

Abstract
Background
Genetics play a significant role in coagulation phenotype and venous thromboembolism risk. Resistance to the anticoagulant activated protein C (APC) is an established risk for thrombosis. Herein, we explored the genetic determinants of thrombin generation (TG) and thrombomodulin (TM)‐modulated TG using plasma from the Human Functional Genomics Project.
Methods
Calibrated TG was measured both in absence and presence of TM using tissue factor as trigger. Genetic determinants of TG parameters and protein C pathway function were assessed using genome‐wide single‐nucleotide polymorphism (SNP) genotyping. Plasma samples were supplemented with purified apolipoprotein A‐IV, prekallikrein, or kallikrein to test their influence on the anticoagulant function of TM and APC in TG.
Results
Thrombin generation data from 392 individuals were analyzed. Genotyping showed that the KLKB1 gene (top SNP: rs4241819) on chromosome 4 was associated with the normalized sensitivity ratio of endogenous thrombin potential to TM at genome‐wide level (nETP‐TMsr, P = 4.27 × 10−8). In vitro supplementation of kallikrein, but not prekallikrein or apolipoprotein A‐IV, into plasma dose‐dependently augmented the anticoagulant effect of TM and APC in TG. Variations of rs4241819 was not associated with the plasma concentration of prekallikrein. Association between rs4241819 and nETP‐TMsr was absent when TG was measured in presence of a contact pathway inhibitor corn trypsin inhibitor.
Conclusions
Our results suggest that kallikrein plays a role in the regulation of the anticoagulant protein C pathway in TG, which may provide a novel mechanism for the previously observed association between the KLKB1 gene and venous thrombosis.
Keywords: genetic association, kallikrein, protein C, thrombin, thrombosis
Essentials.
Function of the anticoagulant protein C pathway is an important determinant of thrombosis risk.
We studied the genetic variations associated with thrombin generation (TG) +/– thrombomodulin.
The KLKB1 gene was significantly associated with the sensitivity of TG to thrombomodulin.
Kallikrein supplementation augmented the anticoagulant effect of thrombomodulin in TG.
1. INTRODUCTION
The coagulation system, a complex machinery consisting of a serial of pro‐ and anti‐coagulant (pro)enzymes and cofactors, is responsible for the prevention of excessive blood loss and the formation of unnecessary thrombi. Kallikrein (PKa) is a component of the contact activation system, together with factor XII (FXII) and high molecular weight kininogen (HK). Prekallikrein (PK), encoded by the KLKB1 gene, is the precursor of the trypsin‐like plasma protease PKa and circulates in plasma in complex with HK at a concentration of approximately 580 nM. 1 PK is activated to PKa by FXIIa, which then results in the reciprocal activation of both enzymes and subsequently leads to the activation of factor XI (FXI) and downstream thrombin generation (TG). 2 Furthermore, PKa has also been recently found to directly activate factor IX (FIX). 3 , 4 , 5 Previous genetic association studies showed that the KLKB1 gene was associated with venous thrombosis, 6 , 7 but the mechanism is unknown.
The protein C system is an important anticoagulant pathway. 8 During coagulation, thrombin binds to the endothelial receptor thrombomodulin (TM) and activates protein C to its active form (APC). Together with its cofactor protein S, APC proteolytically inactivates factor V (FVa) and FVIIIa, two important cofactors in the tenase and prothrombinase complexes, respectively. Cleavages at Arg306, Arg506, and Arg679 in FVa, as well as Arg336 and Arg562 in FVIIIa result in the loss of activity of these cofactors and consequently reduced thrombin formation. 9 , 10 , 11 Dysfunction of the protein C pathway is a common cause of thrombophilia. 12 For example, the FV Arg506Gln mutation (FVLeiden) causes a resistance to APC cleavage; 13 as a consequence, the risk of venous thrombosis is 7‐fold increased in heterozygous carriers of this mutation and 80‐fold in homozygous carriers. 14 Similarly, deficiencies of protein S and protein C have been related to increased thrombosis risk. 15 , 16
The capacity of a plasma to form thrombin, as reflected by the endogenous thrombin potential (ETP) in calibrated thrombin generation tests, 17 is a crucial determinant of blood coagulability and was shown to be predictive of bleeding or thrombosis. 18 , 19 , 20 Modification of TG assays with exogenous APC or TM makes them sensitive to dysfunctions of the protein C pathway, including protein S/C deficiency, FVLeiden, and prothrombin G20210A. 21 , 22
Previous studies demonstrated that TG profiles could serve as an intermediate phenotype to discover genetic variants related to venous thromboembolism (VTE). 23 , 24 , 25 , 26 Segers et al. showed that several established thrombophilic mutations including FVLeiden and prothrombin G20210A were related to enhanced TG or APC‐modified TG in a group of healthy donors, 25 which was also confirmed in 188 FVLeiden heterozygote carriers. 24 Importantly, using this approach, Rocanin‐Arjo et al. discovered in the MARTHA study and two replication studies that the ORM1 gene is a locus associated with the lag time of TG, and further functional studies confirmed that increased orosomucoid concentrations impaired TG. 26
In this study, we sought to use TM‐modified TG profiles to discover novel genetic determinants of TG and the protein C pathway function.
2. MATERIALS AND METHODS
2.1. Study population
The study population is part of the previously described 500 Human Functional Genomics Project (500FG). 27 , 28 This study was approved by the local ethical committee (NL42561.091.12, 2012/550) and was conducted according to the Declaration of Helsinki and in accordance with the Dutch Medical Research involving Human Subjects Act. Between August 2013 and December 2014 healthy adult individuals of Western‐European origin were recruited at Radboud University Medical Centre, Nijmegen, the Netherlands. Exclusion criteria were pregnancy, breastfeeding, the use of any medication in the past month, and acute or chronic diseases. All participants gave written informed consent before blood was drawn into 3.2% sodium citrate Vacutainer tubes (Becton Dickinson). Platelet poor plasma (PPP) was prepared by centrifugation (2000 g for 10 min) within 2 h post blood draw and stored at −80°C.
