

Abstract
Background
The administration of L‐glutamine (Gln) suppresses allergic airway inflammation via the rapid upregulation of MAPK phosphatase (MKP)‐1, which functions as a negative regulator of inflammation by deactivating p38 and JNK mitogen‐activated protein kinases (MAPKs). However, the role of endogenous Gln remains to be elucidated. Therefore, we investigated the mechanism by which endogenous Gln regulates MKP‐1 induction and allergic airway inflammation in an ovalbumin‐based murine asthma model.
Methods
We depleted endogenous Gln levels using L‐γ‐glutamyl‐p‐nitroanilide (GPNA), an inhibitor of the Gln transporter ASCT2 and glutamine synthetase small interfering siRNA. Lentivirus expressing MKP‐1 was injected to achieve overexpression of MKP‐1. Asthmatic phenotypes were assessed using our previously developed ovalbumin‐based murine model, which is suitable for examining sequential asthmatic events, including neutrophil infiltration. Gln levels were analyzed using a Gln assay kit.
Results
GPNA or glutamine synthetase siRNA successfully depleted endogenous Gln levels. Importantly, homeostatic MKP‐1 induction did not occur at all, which resulted in prolonged p38 MAPK and cytosolic phospholipase A2 (cPLA2) phosphorylation in Gln‐deficient mice. Gln deficiency augmented all examined asthmatic reactions, but it exhibited a strong bias toward increasing the neutrophil count, which was not observed in MKP‐1‐overexpressing lungs. This neutrophilia was inhibited by a cPLA2 inhibitor and a leukotriene B4 inhibitor but not by dexamethasone.
Conclusion
Gln deficiency leads to the impairment of MKP‐1 induction and activation of p38 MAPK and cPLA2, resulting in the augmentation of neutrophilic, more so than eosinophilic, airway inflammation.
Keywords: cPLA2 , endogenous glutamine, MAPK phosphatase‐1, neutrophilic airway inflammation, p38 MAPK
Glutamine depletion was achieved by using the glutamine transporter ASCT2 inhibitor l‐γ‐glutamyl‐p‐nitroanilide or glutamine synthetase siRNA. In glutamine deficiency, no MKP‐1 induction and prolonged p38 and cPLA2 phosphorylation were observed in response to airway ovalbumin challenge. As a result, dominant neutrophilic Th1 response, rather than eosinophilic Th2 response occurred.
Abbreviations: ASCT2, solute carrier 1A5; cPLA2, cytosolic phospholipase A2; EOS, eosinophil; Gln, glutamine; GPNA, l‐γ‐glutamyl‐p‐nitroanilide; GS, glutamine synthetase; MKP‐1, MAPK phosphatase‐1; NEU, neutrophil; OVA, ovalbumin; siRNA, small interfering RNA.

Abbreviations
- ASCT2
solute carrier 1A5
- BALF
bronchoalveolar lavage fluid
- BLT1
LTB4 receptor 1
- COPD
chronic obstructive pulmonary disease
- cPLA2
cytosolic phospholipase A2
- DEX
dexamethasone
- ELISA
enzyme‐linked immunosorbent assay
- Gln
L‐glutamine
- GPNA
L‐γ‐glutamyl‐p‐nitroanilide
- GS
glutamine synthetase
- IL
interleukin
- LO
lipoxygenase
- LPS
lipopolysaccharide
- LT
leukotriene
- MAPK
mitogen‐activated protein kinase
- MKP‐1
MAPK phosphatase‐1
- OVA
ovalbumin
- SEM
standard error of the mean
- siRNA
small interfering RNA
- SLC
solute carrier
- T2
type 2
- TNF‐α
tumor necrosis factor‐alpha
1. INTRODUCTION
Asthma, a chronic inflammatory disorder of the airway, is characterized by bronchial hyperresponsiveness and variable airflow obstruction. 1 Two major endotypes, type 2 (T2)‐high and T2‐low asthma, have been established. T2‐high endotypes are primarily characterized by the presence of eosinophilic inflammation and increased airway expression of T2 cytokines, such as interleukin (IL)‐4, IL‐5, and IL‐13. 2 Corticosteroids and biological therapeutics targeting IL‐4, IL‐5, and IL‐13 signaling can suppress the T2‐high phenotype in most patients. 3 , 4 Conversely, the T2‐low endotype is usually characterized by neutrophilic infiltration in the airways, which results in the activation of T1 and T17 cells. 5 T2‐low asthmatics respond poorly to the standard therapy for severe asthma, i.e., high‐dose inhaled corticosteroids and long‐acting bronchodilators. 6 Biological therapeutics are not recommended in patients with the T2‐low endotype. 7
L‐glutamine (Gln) is the most abundant amino acid in the human blood, skeletal muscle, and free amino acid pool. 8 Gln plays physiologically important roles in various metabolic processes: as an intermediary in energy metabolism and as a substrate for the synthesis of peptides and non‐peptides, such as nucleotide bases, glutathione, and neurotransmitters. 9 , 10 Gln is an important energy substrate for most cells, including immune cells, enterocytes, and hepatocytes, and is regarded as a “conditionally essential” amino acid, especially during intense exercise 11 and critical illness. 12 We have previously reported the beneficial effects of exogenous Gln in several experimental inflammatory diseases, including allergic asthma in mice. 13 , 14 , 15 , 16 , 17 , 18 , 19 We demonstrated that the anti‐inflammatory mechanism of Gln entails the effective deactivation of not only p38 and JNK mitogen‐activated protein kinases (MAPKs) but also one of the p38 substrates, cytosolic phospholipase A2 (cPLA2), via the rapid induction (within 5 min) of MAPK phosphatase (MKP)‐1. 19
MKPs represent a distinct subfamily within a larger group of dual‐specificity protein phosphatases, which dephosphorylate MAPK. MKP‐1 dephosphorylates and inactivates both p38 and JNK MAPKs: it is substrate‐specific and dependent on cell type and context (please refer to 20 for an overview). Furthermore, MKP‐1 deactivates cPLA2 by either dephosphorylating p38, since p38 is a major upstream pathway for cPLA2 phosphorylation 21 or directly dephosphorylating cPLA2, owing to the enhanced physical interaction between MKP‐1 and phosphorylated cPLA2. 22 Given that p38 MAPK and cPLA2 are among the most pro‐inflammatory kinases, 23 , 24 which play key roles in the pathogenesis of asthma, 25 , 26 MKP‐1 plays a key role in the resolution of inflammation, is a critical negative feedback effector, and performs homeostatic functions in cellular signaling. We have shown that Gln upregulates MKP‐1 protein levels by increasing ERK activity via the activation of the pathway involving Ca2+/Ras/c‐Raf/MEK/ERK (ERK cascade), resulting in increased MKP‐1 phosphorylation of the serine residues and their stabilization. 27
Therefore, we hypothesized that Gln deficiency would lead to the impairment of MKP‐1 induction and prolonged MAP kinase activation, resulting in the development of augmented inflammation. In this study, we investigated the mechanism by which endogenous Gln regulates the equilibrium between MKP‐1/p38 MAPK and cPLA2 and inflammation using an ovalbumin (OVA)‐based murine asthma model.
