

Abstract
The nuclear farnesoid X receptor (FXR) regulates bile acid homeostasis and is a drug target for metabolic liver diseases. FXR also plays an important role in hepatitis B virus (HBV) DNA transcription. In vitro and in mice, FXR agonist treatment leads to inhibition of viral replication and a decline in viral proteins, pregenomic RNA (pgRNA) and HBV DNA levels. We aimed to translate this to a clinical use by primarily evaluating the safety and secondary the anti‐viral effect of Vonafexor, a FXR agonist, in chronic hepatitis B (CHB) patients. In total, 73 CHB patients were enrolled in a two‐part Phase Ib double‐blind, placebo‐controlled trial. Patients were randomized to receive oral Vonafexor (100, 200 and 400 mg once daily, or 200 mg twice daily), placebo, or entecavir (Part A, n = 48) or to receive Vonafexor (300 mg once daily or 150 mg twice daily), or placebo, combined with pegylated‐interferon‐α2a (Part B, n = 25) for 29 days. Patients were followed up for 35 days. Enrolled CHB patients were mostly HBeAg‐negative. Vonafexor was overall well tolerated and safe. The most frequent adverse events were moderate gastrointestinal events. Pruritus was more frequent with twice‐daily compared with once‐daily regimens (56%–67% vs. 16%, respectively, p < 0.05). Vonafexor monotherapy of 400 mg once daily decreased HBsAg concentrations (–0.1 log10 IU/mL, p < 0.05), and Vonafexor/pegylated‐IFN‐α2a combination therapy decreased HBcrAg and pgRNA. In conclusion, Vonafexor was safe with a decline in HBV markers observed in CHB patients suggesting a potential anti‐viral effect the therapeutic potential of which has to be evaluated in larger trials.
Keywords: cccDNA transcription, farnesoid X receptor, HBV therapy, phase Ib, Vonafexor
Abbreviations
- ALT
alanine aminotransferase
- BA
bile acid
- cccDNA
covalently closed circular DNA
- CHB
chronic hepatitis B
- FXR
farnesoid X receptor
- HBcrAg
hepatitis B core‐related antigen
- HBsAg
hepatitis B surface antigen
- HBV
hepatitis B virus
- LDL
low‐density lipoprotein
- pgRNA
pregenomic RNA
Significance Statement.
Available treatments for chronic hepatitis B (CHB) may control, but rarely lead to a cure. The virus interacts in the hepatocyte with the bile acid homeostasis, which could become an important target for treatment. Here, we studied the investigational drug Vonafexor that interferes with the farnesoid X receptor, a main bile acid regulator. We evaluated for the first time Vonafexor in patients with CHB and found that it was well tolerated and reduced blood HBsAg levels. The potential of this new target for the treatment of CHB must be further determined in a larger trial with longer treatment durations.
1. INTRODUCTION
Despite mass efforts in preventive vaccination, there are more than 250 million hepatitis B virus (HBV) carriers globally, with more than 880,000 deaths due to HBV‐related liver disease every year. 1 , 2 Functional cure of HBV is characterized by a sustained loss of the hepatitis B surface antigen (HBsAg) and undetectable HBV DNA after treatment. 3 Currently, HBV treatments include suppressing viral replication with nucleoside or nucleotide analogues or interferon, but rarely lead to a functional cure.
Hepatitis B virus covalently closed circular DNA (cccDNA) in the nucleus of infected hepatocytes serves as the template for HBV replication. Targeting HBV cccDNA transcription is a potential route for disrupting viral replication, 4 reducing the host viral load and circulating viral proteins. 5 Several studies provide evidence that the farnesoid X receptor (FXR) in the nucleus of hepatocytes, which is the main bile acid (BA) regulator, 6 , 7 may also promote cccDNA transcription. 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 Furthermore, the BA transporter, sodium taurocholate co‐transporting polypeptide (NTCP), acts as the HBV cell entry receptor through which HBsAg and BA compete for cellular entry. 16 As more viruses enter via the NTCP transporter, BA transport into hepatocytes is inhibited through a negative feedback process, leading to an increase in plasma BA levels and a decrease in the intrahepatic BA pool and resulting in alterations in FXR expression. 8 , 17
Previous studies have shown that FXR may be directly involved in HBV transcription and replication via viral gene regulation. 13 , 14 , 18 FXR has been shown to interact with the viral protein HBx which affects HBV transcriptional regulation 15 and several HBV regulating host factors. 19 , 20 , 21 , 22 Finally, FXR was observed to be an HBV pro‐viral factor whose activity is reversed by FXR agonists, leading to inhibition of viral transcription and subsequently reduction of viral proteins, pgRNA and HBV DNA both in vitro and in vivo. 11
We hypothesized that Vonafexor, a novel synthetic, orally active, non‐bile salt, non‐steroidal, carboxylic acid selective FXR agonist, may reduce the viral transcription activity in CHB patients. Vonafexor was identified in vitro as highly selective for FXR relative to other FXR agonists (eg GS9674, GW4064 and Ocaliva) and with properties better suited to clinical development. Moreover, Vonafexor has been shown to be less potent than alternative compounds, 23 which would support the safe long‐term use in patients while maintaining an appropriate balance between its anti‐HBV activity and its metabolic effects. Accordingly, we further explored the safety and anti‐viral potential of Vonafexor in CHB patients in a Phase Ib clinical trial.