2.2. PPP‐TG measurement
A modified Calibrated Automated Thrombography (MidiCAT) assay 29 was employed for TG measurement using 5pM recombinant tissue factor (Innovin®), 16.7 mM CaCl2, 416.7 µM ZGGR‐AMC (Bachem), and 4 µM synthetic phospholipid vesicles (prepared as described 17 by mixing 20 mol% phosphatidylserine, 20 mol% phosphatidylethanolamine, and 60 mol% phosphatidylcholine; the lipids were all from Avanti Polar Lipids). TG was tested both in the absence and presence of 7 nM recombinant TM (prepared by U‐protein Express). This TM concentration inhibits the ETP of a normal pooled plasma (NPP) by 50%. In each run the 500FG samples were tested beside an NPP, which was prepared by mixing the plasma of 116 healthy adult volunteers. In a subset of the 500FG sample, 1.45 µM (final concentration) corn trypsin inhibitor (CTI, from Haematologic Technologies) was also added in TG trigger solutions, both with and without TM, to prevent activation of the contact system during reaction. 30
Thrombin generation parameters in presence of TM are denoted as lagtimeTM+, time to peak (TTP TM+), peakTM+, and ETPTM+, respectively. The peak and ETP, both in absence and presence of TM, were normalized as percentage of that of NPP tested without TM.
A normalized thrombomodulin sensitivity ratio of ETP (nETP‐TMsr) was determined by dividing ETP values in presence and absence of TM, then normalized against the same ratio determined in NPP (Equation 1). nPeak‐TMsr was calculated similarly. A n‐TMsr >1 means an increased resistance to TM compared to NPP. A higher nETP‐TMsr value indicates a higher residual ETP in the presence of TM, therefore a weaker anticoagulant effect of TM.
| (1) |
2.3. Functional validation
Purified human PK, PKa (both from Enzyme Research Laboratories), or apolipoprotein A‐IV (APOA4; from Sigma‐Aldrich) were titrated into NPP to designated concentrations before TG was initiated with tissue factor (TF; 2, 5, or 15 pM), in the presence or absence of TM (1.5, 7, or 24 nM for different TF concentrations, respectively) or APC (0.8, 2.5, or 6 nM, respectively; prepared as described previously 17 ).
2.4. PK concentration measurement
Prekallikrein concentration was measured in part of the 500FG samples using enzyme‐linked immunosorbent assay (ELISA) kit (#ab202405 from Abcam) following manufacturer’s instructions. In brief, 50 µL samples (pre‐diluted 6400 times) or PK standard solutions were added into pre‐coated 96‐well plates, followed by the addition of 50 µL mixture of primary and secondary (horseradish peroxidase [HRP] conjugated) detection antibodies. After 1 h incubation on a plate shaker, the plate was washed three times and 3,3'5,5'‐tetramethylbenzidine (TMB) was added to allow color development. Absorbance at 450 nm was recorded on an ELx808 Absorbance Microplate Reader (Biotek) after adding a stopping solution. PK concentrations in the samples were calculated after comparing with the standard curve.
2.5. Genotyping
The genotype data of 500FG cohort were generated previously 28 using the single‐nucleotide polymorphism (SNP) chip Illumina HumanOmniExpressExome‐8 and was imputed to obtain approximately 7 million SNPs. The strands and variant identifiers were aligned to the reference Genome of the Netherlands (GoNL) dataset and other public datasets 31 , 32 using Genotype Harmonizer v1. 33 Data were phased and imputed by SHAPEIT2 v2.r64424 34 and IMPUTE225, 35 respectively.
2.6. Statistics
Statistical analyses were performed with R and GraphPad Prism. Comparisons of the TG parameters between females and males were performed with the Mann‐Whitney U test. A two‐sided P < .05 was considered statistical significance.
Inverse ranked based transformations were performed to normalize distribution in each TG trait before quantitative trait loci mapping were performed using an additive linear regression model with age and sex as covariates. Systematic inflation or deflation in test statistics over all loci was assessed through the quantile‐quantile (Q‐Q) plot (Figure S1 in supporting information) for TG parameters. A P‐value <5 × 10−8 was considered genome‐wide significance and a P‐value <5 × 10−6 was defined as suggestive significance. Of note, only SNPs with allele frequency >5% were analyzed for associations.
3. RESULTS
In total, 392 participants had both demographic data and TG parameters available, as shown in Figure 1A. The participants were of Western‐European origin and had a median age of 24. Among the participants, 194 (49.5%) were female. Females and males were comparable in age (median and interquartile range [IQR]: 23 [21–27] vs. 24 [21–27], P = .376) but the body mass index (BMI) was lower in the females (21.7 [20.5–23.7] vs. 23 [21.6–24.6], P < .001).
FIGURE 1.
