

Abstract
The NRF2 transcription factor is a key regulator in cellular oxidative stress response, and acts as a tumor suppressor. Aberrant activation of NRF2 has been implicated in promoting chemo‐resistance, tumor growth, and metastasis by activating its downstream target genes. Hence, inhibition of NRF2 promises to be an attractive therapeutic strategy to suppress cell proliferation and enhance cell apoptosis in cancer. Direct targeting of NRF2 with small‐molecules to discover protein‐DNA interaction inhibitors is challenging as it is a largely intrinsically disordered protein. To discover molecules that bind to NRF2 at the DNA binding interface, we performed an NMR‐based fragment screen against its DNA‐binding domain. We discovered several weakly binding fragment hits that bind to a region overlapping with the DNA binding site. Using SAR by catalogue we developed an initial structure‐activity relationship for the most interesting initial hit series. By combining NMR chemical shift perturbations and data‐driven docking, binding poses which agreed with NMR information and the observed SAR were elucidated. The herein discovered NRF2 hits and proposed binding modes form the basis for future structure‐based optimization campaigns on this important but to date ‘undrugged’ cancer driver.
Keywords: NRF2, protein-DNA interactions, fragment screening, structure-activity relationships, ligand docking, NMR solution structures
Key regulator modulation: In a fragment‐based ligand screening approach two small‐molecule binders to the key pro‐oncogenic cancer driver NRF2 were discovered. The structure‐activity relationship of the initial fragment hits was established by a SAR‐by‐catalogue exploration. The binding pose of one the compounds, which binds to a region overlapping with the DNA binding site, was determined by a chemical shift perturbation restrained docking simulation which can serve as a starting point in a hit‐to‐lead campaign.
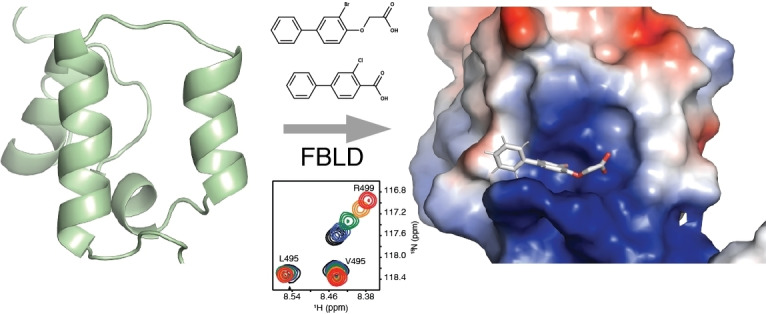
Introduction
Nuclear factor erythroid 2‐related factor 2 (NRF2) a cap'n’ collar (CNC), basic leucine zipper (bZIP) transcription factor is one of the main regulators of cellular antioxidant response, thereby mitigating cell damage during chemical and oxidative stress. During basal conditions NRF2 is bound to its cytosolic repressor Kelch‐like ECH‐associated protein 1 (KEAP1), which is involved in marking NRF2 for proteasomal degradation, thereby maintaining low levels of NRF2. However, increased levels of oxidative or electrophilic stress lead to the dissociation of the KEAP1‐NRF2 complex and NRF2 translocates to the nucleus where it forms a heterodimer with the small musculoaponeurotic fibrosarcoma oncogene homologue (Maf) proteins. Dimerization enables binding to the antioxidant‐response elements (ARE) sequence within the promoter regulatory regions of cytoprotective genes. Binding to the promoter region induces transactivation of proteins involved in a diverse set of cellular functions such as xenobiotic metabolism and excretion, autophagy, protein homeostasis, DNA repair, cell survival, and mitochondrial function, giving activation of the NRF2 pathway a direct role in cancer prevention. [1]
In recent years it has become apparent that high levels of NRF2 can have a prooncogenic effect and lead to chemo‐resistance; elevated NRF2 activity is mostly caused through genetic alterations in the KEAP1/NRF2 pathway. [2] Constitutive activation of NRF2 can be caused by promoter demethylation, copy number amplification of its gene, oncogene‐induced transcription of NRF2 via c‐MycERT2, BRAFV600E, and KRASG12D, or by gain‐of‐function somatic mutations in the regions responsible for KEAP1 interactions.[ 2b , 3 ] Direct alterations of NRF2 have been documented for lung, colorectal, ovarian, pancreatic, and head and neck cancers.[ 1 , 4 ] Further, an increase of NRF2 through deletion, decreased transcription, and loss‐of‐function mutations for KEAP1 has been described in the literature.[ 4a , 5 ] High levels of NRF2 in cancer cells enables increased proliferation and chemo‐ and radio resistance, making NRF2 inactivation a promising intervention strategy to enhance therapeutic effects for certain types of cancer. [6]
Human isoform 1 NRF2 consists of 605 amino acids which form seven conserved NRF2‐ECH homology (Neh1‐7) domains (Figure 1A). [7] The N‐terminal Neh2 domain is responsible for KEAP1 binding; Neh6 functions as an additional negative regulatory domain through its interaction with beta‐transducin repeat‐containing domain (β‐TrCP). The domains Neh3 to Neh5 are responsible for target gene transactivation, whereas the Neh7 domain has been shown to be involved in the inhibition of the NRF2‐ARE‐signaling pathway. [4b] Our study focuses on the Neh1 domain which forms a CNC basic leucine zipper DNA binding domain and is crucial for heterodimer formation with the small Maf (sMaf) proteins. NRF2 is predicted to be largely an intrinsically disordered protein in the absence of its binding partners with the exception of the CNC and parts of the basic leucine zipper region of Neh1 (Figure S1).
Figure 1.

NMR‐based fragment screen identified Neh1 binders. (A) Schematic representation of the organization of the seven NRF2 domains, the Neh1 domain is highlighted in red. (B) Chemical structures of the biphenyl carboxylic acid compounds identified in the fragment screen.
So far, only a few small molecule inhibitors of NRF2 have been reported, and only one of them, ML385, has been shown to directly interact with NRF2. ML385 is reported to interfere with small MafG heterodimer formation through binding to the leucine zipper region (LZIP), [8] although concern has been raised about ML385 acting as a broad inhibitor of various transcription factors.[ 1 , 4b ]
The paucity of NRF2 inhibitors is mostly due to its lack of a well‐defined three‐dimensional structure. This, and its activation through protein‐protein interactions (PPI) make NRF2 a challenging target for small‐molecule drug discovery. [9] These circumstances apply to transcription factors (TFs) and intrinsically disordered proteins (IDPs) in general. Consequently, they were historically deemed to be ‘undruggable’. Direct targeting of TFs, proposed by James Darnell almost 20 years ago, [10] with small‐molecules that modulate deregulated TFs in cancer, requires the disruption of protein‐protein or protein‐DNA interactions. The tractability of modulating or disrupting PPIs by direct targeting of the frequently large and solvent exposed surface areas between TFs and their cofactors, as well as activators/repressors with drug‐like small‐molecules has been established in recent years. [11] The discovery of small‐molecule binders to the typically convex and positively charged protein surface at the DNA binding site for direct modulation of their interactions, has however proven to be an even more challenging task. [12] Hence, only a very small number of molecules interfering competitively with DNA binding have been described in the literature. [13] Protein‐protein and protein‐DNA interfaces tend to have flat discontinuous cavities more amenable to fragment sized compounds which can be missed in HTS campaigns. [14]
Commercial so‐called virtual make‐on‐demand chemical libraries can be an efficient way to widen and tailor the chemical space around a small‐molecule hit, successfully demonstrated in several recent structure‐based ligand docking screens using Enamine's virtual compound library (https://enamine.net/). [15] Therefore, we set out to discover small‐molecule hits to NRF2 with a protein NMR‐based fragment screen ‐ a method that has proven to be fruitful in the past for challenging drug targets [16] – to identify potential small‐molecule binding pockets. In a second step we explore the structure‐activity relationships (SARs) of the initial hits, for which we search the Enamine virtual make‐on‐demand compound library [17] to increase the number of fragment hits and to assess the feasibility of obtaining higher affinity binders without tying up in‐house medicinal chemistry resources. Subsequently, to rationalize the SAR results and guide future structure‐based analogous optimization we determined the NMR solution protein‐ligand complex structure in a chemical shift perturbation driven docking simulation.