2. MATERIALS AND METHODS
2.1. Trial design
A phase Ib, multicentric (Australia, the Netherlands, Poland and Thailand), randomized, double‐blind (Vonafexor or placebo. Entecavir and peg‐IFN were not blinded), placebo‐controlled, sequential two‐part trial was conducted in CHB patients to explore the safety, tolerability and anti‐viral activities of Vonafexor. The trial protocol and 3 amendments were approved by the institutional ethics committees of each trial centre, and the conduct of the trial was consistent with the Declaration of Helsinki and compliant with the International Council for Harmonization guidelines for Good Clinical Practice as well as with all local and national regulations. An informed consent form was signed by each patient before enrolment into the trial. The trial was conducted between December 2017 and August 2018.
2.2. Participants
Patients aged 18–65 years with CHB for ≥6 months with a positive HBsAg test, HBV DNA >750 IU/ml, HBeAg‐positive or ‐negative, alanine aminotransferase (ALT) ≤5× upper limit of normal (ULN) at screening were eligible for inclusion. Furthermore, patients had to be treatment‐naïve or off treatment for more than 30 days before inclusion. Patients were excluded if they had relevant hepatic comorbidities or cirrhosis (details in Supplementary protocol).
2.3. Procedures
In Part A, patients were randomly assigned using a block randomization algorithm to receive blinded oral Vonafexor (100 mg once daily (o.d.), 200 mg o.d., 200 mg twice daily (b.d.), or 400 mg o.d.), placebo, or open‐label entecavir (0.5 mg o.d., Baraclude®, Bristol‐Myers Squibb, Clinton, NY) for 29 days (Figure 1). After establishing safety in Part A, patients in Part B were randomized to receive blinded Vonafexor (300 mg o.d. or 150 mg b.d.) or placebo for 29 days, combined with weekly subcutaneous injections of 180 µg pegylated‐interferon‐α2a (Peg‐IFNα2a; Pegasys®; F Hoffman‐La Roche, Basel, Switzerland). Patients had the option to participate in both Part A and Part B after a minimum washout period of 14 days between the two parts and complete eligibility assessment prior to each part. In both parts, safety assessments and blood collections were performed on days 1, 8, 15, 22, 29 and on the follow‐up at day 35. Adverse events were monitored throughout the treatment period and follow‐up and registered as MedDRA terms. Liver function tests (ALT, aspartate transaminase, total bilirubin and alkaline phosphatase) were scored using the AIDS‐CTG severity grading scale. 24 Fibroblast growth factor‐19 (FGF19), an intestinal BA regulatory protein and the BA precursor C4 (7αhydroxy‐4‐cholesten‐3‐one), both important pharmacodynamic indicators of FXR modulation, were evaluated in all patients. Moreover, Vonafexor pharmacokinetics and HBV markers were evaluated in plasma samples (Table S1).
FIGURE 1.
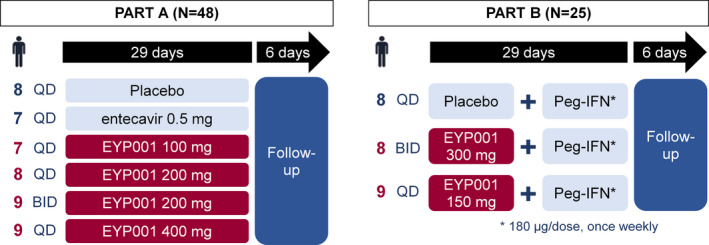
Study design. EYP001, code for the international non‐proprietary name Vonafexor; QD, once daily; B.D., twice daily
2.4. Measurements
Hepatitis B surface antigen was measured with CLIA, Liaison XL HBsAg Quant (Diasorin, Saluggia, Italy), and HBV DNA levels were measured with COBAS 4800 PCR assay (F Hoffmann‐La Roche, Rotkreuz Switzerland). HBV pgRNA levels were quantified using an in‐house, PCR assay (DDL Diagnostic Laboratory, Rijswijk, The Netherlands) (range, 2.49–9.54 log10 copies/ml) and hepatitis B core‐related antigen (HBcrAg) levels using a chemiluminescent immunoassay (Lumipulse, Fujirebio, Tokyo, Japan), quantification range, 3–7 log10 U/ml. Outcomes were compared with placebo or standard treatment (entecavir or Peg‐IFNα2a).
2.5. Statistical analyses
Baseline and on‐treatment variables were compared between groups using repeated measures linear model with fixed terms for treatment, visit and visit‐by‐treatment. An unstructured variance‐covariance matrix was generated using SAS® Proc Glimmix (SAS Institute, Cary N, USA). A paired sample t test or Wilcoxon signed‐rank test was used for paired analyses. All p‐values were two‐sided, and p‐values <0.05 were considered statistically significant. All statistical analyses were performed with SAS®, Version 9.4 (SAS Institute, Cary, NC, USA).
3. RESULTS
3.1. Patient disposition and baseline characteristics
A total of 73 subjects were enrolled, of whom 48 were included in Part A and 25 in Part B (Figure S1). Ten patients participated in both parts. Four patients (3 in Part A and 1 in Part B) withdrew prematurely because of adverse events: urticaria, rash, pruritus and pre‐existing borderline QT prolongation missed during screening. Overall, male and female patients were evenly distributed the mean age of patients was 39.7 years (range, 19–63 years) (Table 1). Most (91.8%) patients were HBeAg‐negative and 70% were treatment‐naive. Mean HBV DNA baseline (log10 IU/ml) was 4.2 (SD, 1.5) and HBsAg (log10 IU/ml) 3.5 (SD, 0.8). HBV genotype A was most common (overall 31.5%).