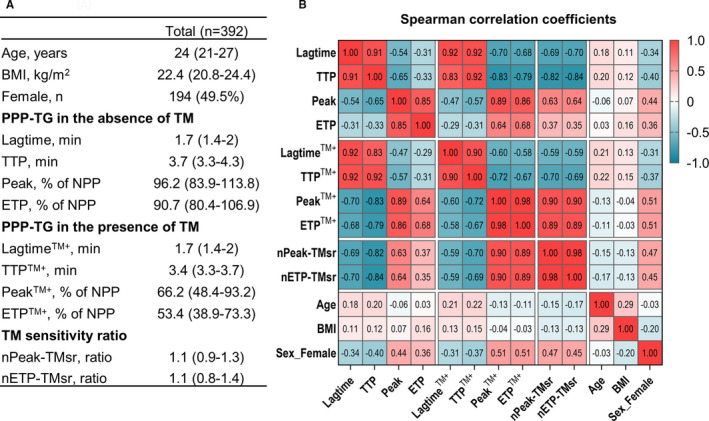
Demographic and thrombin generation parameters of the cohort. A, Demographic and TG parameters of the cohort. Data are presented as number (percentage) or median (interquartile range). TG parameters, both in the absence and presence of TM, were normalized as the percentage of the TG parameters of NPP tested without TM in the same run. B, Spearman correlation coefficients between the TG parameters, markers of protein C pathway function (nPeak‐TMsr and nETP‐TMsr), and demographics. BMI, body mass index; ETP, endogenous thrombin potential; nETP‐TMsr, normalized sensitivity ratio of ETP to thrombomodulin; nPeak‐TMsr, normalized sensitivity ratio of peak to thrombomodulin; NPP, normal pooled plasma; TG, thrombin generation; TM, thrombomodulin; TTP, time to peak
Correlation coefficients among demographics, TG parameters, and markers of the protein C pathway function (i.e., nETP‐TMsr and nPeak‐TMsr) are shown in Figure 1B. The lagtimeTM+ and TTPTM+ correlated strongly with their counterparts in absence of TM (i.e., lagtime and TTP; Spearman r > 0.83). The PeakTM+ and ETPTM+ correlated moderately to strongly with their counterparts in absence of TM (r > 0.64), and strongly with the normalized sensitivity ratio of peak and ETP (r > 0.89).
Compared to males, females had stronger TG, as reflected by their shorter lagtime and TTP, higher peak and ETP; the TG of females were also more resistant to TM, as shown by the higher nETP‐TMsr and nPeak‐TMsr values (P < .001 for all comparisons, Figure S2 in supporting information). These differences between males and females were partially explained by the hormonal oral contraceptive use in females (n = 74 [38.7% of the females]), which cause stronger TG and a reduced response to the anticoagulant effect of TM, in line with previous reports. 22 , 36
3.1. Genome‐wide significant loci for TG and the anticoagulant effect of TM
We explored SNPs that are associated with the TG parameters after adjusting for age and sex. A total of 327 samples were available for genetic analyses. As shown Table 1, Figure 2, and Figure S3 in supporting information, a locus on chromosome 4 at the KLKB1 gene (top SNP: rs4241819) was significantly associated with both the nETP‐TMsr and nPeak‐TMsr (P < 5 × 10−8 for both). The rs4241819‐T allele (minor allele frequency 0.48) was associated with a higher nETP‐TMsr value (β = 0.36), that is, a TM‐resistant prothrombotic tendency. Interestingly, the KLKB1 gene has been found to be associated with venous thrombosis in previous studies. 6 , 7
TABLE 1.
Genome‐wide significant (P‐value <5 × 10−8) loci for thrombin generation traits
| Trait | Top SNP | Gene | Chr | Position (bp) | Effect/alternative allele | MAF | Beta | P‐value |
|---|---|---|---|---|---|---|---|---|
| nETP‐TMsr | rs4241819 | KLKB1 a , b , c | 4 | 186235986 | T/C | 0.485 | 0.36 | 4.27 × 10−8 |
| nPeak‐TMsr | rs4241819 | KLKB1 a , b , c | 4 | 186235986 | T/C | 0.484 | 0.37 | 1.97 × 10−8 |
| ETPTM+ | rs404479 | RP11‐315I14.2 a | 9 | 23479588 | T/G | 0.433 | 0.46 | 4.03 × 10−8 |
Abbreviations: Chr, chromosome; ETPTM+, endogenous thrombin potential in presence of thrombomodulin; MAF, minor allele frequency; nETP‐TMsr, normalized sensitivity ratio of endogenous thrombin potential to thrombomodulin; SNP, single‐nucleotide polymorphism.
Genes close to thrombin generation associated SNPs.
Missense in linkage disequilibrium variants (rs3733402).
FIGURE 2.
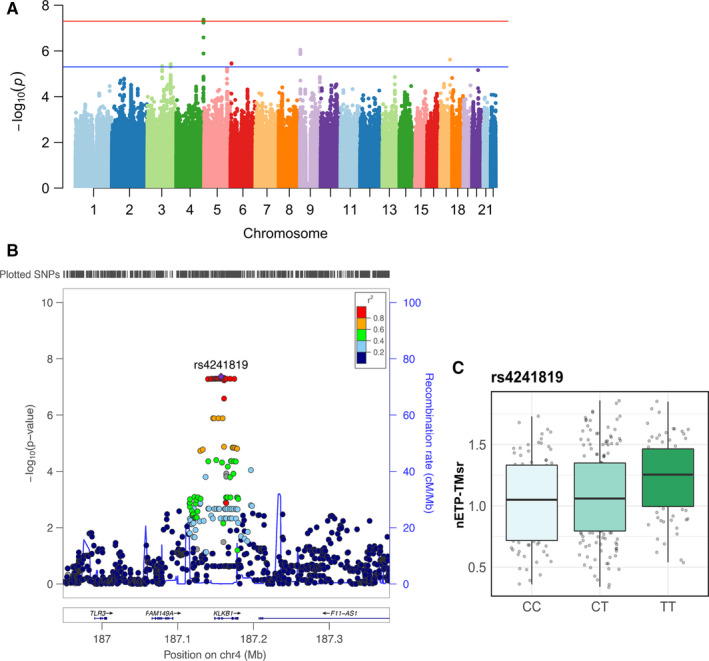
Genome‐wide significant loci associated with the normalized sensitivity ratio of endogenous thrombin potential to thrombomodulin (nETP‐TMsr). A, Manhattan plot of single nucleotide polymorphisms (SNPs) associated with nETP‐TMsr. The red line indicates the threshold for genome‐wide significance (5 × 10−8). B, Regional association plot of genome‐wide significant SNPs at chromosome 4. C, Genotype‐stratified nETP‐TMsr
In addition, a locus at chromosome 9:23479590 (top SNP: rs404479) was significantly associated with ETPTM+ (P = 4.03 × 10−8; Table 1 and Figure 3).