Results and Discussion
Fragment‐based screen. We set out to identify compounds that bind the Neh1 (residues 445–523, denoted by Neh1‐ΔLZIP) domain of NRF2, a construct that does not include the LZIP part, by a fragment‐based screening approach. Initially a library of 1800 commercial and internal fragments was screened by observing cross‐peak shifts of uniformly 15N labelled Neh1‐ΔLZIP, indicative of ligand binding, in a 2 dimensional 1H‐15N heteronuclear single‐quantum correlation (HSQC) experiment. Two fragments (Figure 1B) found in the screen were characterized in more detail, where compound 2 showed significantly larger chemical shift perturbations (CSPs) (Figure 2A, B and Figure S2A) and the dissociation constant (KD) was determined to be 1.7±0.1 mM (average over R499, D500, R503) as obtained from the CSP values (Figure 2C). For several residues CSP values larger than 0.10 ppm were observed, namely A452, M484, R499, D500, and R503, with a standard deviation of 0.05 ppm for all assigned backbone amide chemical shift changes in the 1H‐15N HSQC titration experiment for compound 2. Furthermore, the side chain amide of N482 had similar dose‐response to 2; a chemical shift change of 0.3 ppm was recorded for the side chain and 0.04 ppm for the backbone amide, respectively. Due to the small magnitude of CSPs for compound 1 (Figure S2A) we focused our follow‐up screen of commercial and internal compounds on molecules structurally similar to compound 2.
Figure 2.
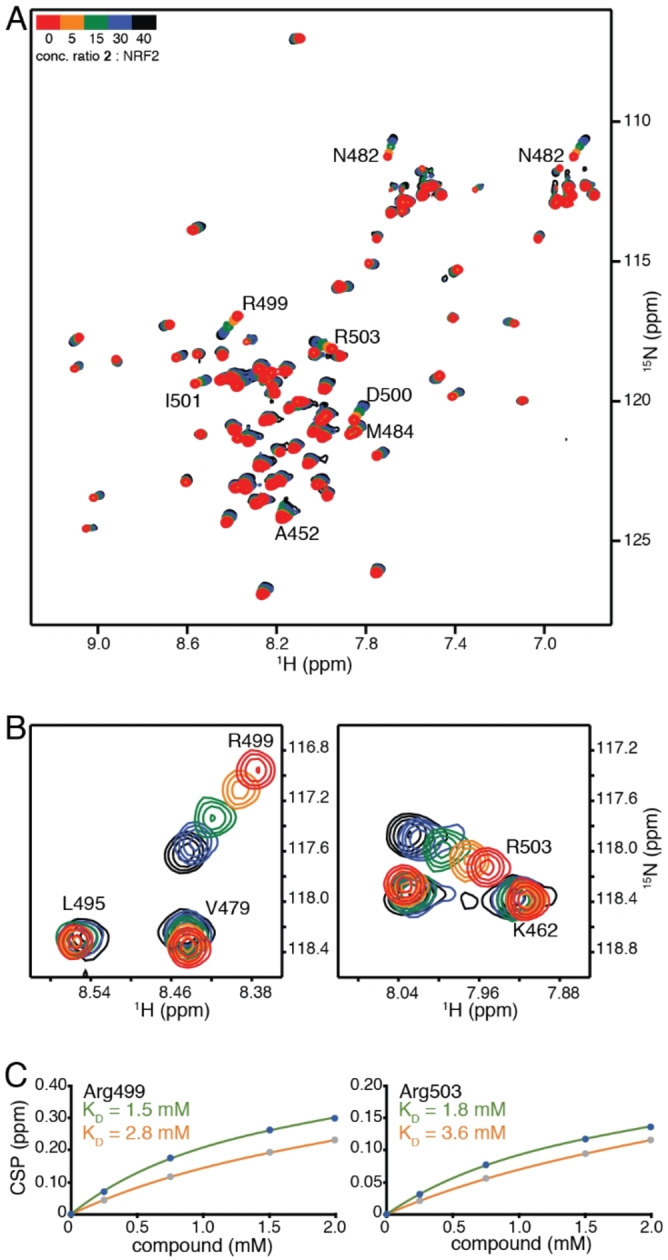
NMR experiments of Neh1 binders. (A) Overlay of 1H‐15N HSQC spectra of uniformly labeled 50 μM 15N‐Neh1‐ΔLZIP (red) titrated with 250, 750, 1500, and 2000 μM compound 2 (from orange to black). Residues with normalized chemical shift perturbations larger than 0.1 ppm are indicated with residue numbers. (B) Selected regions of the 1H‐15N HSQC spectra overlay showing close up views of R499 and R503 and their concentration‐dependent chemical shift changes upon increasing amounts of compound 2. (C) Titration curves obtained from the 1H‐15N HSQC titration measurements for residues R499 and R503. KDs were derived by fitting experimental CSP values to equation (1); experimental CSP values are shown as blue and grey points for compound 2 and 3, respectively. The lines represent an individual residue fit to equation (1); fitted lines for compound 2 and 3 are colored green and orange, respectively.
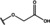
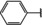
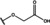
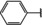
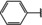
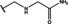
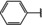
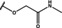
Because of the low hit rate in the initial screen, often seen for proteins lacking an easily druggable binding pocket, [18] a close analogs search was carried out. Specifically, we searched for compounds with a core phenyl carboxylic acid scaffold with variable side chains branching of the phenyl ring(s) as well as derivatives of the linker connecting the aromatic ring and the carboxylic acid. The search resulted in 27 analogs from internal and commercial sources (e. g. compounds 3, 4, 10, 11, 12, 13, 14, and 15 in Table 1). To further expand the SAR we searched the virtual make‐on‐demand library from Enamine, which contains 15.5 billion virtual compounds that can be synthesized out of 111 500 building blocks (https://enamine.net/library‐synthesis/real‐compounds/real‐space‐navigator). Compound 2 served as query structure in a REAL Space Navigator (bioSolveIT) search, a software tool (now infiniSee) that allows the exploration – based on pharmacophore similarity – of the virtual chemical space of the Enamine Real Space compounds. [17a] The chosen make‐on‐demand compounds were all composed of three Enamine building blocks; in total 8 compounds were delivered (e. g. compounds 5, 6, 7, 8, and 9 in Table 1). All 35 analogs of the follow‐up search were tested by 1H‐15N HSQC against Neh1‐ΔLZIP. 15 representative analogs are shown in Table 1, four of which (3, 5, 6, 9) showed a similar CSP pattern to 2 (Figure S2B, C, D); compound 5 caused significantly larger CSP values than 2, which could be due to the additional hydroxy group. The remaining compounds showed no or very small CSPs, furthermore compound 7 and 8 showed dose response as well. However, they lead to the precipitation of Neh1 at higher compound concentrations. Analysis of the CSP patterns indicated that the compounds most likely have similar binding sites and modes, however, they differ in affinities (Table 1). In a subsequent step we assessed the binding contribution of the negatively charged carboxyl group with four commercial and internal compounds lacking this functional group (e. g. compounds 16, 17, and 18 in Table 1). These molecules contained an amine or an amide instead of the carboxylic acid, none of which showed CSPs in 1H‐15N HSQC titration experiments against Neh1‐ΔLZIP in the structured part of NRF2. Small chemical shift changes upon ligand addition were observed for an unassigned residue in the unstructured C‐terminal part of the protein which are most likely caused by small changes in pH and/or ionic strength of the protein solution (Figure S2H, I).
Table 1.
Structure‐activity relationships of analogs tested by 1H‐15N HSQC titration.
|
| |||||
|---|---|---|---|---|---|
|
Compound |
R1 |
R2 |
R3 |
HSQC[a] |
KD [mM][b] |
|
1 |
|
Cl |
|
+ |
− |
|
2 |
|
Br |
|
+ |
1.7±0.1 |
|
3 |
|
Cl |
|
+ |
3.0±0.5 |
|
4 |
|
H |
|
− |
− |
|
5 |
|
Cl |
|
+ |
1.5±0.1 |
|
6 |
|
Cl |
|
+ |
2.8±0.4 |
|
7 |
|
Cl |
|
+ |
precipitation |
|
8 |
|
Cl |
|
+ |
precipitation |
|
9 |
|
Cl |
|
+ |
3.2±0.7 |
|
10 |
CH3 |
Br |
|
− |
− |
|
11 |
|
Br |
|
− |
− |
|
12 |
|
Br |
|
− |
− |
|
13 |
|
|
|
− |
− |
|
14 |
|
H |
|
− |
− |
|
15 |
|
H |
|
− |
− |
|
16 |
|
F |
|
− |
− |
|
17 |
|
H |
|
− |
− |
|
18 |
|
Br |
|
− |
− |
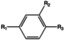
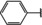
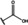
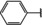
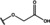
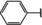
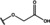
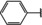

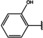
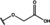
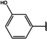
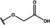
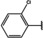
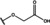
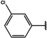
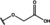
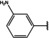
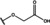
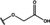
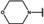
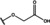
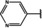

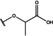
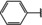
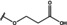
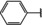

[a] +indicates chemical shift perturbations in 1H‐15N HSQC spectra in presence of the compound. [b] KD values were determined from CSP values using equation (1).