TABLE 1.
Baseline characteristics of CHB patients
|
Total (N = 73) |
Part A (N = 48) |
Part B (N = 25) |
|
|---|---|---|---|
| Sex, n (%) | |||
| Female | 34 (46.6) | 24 (50) | 10 (40) |
| Male | 39 (53.4) | 24 (50) | 15 (60) |
| Age, years (SD) | 39.7 (9.8) | 39.8 (9.9) | 39.3 (9.8) |
| Race, n (%) | |||
| Asian | 22 (28.2) | 15 (31.3) | 7 (28.0) |
| Black | 11 (15.1) | 8 (16.7) | 3 (12.0) |
| White | 40 (54.8) | 25 (52.1) | 15 (60.0) |
| HBeAg status, n (%) | |||
| Negative | 64 (91.8) | 45 (93.8) | 19 (76) |
| Positive | 9 (8.2) | 3 (6.2) | 6 (24) |
| Anti‐HBeAg status, n (%) | |||
| Negative | 8 (11.0) | 3 (6.3) | 5 (20.0) |
| Positive | 62 (84.9) | 43 (89.6) | 19 (76.0) |
| Unknown | 3 (4.1) | 2 (4.2) | 1 (4.0) |
| HBV genotype, n (%) | |||
| A | 23 (31.5) | 15 (31.3) | 8 (32.0) |
| B | 8 (11.0) | 5 (10.4) | 3 (12.0) |
| C | 10 (13.7) | 7 (14.6) | 3 (12.0) |
| D | 7 (9.6) | 5 (10.4) | 2 (8.0) |
| E | 4 (5.5) | 3 (6.3) | 1 (4.0) |
| Unknown | 21 (28.8) | 13 (27.1) | 8 (32.0) |
| Alanine transaminase, U/L (SD) | 29 (16) | 28 (12) | 31 (23) |
| HBV DNA, log10 IU/ml (SD) | |||
| HBeAg‐positive | 7.42 (1.56) | 7.55 (0.78) | 7.36 (1.91) |
| HBeAg‐negative | 3.71 (0.77) | 3.80 (0.79) | 3.50 (0.68) |
| HBsAg, IU/ml (SD) | |||
| HBeAg‐positive | 4.02 (1.12) | 4.09 (0.40) | 3.98 (1.39) |
| HBeAg‐negative | 3.47 (0.67) | 3.41 (0.68) | 3.61 (0.63) |
| HBV RNA, copies/ml (SD) | |||
| HBeAg‐positive | 6.06 (1.19) | 6.15 (0.75) | 6.02 (1.42) |
| HBeAg‐negative | 0.85 (1.79) | 1.10 (1.74) | 0.24 (1.80) |
| HBcrAg, log10 U/ml (SD) | |||
| HBeAg‐positive | 7.48 (0.65) | 7.77 (0.23) | 7.34 (0.77) |
| HBeAg‐negative | 2.17 (1.66) | 2.36 (1.68) | 1.72 (1.56) |
Abbreviations: HBcrAg, hepatitis B core‐related antigen; HBeAg, hepatitis B envelope antigen; HBsAg, hepatitis B surface antigen; HBV, hepatitis B virus; SD, standard deviation.
3.2. Safety and adverse events
The proportion of patients with at least one treatment emergent adverse event (TEAE) was similar between patients treated with Vonafexor (70% in Part A, 82% in Part B) and placebo (63% in Part A, 75% in Part B). Most (83% in Part A, 74% in Part B) TEAEs were of mild intensity in both groups. The most frequent TEAEs were headache, pruritus and diarrhoea in Part A, and neutropenia, pyrexia and headache in Part B (Table 2). Adverse events led to early treatment withdrawal in four patients, 3 of which possibly attributed to Vonafexor treatment: urticaria (Vonafexor 400 mg o.d.), rash (Vonafexor 200 mg b.d.) and pruritus (Vonafexor 150 mg b.d. with Peg‐IFNα2a). In Part A, 3/24 patients treated with Vonafexor once daily had pruritus versus 6/9 patients for the twice‐daily treatment; in Part B, pruritus occurred in 2/8 patients with once daily versus in 5/9 patients with twice‐daily regimens. Overall, pruritus occurred with once‐daily regimens in 15.6% patients (5/32) versus 61.1% patients (11/18) with twice‐daily regimens (p < 0.05). One patient in each twice‐daily group had a TEAE that was rated by the investigator as severe: one headache (Vonafexor 200 mg b.d. and one ALT elevation, Vonafexor 150 mg b.d. with Peg‐IFNα2a).
TABLE 2.
Frequencies of patients with treatment‐related adverse events.