FIGURE 3.
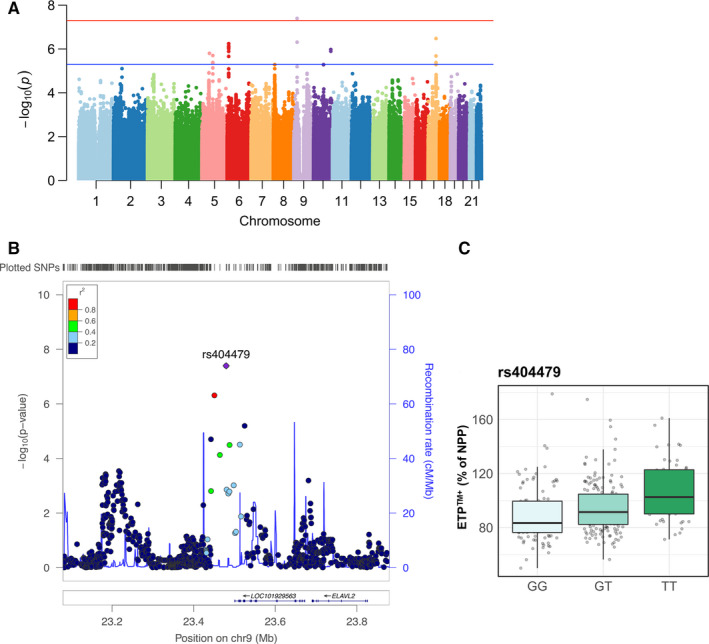
Genome‐wide significant loci associated with endogenous thrombin potential (ETP) in presence of thrombomodulin (TM; ETPTM+). A, Manhattan plot of single nucleotide polymorphisms (SNPs) associated with ETPTM+. The red line indicates the threshold for genome‐wide significance (5 × 10−8). B, Regional association plot of genome‐wide significant SNPs at chromosome 9. C, Genotype‐stratified ETPTM+. NPP, normal pooled plasma
Furthermore, eight suggestive loci were identified for TG parameters and protein C pathway function (Table S1 in supporting information). For example, the ETP was associated positively with rs1767776‐C on chromosome 6 and rs1985749‐A on chromosome 17, whereas ETPTM+ was negatively associated with rs610551‐A on chromosome 9 (P < 6*10−7 for all correlations). However, none of these eight loci have previously been found to influence plasmatic coagulation.
We also explored the association between TG parameters and some previously known TG‐related genes. A list of candidate genes was extracted from literature, 23 , 24 , 25 , 26 , 37 , 38 , 39 in particular those that encoded coagulation factors, including F5, HAAT, TFPI, PROC, PROS1, FGA, F12, ORM1, F2, MYBPC3, PTPRJ, F10, THBD, PROCR, and SERPIN C1 genes. All variants located within 150kb of either end of the full‐length transcript were examined to determine the strongest SNP association against each TG and protein C pathway function traits. Forty SNPs associated with at least one TG parameter with P‐values <.01 are shown in Figure S4 in supporting information. Of note, we only analyzed SNPs with allele frequency >5%, whereas the frequency of FVLeiden in the Dutch population is approximately 3%; therefore, it was not included in the genetic association analyses.
3.2. The anticoagulant effects of TM or APC were hardly affected by APOA4 supplementation
Single‐nucleotide polymorphism rs4241819 was previously shown to influence the concentration of several plasma proteins including apolipoprotein A‐IV (APOA4), 40 , 41 a protein that was shown to influence arterial thrombosis risk 42 , 43 and the rs4241919‐C allele was related to an increased APOA4 concentration. 40 APOA4 circulates in blood at a concentration of approximately 150 µg/ml, in both a free form (75% of all) and another form that binds to high density lipoprotein particles. 44 However, it was shown that the SNP rs4241819 only accounts for a very small fraction (0.19% to 0.44%) of the variations of APOA4 concentration (β = 1.47 ± 0.7 µg/ml for the C allele). 40
We tested the effect of APOA4 supplementation in a range between 0 to 40 µg/ml on the inhibitory effect of TM and APC in TG. As shown in Figure S5 in supporting information, the inhibition rate of ETP by TM and APC remained largely unchanged in response to different APOA4 concentrations.
In addition, the rs42418189‐T allele was also associated with decreased circulating concentrations of endothelin‐2, protachykinin‐1, and acidic leucine‐rich nuclear phosphoprotein 32 family member B, as well as increased interleukin‐2 concentration. 41 There are, however, no data on the relation of these proteins to the protein C pathway.
3.3. Kallikrein but not prekallikrein augmented the anticoagulant effect of TM and APC
The KLKB1 gene encodes plasma kallikrein of the contact activation system. We first tested if concentrations of PK, the precursor of PKa, could affect TM‐ and APC‐ modified TG. As shown in Figure S6 in supporting information, the supplementation of human PK at doses from 0 to 580 nM into NPP hardly affected the inhibitory effect of TM and APC on ETP. The concentration of PK was also measured in part of the cohort (n = 71, 35 males and 36 females). PK concentrations were not significantly different in individuals with rs4241819‐CC, ‐CT, or ‐TT alleles (analysis of variance [ANOVA] P = .29; Figure S7 in supporting information).