2. MATERIALS AND METHODS
2.1. Animals
Specific pathogen‐free female C57BL/6 mice were purchased from NARA Biotech (Pyeongtaek, Republic of Korea) and housed in clean, pathogen‐free rooms in an environment with controlled temperature (23°C), humidity (55%), and a 12‐h light/dark cycle. All mice were 7–8‐week‐old at the beginning of each experiment. All animals were handled as per the protocol approved by the Institutional Animal Care and Use Committee of the Jeonbuk National University Medical School, Jeonbuk, Republic of Korea (approval number: CBNU 2018‐0049).
2.2. Reagents
Gln (Biotechnology performance certified, G‐8540), L‐γ‐glutamyl‐p‐nitroanilide (GPNA‐HCl; G‐6133), and lipopolysaccharide (LPS, Escherichia coli serotype O55:B5) were purchased from Sigma‐Aldrich. The p38 MAPK inhibitor SB202190 was purchased from Calbiochem. The 5‐lipoxygenase (5‐LO) inhibitor MK‐886 and the leukotriene (LT) B4 receptor 1 (BLT1) antagonist U75302 were obtained from Tocris Biotechne. The cPLA2 inhibitor pyrrophenone was obtained from Cayman Chemical Company. Dexamethasone (DEX) was obtained from Enzo Life Sciences. Antibodies against p‐cPLA2, p‐p38, and MKP‐1 were purchased from Cell Signaling Technology. The antibody against glutamine synthetase (GS) was obtained from Abcam. Enhanced green fluorescent protein (eGFP) antibodies were obtained from Thermo Fisher Scientific (Dreieich, Germany). Glyceraldehyde 3‐phosphate dehydrogenase (GAPDH), which was used as the loading control, was obtained from Santa Cruz Biotechnology.
Gln or sham (isovolumic sterile water) solution was administered via oral gavage. SB202190 (5 mg/kg) was dissolved in dimethyl sulfoxide and injected intraperitoneally 48 and 24 h before the second airway challenge. GPNA (50 mg/kg in saline) was injected intraperitoneally 1 h before the second airway challenge. MK‐866 (1 mg/kg) and U75302 (1 mg/kg, both in saline) were administered intraperitoneally 1 h before the second airway challenge. Pyrrophenone (20 mg/kg in saline) was injected intraperitoneally 30 min before the second airway challenge. DEX (2.5 mg/kg in saline) was administered intraperitoneally 24 h and 30 min before the second airway challenge. LPS (10 μg) was immediately administered intranasally after the end of the second airway OVA challenge to induce Th1 cytokines. 28 , 29
2.3. Immunization and OVA challenge
The mice were immunized and challenged with OVA to achieve the induction of neutrophils, eosinophils, and Th2 cytokines in the airway, as described previously. 30 In brief, mice were immunized intraperitoneally with 20 mg chicken egg OVA (OVA, grade V; Sigma‐Aldrich) plus 1.0 mg aluminum hydroxide adjuvant (Imject Alum; Pierce) on day 0 and with OVA alone on day 14. The immunized mice were exposed to 1% aerosolized OVA for 20 min on days 28 and 35. The control saline‐treated group received the same immunization and the first OVA airway challenge but were exposed to aerosolized saline instead of OVA during the second airway challenge.
2.4. Determination of airway hyperresponsiveness
Invasive airway measurements were performed 48 h after the second airway challenge using previously described methods. 31
2.5. Bronchoalveolar lavage fluid (BALF)
Bronchoalveolar lavage was performed at the time indicated after the second airway challenge, as described previously. 18 , 26
2.6. Immunoblotting
The mice were sacrificed by cervical dislocation. Their lungs were immediately harvested, briefly washed with cold phosphate‐buffered saline, and dried with blotting paper. The isolated lung tissues were frozen in liquid nitrogen and stored at −80°C until further analysis. Small lung tissue specimens were homogenized in PhosphoSafe Extraction Reagent (Novagen, Madison, WI) and subjected to immunoblotting, as described previously. 16 , 17
2.7. Immunohistochemistry
Immunohistochemical detection of eGFP and MKP‐1 expression in the lungs was performed with the immunoperoxidase method using the streptavidin‐biotinylated horseradish peroxidase complex (DAKO), as described in a previous study. 32
2.8. Construction of the lentiviral vector and infection
The cLV‐CMV‐DUSP1‐EF1a‐eGFP‐T2A‐Puro‐WPRE lentiviral vector and control vector were purchased from Sirion Biotech. A recombination reaction was conducted between the entry clone (mDUPS1) and destination vector (pcLV‐CMV‐DEST‐EF1a‐eGFP‐T2A‐Puro‐WPRE) according to the manufacturer's instruction. The titration of the viruses was determined using the Lenti‐X™ Titration Kit: the infectious titer (IU) was 1.7 × 109 IU/ml. The viral and control vectors (0.5–4.0 × 107 IU) were administered intravenously 2 days before the second airway challenge. The optimum dosage and injection time were determined by a preliminary experiment.
2.9. Cytokine assay
Enzyme‐linked immunosorbent assays (ELISAs) were used to quantify the levels of tumor necrosis factor (TNF)‐α, IL‐4, IL‐5, IL‐6, and IL‐1β in BALF according to the manufacturer's instructions (R&D Systems, Minneapolis, MN). The LTB4 ELISA kit was purchased from Cayman Chemical Company (520111). The lower limits of detection for the cytokines were as follows: TNF‐α > 2 pg/ml, IL‐4 > 2 pg/ml, IL‐5 > 7 pg/ml, IL‐6 > 1.6 pg/ml, IL‐13 > 1.5 pg/ml, IL‐1β/IL1‐1F2 > 2.31 pg/ml, and LTB4 > 13 pg/ml.
2.10. Measurement of cPLA2 activity
cPLA2 activity in the lung homogenates was measured using a cPLA2 assay kit, according to the manufacturer's instructions (765021, Cayman Chemical Company). The sensitivity of detection was as follows: 3.5–42 nmol/min/ml.
2.11. Small interfering RNA interference
Small interfering RNA (siRNA) strands for mouse GS and the corresponding controls were purchased from OriGene Technologies (SR412598A, SR412598B, SR412598C) and Sigma‐Aldrich (SASI_Mm01_00044456). Control siRNA was provided by OriGeneTech. GS siRNA or control siRNA was transfected using the in vivo‐jetPEI™ (Polyplustransfection) delivery system according to the manufacturer's instructions. Briefly, GS siRNA and PEI were dissolved in 5% glucose and mixed for 20 min to obtain a solution with a volume of 200 µl. The mixture was administered intravenously two times (on days 5 and 3) before the second airway challenge. The mixture containing control siRNA and PEI dissolved in 5% glucose was used as the control.