The SAR analysis showed that the potency of the Neh1 binder 2, 2‐((3‐bromo‐[1,1’‐biphenyl]‐4‐yl)oxy)acetic acid, is very sensitive to structural changes at several positions; changes of the bromo substituent at the three‐position on the phenyl ring to either a bulky cyclohexane (13) or to a small hydrogen (4) led to no detectable chemical shift changes upon compound titration. Furthermore, substituting the bromine with a chlorine led to an approximately 2‐fold decrease in ligand potency determined by 1H‐15N HSQC titration, confirming the importance of the halogen substituent in the binding mode (Figure 2C, Table 1). The contribution of the halogen bond could not be further investigated by an iodine containing compound due to the lack of availability. Special care must be taken as this is a KD difference close to the limit of distinguishability, therefore the titration experiments were carried out using the same protein stock solution and the ligand concentration was determined by 1D peak integration. On the one hand the 2‐fold potency decrease could be reversed by changing the R3 phenyl group to an ortho phenol group (2’‐hydroxy‐phenyl) 5 but not by a meta phenol (3’‐hydroxy‐phenyl) group 6. On the other hand substituting the R3 phenyl group to a methyl (10), morpholine (11), and a pyrimidine (12) group caused the compounds to lose their potency (Figure S2F, G, Table 1).
Based on our structure‐activity relationship we derived a pharmacophore for small‐molecule Neh1 binders; all of the better binders in this study contained three features, a biphenyl motif, a halogen at the three‐position on the first phenyl ring and additionally an acetic acid para to the second aryl ring.
As none of the fragments in the follow up screen resulted in higher affinity binders we set out to determine the three‐dimensional structure of the protein‐small‐molecule complex. This is key to advance the initial fragments hits in a structure‐based design approach.
Structure determination of Neh1. We determined the binding pose of fragment hit 2 in a two‐step approach. In the first step we solved the ligand free Neh1 domain structure by solution state NMR spectroscopy and in a second step determined the binding pose of 2 by data‐driven docking using the program HADDOCK. [19] The NMR solution structure of the Neh1‐ΔLZIP domain of NRF2 (residues 445–523, PDB ID 7O7B) was determined using triple‐resonance‐resolved NMR spectroscopic techniques; backbone chemical shifts and NOEs were used as input restraints for the structure determination (Table 2).
Table 2.
NMR and refinement statistics.
|
NMR distance and dihedral restraints |
Neh1‐ΔLZIP (BMRB 34617, PDB 7O7B) |
|---|---|
|
Distance restraints |
|
|
Total NOE |
337 |
|
Intraresidual (i=j) |
182 |
|
Sequential (i–j=1) |
77 |
|
Medium range (1<i–j<5) |
34 |
|
Long range (i–j≥5) |
44 |
|
Total Dihedral restraints |
92 |
|---|---|
|
ϕ |
46 |
|
ψ |
46 |
|
Structure Statistics |
|
|---|---|
|
Violations (mean±SD) |
|
|
Distance restraints (Å) |
0.09±0.01 |
|
Dihedral restraints (°) |
0.97±0.12 |
|
Deviation from idealized geometry |
|
|---|---|
|
Bond length (Å) |
0.0034±0.0001 |
|
Bond angles (°) |
0.50±0.02 |
|
Ramachandran statistics |
|
|---|---|
|
Residues in allowed regions |
96.1 % |
|
Residues in disallowed regions |
3.9 % |
|
Average pairwise rmsd (Å) |
|
|---|---|
|
Heavy atoms (2° structure) |
1.07±0.08 |
|
Backbone atoms (2° structure) |
0.42±0.07 |
The central scaffold of the Neh1‐ΔLZIP domain is formed by a bundle of four α‐helices (residues R456‐L464, α1; residues V470‐N475, α2; residues V478‐K487, α3; residues E492‐G505, α4) (Figure 3A, B). The structural orientation of the central scaffold is well defined by the 44 long range NOEs, which were determined in this study, as there are several NOEs between the structural elements that determine their relative arrangement to one another (Figure S3A, B). The C‐terminal part of helix 1, the loop connecting helix 1 and helix 2 as well as terminal parts of helices 3–4 and their connecting loop pack together to form a hydrophobic core, which is capped by helix 2 on the C‐terminal side of the protein. Helix 4 contains the basic region, starting from D500 and ending at K518, and is unstructured beyond residue G505 with the remaining residues likely only folded when DNA is bound, as seen for the DNA‐binding domain of the CNC transcription factor Skn‐1. [20]
Figure 3.

NMR solution structure of the Neh1 domain of NRF2. (A) NMR ensemble of 20 representative structures of the Neh1‐ΔLZIP (PDB ID 7O7B) domain of NRF2; helices are displayed as ribbons. Secondary structure elements are labeled with increasing numbers from N‐ to C‐terminus. (B) Lowest energy structure of Neh1‐ΔLZIP (PDB ID 7O7B) shown in a surface representation colored by its electrostatic potential with red being negative and blue positive. The white dotted line outlines the binding area of the fragment hits.
The solution structure of a similar construct of the Neh1 domain was previously determined by the Northeast Structural Genomics Consortium (NESG). The NESG structure calculation and ours were conducted with a similar number of long range NOEs between unique residue pairs, 31 and 25, respectively. The here reported Neh1‐ΔLZIP (PDB ID 7O7B) backbone structure can be superimposed with the Neh1 solution NMR structure (PDB ID 2LZ1) determined by NESG with a root mean square deviation for Cα atoms (rmsdCα) of 1.8 Å (Figure S4A). Helix 3 differs significantly between the two structures, in our structure helix 3 is in a straight conformation and lacks the bend that is observed in 2LZ1. This bend is also absent in the sMaf:DNA (PDB ID 3 A5T) and Skn‐1:DNA (PDB ID 1SKN) complex structure, with the three proteins showing high sequence identity and presumably a conserved fold of their DNA‐binding domains (Figure S4B, C).[ 20b , 21 ] Structural alignment of the solution Neh1‐ΔLZIP (PDB ID 7O7B) structure and the X‐ray structures of the sMaf and Skn‐1 DNA binding domains (PDB ID 3 A5T, 1SKN) yield a rmsdCα of 1.6 and 1.3 for the helical regions, respectively.
Data‐driven docking of compound 2. Mapping the chemical shift changes of the fragment hits onto the Neh1‐ΔLZIP structure revealed that the largest changes occur in a well‐defined surface patch, formed by the side chain amide of N482 on helix 3, and the C‐terminal part of helix 4 for which a very shallow surface pocket formed by the arginine and lysine side chains can be delineated on several of the lowest energy Neh1 NMR solution structures (Figure 3B, indicated by a white dotted circle). The side chain amide of N482 showed CSP values of approximately 0.3 ppm, for R499, D500, and R503 values of 0.30, 0.23, and 0.14 ppm were observed upon addition of compound 2 (Figure 4A, B).
Figure 4.

NMR determination of the fragment hits binding site. (A) Ribbon and surface representation of Neh1‐ΔLZIP (PDB ID 7O7B) with nitrogen atoms shown as blue spheres for residues that showed CSP values larger than 2 standard deviations of the mean value of all CSPs in the NMR titration experiment. (B) Histogram showing CSP values for backbone amide groups of 15N labelled Neh1‐ΔLZIP in the presence of 2 mM compound 2. Horizontal dotted grey and black lines indicate one and two standard deviations of the CSP values, respectively. Residues with backbone amide CSP values greater than two standard deviations are indicated with labels. Residues with without a bar were not assigned.