| MedDRa preferred term | Part A | Part B | |||||||
|---|---|---|---|---|---|---|---|---|---|
|
Vonafexor 100 mg o.d. |
Vonafexor 200 mg o.d. |
Vonafexor 200 mg b.d. |
Vonafexor 400 mg o.d. |
Entecavir 0.5 mg o.d. |
Placebo |
Vonafexor 300 mg o.d. +Peg‐IFNα2a |
Vonafexor 150 mg b.d. +Peg‐IFNα2a |
Placebo +Peg‐ IFNα2a |
|
| (N = 7) | (N = 8) | (N = 9) | (N = 9) | (N = 7) | (N = 8) | (N = 8) | (N = 9) | (N = 8) | |
| Patients with at least one TEAE, n (%) | 4 (57) | 2 (25) | 9 (100) | 8 (89) | 4 (57) | 5 (63) | 6 (75) | 8 (89) | 6 (75) |
| Headache | 3 (43) | 1 (13) | 1 (11) | 4 (44) | 0 | 1 (13) | 3 (38) | 3 (33) | 4 (50) |
| Pruritus | 1 (14) | 1 (13) | 6 (11) | 1 (44) | 0 | 0 | 2 (25) | 5 (56) | 0 |
| Diarrhoea | 1 (14) | 0 | 1 (11) | 4 (44) | 1 (14) | 1 (13) | 3 (38) | 1 (11) | 1 (13) |
| Transaminase elevated | 2 (29) | 2 (25) | 1 (11) | 0 | 0 | 0 | 1 (13) | 2 (22) | 2 (25) |
| Abdominal pain | 2 (29) | 0 | 1 (11) | 1 (11) | 0 | 0 | 0 | 0 | 0 |
| Myalgia | 0 | 0 | 1 (11) | 0 | 0 | 2 (25) | 3 (38) | 2 (22) | 4 (50) |
| Insomnia | 0 | 0 | 0 | 0 | 2 (25) | 0 | 0 | 0 | 0 |
| Fatigue | 0 | 0 | 2 (22) | 0 | 0 | 0 | 1 (13) | 2 (22) | 0 |
| Neutropenia | 0 | 0 | 0 | 0 | 0 | 0 | 4 (50) | 5 (56) | 0 |
| Leukopenia | 0 | 0 | 0 | 0 | 0 | 0 | 2 (25) | 2 (22) | 1 (13) |
| Pyrexia | 0 | 0 | 0 | 0 | 0 | 0 | 5 (63) | 3 (33) | 4 (50) |
| Nausea | 0 | 1 (13) | 1 (11) | 0 | 0 | 0 | 2 (25) | 1 (11) | 1 (13) |
| Decreased appetite | 0 | 0 | 0 | 0 | 0 | 0 | 2 (25) | 0 | 0 |
| Musculoskeletal pain | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (22) | 0 |
| Asthenia | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (11) | 2 (25) |
Only the most frequent treatment‐related adverse events are shown (>20% of patients in any treatment group). B.D., twice daily; Peg‐IFNα2a; pegylated‐interferon α2a; O.D., once daily.
The dynamics and maximum (mean G/L, SD) neutrophil decrease was similar in all Part B groups: ‒2.1 (1.3) with Vonafexor 300 mg o.d. +Peg‐IFNα2a, ‒3.3 (1.5) with Vonafexor 150 mg BID +Peg‐IFNα2a, and ‒2.4 (1.3) with placebo +Peg‐IFNα2a.
No serious TEAEs or deaths occurred during the study.
3.3. Change in transaminases
In Part A, 15/33 patients treated with Vonafexor showed elevated ALT levels, most of which were of mild intensity per AIDS‐CTG grading (grade 1 [1.25‒2.5× ULN], n = 11). One patient with grade 3 (5‒10× ULN) ALT increases with a total bilirubin of 1.4× ULN, which was similar to the baseline value. In Part B, 19 patients had elevated ALT levels: grade 1 [n = 9], grade 2 [2.5‒5× ULN; n = 8], and grade 3 [n = 2], all of whom had normal bilirubin and INR levels. Transient elevations (n = 2) or decline (n = 2) in HBV DNA levels ranging approximately between +1 and −2 log10 IU/ml during treatment was seen in the patients with ALT elevations (Figure 2). The ALT elevations in these patients were not dose‐related and ALT declined despite continuous Vonafexor administration in two of the four patients.
FIGURE 2.
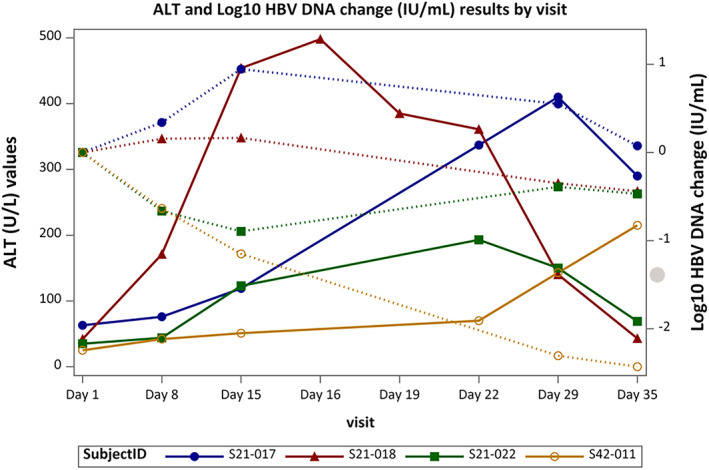
ALT and virology dynamics in patients with grade 3 or 4 ALT elevation. Treatment allocations per patient: S21–017, 100 mg Vonafexor once daily; S21–018, 200 mg Vonafexor once daily; S21–022, 150 mg Vonafexor twice daily +Peg‐IFNα2a; S42–011, 150 mg Vonafexor twice daily +Peg‐IFNα2a. ALT, alanine transaminase; Peg‐IFNα2a, pegylated‐interferon α2a. Dotted lines are HBV DNA changes and filled lines are ALT changes
3.4. Pharmacokinetics and pharmacodynamics
Vonafexor Cmax increased less the dose proportional and Tmax was 2 h (Table S2). The systemic drug exposure over time (AUC0–6h) was also dose‐dependent in patients who received Vonafexor once daily. Twice‐daily dosing did not increase AUC0–6h or Cmax compared with once‐daily regimens. Both AUC and Cmax decreased on days 8 and 15 compared with day 1, with lower concentrations at days 15 and 29 than at days 1 and 8 for each dosing regimen. No statistical significance for most values was reached because of high inter‐individual variation (CV 50%–70%).