We next explored if PKa supplementation could influence the anticoagulant effect of TM or APC in PPP‐TG initiated by 5 pM TF. As shown in Figure 4A, the addition of PKa dose‐dependently enhanced the inhibition rate of ETP by TM and APC from 54.1% (no PKa addition) to 69.9% (when PKa was supplemented at 120 nM; P < .001). Meanwhile the ETP in absence of TM was not influenced by PKa addition (P > .05), indicating that kallikrein did not significantly promote TG when 5 pM TF was used. The inhibitory effect of APC on TG was similarly augmented by PKa addition.
FIGURE 4.
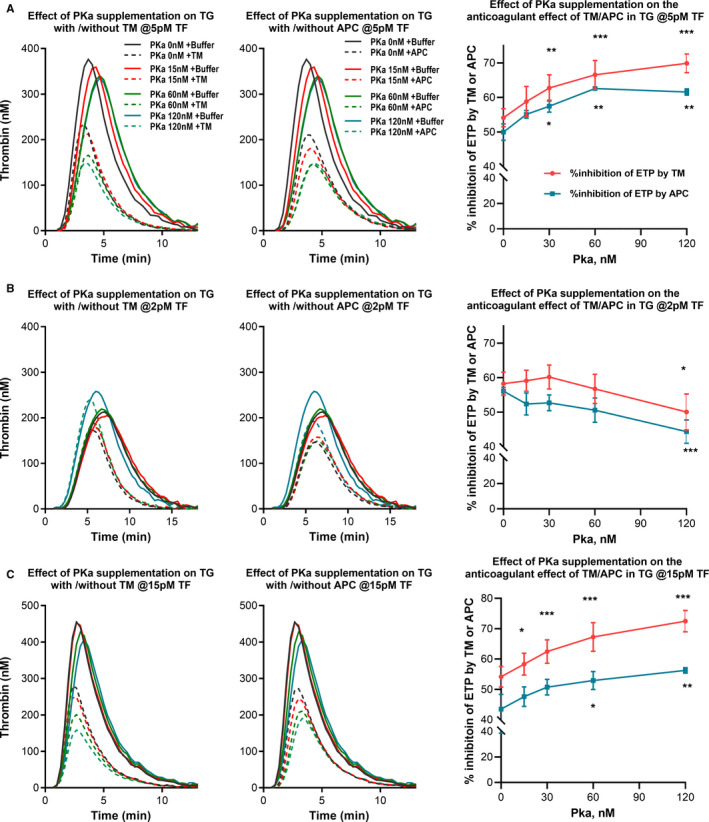
Effect of kallikrein (PKa) supplementation on the anticoagulant effect of thrombomodulin (TM) or activated protein C (APC) on thrombin generation (TG). TG was initiated with 5 pM (A), 2pM (B), or 15pM (C) tissue factor (TF), in the absence or presence of TM (7, 1.5, or 24 nM for the different TF concentrations, respectively) or APC (2.5, 0.8, or 6 nM, respectively) in normal pooled plasma (NPP) supplemented with varying doses of human PKa (0, 7.5, 15, 30, 60, or 120 nM). Representative TG curves are shown in the left and middle columns. The %inhibition of endogenous thrombin potential (ETP) by TM or APC, as an indication of their inhibitory effects, are shown in the right column. Paired repeated measures one‐way analysis of variance and Dunnett’s multiple comparison test were used to compare conditions with and without PKa addition. *P < .05; **P < .01; ***P < .001
We also tested the effect of PKa addition in TG reactions triggered by 2 and 15pM TF. For TG triggered by 2 pM TF, the ETP was increased from 1322 nM*min to 1472 nM*min when 120 nM PKa was added. This ETP‐enhancing effect of PKa at low TF is expected because PKa can activate contact pathway factors FXII and FIX and enhance downstream TG. A stronger TG would also make it more difficult for a given concentration of TM or APC to deliver 50% inhibition on ETP. Indeed, the anticoagulant effect of TM or APC was not enhanced by supplemented PKa (Figure 4B). Contrarily, for TG triggered by 15 pM TF, the ETP inhibition by TM/APC increased dose dependently with the increasing PKa concentration added, from 54.1% with no PKa supplementation to 72.5% at 120 nM PKa (Figure 4C).
The use of CTI could eliminate activation of the contact pathway during TG; 30 we therefore also tested TG (both with and without TM) in part of the cohort (n = 79, 41 males and 38 females). As shown in Figure S8 in supporting information, in the presence of CTI the TM sensitivity ratio was not significantly influenced by the variations of the SNP rs4241819 (ANOVA P > .06 for both nETP‐TMsr and nPeak‐TMsr).
4. DISCUSSION
In the present study, we found several genome‐wide significant loci for TG parameters and protein C pathway function in a population of healthy individuals of Western‐European origin. In particular, the KLKB1 gene that encodes plasma PK was associated with the anticoagulant function of TM in TG. We provided a possible explanation for the observed genetic associations by demonstrating that in vitro supplementation of PKa augmented the anticoagulant effect of TM and APC in TG.
In addition to our observation of a genome‐wide association between the KLKB1 gene (top SNP: rs4241819) and the protein C pathway function, several previous studies also found that this gene was associated with the risk of VTE. 6 , 7 , 45 Thus, our study provides a possible explanation that the KLKB1 gene might mediate thrombotic risk through its influence on protein C pathway function. Our validation experiments further showed that in vitro supplementation of PKa augments the anticoagulant effect of TM and APC by approximately 30% in TG reactions triggered by both 5 and 15pM TF. A possible mechanism might be that kallikrein binds to protein C inhibitor (PCI) 46 and alleviate APC from inhibition by PCI. This mechanism might also protect people with excessive kallikrein activity (e.g., due to C1‐inhibitor deficiency) from having excessive TG or clot formation.