We sought to obtain the complex structure of compound 2 bound to Neh1‐ΔLZIP, to determine key interactions that are responsible for ligand binding. Due to the weak affinity and surface location of the binding site no definite unambiguous NOE could be assigned, therefore, CSP values obtained from the 1H‐15N HSQC titration experiments, were converted into ambiguous restraints and used in a data‐driven docking approach using HADDOCK. [19] Residues with CSP values equal or larger than two standard deviations of the mean value were defined as residues actively involved in ligand binding, hence so‐called ambiguous interaction restraints (AIRs) were generated for residues N482, R499, D500, and R503 to all polar atoms of the ligand. [19] HADDOCK docking calculations generated three clusters of binding poses with a similar HADDOCK score, in the three clusters compound 2 is oriented with its carboxylic acid toward helix 3 and the aryl rings toward helix 4 (Figure 5 and Figure S5). The orientation of compound 2 in the first and second cluster is rather perpendicular to helix 3/4 (Figure 5A, B and Figure S5A, B), whereas in cluster three the ligand is in an approximately 45 degree angle to the two helices (Figure 5C, D and Figure S5C, D). Compound 2 in cluster two is translated 1.5–2 Å away from helix 3 in comparison to the ligand binding pose in cluster one (Figure 5A, B, D). To determine if one of the three conformations is in better agreement with the experimental data, we applied post‐docking filtering by comparing back‐calculated ligand induced chemical shift changes of the backbone amide protons from our HADDOCK generated models to the experimental CSPs. This reintroduces the CSPs sign information which is otherwise lost in the HADDOCK calculations, as in HADDOCK CSPs are transformed into upper limit distance constraints,[ 19 , 22 ] and it was first shown by McCoy and Wyss [23] and later by others[ 22a , 24 ] that this information can be crucial to identify the correct binding mode. In our case the sign of the CSPs constrain the relative orientation of the affected protein backbone amide protons and the two aromatic rings in the ligand, as it can be safely assumed due to the weak binding affinity that no or little protein backbone rearrangements occur upon ligand binding, thus the major contribution to the CSPs is caused by the direct ligand binding event. Hence, for the back‐calculation in a first approximation the sign of a proton chemical shift perturbation is a function of the orientation of the ligand aromatic rings, and it has been shown that they are to a lesser extent a function of the relative orientation between the protein proton and charged ligand groups.[ 22b , 25 ]
Figure 5.
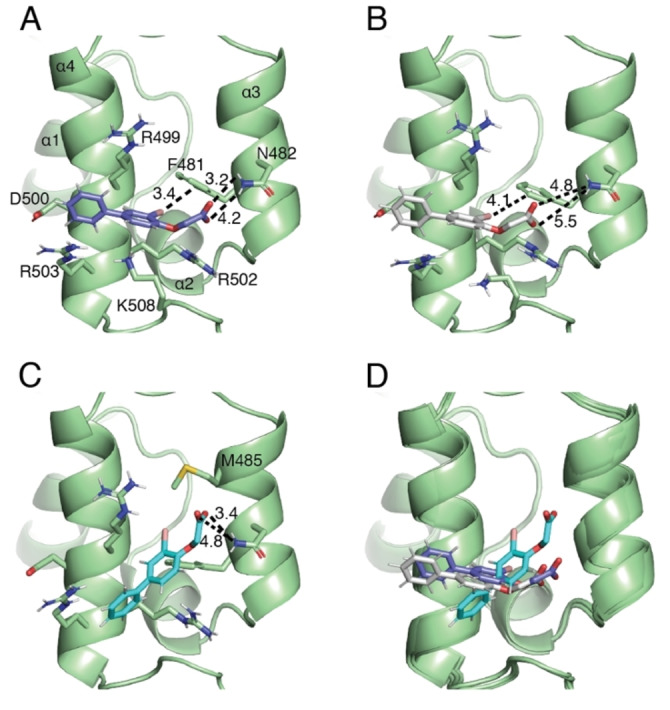
Data‐driven docking structures of fragment 2; ribbon representation of NRF2 (residues R456‐A510 are shown with the remaining residues omitted for clarity, PDB ID 7O7B) complexed to compound 2. (A) Shown in sticks representation the binding pose of 2 with lowest Q‐score in cluster 1. (B) Shown in sticks representation the binding pose of 2 with lowest Q‐score in cluster 2. (C) Shown in sticks representation the binding pose of 2 with lowest Q‐score in cluster 3. Black dotted lines indicate distances in Angstrom. (D) Superimposition of these docking poses in the binding site.
In this study we simulated the sign and magnitude of backbone amide 1HN chemical shift changes, which were caused by the ring current effect induced by the two aromatic rings, with the empirical point‐dipole model derived by Pople. [26] The electric field effect generated by electric charges on the carboxylic acid of the ligand was simulated with the empirical equation introduced by Buckingham. [27] The three clusters were assessed with the Q‐score used by McCoy and Wyss [23] to quantify the difference between the experimental and calculated chemical shifts – with smaller Q‐scores indicative of better agreement. This analysis showed that several ligand conformations can equally well reproduce the observed experimental chemical shift changes. Binding poses in cluster 1 and 2 had an average (five structures) Q‐score for the ring current effect of 4.6±1.1 and 4.9±0.8, respectively, whereas the Q‐score was 6.1±1.5 for cluster 3. These values slightly increased when the electric field effect was additionally taken into account to 5.2±0.6, 5.4±0.8, and 8.1±0.7. Furthermore, the predicted average chemical shift change for the side chain amide nitrogen Nδ2 of N482, assumed to be solely caused by the electric field effect, was 2.54±0.18, 1.04±0.13, and 1.12±0.26 ppm respectively, with a measured value of 0.64 ppm at approximately 50–60 % protein fraction bound. Thus, the pattern of calculated chemical shift perturbations agree reasonably well with the experimentally determined values (Figure S6), with the discrepancy potentially coming from not including the bromine and the ether oxygen in the calculations, which were omitted from the calculation as the partial charges for these atoms are not readily described. Overall, the binding pose of compound 2 in cluster two showed the best agreement with the experimental CSP data when taking the chemical shift changes of the Nδ2 atom of N482 into consideration. However, due to the similar Q‐values of the three binding poses a dynamic binding mode of the fragments cannot be ruled out, a phenomenon frequently seen for fragments even in less shallow binding sites. [28]
Based on the docking structures the structure activity relationship observed for the majority of screening hits can be rationalized. In cluster 1 and 2 the second aromatic ring is positioned above R503 allowing for a cation‐π interaction between the protein and the ligand (Figure 5A, B and Figure S5A, B). This interaction explains how the absence of the second aromatic ring or an electron poor (π‐deficient) heterocycle such as pyrimidine decreases affinity to the Neh1 domain of NRF2 (Table 1 and Figure S2F). The halogen in ortho position on the first phenyl ring, was shown by the SARs series to be crucial for ligand binding and is positioned in a way that it can favorably interact with F481, where presumably the positively charged sigma hole interacts with the partial negative charge on the aromatic ring plane forming a putative halogen bond. [29] This rationalizes the approximately 2‐fold increase in affinity when chlorine is substituted with bromine at ring position three as the strength of interaction increases with the size of the halogen (Table 1 and Figure 2C).[ 29b , 30 ] The carboxylic acid is in an orientation that allows for the acceptance of hydrogen bonds from the amide side chain of N482 and for a charge‐charge interaction with the guanidinium group of R502 (Figure 5A, B). The ether oxygen could potentially function as a hydrogen bond acceptor donated from R502 and/or K508 and can additionally decrease the distance between the carboxyl group and N482, explaining the significant increase in chemical shift changes and binding affinity of compound 3 compared to its matched pair 1, which lacks this oxygen atom (Table 1 and Figure S2A, B). Furthermore, the docking structures rationalize how the introduction of a phenolic hydroxy group, compound 5, can lead to a 2‐fold increase in affinity by functioning as a hydrogen donor to the backbone carbonyl of R499 compared to its nonphenolic matched molecular pair 3.
In cluster 3 the carboxylic acid of compound 2 engages similarly in a hydrogen bonding network with N482. However, in this binding pose the bromine is positioned between F481 and M485 where it can participate in a hydrophobic core. The two aryl rings are potentially in an orientation which allows them to interact in a cation‐π type interaction with the arginine residues of the basic region and a π‐π interaction with F481 (Figure 5C).
From the binding models it becomes clear that the fragments bind at the putative Neh1 ARE DNA binding site (Figure S7). To further corroborate this finding we performed ARE DNA binding titration experiments.