Plasma C4 concentrations decreased at day 1, 6 h after dosing in all Vonafexor groups, but increased at all visits in the placebo and entecavir groups (Figure S2). The BA absorption response marker, FGF19, increased significantly at 6 h post‐dose in all groups but to a larger extent for Vonafexor compared with control groups (Figure S3). Remarkably, the Vonafexor‐induced FGF19 increase was more pronounced during the last two weeks of treatment compared with the first two weeks of treatment (p‐value < 0.05).
3.5. Impact on LDL
The low‐density lipoprotein (LDL) increased by 39% (p = 0.01) from baseline to day 29 with Vonafexor 200 mg b.d., but did not significantly change over time for patients who received Vonafexor o.d. (6%–13%), entecavir (14%) or placebo (–2.8%). Compared with baseline, LDL concentrations did not change significantly at day 29 in the Vonafexor +Peg‐IFNα2a group (6%–8%, p‐value > 0.05) but decreased in the placebo +Peg‐IFNα2a group (–24%, p < 0.01).
3.6. Vonafexor's effect on HBV
Plasma HBsAg concentrations significantly and steadily decreased overtime between day 1 and day 29 in the Vonafexor 400 mg monotherapy group (–0.10 log10 IU/ml, p = 0.002) (Figure 3). No significant changes were seen with placebo (–0.04 log10), entecavir (–0.04 log10), Vonafexor 100 mg o.d. (–0.02 log10), Vonafexor 200 mg o.d. (–0.02 log10) or Vonafexor 200 mg b.d. (–0.02 log10) (all p > 0.05). In Part B on day 29, Vonafexor with Peg‐IFNα2a‐treated patients had statistically lower HBsAg concentrations compared with placebo with Peg‐IFNα2a: Vonafexor 150 mg b.d. (−0.17 log10 IU/ml, p = 0.004) and Vonafexor 300 mg O.D. (−0.12 log10 IU/ml, p = 0.040) (Figure 3). The difference between HBsAg concentrations in the Vonafexor‐treated groups compared with placebo was due to the HBsAg increase between day 1 and day 29 (0.12 log10 IU/ml, p = 0.004) seen with placebo +Peg‐IFNα2a‐treated patients only (Figure 3). HBV DNA levels did not change over time in any of the Vonafexor monotherapy groups (Figure 4) but decreased significantly and similarly in all patient groups treated with Peg‐IFNα2a (–1.80 log10 IU/ml with Vonafexor 150 mg twice daily, –2.23 log10 IU/ml with Vonafexor 300 mg once daily, and –2.35 log10 IU/ml with placebo [all p < 0.001]) (Figure 4). HBV pgRNA (Figure S4) and HBcrAg (Figure S5) were measured as surrogate markers for cccDNA transcription. At baseline, most patients had detectable serum concentrations of HBV pgRNA and HBcrAg, respectively 38/48 [79.2%] and 35/48 [72.3%] patients in Part A, and 15/25 [60.0%]) and 17/25 [68.0%] patients in Part B. In all monotherapy groups of Part A, pgRNA and HBcrAg levels did not change significantly. In Part B, HBcrAg and pgRNA levels in the Vonafexor 300 mg once daily +Peg‐IFNα2a group declined significantly (p‐value < 0.05) at day 29 compared with baseline, and HBcrAg significantly (p‐value < 0.05) compared with placebo, suggesting a possible synergy between Vonafexor and Peg‐IFNα2a on HBcrAg and HBV pgRNA (Figure S5).
FIGURE 3.
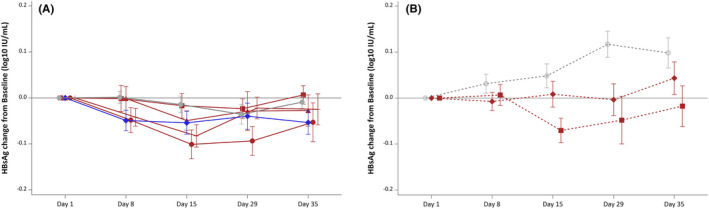
Change in circulating HBsAg in patients after a 4‐week Vonafexor treatment. Mean changes (SD) from day 1 baseline to day 35 of HBsAg (log10 IU/ml). Groups were treated with Vonafexor (red lines), placebo (grey, straight line, circle) or entecavir (blue, straight line, rhombus). Vonafexor 100 mg once daily (straight line square); Vonafexor 200 mg once daily (straight line, triangle); Vonafexor 400 mg once daily (straight line, circle); Vonafexor 200 mg twice daily (straight line, cross); Vonafexor 300 mg daily combined with Peg‐IFNα2a (dotted line, rhombus); Vonafexor 150 mg twice daily combined with Peg‐IFNα2a (dotted line, square); placebo with Peg‐IFNα2a (dotted line, circle). HBsAg, hepatitis B surface antigen. Significance: *indicates p < .05 for change from baseline. # indicates p < .05 vs. placebo. o indicates p < .05 for change from baseline and vs. Placebo
FIGURE 4.