2.12. Histological analysis
The lungs were fixed with 10% formalin, and the tissues were embedded in paraffin. The fixed tissue was sectioned to a thickness of 4 μm, and the paraffin was removed. The sections were stained with hematoxylin and eosin.
2.13. Glutamine extraction and measurement
Immediately after euthanizing the animals using inhalation anesthesia (isoflurane), the animals were perfused with 10 ml of cold saline through the heart. The whole lung tissue was homogenized in 80% ethanol 33 using a Dounce homogenizer chilled on ice. After spinning down the debris, the supernatant was lyophilized and dissolved in distilled water (1 μl water per mg of tissue). The Gln concentration was determined using the glutamine assay kit (ab197011, Abcam) according to the manufacturer's protocol.
2.14. Statistical analysis
All data are presented as the mean ± standard error of the mean (SEM). Statistical differences were determined using one‐way ANOVA or two‐way ANOVA. Tukey's multiple comparisons were performed if significant interactions were observed. Univariate analyses were performed using GraphPad Prism software (version 9.1).
3. RESULTS
3.1. Gln deficiency leads to the impairment of MKP‐1 induction and prolonged activation of p38
We attempted to deplete the endogenous Gln levels using two strategies in order to determine the role of endogenous Gln in MKP‐1 induction. The first method entailed blocking Gln transportation into the cells using a Gln transporter inhibitor. Amino acid transporters are membrane‐bound transport proteins that mediate the transfer of amino acids into and out of cells or cellular organelles. Gln transporters are found in four gene families: Solute carrier‐type transporters (SLC) 1, SLC6, SLC7, and SLC38. 34 We inhibited Gln transport using L‐γ‐glutamyl‐p‐nitroanilide (GPNA), an inhibitor of ASCT2, which is a Na+‐dependent neutral amino acid transporter encoded by SLC1A5. The second method entailed blocking Gln synthesis by knocking down the Gln‐producing enzyme Gln synthetase using GS siRNA. The pretreatment of GS siRNA resulted in the knockdown of GS (Figure 1A). We found that pretreatment with GPNA or GS siRNA reduced Gln levels by more than 80% in the lungs of normal mice (Figure 1B). MKP‐1 protein levels at 10–90 min and p38 phosphorylation at 10 min were both increased in response to the OVA challenge (Figure 1C,D). Importantly, MKP‐1 induction was markedly reduced and p38 phosphorylation was sustained throughout the observation period (till 90 min) in both GPNA‐ and GS siRNA‐treated mice (Figure 1C,D, respectively). Control siRNA neither reduced MKP‐1 induction nor increased p38 phosphorylation (data not shown).
FIGURE 1.
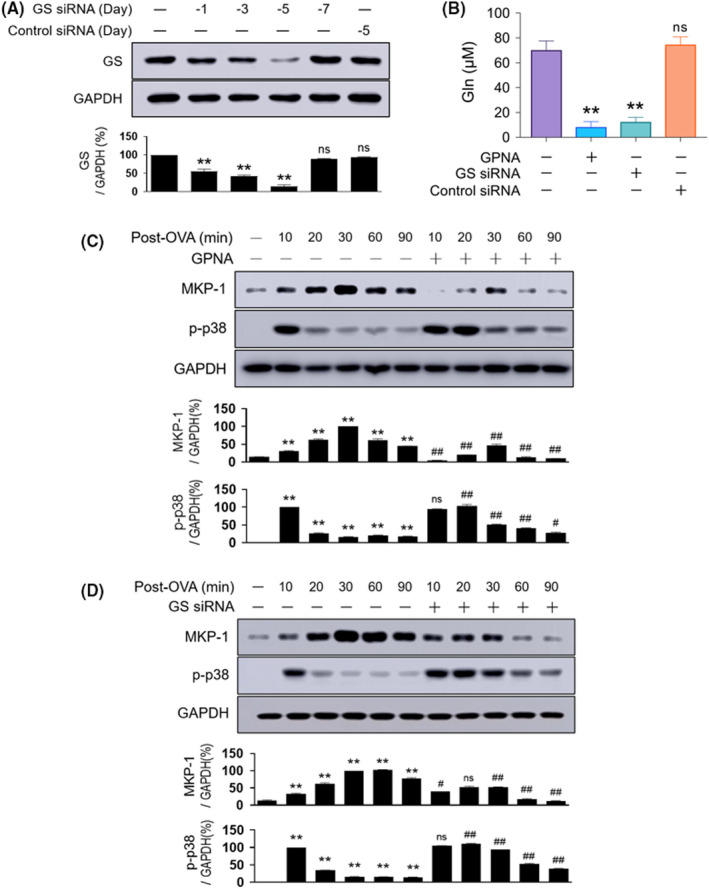
Effects of Gln deficiency on MKP‐1 induction and p38 phosphorylation. (A) Representative immunoblots and densitometric analyses of GS. GS siRNA was injected intravenously from days 7 to 1, and control siRNA was administered at day 5 (n = 6, three independent experiments). (B) Measurement of Gln levels in the lungs using a Gln assay kit. Normal mice were treated either with siRNAs 5 and 3 days before or with GPNA 1 h before sacrifice (n = 12–15, three independent experiments). (C–D) Representative immunoblots and densitometric analyses of the MKP‐1 protein and p38 phosphorylation in the lungs of mice pretreated with GPNA (1 h before the second airway challenge; (C) n = 10–13, three independent experiments) or GS siRNA (5 and 3 days before the second airway challenge; (D) n = 10–13, three independent experiments). Data are presented as the mean ± SEM. A‐B, ns p > .05 vs. normal control, *p < .05 vs. normal control, **p < .01 vs. normal control. (C–D) *p < .05 vs. saline control, **p < .01 vs. saline control, ns p > .05 vs. OVA group, #p < .05 vs. OVA group, ##p < .01 vs. OVA group. GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; Gln, glutamine; GPNA, L‐γ‐glutamyl‐p‐nitroanilide; GS, glutamine synthetase; MKP‐1, MAPK phosphatase‐1; ns, not significant; OVA, ovalbumin; SEM, standard error of the mean; siRNA, small interfering RNA
3.2. Gln deficiency results in the development of neutrophilic airway inflammation
We subsequently assessed the mechanism by which Gln deficiency affected airway inflammation. GPNA and GS siRNA also augmented the eosinophil count (approximately 50%–100%; Figure 2A) and Th2 responses (IL‐4: 50%–80%, IL‐5: 90%–110%, and IL‐13: 40%–70%; Figure 2B) in BALF. However, their effects on the neutrophil count and Th1 response were more prominent. Most strikingly, the neutrophil count increased markedly (4–7‐fold; Figure 2C), and the levels of TNF‐α, IL‐1β, and IL‐6 increased by 120%–170%, 80%–120%, and 100%–180%, respectively (Figure 2D). Respiratory system resistance (Figure 2E), histological lung inflammation (Figure 2F), and ELISA‐based detection of mucus secretion (Figure 2G) were analyzed, revealing elevations in each of the three parameters in GPNA‐ and GS siRNA‐treated mice compared with the OVA‐challenged control group.