The rs4241819‐T allele is likely linked to a qualitative mutation because (1) variation of this SNP was not associated with plasma concentrations of PK, and (2) PK supplementation hardly influences the inhibitory effect of TM/APC in TG. Interestingly, a recent study showed that a recombinant un‐activatable form of PK can still activate FXII, suggesting that PK also possesses proteolytic activity, although at a level 23,000‐fold lower than PKa. 47 It is possible that the rs4241819‐T allele results in a PK mutation that has a lower activation rate. Interestingly, when TG was tested in the presence of CTI to prevent activation of the contact pathway, the association between the rs4241819 variations and the anticoagulant effect of TM was lost. This observation, together with the stimulating effect of PKa on the anticoagulant effect APC, supports the idea that PKa mediates the association between the SNP rs4241819 and the anticoagulant effect of TM.
The SNP rs42418189 has also been shown to influence the plasma concentration of APOA4, 40 a lipoprotein that is involved in many physiological processes such as lipid absorption and metabolism, glucose homeostasis, anti‐atherosclerosis, platelet aggregation, and thrombosis. 43 It was shown that the SNP rs42418189 only accounts for a very small fraction (0.19% to 0.44%) of the variations of APOA4 concentration (β = 1.47 ± 0.7 µg/ml for the C allele). 40 Our results showed that APOA4 supplementation at 40 µg/ml had negligible influence on the function of TM and APC in TG; therefore, APOA4 does not explain the association between the above‐observed association.
We also found that the TG capacity in presence of TM (ETPTM+) was significantly associated with a locus at chromosome 9: 23479588 (top SNP rs404497), which is transcribed to a long intergenic non‐coding RNA. The effect of this variation on TG and coagulation requires further study.
NPP was used to normalize the peak and ETP of the study subjects in each run, which is a major strength of our study approach. This approach could reduce the influence of inter‐assay variations introduced by different measurement machines and different operators. 48
This study also has a few limitations that need to be delineated. First, this study has a relatively small sample size and is mainly comprised of healthy donors, which limited our ability to identify novel variants with low allele frequency. However, we were still able to show that the protein C pathway function was significantly associated with the KLKB1 gene, a known VTE‐related locus, implying that the PKa‐‐protein C axis might be a common moderator of this anticoagulant pathway in this population. This also reinforced that the TM‐modified TG assay could serve as a useful tool to discover novel mutations related to the protein C pathway function. Second, the volunteers included in this study are mainly of Western‐European background, so the applicability of these results in other ethnic origins is unknown. Third, although our functional experiments with PKa supplementation in TG assay provided a possible explanation for the association between KLKB1 gene variation and protein C pathway function, the exact mechanism remains unclear. It is likely that this variation leads to a qualitative mutation of PK, but further studies are required to assess the exact mechanism.
In conclusion, this study shows that kallikrein regulates the anticoagulant protein C pathway in TG, which may provide a novel mechanism for the previously observed association between the KLKB1 gene and thrombosis.
CONFLICT OF INTEREST
J. Wan, J. Konings, B. de Laat, and M. Roest are employed by Synapse Research Institute, which is a research branch of the Stago group that markets the Calibrated Automated Thrombography assay. M.G.N. is a scientific founder of Trained Therapeutix Discovery. Other authors have nothing to disclose.
AUTHOR CONTRIBUTIONS
M. Roest, Q. de Mast, M. G. Netea, L Joosten, A. V. Kumar, B. de Laat, and J. van der Ven conceptualized the study and provided essential resources. J. Wan, J. Konings, M. Roest, P. de Groot, and Q. de Mast did the measurements and/or analyzed data; N. Vadaq performed genetic analyses. J. Wan, N. Vadaq, and M. Roest wrote the manuscript with comments from all authors.
Supporting information
App S1
ACKNOWLEDGMENTS
The authors thank all participants of the study. J. Wan is sponsored by a non‐restricted scholarship from the China Scholarship Council. N. Vadaq is supported by Indonesia Endowment Fund for Education (LPDP) given by the Ministry of Finance of the Republic of Indonesia. M. G. Netea was supported by an ERC Advanced Grant (#833247) and Spinoza Grant of the Netherlands Organization for Scientific Research. V. Kumar was supported by Radboud UMC Hypatia tenure track grant.