DNA binding to Neh1. In order to determine if Neh1 as a monomer possesses affinity for the ARE consensus sequence we performed a NMR titration experiment of unlabelled ARE (19bp) to 15N labelled Neh1‐ΔLZIP (Figure 6A). Indeed, Neh1‐ΔLZIP showed specific chemical shift changes in the α‐helical and unstructured part of the basic region, several unstructured residues starting from G505 showed strong line broadening at higher DNA concentrations. In detail, the peaks of V479, D500, I501, R502, and R503 show CSPs larger than one standard deviation than the mean of all CSPs at a 4‐fold excess of DNA in agreement with specific DNA binding in the basic region (Figure 6B). Residues G505, K506, and V509 as well as basic residues R512 and K518, which based on sequence alignment to Skn‐1 are interacting with the phosphate groups of the DNA, [31] are broadened beyond detection at a 1 : 2 protein DNA ratio (Figure 6A). These findings are indicative of conformational rearrangement of the basic region upon DNA binding. [32] From the CSP data the dissociation constant of monomeric Neh1‐ΔLZIP to ARE was determined to be in the higher micro‐molar range, approximately 200±70 μM (average over R499, D500, R503), assuming a 1 : 1 stoichiometry (Figure 6C). In an earlier surface plasmon resonance (SPR) experiment by Yamamoto T et al. no binding could be observed to monomeric Neh1, whereas for the Neh1‐sMafG heterodimer a KD of 20 nM was reported for the ARE consensus sequence (TGCTGACTCAGCA). [31] However, our measurements indicate that NRF2 can already preform a protein‐DNA complex which could facilitate formation of the NRF2‐sMaf heterodimer on the DNA.
Figure 6.
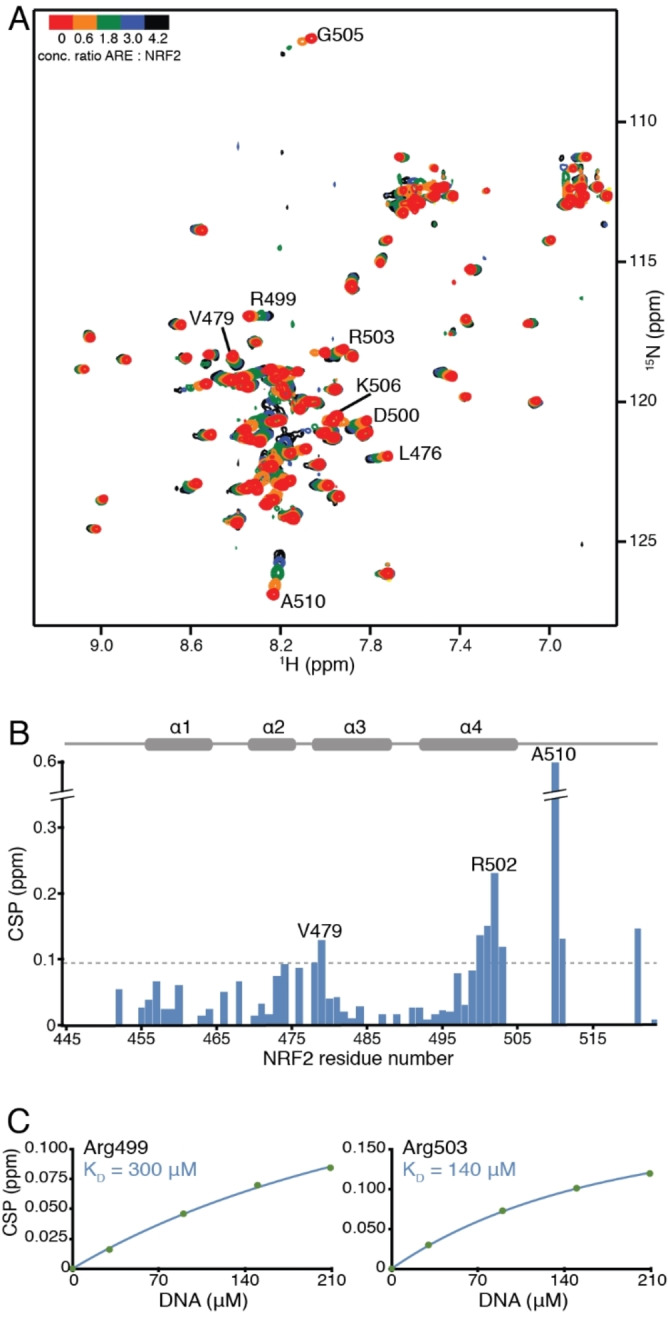
ARE DNA binding to NRF2 (residues 445–523). (A) Overlay of 1H‐15N HSQC spectra of uniformly labeled 50 μM 15N‐Neh1‐ΔLZIP (red) titrated with 30, 90, 150, and 210 μM consensus ARE DNA (from orange to black). (B) Histogram showing chemical shift perturbation values for backbone amide groups of 15N‐Neh1‐ΔLZIP in the presence of 210 μM ARE DNA; the largest CSP value is 0.61 ppm (A510) and is truncated in the plot. Horizontal dotted grey line indicates the standard deviation (0.097 ppm) of the mean value of the CSP values. (C) Titration curves obtained from the 1H‐15N HSQC titration measurements for residues R499, and R503. KDs were derived by fitting experimental CSP values to equation (1). The lines represent an individual residue fit to equation (1).
Furthermore, comparison of the chemical shift perturbations caused by the ARE element and the fragment hits revealed an overlap in binding regions in the structured part of the basic region, which agrees with the superimposition of the Neh1‐ΔLZIP:2 docking structure and the Skn‐1:ARE complex X‐ray structure (Figure S7). To confirm this finding, we performed a competition binding experiment. In order to show specific replacement of 2 by ARE DNA, 15N labelled Neh1‐ΔLZIP in the presence of 2 was titrated with unlabeled ARE DNA. This 1H‐15N HSQC titration experiment resulted in a final spectrum which is very close to the spectrum of Neh1‐ΔLZIP bound to ARE (Figure S8), indicating that tighter binding ligands are expected to modulate DNA binding.
Conclusion
The role of NRF2 as a master regulator of cellular response to oxidative and electrophilic stresses bestows upon it a cancer suppressive function. This protective role is reversed in several types of cancer for which constitutive NRF2 activation has been found to cause chemo‐resistance and tumor progression. [33] Therefore, several previous studies sought to find small molecule inhibitors of NRF2 to improve treatment of NRF2 maintained tumors. However, only a handful of small molecule NRF2 inhibitors, such as Brusatol, [34] Luteolin, [35] AEM1, [36] and ML385, [8] have been described in the literature to date. Furthermore, besides ML385, none of these compounds’ putative mechanisms of action involve direct binding to NRF2.[ 1 , 4b , 33 ] ML385 has been proposed to bind to the leucine zipper region of NRF2, however, no structural characterization of the interaction has been undertaken. [8] The leucine zipper motif is a common feature for dimer formation in many transcription factors, hence, binding to this region could potentially cause inhibition of a large number of transcription factors.[ 1 , 4b ] In this study we explored alternative binding sites on NRF2 by a fragment‐based NMR screening approach, which led to the discovery of several small‐molecule hits with a biphenyl phenoxy‐acetic acid scaffold, which have binding affinities in the milli‐molar range. In a ‘SAR by catalogue’ approach based on a pharmacophore similar to 2, we searched for existing compounds (internal and commercial), and novel make‐on‐demand compounds from Enamine. In this way we exhaustively searched the commercially available chemical space for NRF2 binders in order to discover hits to the Neh1 domain. This resulted in compounds which allowed us to determine the initial SAR of our first fragment hits. The SAR study deciphered subtle differences in binding affinities due to ring substituents on the two phenyl rings; for example we determined that a 2’‐hydroxy substituent in compound 5 decreased the dissociation constant 2‐fold to its respective matched pair compound 3. Additionally, we could show that substituting the second phenyl ring with an electron poor (π‐deficient) pyrimidine abrogated ligand binding. The SAR data further established that the 3‐halo group is crucial for anchoring the compounds in their binding site. Furthermore, the NMR data allowed us to elucidate the small‐molecule binding site, which was composed of the CNC and basic region of NRF2; a region that is specific to transcription factors such as p45 NF−E2, and BACH proteins, as well as Maf transcription factors where the extended homology region has a similar fold. This likely decreases the probability of off‐target binding to other transcription factor families. In a data‐driven docking run we established three possible binding modes of compound 2. These binding poses agree well with our experimentally measured chemical shift perturbations and our SAR data, with simulated CSPs for cluster 2 having the best agreement with the experimental values. In more detail, cluster 1 and 2 can equally well describe chemical shift changes which occur due to the ligand ring current effect on helix 4 with simulated values from cluster 3 being somewhat worse. However, the simulated CSP for the Nδ2 atom of N482 caused by the electric field effect of the ligand carboxyl group correlates best with the orientation of the acid in cluster 2 and 3, with predicted values in cluster 1 being larger than experimentally observed. Although the possibility of dynamic sampling of all three binding poses cannot be ruled out either, a feature rather common for low affinity binders. The binding conformations provided the basis for rationalizing the contribution of different ligand features to compound binding. Based on sequence alignment to Skn‐1 and Maf proteins, it is apparent that this small and very shallow positively charged binding site is potentially engaged in DNA binding as well. We experimentally corroborated, by NMR ARE DNA titration measurements, that indeed the hit binding region coincides with the ARE promoter binding region on NRF2, therefore in theory high affinity binders could modulate NRF2 promoter binding.