FIGURE 2.
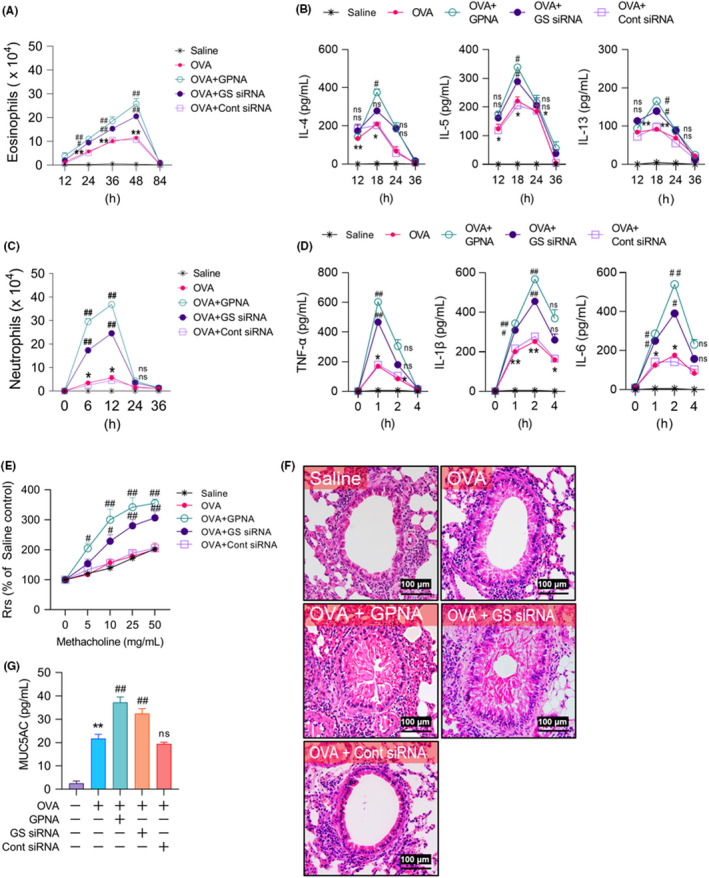
Effects of Gln deficiency on the development of airway inflammation. (A–D) Time kinetics of the levels of neutrophils from 0 to 36 h (A, n = 15–25, three independent experiments), Th1 cytokines from 0 to 4 h (B, n = 15–20, three independent experiments), eosinophils from 24 to 84 h (C, n = 15–25, three independent experiments), and Th2 cytokines from 12 to 36 h (D, n = 15–25, three independent experiments) in BALF after the second airway challenge. (E) Airway responsiveness assessed by invasive measurements (n = 60–75, three independent experiments). (F) Representative H&E‐stained sections of the lung (n = 10–15, three independent experiments). The bars indicate 100 μm. (G) Mucus secretion revealed by ELISA (n = 15–25, three independent experiments) (F–G) The parameters were assessed at 48 h after the second airway challenge. Data are presented as the mean ± SEM. *p < .05 vs. saline control, **p < .001 vs. saline control, ns p > .05 vs. OVA group, #p < .05 vs. OVA group, ##p < .001 vs. OVA group. BALF, bronchoalveolar lavage fluid; ELISA, enzyme‐linked immunosorbent assay; Gln, glutamine; GPNA, L‐γ‐glutamyl‐p‐nitroanilide; GS, glutamine synthetase; H&E, hematoxylin and eosin; IL, interleukin; MUC5AC, musin‐5 subtype A and C; ns, not significant; OVA, ovalbumin; Rrs, resistance of the respiratory system; SEM, standard error of the mean; siRNA, small interfering RNA; TNF, tumor necrosis factor
We overexpressed MKP‐1 by injecting lentivirus‐expressing MKP‐1 (lentiviral MKP‐1) to verify that MKP‐1 downregulation was responsible for neutrophilic inflammation in Gln deficiency. Western blot analysis showed that the MKP‐1 protein was successfully overexpressed in a dose‐dependent manner in the lung tissues, but the control virus (lentivirus‐expressing eGFP) had no such effect on target gene expression. The expression of MKP‐1 was peaked (Figure 3A) 2–3 days (data not shown) after the injection of 4 × 107 plaque‐forming units of lentiviral MKP‐1. Immunohistochemical staining revealed a strong eGFP positivity in the airway epithelial cells in both lentiviral MKP‐1‐ and control virus‐injected normal mice, and MKP‐1 expression in lentiviral MKP‐1‐, but not in control virus‐, injected normal mice (Figure 3B). Based on these results, we injected 4 × 107 units of lentiviral MKP‐1 2 days before the second airway challenge. In these mice, GPNA‐induced MKP‐1 downregulation/increase in p38 and cPLA2 phosphorylation (Figure 3C) increased the neutrophil count (Figure 3D), and Th1 cytokine levels (Figure 3E) returned to the baseline levels of the OVA‐exposed control group.
FIGURE 3.