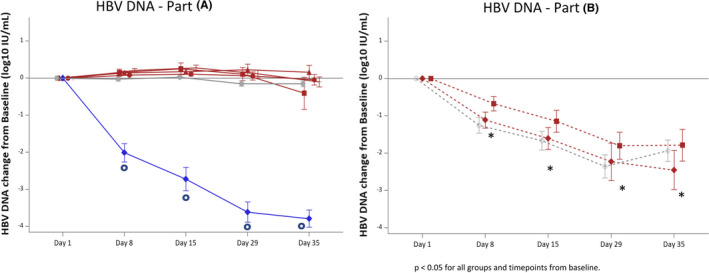
Change in circulating HBV DNA in patients after a 4‐week Vonafexor treatment. Mean changes (SD) from day 1 baseline to day 35 of HBV DNA (log10 IU/ml). Groups were treated with Vonafexor (red lines), placebo (grey, straight line, circle) or entecavir (blue, straight line, rhombus). Vonafexor 100 mg once daily (straight line square); Vonafexor 200 mg once daily (straight line, triangle); Vonafexor 400 mg once daily (straight line, circle); Vonafexor 200 mg twice daily (straight line, cross); Vonafexor 300 mg daily combined with Peg‐IFNα2a (dotted line, rhombus); Vonafexor 150 mg twice daily combined with Peg‐IFNα2a (dotted line, square); placebo with Peg‐IFNα2a (dotted line, circle). HBV, hepatitis B virus. Significance: *indicates p < .05 for change from baseline. #indicates p < .05 vs. placebo. o indicates p < .05 for change from baseline and vs. Placebo
4. DISCUSSION
This study explored the safety and for the first time the anti‐viral effect of FXR targeting agonist Vonafexor in CHB patients. Overall, Vonafexor was well tolerated. The safety profile showed no significant unexpected adverse events in CHB patients over the 4‐week treatment course as monotherapy or in combination with Peg‐IFNα2a. Furthermore, signs of a possible anti‐viral effect of Vonafexor were observed as HBsAg decline in part A and changes in HBcrAg and pgRNA levels in part B of the study.
An adverse event of special interest was the prevalence of pruritus which was low to similar in once‐daily regimens (16%) compared observations with other FXR agonists (14%–40%). 25 , 26 , 27 Interestingly, however, pruritus was seen significantly more frequently when Vonafexor was administered twice daily (56%–67%). This difference could not be explained by plasma concentrations of Vonafexor, since the peak levels were lower in the twice‐daily groups compared with the equivalent once‐daily groups and the AUCs were similar. In addition, no dose‐related effect was observed for pruritus. When analysing bile acid changes, we found no association with pruritus. C4, the pharmacological marker reflecting FXR target engagement and key bile acid precursor metabolite, had similar reduced plasma levels after twice‐daily compared with once‐daily dosing regimens. Increase of the BA absorption response marker FGF19 after dosing was also similar for the two dosing groups. As pruritus is not directly associated with specific serum BA thresholds, a possible hypothesis for the increased pruritus might be related to the BA circadian physiology. Pruritus may potentially decrease medication adherence; therefore, twice‐daily regimens of Vonafexor will not be tested further in future trials.
In four subjects, asymptomatic transient elevations of ALT grade 3/4 were seen. These flares were considered as possibly therapeutic transaminase flares. 28 , 29 Two of the subjects receiving Vonafexor in monotherapy experienced an initial transient elevation of HBV DNA followed by ALT elevation; however, the increase was followed by a decrease in both ALT and HBV DNA By contrast, two patients in the Peg‐IFNα2a combination group had a less recognizable pattern, where HBV DNA decrease occurred randomly. ALT elevation in Peg‐IFNα2a treatment for 48 weeks is associated with favourable treatment outcome. 30 Speculation on what this means for Peg‐IFNα2a treatment in combination with Vonafexor is not possible due to the limited number of patients and short treatment duration in this phase 1 study. No association between the observed ALT, HBV DNA changes and hepatotoxicity in relation to different Vonafexor doses or treatment combinations was seen. Importantly, no Hy's law criteria were met 31 and all patients did not show clinical or other laboratory abnormalities.
Although this trial was not powered to assess a clinically relevant decrease in viral markers, we observed a significant decrease of HBsAg levels in the highest monotherapy dose. Most treated patients were HBeAg‐negative, a status correlated with high levels of integrated HBV DNA that can also produce HBsAg. 32 As a consequence, the HBsAg decrease induced by Vonafexor could partially be masked by HBsAg production from the integrated genome. The observed HBsAg decline was small and occurred in a very short treatment period; also, the outcomes were of the same order of magnitude as HBsAg changes observed with other exploratory anti‐HBV agents. 33 Further HBsAg decline may be expected with a longer treatment duration. HBsAg levels at day 29 in patients treated with Vonafexor and Peg‐IFNα2a were not decreased compared with baseline but interestingly were lower compared with the placebo group. It is known that Peg‐IFNα2a can induce an HBsAg increase in the first few weeks of treatment, 34 in particular known to occur in CHB patients infected with HBV genotype D. Such an increase was observed in the placebo group but not in patients treated with Vonafexor +Peg‐IFNα2a. This difference may be attributed to an anti‐viral effect of Vonafexor on top of Peg‐IFNα2a.