Wan J, Vadaq N, Konings J, et al. Kallikrein augments the anticoagulant function of the protein C system in thrombin generation. J Thromb Haemost. 2021;20:48–57. doi: 10.1111/jth.15530
Manuscript handled by: Alan Mast
Final decision: Alan Mast, 13 September 2021
REFERENCES
- 1. Schmaier AH. The contact activation and kallikrein/kinin systems: pathophysiologic and physiologic activities. J Thromb Haemost. 2016;14:28‐39. doi: 10.1111/jth.13194 [DOI] [PubMed] [Google Scholar]
- 2. Visser M, Heitmeier S, ten Cate H, Spronk HMH. Role of factor XIa and plasma kallikrein in arterial and venous thrombosis. Thromb Haemost. 2020;120:883‐993. doi: 10.1055/s-0040-1710013 [DOI] [PubMed] [Google Scholar]
- 3. Kearney KJ, Butler J, Posada OM, et al. Kallikrein directly interacts with and activates factor IX, resulting in thrombin generation and fibrin formation independent of factor XI. Proc Natl Acad Sci USA. 2021;118:e2014810118. doi: 10.1073/pnas.2014810118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Visser M, van Oerle R, Ten Cate H, et al. Plasma kallikrein contributes to coagulation in the absence of factor XI by activating factor IX. Arterioscler Thromb Vasc Biol. 2020;40:103‐111. doi: 10.1161/ATVBAHA.119.313503 [DOI] [PubMed] [Google Scholar]
- 5. Noubouossie DF, Henderson MW, Mooberry M, et al. Red blood cell microvesicles activate the contact system, leading to factor IX activation via 2 independent pathways. Blood. 2020;135:755‐765. doi: 10.1182/blood.2019001643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Austin H, De Staercke C, Lally C, Bezemer ID, Rosendaal FR, Hooper WC. New gene variants associated with venous thrombosis: a replication study in White and Black Americans. J Thromb Haemost. 2011;9:489‐495. doi: 10.1111/j.1538-7836.2011.04185.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bezemer ID, Bare LA, Doggen CJ, et al. Gene variants associated with deep vein thrombosis. JAMA. 2008;299:1306‐1314. doi: 10.1001/jama.299.11.1306 [DOI] [PubMed] [Google Scholar]
- 8. Dahlbäck B, Villoutreix BO. The anticoagulant protein C pathway. FEBS Lett. 2005;579:3310‐3316. doi: 10.1016/j.febslet.2005.03.001 [DOI] [PubMed] [Google Scholar]
- 9. Fulcher CA, Gardiner JE, Griffin JH, Zimmerman TS. Proteolytic inactivation of human factor VIII procoagulant protein by activated human protein C and its analogy with factor V. Blood. 1984;63:486‐489. [PubMed] [Google Scholar]
- 10. Fay PJ, Smudzin T, Walker F. Activated protein C‐catalyzed inactivation of human factor VIII and factor VIIIa. Identification of cleavage sites and correlation of proteolysis with cofactor activity. J Biol Chem. 1991;266:20139‐20145. [PubMed] [Google Scholar]
- 11. Kalafatis M, Rand MD, Mann KG. The mechanism of inactivation of human factor V and human factor Va by activated protein C. J Biol Chem. 1994;269:31869‐31880. [PubMed] [Google Scholar]
- 12. Castoldi E, Rosing J. APC resistance: biological basis and acquired influences. J Thromb Haemost. 2010;8:445‐453. doi: 10.1111/j.1538-7836.2009.03711.x [DOI] [PubMed] [Google Scholar]
- 13. Zöller B, Dahlbäck B. Linkage between inherited resistance to activated protein C and factor V gene mutation in venous thrombosis. Lancet. 1994;343:1536‐1538. doi: 10.1016/s0140-6736(94)92940-8 [DOI] [PubMed] [Google Scholar]
- 14. Rosendaal FR, Koster T, Vandenbroucke JP, Reitsma PH. High risk of thrombosis in patients homozygous for factor V Leiden (activated protein C resistance). Blood. 1995;85:1504‐1508. [PubMed] [Google Scholar]
- 15. Lijfering WM, Mulder R, ten Kate MK, Veeger NJ, Mulder AB, van der Meer J. Clinical relevance of decreased free protein S levels: results from a retrospective family cohort study involving 1143 relatives. Blood. 2009;113:1225‐1230. doi: 10.1182/blood-2008-08-174128 [DOI] [PubMed] [Google Scholar]
- 16. Allaart CF, Poort SR, Rosendaal FR, Reitsma PH, Bertina RM, Briët E. Increased risk of venous thrombosis in carriers of hereditary protein C deficiency defect. Lancet. 1993;341:134‐138. doi: 10.1016/0140-6736(93)90003-y [DOI] [PubMed] [Google Scholar]
- 17. Hemker HC, Giesen P, Al Dieri R, et al. Calibrated automated thrombin generation measurement in clotting plasma. Pathophysiol Haemost Thromb. 2003;33:4‐15. doi: 10.1159/000071636 [DOI] [PubMed] [Google Scholar]
- 18. Bosch Y, Al Dieri R, ten Cate H, et al. Preoperative thrombin generation is predictive for the risk of blood loss after cardiac surgery: a research article. J Cardiothorac Surg. 2013;8:154. doi: 10.1186/1749-8090-8-154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hron G, Kollars M, Binder BR, Eichinger S, Kyrle PA. Identification of patients at low risk for recurrent venous thromboembolism by measuring thrombin generation. JAMA. 2006;296:397‐402. doi: 10.1001/jama.296.4.397 [DOI] [PubMed] [Google Scholar]
- 20. Lutsey PL, Folsom AR, Heckbert SR, Cushman M. Peak thrombin generation and subsequent venous thromboembolism: the longitudinal investigation of thromboembolism etiology (LITE) study. J Thromb Haemost. 2009;7:1639‐1648. doi: 10.1111/j.1538-7836.2009.03561.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Curvers J, Thomassen MC, Rimmer J, et al. Effects of hereditary and acquired risk factors of venous thrombosis on a thrombin generation‐based APC resistance test. Thromb Haemost. 2002;88:5‐11. [PubMed] [Google Scholar]
- 22. Dargaud Y, Trzeciak MC, Bordet JC, Ninet J, Negrier C. Use of calibrated automated thrombinography +/‐ thrombomodulin to recognise the prothrombotic phenotype. Thromb Haemost. 2006;96:562‐567. [PubMed] [Google Scholar]