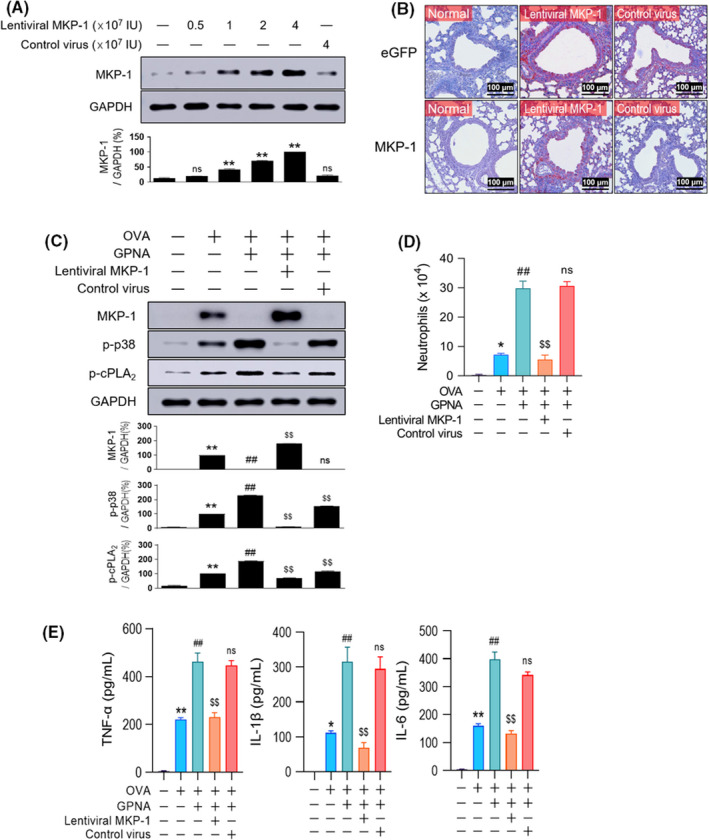
Lentiviral overexpression of MKP‐1 abrogates Gln deficiency‐induced neutrophilic inflammation. (A–B) Representative immunoblots (A) and immunohistochemical stainings (B) for MKP‐1 protein. Normal mice were injected with varying virus doses and sacrificed 2 days thereafter for western blots (A, n = 9–12, three independent experiments) or immunohistochemistry of the lung (B, n = 6, three independent experiments). The bars indicate 100 μm. C, MKP‐1 and phosphorylation of p38 and cPLA2 (n = 10–15, three independent experiments) in the lungs of mice pretreated with lentiviral MKP‐1. (D–E) The levels of neutrophils at 10 h (D, n = 15–20, three independent experiments) and Th1 cytokines at 1.5 h (E, n = 15–20, three independent experiments) in BALF. (C–E) GPNA and viruses (4 × 107 IU) were injected 1 h and 2 days before the second airway challenge, respectively. Data are presented as the mean ± SEM. (A) ns p > .05 vs. normal control, *p < .05 vs. normal control, **p < .001 vs. normal control. (C–E) *p < .05 vs. saline control, **p < .001 vs. saline control, #p < .05 vs. OVA group, ##p < .001 vs. OVA group, ns p > .05 vs. OVA + GPNA group, $p < .05 vs. OVA + GPNA group, $$p < .001 vs. OVA + GPNA group. BALF, bronchoalveolar lavage fluid; cPLA2, cytosolic phospholipase A2; eGFP, enhanced green fluorescent protein; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; Gln, glutamine; GPNA, L‐γ‐glutamyl‐p‐nitroanilide; IL, interleukin; MKP‐1, MAPK phosphatase‐1; ns, not significant; OVA, ovalbumin; SEM, standard error of the mean; TNF, tumor necrosis factor
3.3. Gln deficiency‐induced neutrophilia is cPLA2/LTB4‐dependent but steroid‐resistant
We previously reported that the MKP‐1‐dependent cPLA2/5‐LO/LTB4 pathway plays a major role in airway neutrophilia 18 in a similar model of murine asthma. Among the 5‐LO metabolites, LTB4 is a well‐known potent neutrophil chemoattractant. 35 , 36 Therefore, we examined the link between Gln deficiency‐induced neutrophilia and cPLA2. GS siRNA pretreatment resulted in the augmentation and prolongation of not only cPLA2 phosphorylation (Figure 4A) and activity (Figure 4B) but also the production of the cPLA2 metabolite LTB4 (Figure 4C). Control siRNA failed to increase cPLA2 phosphorylation (data not shown). Administration of a p38 inhibitor (SB202190), cPLA2 inhibitor (Pyrrophenone), or 5‐LO inhibitor (MK‐886) all restricted GPNA‐induced neutrophilia by up to 75%–80% (Figure 4D). In particular, the LTB4 receptor BLT1 antagonist U75302 reduced the neutrophil count in a dose‐dependent manner and nearly completely at a concentration of 1 mg/kg (Figure 4E).
FIGURE 4.
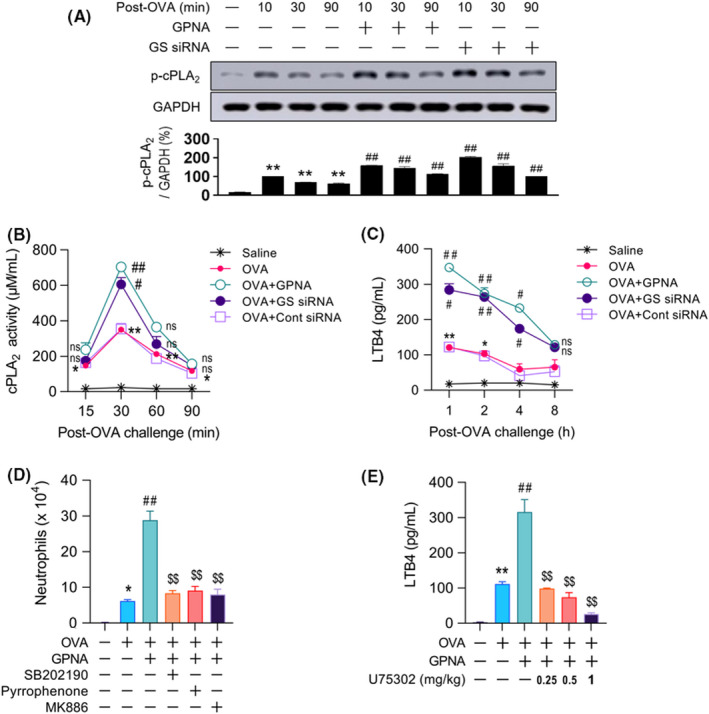
Association between the p38/cPLA2/LTB4 pathway and neutrophilia in Gln deficiency. (A–C) cPLA2 phosphorylation (A, n = 11, three independent experiments), activity (B, n = 10–15, three independent experiments), and levels of LTB4 in BALF from 1 to 8 h (C, n = 10–15, three independent experiments). (D) Number of neutrophils in BALF at 12 h. (E) Levels of LTB4 in BALF at 1 h post‐challenge (D and E, n = 15–18, three independent experiments). Data are presented as the mean ± SEM. ns p > .05 vs. saline control, *p < .05 vs. saline control, **p < .001 vs. saline control, #p < .05 vs. OVA group, ##p < .001 vs. OVA group, ns p > .05 vs. OVA + GPNA group, $p < .05 vs. OVA + GPNA group, $$p ≤ .001 vs. OVA + GPNA group. BALF, bronchoalveolar lavage fluid; cPLA2, cytosolic phospholipase A2; Gln, glutamine; GPNA, L‐γ‐glutamyl‐p‐nitroanilide; GS, glutamine synthetase; LTB4, leukotriene B4; ns, not significant; OVA, ovalbumin; SEM, standard error of the mean; siRNA, small interfering RNA
Clinically, patients with neutrophilic asthma are unresponsive to high‐dose inhaled corticosteroids. 7 We examined how DEX affects the alterations characteristic of Gln deficiency, such as neutrophilia and the MKP‐1/p38/cPLA2 and LTB4 levels. DEX failed to upregulate MKP‐1 protein levels or decrease p38 and cPLA2 phosphorylation (Figure 5A). DEX inhibited the GS siRNA‐induced elevation in the eosinophil count (Figure 5B) and Th2 cytokine levels in BALF (Figure 5C) (both >80%), but it did not significantly affect the neutrophil count (Figure 5D) and LTB4 levels (Figure 5E). In contrast to DEX, oral Gln (2 g/kg) restored GS siRNA‐induced alterations, such as the MKP‐1 level, p38, and cPLA2 phosphorylation (Figure 5A), neutrophil count (Figure 5D), and LTB4 levels (Figure 5E).
