Mol Med Berl. 2017;95(4):353‐360. doi: 10.1007/s00109-017-1520-x [DOI] [PubMed] [Google Scholar]
- 59. Marquis M, Beaubois C, Lavallee VP, et al. High expression of HMGA2 independently predicts poor clinical outcomes in acute myeloid leukemia. Blood Cancer J. 2018;8(8):68. doi: 10.1038/s41408-018-0103-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tremblay G, Rousseau B, Marquis M, Beaubois C, Sauvageau G, Hebert J. Cost‐effectiveness analysis of a HMGA2 prognostic test for acute myeloid leukemia in a Canadian setting. Appl Health Econ Health Policy. 2019;17(6):827‐839. doi: 10.1007/s40258-019-00503-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Malard F, Mohty M. Acute lymphoblastic leukaemia. Lancet. 2020;395(10230):1146‐1162. doi: 10.1016/S0140-6736(19)33018-1 [DOI] [PubMed] [Google Scholar]
- 62. Pierantoni GM, Agosti V, Fedele M, et al. High‐mobility group A1 proteins are overexpressed in human leukaemias. Biochem J. 2003;372(Pt 1):145‐150. doi: 10.1042/BJ20021493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Xu Y, Sumter TF, Bhattacharya R, et al. The HMG‐I oncogene causes highly penetrant, aggressive lymphoid malignancy in transgenic mice and is overexpressed in human leukemia. Cancer Res. 2004;64(10):3371‐3375. doi: 10.1158/0008-5472.CAN-04-0044 [DOI] [PubMed] [Google Scholar]
- 64. Roy S, Di Cello F, Kowalski J, et al. HMGA1 overexpression correlates with relapse in childhood B‐lineage acute lymphoblastic leukemia. Leuk Lymphoma. 2013;54(11):2565‐2567. doi: 10.3109/10428194.2013.782610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Di Cello F, Dhara S, Hristov AC, et al. Inactivation of the Cdkn2a locus cooperates with HMGA1 to drive T‐cell leukemogenesis. Leuk Lymphoma. 2013;54(8):1762‐1768. doi: 10.3109/10428194.2013.764422 [DOI] [PubMed] [Google Scholar]
- 66. Santulli B, Kazmierczak B, Napolitano R, et al. A 12q13 translocation involving the HMGI‐C gene in Richter transformation of a chronic lymphocytic leukemia. Cancer Genet Cytogenet. 2000;119(1):70‐73. doi: 10.1016/s0165-4608(99)00205-8 [DOI] [PubMed] [Google Scholar]
- 67. Fedele M, Paciello O, De Biase D, et al. HMGA2 cooperates with either p27(kip1) deficiency or Cdk4(R24C) mutation in pituitary tumorigenesis. Cell Cycle. 2018;17(5):580‐588. doi: 10.1080/15384101.2017.1403682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dierlamm J, Wlodarska I, Michaux L, et al. FISH identifies different types of duplications with 12q13‐15 as the commonly involved segment in B‐cell lymphoproliferative malignancies characterized by partial trisomy 12. Genes Chromosom Cancer. 1997;20(2):155‐166. [PubMed] [Google Scholar]
- 69. Pierantoni GM, Santulli B, Caliendo I, et al. HMGA2 locus rearrangement in a case of acute lymphoblastic leukemia. Int J Oncol. 2003;23(2):363‐367. [PubMed] [Google Scholar]
- 70. Kottickal LV, Sarada B, Ashar H, Chada K, Nagarajan L. Preferential expression of HMGI‐C isoforms lacking the acidic carboxy terminal in human leukemia. Biochem Biophys Res Commun. 1998;242(2):452‐456. doi: 10.1006/bbrc.1997.7984 [DOI] [PubMed] [Google Scholar]
- 71. Efanov A, Zanesi N, Coppola V, et al. Human HMGA2 protein overexpressed in mice induces precursor T‐cell lymphoblastic leukemia. Blood Cancer J. 2014;4:e227‐e227. doi: 10.1038/bcj.2014.46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Patel HS, Kantarjian HM, Bueso‐Ramos CE, Medeiros LJ, Haidar MA. Frequent deletions at 12q14.3 chromosomal locus in adult acute lymphoblastic leukemia. Genes Chromosom Cancer. 2005;42(1):87‐94. doi: 10.1002/gcc.20116 [DOI] [PubMed] [Google Scholar]