This first high‐resolution characterization of a NRF2 binder could serve as a starting point in a hit‐to‐lead campaign for optimizing binding affinity and selectivity for NRF2 protein‐DNA inhibitors. The protein–ligand structure obtained by data‐driven docking has potential for linker length optimization between the biphenyl scaffold and the carboxylic acid as well as its shape complementary to the binding pocket. Additionally, it could be envisioned that an optimal combination of phenyl ring side chains could lead to higher ligand potency. Optimized ligands could directly modulate NRF2 ARE promoter binding, and thereby modulate the transcription of NRF2 target genes. Mitigating the effects of constitutive NRF2 activation in several cancer types and help increase effectiveness of cancer treatment.
Experimental Section
Protein expression and purification. Uniformly 15N and 13C, 15N labeled samples of Neh1 (residues 445–523, denoted by Neh1‐ΔLZIP) of human NRF2 (isoform 1) were overexpressed in Escherichia coli BL21 (DE3) as a TEV cleavable N‐terminal fusion to MBP, and additionally contained a C‐terminal TEV cleavable hexa‐histidine tag. Cells were grown at 37 °C in M9 minimal media containing 15NH4Cl and 13C6‐glucose as sole nitrogen and carbon sources in presence of ampicillin until OD600≈0.6, then the temperature was lowered to 18 °C, and after 45 min protein synthesis was induced by adding isopropyl‐β‐D‐thiogalactopyranoside (IPTG) to a final concentration of 0.4 mM. Expression was carried out overnight.
Cells were lysed by sonication in lysis buffer containing 50 mM Tris‐HCl (pH 7.5), 500 mM NaCl, 1 mM dithiothreitol (DTT) and afterward clarified by centrifugation. Subsequently, the supernatant was loaded onto a MBPTrap HP (cytiva) column. In the next step, MBP‐Neh1 fusion protein was loaded onto a HisTrap FF crude (cytiva) column and after elution the MBP – and His – tag was cleaved by incubation with TEV. MBP and the His‐tag were removed by running a Resource S column (cytiva). In a final purification step, Neh1 was purified to homogeneity by size exclusion chromatography using a Superdex 75 (cytiva).
NMR spectroscopy. NMR spectra were recorded at 25 °C on a Bruker AVANCE NEO 600 MHz (14.1 T) spectrometer and an Avance III HD 800 MHz (18.8 T) spectrometer. Data were processed using NMRPipe [37] and analyzed using CcpNmr. [38] Protein assignment and NOE experiments were carried out on a 0.5 mM 15N, 13C, uniformly labeled Neh1‐ΔLZIP solution in 50 mM sodium phosphate buffer, pH 6.5, 150 mM NaCl, 1 mM DTT in 5 % D2O/95 % H2O. Backbone and side chain 1H, 13C, and 15N chemical shift assignment of Neh1‐ΔLZIP was obtained with standard triple‐resonance experiments: HNCA/HN(CO)CA, HNCO/HN(CA)CO, CBCA(CO)NH/HNCACB, (H)CCONNH‐TOCSY, and HCCH‐TOCSY. Intramolecular distance restraints were obtained from 3D 1H−1H‐NOESY−15N/13C‐HSQC (mixing time 120 ms) experiments.
Small‐molecule titration experiments were carried out on a 50 μM 15N uniformly labeled Neh1‐ΔLZIP solution in 50 mM sodium phosphate buffer, pH 6.5, 150 mM NaCl, 1 mM DTT in 10 % D2O/95 % H2O with 50 mM ligand stock solutions in dimethyl sulfoxid‐d6 (DMSO‐d6). 1D NMR spectroscopy was used to determine concentrations. For all titration experiments the same protein stock solution was used, the ligands were titrated to final concentrations of 0.250, 0.750, 1.5, and 2.0 mM. At each titration step chemical shift changes upon ligand addition were monitored with 1H‐15N HSQC experiments. The combined chemical shift perturbation of 1HN and 15NH was calculated as .
Chemical shift perturbations at each titration point were used to calculate the dissociation constants (KD) by nonlinear least square fit to equation 1.
| (1) |
where is the observed chemical shift perturbation, the maximum chemical shift perturbation and and the protein and ligand concentrations, respectively.
For DNA titration measurements double stranded ARE DNA was obtained by mixing complementary oligonucleotides (5’‐CCGGTGCTGAGTCAGCAGG and 5’‐CCCTGCTGACTCAGCACCG) which were denatured at 95 °C and slowly annealed at 45 °C. The titration experiments were conducted with a 5 mM ARE DNA stock solution in 50 mM sodium phosphate buffer, pH 6.5, 150 mM NaCl, 1 mM DTT in 5 % D2O/95 % H2O. DNA was added to final concentrations of 0.03, 0.09, 0.150, and 0.210 mM to a 50 μM 15N uniformly labeled Neh1‐ΔLZIP solution. The KD was obtained by fitting the CSP data to equation 1.
The competition experiments were conducted with a 5 mM ARE DNA stock solution in 50 mM sodium phosphate buffer, pH 6.5, 150 mM NaCl, 1 mM DTT in 5 % D2O/95 % H2O. DNA was added to final concentrations of 0.10, 0.20, and 0.30 mM to a 50 μM 15N uniformly labeled Neh1‐ΔLZIP solution in the presence of 0.750 mM compound 2.
Structure calculation and refinement. Backbone dihedral angle restraints were set to ϕ=−60 (±20)° and ψ=−45 (±20)° for residues that were predicted to be α‐helical based on 13Cα, 13Cβ, 13C’, 1Hα, 15N, and 1HN chemical shifts using the software TALOS−N. [39] For residues with a TALOS−N classification of good, outside of the α‐helices, the respective predicted dihedral angles were used in the structure calculation. Intramolecular distance restraints were obtained from 3D 1H‐1H‐NOESY‐15N/13C‐HSQC experiments. Ambiguity of NOE assignments were partially resolved, and distance restraints were calibrated, with the program Aria2.3. [40] The experimentally determined distance, and TALOS−N predicted dihedral restraints were used in a torsion angle simulated annealing protocol using CNS1.2/Aria2.3[ 40 , 41 ] to solve the solution structure of the Neh1 (residues 445–523) domain of NRF2. The final NMR ensemble was refined in an explicit water shell. [42] The 20 lowest‐energy solution structures (out of 100 calculated) were selected as a final representative ensemble of Neh1‐ΔLZIP (PDB ID 7O7B).
Data‐driven docking protocol. The docking conformation of compound 2 was generated by minimizing the MMFF94 [43] energy of the free molecule in LigandScout (version 4.4) [44] using default parameters. CNS parameter and topology files for compound 2 were generated using PRODRG [45] and the dihedral angles in the parameter file were modified manually to reflect the energy minimized ligand conformation. The lowest energy structure of Neh1‐ΔLZIP was used as protein starting structure for the docking run. Ambiguous Interaction Restraints (AIRs) were generated for residues N482, R499, D500 and R503, as they showed CSP values equal or larger than two standard deviations of the mean value, upon compound 2 addition. Compound 2 was docked using the software HADDOCK (version 2.2)/CNS (version 1.3),[ 19 , 41 , 46 ] with recommended settings for small‐molecule docking. Briefly, in a rigid body docking step 1000 structures were calculated, subsequently the 200 structures with the lowest HADDOCK scores were used in a final docking step with a fully flexible binding interface and flexible ligand. For each of the three HADDOCK clusters, with similar HADDOCK scores, five representative conformers with the lowest energies and the least experimental restraint violations were picked for chemical shift perturbations back‐calculation.
CSP calculation. There are two major through space contributions to protein 1HN chemical shift changes upon ligand binding, namely, the ring current (RC) and the electric filed (EF) effect, which are caused by aromatic rings and electric charges on the ligand. [47] The RC effect of the two aromatic rings of 2 was calculated by the empirical point‐dipole model derived by Pople:[ 26 , 48 ]
where is the change in the isotropic nuclear shielding constant, is the number of circulating aromatic ring electrons, is the elementary charge, is the radius of the aromatic ring, is the electron mass, is the speed of light, is the angle between the ring normal and the proton to ring center vector, and r is the distance from the proton to the ring center.