FIGURE 5.
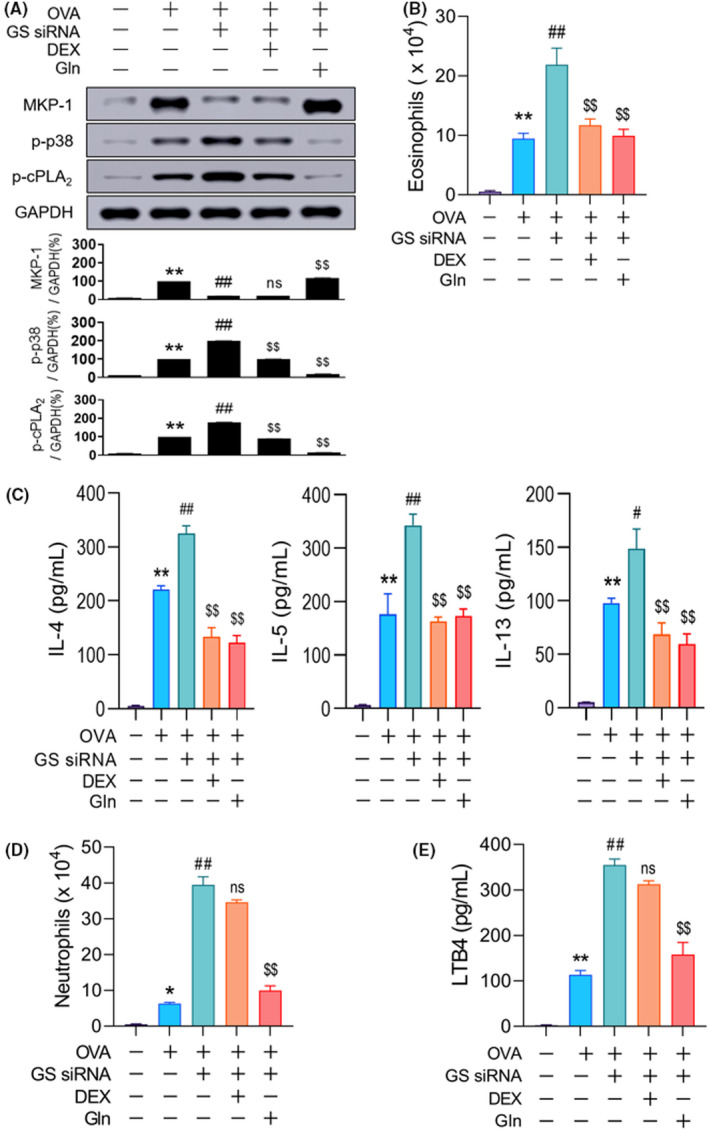
Steroid resistance of neutrophilia in Gln deficiency. (A) Representative immunoblots and densitometric analyses of MKP‐1 protein and phosphorylation of p38 and cPLA2 (n = 5–10, three independent experiments) at 1 h post‐challenge. (B–C) Number of eosinophils at 48 h (B, n = 10–15, three independent experiments) and Th2 cytokine levels at 18 h (C, n = 10–15, three independent experiments) in BALF. (D) Number of neutrophils in BALF at 12 h (n = 10–15, three independent experiments). (E) Levels of LTB4 in BALF at 1 h (n = 10–15, three independent experiments). A‐E, Gln (2 g/kg) was orally administered 30 min before the second airway challenge. Data are presented as the mean ± SEM. *p < .05 vs. saline control, **p < .001 vs. saline control, #p < .05 vs. OVA group, ##p < .001 vs. OVA group, ns p > .05 vs. OVA + GS siRNA group, $p < .05 vs. OVA + GS siRNA group, $$p < .001 vs. OVA + GS siRNA group. BALF, bronchoalveolar lavage fluid; cPLA2, cytosolic phospholipase A2; DEX, dexamethasone; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; Gln, glutamine; GS, glutamine synthetase; IL, interleukin; LTB4, leukotriene B4; MKP‐1, MAPK phosphatase‐1; ns, not significant; OVA, ovalbumin; SEM, standard error of the mean; siRNA, small interfering RNA
4. DISCUSSION
In this study, we investigated how endogenous Gln regulated MKP‐1 induction and asthmatic inflammation. We successfully depleted endogenous Gln levels using a Gln transporter (ASCT2) inhibitor, GPNA, and GS siRNA. We arrived at two important findings with the aid of the Gln‐deficient mouse model. First, in contrast to the early p38 phosphorylation followed by MKP‐1 induction in the OVA‐challenged control group, MKP‐1 induction was negligible in the GPNA‐ and GS siRNA‐treated groups, which resulted in sustained p38 phosphorylation. Furthermore, these phenomena in GS siRNA‐treated mice were completely reversed by exogenous Gln (Figure 5A), indicating that Gln is indeed an endogenous MKP‐1 inducer. To the best of our knowledge, this is the first study to demonstrate the MKP‐1‐inducing action of endogenous Gln and its consequent role as a negative regulator of inflammation in vivo. Our data are in good agreement with several studies demonstrating the relationship between MKP‐1 downregulation and enhanced inflammation, ie, the depletion of MKP‐1 leads to an excessive release of inflammatory cytokines, such as TNF‐α, IL‐6, CCL3, and CCL4, and increased LPS‐induced mortality, 37 , 38 , 39 , 40 further supporting the notion that MKP‐1 is a key negative regulator of inflammation. This suggests that any condition that lowers Gln levels can cause inflammation. Second, neutrophilic airway inflammation was strongly augmented in the OVA‐based murine asthma model. Although both Th2 and Th1 responses were augmented, neutrophil‐dominant Th1 responses (neutrophil count and Th1 cytokines, such as TNF‐α, IL‐1β, and IL‐6) were substantially higher. This augmented neutrophilic inflammation was no longer observed in lungs with lentivirus‐based MKP‐1‐overexpression (Figure 3), indicating that the failure of MKP‐1 induction in Gln deficiency was responsible for the augmented inflammation.