- 73. Brynes RK, McCourty A, Sun NC, Koo CH. Trisomy 12 in Richter's transformation of chronic lymphocytic leukemia. Am J Clin Pathol. 1995;104(2):199‐203. doi: 10.1093/ajcp/104.2.199 [DOI] [PubMed] [Google Scholar]
- 74. Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. EZH2 is downstream of the pRB‐E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003;22(20):5323‐5335. doi: 10.1093/emboj/cdg542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tang X, Milyavsky M, Shats I, Erez N, Goldfinger N, Rotter V. Activated p53 suppresses the histone methyltransferase EZH2 gene. Oncogene. 2004;23(34):5759‐5769. doi: 10.1038/sj.onc.1207706 [DOI] [PubMed] [Google Scholar]
- 76. Simon JA, Kingston RE. Mechanisms of polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol. 2009;10(10):697‐708. doi: 10.1038/nrm2763 [DOI] [PubMed] [Google Scholar]
- 77. Simon JA, Lange CA. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat Res. 2008;647(1‐2):21‐29. doi: 10.1016/j.mrfmmm.2008.07.010 [DOI] [PubMed] [Google Scholar]
- 78. Karanikolas BD, Figueiredo ML, Wu L. Polycomb group protein enhancer of zeste 2 is an oncogene that promotes the neoplastic transformation of a benign prostatic epithelial cell line. Mol Cancer Res. 2009;7(9):1456‐1465. doi: 10.1158/1541-7786.MCR-09-0121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Yan J, Ng SB, Tay JL, et al. EZH2 overexpression in natural killer/T‐cell lymphoma confers growth advantage independently of histone methyltransferase activity. Blood. 2013;121(22):4512‐4520. doi: 10.1182/blood-2012-08-450494 [DOI] [PubMed] [Google Scholar]
- 80. Kanduri M, Sander B, Ntoufa S, et al. A key role for EZH2 in epigenetic silencing of HOX genes in mantle cell lymphoma. Epigenetics. 2013;8(12):1280‐1288. doi: 10.4161/epi.26546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Abd Al Kader L, Oka T, Takata K, et al. Aggressive variants of non‐Hodgkin lymphomas, Ezh2 is strongly expressed and polycomb repressive complex PRC1.4 dominates over PRC1.2. Virchows Arch. 2013;463(5):697‐711. doi: 10.1007/s00428-013-1428-y [DOI] [PubMed] [Google Scholar]
- 82. De Martino M, Nicolau‐Neto P, Ribeiro Pinto LF, et al. HMGA1 induces EZH2 overexpression in human B‐cell lymphomas. Am J Cancer Res. 2021;11(5):2174‐2187. [PMC free article] [PubMed] [Google Scholar]
- 83. Baldassarre G, Fedele M, Battista S, et al. Onset of natural killer cell lymphomas in transgenic mice carrying a truncated HMGI‐C gene by the chronic stimulation of the IL‐2 and IL‐15 pathway. Proc Natl Acad Sci U S A. 2001;98(14):7970‐7975. doi: 10.1073/pnas.141224998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Fedele M, Pentimalli F, Baldassarre G, et al. Transgenic mice overexpressing the wild‐type form of the HMGA1 gene develop mixed growth hormone/prolactin cell pituitary adenomas and natural killer cell lymphomas. Oncogene. 2005;24(21):3427‐3435. doi: 10.1038/sj.onc.1208501 [DOI] [PubMed] [Google Scholar]
- 85. De Martino M, De Biase D, Forzati F, et al. HMGA1‐pseudogene7 transgenic mice develop B cell lymphomas. Sci Rep. 2020;10(1):7057. doi:10.1038/s41598‐0200‐62974‐0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. De Martino M, Palma G, Arra C, Chieffi P, Fusco A, Esposito F. Characterization of HMGA1P6 transgenic mouse embryonic fibroblasts. Cell Cycle. 2020;19:1‐5. doi: 10.1080/15384101.2020.1807080 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.