The EF effect of the carboxylic acid point charges on 2, which can introduce CSP at spatially close atoms, was calculated with an empirical equation derived by Buckingham:[ 27 , 49 ]
Here, is the distance between the ligand atom with the partial charge and the protein 1HN or Nδ2 atom, the angle is between the distance vector and the vector of the HN‐NH amide bond or the side chain Cγ‐Nδ2 bond. The partial charges for the carboxyl group were taken from the Amber force field. [50] The magnitude of the CSP due to the EF effect is proportional to the polarizability of the HN‐NH or Cγ‐Nδ2 bond and is given by the nuclear shielding polarizability , in this study we used the values determined by Jens J. Led and co‐workers [49] of 188 ppm au for the 1HN atom and 868 ppm au for the 15NH atom. For the dielectric constant the value 6 was used in our calculations. [51] The CSP values were back‐calculated for helices 3 and 4 as these are, based on the 1H‐15N HSQC titration experiments, directly involved in ligand binding. The quality of agreement between experimentally measured and simulated CSPs was quantified with the Q‐score suggested by McCoy and Wyss: [23]
where and are the experimental and calculated CSP value for the amide backbone proton i, respectively. and are the largest experimentally observed and simulated CSP value.
Accession codes
The NMR structure ensemble determined in this article has been deposited in the Protein Data Bank under accession ID 7O7B. The chemical shift assignments have been deposited in the Biological Magnetic Resonance Bank (BMRB) under accession code 34617. The authors will release the atomic coordinates and experimental data upon article publication.
Conflict of interest
The authors declare the following competing financial interest(s): Julian E. Fuchs, Gerd Bader, Darryl B. McConnell, and Moriz Mayer were fulltime employees for Boehringer Ingelheim at the time the data presented herein were collected.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
S. B. was funded by the Christian Doppler Laboratory for High‐Content Structural Biology and Biotechnology, Austria. The financial support by the Austrian Federal Ministry for Digital and Economic Affairs, the National Foundation for Research, Technology and Development and the Christian Doppler Research Association is gratefully acknowledged. We also thank Dirk Kessler for project support and coordinating the initial fragment screens.
S. Brüschweiler, J. E. Fuchs, G. Bader, D. B. McConnell, R. Konrat, M. Mayer, ChemMedChem 2021, 16, 3576.
References
- 1. Dodson M., de la Vega M. R., Cholanians A. B., Schmidlin C. J., Chapman E., Zhang D. D., Annu. Rev. Pharmacol. Toxicol. 2019, 59, 555–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Cancer Genome Atlas Research N., Nature 2012, 489, 519–525;22960745 [Google Scholar]
- 2b. Panieri E., Saso L., Oxid Med Cell Longev 2019, 2019, 8592348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Campbell J. D., Alexandrov A., Kim J., Wala J., Berger A. H., Pedamallu C. S., Shukla S. A., Guo G., Brooks A. N., Murray B. A., Imielinski M., Hu X., Ling S., Akbani R., Rosenberg M., Cibulskis C., Ramachandran A., Collisson E. A., Kwiatkowski D. J., Lawrence M. S., Weinstein J. N., Verhaak R. G., Wu C. J., Hammerman P. S., Cherniack A. D., Getz G., Cancer Genome Atlas Research N., Artyomov M. N., Schreiber R., Govindan R., Meyerson M., Nat. Genet. 2016, 48, 607–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Cloer E. W., Goldfarb D., Schrank T. P., Weissman B. E., Major M. B., Cancer Res. 2019, 79, 889–898; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Qin J. J., Cheng X. D., Zhang J., Zhang W. D., Cell Commun. Signaling 2019, 17, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Padmanabhan B., Tong K. I., Ohta T., Nakamura Y., Scharlock M., Ohtsuji M., Kang M. I., Kobayashi A., Yokoyama S., Yamamoto M., Mol. Cell 2006, 21, 689–700; [DOI] [PubMed] [Google Scholar]
- 5b. Singh A., Misra V., Thimmulappa R. K., Lee H., Ames S., Hoque M. O., Herman J. G., Baylin S. B., Sidransky D., Gabrielson E., Brock M. V., Biswal S., PLoS Med. 2006, 3, e420; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5c. Martinez V. D., Vucic E. A., Pikor L. A., Thu K. L., Hubaux R., Lam W. L., Mol. Cancer 2013, 12, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang X. J., Sun Z., Villeneuve N. F., Zhang S., Zhao F., Li Y., Chen W., Yi X., Zheng W., Wondrak G. T., Wong P. K., Zhang D. D., Carcinogenesis 2008, 29, 1235–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Moi P., Chan K., Asunis I., Cao A., Kan Y. W., Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Singh A., Venkannagari S., Oh K. H., Zhang Y. Q., Rohde J. M., Liu L., Nimmagadda S., Sudini K., Brimacombe K. R., Gajghate S., Ma J., Wang A., Xu X., Shahane S. A., Xia M., Woo J., Mensah G. A., Wang Z., Ferrer M., Gabrielson E., Li Z., Rastinejad F., Shen M., Boxer M. B., Biswal S., ACS Chem. Biol. 2016, 11, 3214–3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Arkin M. R., Wells J. A., Nat. Rev. Drug Discovery 2004, 3, 301–317. [DOI] [PubMed] [Google Scholar]
- 10. Darnell J. E., Nature Reviews Cancer 2002, 2, 740–749. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Arkin M. R., Tang Y., Wells J. A., Chem. Biol. 2014, 21, 1102–1114; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Bhagwat A. S., Vakoc C. R., Trends Cancer 2015, 1, 53–65; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11c. Chen A., Koehler A. N., Trends Mol. Med. 2020, 26, 508–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bushweller J. H., Nat. Rev. Cancer 2019, 19, 611–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Huang W., Dong Z., Wang F., Peng H., Liu J. Y., Zhang J. T., ACS Chem. Biol. 2014, 9, 1188–1196; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b. Li H., Ban F., Dalal K., Leblanc E., Frewin K., Ma D., Adomat H., Rennie P. S., Cherkasov A., J. Med. Chem. 2014, 57, 6458–6467. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Kuenemann M. A., Bourbon L. M., Labbe C. M., Villoutreix B. O., Sperandio O., J. Chem. Inf. Model. 2014, 54, 3067–3079; [DOI] [PubMed] [Google Scholar]
- 14b. Laraia L., McKenzie G., Spring D. R., Venkitaraman A. R., Huggins D. J., Chem. Biol. 2015, 22, 689–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Lyu J., Wang S., Balius T. E., Singh I., Levit A., Moroz Y. S., O′Meara M. J., Che T., Algaa E., Tolmachova K., Tolmachev A. A., Shoichet B. K., Roth B. L., Irwin J. J., Nature 2019, 566, 224–229; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15b. Stein R. M., Kang H. J., McCorvy J. D., Glatfelter G. C., Jones A. J., Che T., Slocum S., Huang X. P., Savych O., Moroz Y. S., Stauch B., Johansson L. C., Cherezov V., Kenakin T., Irwin J. J., Shoichet B. K., Roth B. L., Dubocovich M. L., Nature 2020, 579, 609–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.
- 16a. Maurer T., Garrenton L. S., Oh A., Pitts K., Anderson D. J., Skelton N. J., Fauber B. P., Pan B., Malek S., Stokoe D., Ludlam M. J., Bowman K. K., Wu J., Giannetti A. M., Starovasnik M. A., Mellman I., Jackson P. K., Rudolph J., Wang W., Fang G., Proc. Natl. Acad. Sci. USA 2012, 109, 5299–5304; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16b. Sun Q., Burke J. P., Phan J., Burns M. C., Olejniczak E. T., Waterson A. G., Lee T., Rossanese O. W., Fesik S. W., Angew. Chem. Int. Ed. 2012, 51, 6140–6143; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 6244–6247; [Google Scholar]
- 16c. Erlanson D. A., Fesik S. W., Hubbard R. E., Jahnke W., Jhoti H., Nat. Rev. Drug Discovery 2016, 15, 605–619; [DOI] [PubMed] [Google Scholar]
- 16d. Kessler D., Gmachl M., Mantoulidis A., Martin L. J., Zoephel A., Mayer M., Gollner A., Covini D., Fischer S., Gerstberger T., Gmaschitz T., Goodwin C., Greb P., Haring D., Hela W., Hoffmann J., Karolyi-Oezguer J., Knesi P., Kornigg S., Koegl M., Kousek R., Lamarre L., Moser F., Munico-Martinez S., Peinsipp C., Phan J., Rinnenthal J., Sai J. Q., Salamon C., Scherbantin Y., Schipany K., Schnitzer R., Schrenk A., Sharps B., Siszler G., Sun Q., Waterson A., Wolkerstorfer B., Zeeb M., Pearson M., Fesik S. W., McConnell D. B., P Natl Acad Sci USA 2019, 116, 15823–15829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.