A significant decrease in HBcrAg and pgRNA levels when combining Vonafexor with Peg‐IFNα2a was also observed. This observation suggests a direct impact of Vonafexor on viral transcription in the hepatocytes of CHB patients. However, because of the small sample size, the heterogeneity of patients participating in this trial and the relatively high limit of quantification of these viral markers, our results must be interpreted with caution. It is furthermore noteworthy that, unlike with Peg‐IFNα2a and entecavir, we did not observe a change in the viral load with Vonafexor monotherapy. A possible explanation is that the Vonafexor mechanism of action includes disruption of HBV pgRNA transcription not translating into relevant viral particle reduction because of the short treatment duration, while interferon's multifaceted mechanism of action includes a direct interaction with the HBV cccDNA more rapidly translating into viral load decrease. Accordingly, a longer duration of treatment with Vonafexor monotherapy may eventually reduce the viral load. Based on the pgRNA and HBcrAg reduction, which positively correlates with intrahepatic transcriptional activity of liver cccDNA, Vonafexor is to be further studied in combination with Peg‐IFNα2a over a longer duration to achieve clinically meaningful outcomes.
In conclusion, our clinical trial showed that Vonafexor is safe, both as monotherapy and when combined with Peg‐IFNα2a. Signs of a potential anti‐viral effect were seen in the highest dose of monotherapy and when combining Vonafexor with Peg‐IFNα2a. Additional studies with a longer treatment duration may provide further evidence for efficacy. Vonafexor is one of the few compounds which target non‐viral host factors and may therefore be of value in future multi‐therapy efforts aiming to achieve higher functional cure rates in CHB.
CONFLICT OF INTEREST
ER, NB, HG, PRP, RD, JV and PS are employees of Enyo Pharma. PA, CL and HWR have received consulting fees from Enyo Pharma. Financial support from the French Agence Nationale de la Recherche sur le sida et les Hépatites Virales (ANRS) to PA and CR. RE, NK, PT and RF declare no conflict of interest.
AUTHOR CONTRIBUTIONS
Guarantor of article: Henk Reesink. Specific author contributions: PA, CL, JV, PS, PT, RF and HWR conceived and designed the study. PA, ER, NB and PS supervised the study. RE, PA, ER, NB, PRP, RD, PT, RF and HWR contributed to the acquisition of data. RE, PA, NK, HG, CL, PRP, RD, JV, PS and HWR contributed to the analysis and interpretation of data. ER and NB provided administrative, technical or material support. HG performed the statistical analysis. RE, PA, NK, HG and RD drafted the manuscript. All authors contributed to the critical revision of the manuscript for important intellectual content and approved the final version. PS had access to all the data and vouches for the integrity of the data analyses.
Supporting information
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank the patients and their families, the investigators and site staff (Pr Stephen Riordan, Dr Wendy Cheng, Pr Kittyod Poovorawan, Pr Krzysztof Tomasiewicz, Dr Paweł Pabjan and Pr Włodzimierz Mazur), the Data Safety Monitoring Committee (Pr Peter LM Jansen, Dr Edmund Tse, Mr Patrick Lim and Ms Barbara Francis) and the clinical operations team (Gabriel Kremmidiotis, Sandrien Louwaars, Annemieke Hatzmann, Agniezska Olek, Małgorzata Łokociejewska, Khajirat Netnee and Chayanont Chaimongkol). Medical writing support was provided by Dr. Julie Harriague of 4clinics and Dr. Artin Karapet of LyonaPharm and funded by Enyo Pharma.
Erken R, Andre P, Roy E, et al. Farnesoid X receptor agonist for the treatment of chronic hepatitis B: A safety study. J Viral Hepat. 2021;28:1690–1698. 10.1111/jvh.13608
Funding information
The trial described in this manuscript was funded by Enyo Pharma
REFERENCES
- 1. WHO . Global hepatitis report World Health Organisation; 2017.
- 2. Razavi‐Shearer D, Gamkrelidze I, Nguyen MH, et al. Global prevalence, treatment, and prevention of hepatitis B virus infection in 2016: a modelling study. Lancet Gastroenterol Hepatol. 2018;3(6):383‐403. [DOI] [PubMed] [Google Scholar]
- 3. European Association for the Study of the Liver 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J Hepatol. 2017;67(2):370‐398. [DOI] [PubMed] [Google Scholar]
- 4. Allweiss L, Dandri M. The role of cccDNA in HBV maintenance. Viruses. 2017;9(6):156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Höner zu Siederdissen C, Maasoumy B, Cornberg M. What is new on HBsAg and other diagnostic markers in HBV infection? Best Pract Res Clin Gastroenterol. 2017;31(3):281‐289. [DOI] [PubMed] [Google Scholar]
- 6. Kim I, Ahn SH, Inagaki T, et al. Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J Lipid Res. 2007;48(12):2664‐2672. [DOI] [PubMed] [Google Scholar]
- 7. Inagaki T, Choi M, Moschetta A, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2(4):217‐225. [DOI] [PubMed] [Google Scholar]
- 8. Oehler N, Volz T, Bhadra OD, et al. Binding of hepatitis B virus to its cellular receptor alters the expression profile of genes of bile acid metabolism. Hepatology. 2014;60(5):1483‐1493. [DOI] [PubMed] [Google Scholar]
- 9. Li L, Oropeza CE, Kaestner KH, McLachlan A. Limited effects of fasting on hepatitis B virus (HBV) biosynthesis in HBV transgenic mice. J Virol. 2009;83(4):1682‐1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Curtil C, Enache LS, Radreau P, et al. The metabolic sensors FXRalpha, PGC‐1alpha, and SIRT1 cooperatively regulate hepatitis B virus transcription. FASEB J. 2014;28(3):1454‐1463. [DOI] [PubMed] [Google Scholar]