We have previously reported on the role of cPLA2 in airway neutrophilia. We demonstrated that the MKP‐1‐dependent cPLA2/5‐LO/LTB4 pathway plays a major role in airway neutrophilia based on the following findings: (1) inhibition of airway neutrophilia by cPLA2 or 5‐LO inhibitors; (2) elevated levels of LTB4, a 5‐LO metabolite and potent neutrophil chemoattractant; and (3) a dose‐dependent increase in the number of neutrophils in the airway after intratracheal administration of LTB4. 18 The present study demonstrated the activation of the p38/cPLA2/5LP/LTB4 pathway due to the impairment of MKP‐1 induction, which in turn, favored the development of neutrophil‐dominant Th1 inflammation over that of the eosinophil‐dominant Th2 response. Regarding the role of the p38/cPLA2/LTB4 pathway in eosinophilia and Th2 response, Gln deficiency‐induced increases in eosinophilia and Th2 response were attenuated by administration of the cPLA2 inhibitor, but not the BLT1 antagonist (data now shown), suggesting that the eosinophil‐dominant Th2 response was less sensitive to the p38/cPLA2/LTB4 pathway compared to the neutrophil‐dominant Th1 response.
Previous studies reported relationships between LTB4 and neutrophilic airway inflammation in asthma. LTB4 may play an important role in neutrophilic asthma, 41 , 42 and high LTB4 levels were found in the sputum of severe asthmatics. 43 , 44 Our data suggest a relationship between Gln deficiency and T2‐low neutrophilic inflammation in humans. This is perhaps the case considering the following: (1) neutrophilic asthma is associated with comorbidities, which may contribute to the severity of the disease and exacerbations, such as very late‐onset (>50–65 years), respiratory infections (viral, bacterial, and fungal), rhinosinusitis, obesity, smoking, air pollution, and occupational asthma, 45 , 46 which are known to cause oxidative stress 47 , 48 ; and (2) the Gln‐producing enzyme GS is particularly sensitive to inactivation due to oxidative stress. 49 , 50 , 51 Therefore, Gln deficiency due to oxidative stress‐induced GS inactivation can lead to neutrophilic asthma. The same mechanism can be applied to chronic obstructive pulmonary disease (COPD) because high oxidative stress levels are recognized in the pathogenesis of COPD 52 and neutrophilic inflammation predominates in COPD as well. 53 This aspect is interesting and worthy of further investigation.
It is well known that neutrophilic T2‐low asthma responds poorly to steroids. 6 In this study, DEX administration suppressed airway neutrophilia to a substantially lesser extent (<20%) compared with eosinophilia (>80%). This finding was attributed to the inability of DEX to induce MKP‐1 upregulation akin to Gln. Hence, DEX failed to inactivate p38 and cPLA2. The in vitro upregulation of MKP‐1 by steroids is considered as one of the anti‐inflammatory mechanisms of steroids. 54 , 55 However, it is not clear whether steroids directly upregulate MKP‐1 based on a posttranscriptional mechanism similar to that of Gln. To the best of our knowledge, Gln is the only agent with the ability to upregulate MKP‐1 in vivo.
In summary, exogenous Gln exhibited potent anti‐inflammatory activity against both Th2 and Th1 allergic lung inflammation via early MKP‐1 induction, followed by the inhibition of two important pro‐inflammatory enzymes, p38 and cPLA2. Gln deficiency leads to the impairment of MKP‐1 induction and activation of the p38/cPLA2 pathway. Thus, we concluded that Gln functions as an endogenous MKP‐1 inducer plays a key role as a negative regulator of inflammation, and facilitates homeostasis by regulating the balance between MKP‐1/p38 and cPLA2 in mice. Therefore, activation of p38 and cPLA2 due to Gln deficiency/failure of MKP‐1 induction can induce and/or augment any inflammation in which p38 and cPLA2 play key roles in their pathogenesis, and the surge in the activities of p38 and cPLA2 favors the development of neutrophilic airway inflammation if Gln deficiency develops in the lungs. However, the mechanism does not seem to be limited to asthmatic inflammation. It is possible that preexisting Gln deficiency due to chronic diseases such as chronic infections, cancers, metabolic diseases, etc., which induce oxidative stress can augment inflammation in any organs or exacerbate the clinical course of infectious diseases such as COVID‐19 in which p38 and cPLA2 play key roles in their pathogenesis. 56 , 57 Our data provided fundamental concepts that will enhance the understanding of the pathogenesis of inflammation including asthma and suggest that Gln could be a promising therapeutic agent for the treatment of such inflammations in humans.
CONFLICT OF INTEREST
The authors declare no commercial or financial conflict of interest.
AUTHOR CONTRIBUTIONS
HKL, MKH, and SYI designed the research and wrote the manuscript. JMK, YNI, and YJC performed experiments. HKL, MKH, and JHY analyzed the data.
ACKNOWLEDGEMENTS
We would like to thank Editage (www.editage.co.kr) for English language editing.
Kim J‐M, Im YN, Chung Y‐J, et al. Glutamine deficiency shifts the asthmatic state toward neutrophilic airway inflammation. Allergy.2022;77:1180–1191. 10.1111/all.15121
June‐Mo Kim and Yoo Na Im contributed equally to this work.
Suhn Young Im, Myung Kwan Han and Hern Ku Lee shared senior authorship.
Funding information
This study was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT: Ministry of Science and ICT) (No. 2017M3A9C8033473 and 2017R1A5A2015061).
Contributor Information
Suhn Young Im, Email: syim@chonnam.ac.kr.
Myung Kwan Han, Email: iamtom@jbnu.ac.kr.
Hern Ku Lee, Email: leeh-k@jbnu.ac.kr.