- 17a. Hoffmann T., Gastreich M., Drug Discovery Today 2019, 24, 1148–1156; [DOI] [PubMed] [Google Scholar]
- 17b. Walters W. P., Journal of Medicinal Chemistry 2019, 62, 1116–1124. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Hajduk P. J., Huth J. R., Fesik S. W., J. Med. Chem. 2005, 48, 2518–2525; [DOI] [PubMed] [Google Scholar]
- 18b. Edfeldt F. N., Folmer R. H., Breeze A. L., Drug Discovery Today 2011, 16, 284–287. [DOI] [PubMed] [Google Scholar]
- 19. Dominguez C., Boelens R., Bonvin A. M., J. Am. Chem. Soc. 2003, 125, 1731–1737. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Lo M. C., Ha S., Pelczer I., Pal S., Walker S., Proc. Natl. Acad. Sci. USA 1998, 95, 8455–8460; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20b. Rupert P. B., Daughdrill G. W., Bowerman B., Matthews B. W., Nat. Struct. Biol. 1998, 5, 484–491. [DOI] [PubMed] [Google Scholar]
- 21. Kurokawa H., Motohashi H., Sueno S., Kimura M., Takagawa H., Kanno Y., Yamamoto M., Tanaka T., Mol. Cell. Biol. 2009, 29, 6232–6244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.
- 22a. Stark J., Powers R., J. Am. Chem. Soc. 2008, 130, 535–545; [DOI] [PubMed] [Google Scholar]
- 22b. Ten Brink T., Aguirre C., Exner T. E., Krimm I., J. Chem. Inf. Model. 2015, 55, 275–283. [DOI] [PubMed] [Google Scholar]
- 23. McCoy M. A., Wyss D. F., J. Biomol. NMR 2000, 18, 189–198. [DOI] [PubMed] [Google Scholar]
- 24. Gonzalez-Ruiz D., Gohlke H., J. Chem. Inf. Model. 2009, 49, 2260–2271. [DOI] [PubMed] [Google Scholar]
- 25. Cioffi M., Hunter C. A., Packer M. J., Spitaleri A., J. Med. Chem. 2008, 51, 2512–2517. [DOI] [PubMed] [Google Scholar]
- 26. Pople J. A., The Journal of Chemical Physics 1956, 24, 1111–1112. [Google Scholar]
- 27. Buckingham A. D., Canadian Journal of Chemistry 1960, 38, 300–307. [Google Scholar]
- 28.
- 28a. van Zundert G. C. P., Hudson B. M., de Oliveira S. H. P., Keedy D. A., Fonseca R., Heliou A., Suresh P., Borrelli K., Day T., Fraser J. S., van den Bedem H., J. Med. Chem. 2018, 61, 11183–11198; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28b. Verteramo M. L., Stenstrom O., Ignjatovic M. M., Caldararu O., Olsson M. A., Manzoni F., Leffler H., Oksanen E., Logan D. T., Nilsson U. J., Ryde U., Akke M., J. Am. Chem. Soc. 2019, 141, 2012–2026. [DOI] [PubMed] [Google Scholar]
- 29.
- 29a. Clark T., Hennemann M., Murray J. S., Politzer P., J. Mol. Model. 2007, 13, 291–296; [DOI] [PubMed] [Google Scholar]
- 29b. Sirimulla S., Bailey J. B., Vegesna R., Narayan M., J. Chem. Inf. Model. 2013, 53, 2781–2791. [DOI] [PubMed] [Google Scholar]
- 30. Scholfield M. R., Zanden C. M., Carter M., Ho P. S., Protein Sci. 2013, 22, 139–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yamamoto T., Kyo M., Kamiya T., Tanaka T., Engel J. D., Motohashi H., Yamamoto M., Genes Cells 2006, 11, 575–591. [DOI] [PubMed] [Google Scholar]
- 32. Dyson H. J., Wright P. E., Curr. Opin. Struct. Biol. 2002, 12, 54–60. [DOI] [PubMed] [Google Scholar]
- 33. Moon E. J., Giaccia A., Free Radical Biol. Med. 2015, 79, 292–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ren D., Villeneuve N. F., Jiang T., Wu T., Lau A., Toppin H. A., Zhang D. D., Proc. Natl. Acad. Sci. USA 2011, 108, 1433–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tang X., Wang H., Fan L., Wu X., Xin A., Ren H., Wang X. J., Free Radical Biol. Med. 2011, 50, 1599–1609. [DOI] [PubMed] [Google Scholar]
- 36. Bollong M. J., Yun H., Sherwood L., Woods A. K., Lairson L. L., Schultz P. G., ACS Chem. Biol. 2015, 10, 2193–2198. [DOI] [PubMed] [Google Scholar]
- 37. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A., J. Biomol. NMR 1995, 6, 277–293. [DOI] [PubMed] [Google Scholar]
- 38. Skinner S. P., Fogh R. H., Boucher W., Ragan T. J., Mureddu L. G., Vuister G. W., J. Biomol. NMR 2016, 66, 111–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shen Y., Bax A., J. Biomol. NMR 2013, 56, 227–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rieping W., Habeck M., Bardiaux B., Bernard A., Malliavin T. E., Nilges M., Bioinformatics 2007, 23, 381–382. [DOI] [PubMed] [Google Scholar]
- 41. Brunger A. T., Nat. Protoc. 2007, 2, 2728–2733. [DOI] [PubMed] [Google Scholar]
- 42. Linge J. P., Williams M. A., Spronk C. A., Bonvin A. M., Nilges M., Proteins 2003, 50, 496–506. [DOI] [PubMed] [Google Scholar]
- 43. Halgren T. A., J. Comput. Chem. 1996, 17, 490–519. [Google Scholar]
- 44. Wolber G., Langer T., Journal of Chemical Information and Modeling 2005, 45, 160–169. [DOI] [PubMed] [Google Scholar]
- 45. Schuttelkopf A. W., van Aalten D. M. F., Acta Crystallogr. Sect. D 2004, 60, 1355–1363. [DOI] [PubMed] [Google Scholar]
- 46. Brunger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L., Acta Crystallogr. Sect. D 1998, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- 47. Williamson M. P., Prog. Nucl. Magn. Reson. Spectrosc. 2013, 73, 1–16. [DOI] [PubMed] [Google Scholar]
- 48. Case D. A., J. Biomol. NMR 1995, 6, 341–346. [DOI] [PubMed] [Google Scholar]
- 49. Hass M. A. S., Jensen M. R., Led J. J., Proteins 2008, 72, 333–343. [DOI] [PubMed] [Google Scholar]
- 50.D. A. Case, S. R. Brozell, D. S. Cerutti, T. E. Cheatham III, V. W. D. Cruzeiro, T. A. Darden, D. G. Duke, M. K. Gilson, H. Gohlke, A. W. Goetz, D. Greene, R. Harris, N. Homeyer, Y. Huang, S. Izadi, A. K. T. Kurtzman, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, D. J. Luo, K. M. M. Mermelstein, Y. Miao, G. Monard, C. Nguyen, H. Nguyen, I. Omelyan, A. Onufriev, R. Pan, D. R. R. Qi, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C. L. Simmerling, J. Smith, R. Salomon Ferrer, J. Swails, R. C. Walker, J. Wang, H. Wei, R. M. Wolf, X. Wu, L. Xiao, D. M. York, P. A. Kollman, University of California, San Francisco, 2018.
- 51. Alford R. F., Leaver-Fay A., Jeliazkov J. R., O′Meara M. J., DiMaio F. P., Park H., Shapovalov M. V., Renfrew P. D., Mulligan V. K., Kappel K., Labonte J. W., Pacella M. S., Bonneau R., Bradley P., Dunbrack R. L., Das R., Baker D., Kuhlman B., Kortemme T., Gray J. J., J. Chem. Theory Comput. 2017, 13, 3031–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information