- 11. Mouzannar K, Fusil F, Lacombe B, et al. Farnesoid X receptor‐α is a proviral host factor for hepatitis B virus that is inhibited by ligands in vitro and in vivo. FASEB J. 2018;33(2):2472‐2483. [DOI] [PubMed] [Google Scholar]
- 12. Radreau P, Porcherot M, Ramiere C, Mouzannar K, Lotteau V, Andre P. Reciprocal regulation of farnesoid X receptor alpha activity and hepatitis B virus replication in differentiated HepaRG cells and primary human hepatocytes. FASEB J. 2016;30(9):3146‐3154. [DOI] [PubMed] [Google Scholar]
- 13. Ramiere C, Scholtes C, Diaz O, et al. Transactivation of the hepatitis B virus core promoter by the nuclear receptor FXRalpha. J Virol. 2008;82(21):10832‐10840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Reese VC, Moore DD, McLachlan A. Limited effects of bile acids and small heterodimer partner on hepatitis B virus biosynthesis in vivo. J Virol. 2012;86(5):2760‐2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Niu Y, Xu M, Slagle BL, et al. Farnesoid X receptor ablation sensitizes mice to hepatitis b virus X protein‐induced hepatocarcinogenesis. Hepatology. 2017;65(3):893‐906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yan H, Peng B, Liu Y, et al. Viral entry of hepatitis B and D viruses and bile salts transportation share common molecular determinants on sodium taurocholate cotransporting polypeptide. J Virol. 2014;88(6):3273‐3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev. 2009;89(1):147‐191. [DOI] [PubMed] [Google Scholar]
- 18. Reese VC, Oropeza CE, McLachlan A. Independent activation of hepatitis B virus biosynthesis by retinoids, peroxisome proliferators, and bile acids. J Virol. 2013;87(2):991‐997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Keasler VV, Hodgson AJ, Madden CR, Slagle BL. Enhancement of hepatitis B virus replication by the regulatory X protein in vitro and in vivo. J Virol. 2007;81(6):2656‐2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Leupin O, Bontron S, Schaeffer C, Strubin M. Hepatitis B virus X protein stimulates viral genome replication via a DDB1‐dependent pathway distinct from that leading to cell death. J Virol. 2005;79(7):4238‐4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Decorsiere A, Mueller H, van Breugel PC, et al. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature. 2016;531(7594):386‐389. [DOI] [PubMed] [Google Scholar]
- 22. van de Klundert M, van den Biggelaar M, Kootstra N, Zaaijer H. Hepatitis B virus protein X induces degradation of Talin‐1. Viruses. 2016;8(10):281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Darteil R, Joly S, Radreau P, et al. In vitro characterization of Eyp001 a novel, potent and selective FXR agonist entering phase 2 clinical trials in chronic hepatitis B. Hepatology. 2019;70(S1). Abstract 87. [Google Scholar]
- 24. Núñez M. Clinical syndromes and consequences of antiretroviral‐related hepatotoxicity. Hepatology. 2010;52(3):1143‐1155. [DOI] [PubMed] [Google Scholar]
- 25. Neuschwander‐Tetri BA, Loomba R, Sanyal AJ, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non‐cirrhotic, non‐alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo‐controlled trial. Lancet (London, England). 2015;385(9972):956‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Harrison SA, Bashir MR, Lee KJ, et al. A structurally optimized FXR agonist, MET409, reduced liver fat content over 12 weeks in patients with non‐alcoholic steatohepatitis. J Hepatol. 2021;75(1):25‐33. [DOI] [PubMed] [Google Scholar]
- 27. Patel K, Harrison SA, Elkhashab M, et al. Cilofexor, a nonsteroidal FXR agonist, in patients with noncirrhotic NASH: A phase 2 randomized controlled trial. Hepatology. 2020;72(1):58‐71. [DOI] [PubMed] [Google Scholar]
- 28. Chang ML, Liaw YF. Hepatitis B flares in chronic hepatitis B: pathogenesis, natural course, and management. J Hepatol. 2014;61(6):1407‐1417. [DOI] [PubMed] [Google Scholar]
- 29. Fontana RJ, Avigan MI, Janssen HLA, et al. Liver safety assessment in clinical trials of new agents for chronic hepatitis B. J Viral Hepat. 2020;27(2):96‐109. [DOI] [PubMed] [Google Scholar]
- 30. Ghany MG, Feld JJ, Chang K‐M, et al. Serum alanine aminotransferase flares in chronic hepatitis B infection: the good and the bad. Lancet Gastroenterol Hepatol. 2020;5(4):406‐417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Robles‐Diaz M, Lucena MI, Kaplowitz N, et al. Use of Hy's law and a new composite algorithm to predict acute liver failure in patients with drug‐induced liver injury. Gastroenterology. 2014;147(1):109‐118.e5. [DOI] [PubMed] [Google Scholar]
- 32. Wooddell CI, Yuen MF, Chan HL, et al. RNAi‐based treatment of chronically infected patients and chimpanzees reveals that integrated hepatitis B virus DNA is a source of HBsAg. Sci Transl Med. 2017;9(409):eaan0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yuen M‐F, Agarwal K, Gane EJ, et al. Safety, pharmacokinetics, and antiviral effects of ABI‐H0731, a hepatitis B virus core inhibitor: a randomised, placebo‐controlled phase 1 trial. Lancet Gastroenterol Hepatol. 2020;5(2):152‐166. [DOI] [PubMed] [Google Scholar]
- 34. Wang K, Huang G, Chen Y, Wang Y. Hepatitis B surface antigen (HBsAg) kinetics in chronic hepatitis B patients during peginterferon treatment. Med Sci Monit. 2020;26:e921487‐1‐e921487‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material