- 23. Martin‐Fernandez L, Ziyatdinov A, Carrasco M, et al. Genetic determinants of thrombin generation and their relation to venous thrombosis: results from the GAIT‐2 project. PLoS One. 2016;11:e0146922. doi: 10.1371/journal.pone.0146922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Segers O, Simioni P, Tormene D, Castoldi E. Influence of single nucleotide polymorphisms on thrombin generation in factor V Leiden heterozygotes. Thromb Haemost. 2014;111:438‐446. doi: 10.1160/th13-05-0360 [DOI] [PubMed] [Google Scholar]
- 25. Segers O, van Oerle R, ten Cate H, Rosing J, Castoldi E. Thrombin generation as an intermediate phenotype for venous thrombosis. Thromb Haemost. 2010;103:114‐122. doi: 10.1160/th09-06-0356 [DOI] [PubMed] [Google Scholar]
- 26. Rocanin‐Arjo A, Cohen W, Carcaillon L, et al. A meta‐analysis of genome‐wide association studies identifies ORM1 as a novel gene controlling thrombin generation potential. Blood. 2014;123:777‐785. doi: 10.1182/blood-2013-10-529628 [DOI] [PubMed] [Google Scholar]
- 27. Ter Horst R, Jaeger M, Smeekens SP, et al. Host and environmental factors influencing individual human cytokine responses. Cell. 2016;167:1111‐1124.e13. doi: 10.1016/j.cell.2016.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li Y, Oosting M, Smeekens SP, et al. A functional genomics approach to understand variation in cytokine production in humans. Cell. 2016;167:1099‐110.e14. doi: 10.1016/j.cell.2016.10.017 [DOI] [PubMed] [Google Scholar]
- 29. Bloemen S, Kelchtermans H, Hemker HC. Thrombin generation in low plasma volumes. Thromb J. 2018;16:10. doi: 10.1186/s12959-018-0164-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dargaud Y, Luddington R, Baglin TP. Elimination of contact factor activation improves measurement of platelet‐dependent thrombin generation by calibrated automated thrombography at low‐concentration tissue factor. J Thromb Haemost. 2006;4:1160‐1161. doi: 10.1111/j.1538-7836.2006.01905.x [DOI] [PubMed] [Google Scholar]
- 31. GTEx Consortium . The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369:1318‐1330. doi: 10.1126/science.aaz1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Westra H‐J, Peters MJ, Esko T, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet. 2013;45:1238‐1243. doi: 10.1038/ng.2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Deelen P, Bonder MJ, van der Velde KJ, et al. Genotype harmonizer: automatic strand alignment and format conversion for genotype data integration. BMC Res Notes. 2014;7:1‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Delaneau O, Zagury J‐F, Marchini J. Improved whole‐chromosome phasing for disease and population genetic studies. Nat Methods. 2013;10:5‐6. [DOI] [PubMed] [Google Scholar]
- 35. Howie B, Marchini J, Stephens M. Genotype imputation with thousands of genomes. G3 (Bethesda). 2011;1:457‐470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bloemen S, Huskens D, Konings J, et al. Interindividual variability and normal ranges of whole blood and plasma thrombin generation. JALM. 2017;2:150‐164. doi: 10.1373/jalm.2017.023630 [DOI] [PubMed] [Google Scholar]
- 37. Yang Q, Kathiresan S, Lin J‐P, Tofler GH, O'Donnell CJ. Genome‐wide association and linkage analyses of hemostatic factors and hematological phenotypes in the Framingham Heart Study. BMC Med Genet. 2007;8:S12. doi: 10.1186/1471-2350-8-S1-S12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tang W, Basu S, Kong X, et al. Genome‐wide association study identifies novel loci for plasma levels of protein C: the ARIC study. Blood. 2010;116:5032‐5036. doi: 10.1182/blood-2010-05-283739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pankow JS, Tang W, Pankratz N, et al. Identification of genetic variants linking protein C and lipoprotein metabolism. Arterioscler Thromb Vasc Biol. 2017;37:589‐597. doi: 10.1161/ATVBAHA.116.308109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lamina C, Friedel S, Coassin S, et al. A genome‐wide association meta‐analysis on apolipoprotein A‐IV concentrations. Hum Mol Genet. 2016;25:3635‐3646. doi: 10.1093/hmg/ddw211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sun BB, Maranville JC, Peters JE, et al. Genomic atlas of the human plasma proteome. Nature. 2018;558:73‐79. doi: 10.1038/s41586-018-0175-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kronenberg F, Stühlinger M, Trenkwalder E, et al. Low apolipoprotein A‐IV plasma concentrations in men with coronary artery disease. J Am Coll Cardiol. 2000;36:751‐757. doi: 10.1016/s0735-1097(00)00775-0 [DOI] [PubMed] [Google Scholar]
- 43. Qu J, Ko CW, Tso P, Bhargava A. Apolipoprotein A‐IV: a multifunctional protein involved in protection against atherosclerosis and diabetes. Cells. 2019;8:319. doi: 10.3390/cells8040319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bisgaier CL, Sachdev OP, Megna L, Glickman RM. Distribution of apolipoprotein A‐IV in human plasma. J Lipid Res. 1985;26:11‐25. [PubMed] [Google Scholar]
- 45. Li Y, Bezemer ID, Rowland CM, et al. Genetic variants associated with deep vein thrombosis: the F11 locus. J Thromb Haemost. 2009;7:1802‐1808. doi: 10.1111/j.1538-7836.2009.03544.x [DOI] [PubMed] [Google Scholar]
- 46. Meijers JC, Kanters DH, Vlooswijk RA, Van Erp HE, Hessing M, Bouma BN. Inactivation of human plasma kallikrein and factor XIa by protein C inhibitor. Biochemistry. 1988;27:4231‐4237. [DOI] [PubMed] [Google Scholar]
- 47. Ivanov I, Verhamme IM, Sun M‐f, et al. Protease activity in single‐chain prekallikrein. Blood. 2020;135:558‐567. doi: 10.1182/blood.2019002224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dargaud Y, Wolberg AS, Gray E, Negrier C, Hemker HC, Subcommittee on Factor Viii FIX, Rare Coagulation D . Proposal for standardized preanalytical and analytical conditions for measuring thrombin generation in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost. 2017;15:1704‐1707. doi: 10.1111/jth.13743 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
App S1