REFERENCES
- 1. To T, Stanojevic S, Moores G, et al. Global asthma prevalence in adults: findings from the cross‐sectional world health survey. BMC Public Health. 2012;12:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stokes JR, Casale TB. Characterization of asthma endotypes: implications for therapy. Ann Allergy Asthma Immunol. 2016;117(2):121‐125. [DOI] [PubMed] [Google Scholar]
- 3. Pepper AN, Renz H, Casale TB, Garn H. Biologic therapy and novel molecular targets of severe asthma. J Allergy Clin Immunol Pract. 2017;5(4):909‐916. [DOI] [PubMed] [Google Scholar]
- 4. Berry M, Morgan A, Shaw DE, et al. Pathological features and inhaled corticosteroid response of eosinophilic and non‐eosinophilic asthma. Thorax. 2007;62(12):1043‐1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Haldar P, Pavord ID. Noneosinophilic asthma: a distinct clinical and pathologic phenotype. J Allergy Clin Immunol. 2007;119(5):1043‐1052; quiz 1053‐1044. [DOI] [PubMed] [Google Scholar]
- 6. Gaga M, Zervas E, Samitas K, Bel EH. Severe asthma in adults: an orphan disease? Clin Chest Med. 2012;33(3):571‐583. [DOI] [PubMed] [Google Scholar]
- 7. Bostantzoglou C, Delimpoura V, Samitas K, Zervas E, Kanniess F, Gaga M. Clinical asthma phenotypes in the real world: opportunities and challenges. Breathe (Sheff). 2015;11(3):186‐193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Calder PC. Glutamine and the immune system. Clin Nutr. 1994;13(1):2‐8. [DOI] [PubMed] [Google Scholar]
- 9. Albrecht J, Sidoryk‐Węgrzynowicz M, Zielińska M, Aschner M. Roles of glutamine in neurotransmission. Neuron Glia Biol. 2010;6(4):263‐276. [DOI] [PubMed] [Google Scholar]
- 10. Amores‐Sánchez MI, Medina MÁ. Glutamine, as a precursor of glutathione, and oxidative stress. Mol Genet Metab. 1999;67(2):100‐105. [DOI] [PubMed] [Google Scholar]
- 11. Rowbottom DG, Keast D, Goodman C, Morton AR. The haematological, biochemical and immunological profile of athletes suffering from the overtraining syndrome. Eur J Appl Physiol Occup Physiol. 1995;70(6):502‐509. [DOI] [PubMed] [Google Scholar]
- 12. Oudemans‐van Straaten HM, Bosman RJ, Treskes M, van der Spoel H, Zandstra DF. Plasma glutamine depletion and patient outcome in acute ICU admissions. Intensive Care Med. 2001;27(1):84‐90. [DOI] [PubMed] [Google Scholar]
- 13. Kim YS, Kim GY, Kim JH, et al. Glutamine inhibits lipopolysaccharide‐induced cytoplasmic phospholipase A2 activation and protects against endotoxin shock in mouse. Shock. 2006;25(3):290‐294. [DOI] [PubMed] [Google Scholar]
- 14. Ayush O, Lee CH, Kim HK, Im SY, Cho BH, Lee HK. Glutamine suppresses DNFB‐induced contact dermatitis by deactivating p38 mitogen‐activated protein kinase via induction of MAPK phosphatase‐1. J Invest Dermatol. 2013;133(3):723‐731. [DOI] [PubMed] [Google Scholar]
- 15. Kim HK, Song CH, Bae YS, Im SY, Lee HK. Glutamine prevents late‐phase anaphylaxis via MAPK phosphatase 1‐dependent cytosolic phospholipase A2 deactivation. Int Arch Allergy Immunol. 2016;171(1):61‐70. [DOI] [PubMed] [Google Scholar]
- 16. Jeong SY, Im YN, Youm JY, Lee HK, Im SY. l‐Glutamine Attenuates DSS‐Induced Colitis via Induction of MAPK Phosphatase‐1. Nutrients. 2018;10(3):288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ko HM, Kang NI, Kim YS, et al. Glutamine preferentially inhibits T‐helper type 2 cell‐mediated airway inflammation and late airway hyperresponsiveness through the inhibition of cytosolic phospholipase A(2) activity in a murine asthma model. Clin Exp Allergy. 2008;38(2):357‐364. [DOI] [PubMed] [Google Scholar]
- 18. Lee CH, Kim HK, Kim JM, et al. Glutamine suppresses airway neutrophilia by blocking cytosolic phospholipase A(2) via an induction of MAPK phosphatase‐1. J Immunol. 2012;189(11):5139‐5146. [DOI] [PubMed] [Google Scholar]
- 19. Ko HM, Oh SH, Bang HS, et al. Glutamine protects mice from lethal endotoxic shock via a rapid induction of MAPK phosphatase‐1. J Immunol. 2009;182(12):7957‐7962. [DOI] [PubMed] [Google Scholar]
- 20. Lang R, Raffi FAM. Dual‐specificity phosphatases in immunity and infection: an update. Int J Mol Sci. 2019;20(11):2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Su B, Karin M. Mitogen‐activated protein kinase cascades and regulation of gene expression. Curr Opin Immunol. 1996;8(3):402‐411. [DOI] [PubMed] [Google Scholar]
- 22. Lee CH, Kim HK, Jeong JS, et al. Mechanism of glutamine inhibition of cytosolic phospholipase a2 (cPLA2): Evidence of physical interaction between glutamine‐Induced mitogen‐activated protein kinase phosphatase‐1 and cPLA2. Clin Exp Immunol. 2015;180(3):571‐580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signalling. Biochem J. 2010;429(3):403‐417. [DOI] [PubMed] [Google Scholar]
- 24. Leslie CC. Cytosolic phospholipase A(2): physiological function and role in disease. J Lipid Res. 2015;56(8):1386‐1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Barnes PJ. Kinases as novel therapeutic targets in asthma and chronic obstructive pulmonary disease. Pharmacol Rev. 2016;68(3):788‐815. [DOI] [PubMed] [Google Scholar]
- 26. Pniewska E, Pawliczak R. The involvement of phospholipases A2 in asthma and chronic obstructive pulmonary disease. Mediators Inflamm. 2013;2013:793505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ayush O, Jin ZW, Kim HK, Shin YR, Im SY, Lee HK. Glutamine up‐regulates MAPK phosphatase‐1 induction via activation of Ca(2+)–> ERK cascade pathway. Biochem Biophys Rep. 2016;7:10‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Eisenbarth SC, Piggott DA, Huleatt JW, Visintin I, Herrick CA, Bottomly K. Lipopolysaccharide‐enhanced, toll‐like receptor 4‐dependent T helper cell type 2 responses to inhaled antigen. J Exp Med. 2002;196(12):1645‐1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim YK, Oh SY, Jeon SG, et al. Airway exposure levels of lipopolysaccharide determine type 1 versus type 2 experimental asthma. J Immunol. 2007;178(8):5375‐5382. [DOI] [PubMed] [Google Scholar]
- 30. Choi IW, Sun K, Kim YS, et al. TNF‐alpha induces the late‐phase airway hyperresponsiveness and airway inflammation through cytosolic phospholipase A(2) activation. J Allergy Clin Immunol. 2005;116(3):537‐543. [DOI] [PubMed] [Google Scholar]
- 31. Kim SR, Kim DI, Kang MR, et al. Endoplasmic reticulum stress influences bronchial asthma pathogenesis by modulating nuclear factor kappaB activation. J Allergy Clin Immunol. 2013;132(6):1397‐1408. [DOI] [PubMed] [Google Scholar]
- 32. Im YN, Lee YD, Park JS, et al. GPCR kinase (GRK)‐2 is a key negative regulator of itch: l‐glutamine attenuates itch via a rapid induction of GRK2 in an ERK‐dependent way. J Invest Dermatol. 2018;138(8):1834‐1842. [DOI] [PubMed] [Google Scholar]
- 33. Ishak Gabra MB, Yang Y, Li H, et al. Dietary glutamine supplementation suppresses epigenetically‐activated oncogenic pathways to inhibit melanoma tumour growth. Nat Commun. 2020;11(1):3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bhutia YD, Ganapathy V. Glutamine transporters in mammalian cells and their functions in physiology and cancer. Biochim Biophys Acta. 2016;1863(10):2531‐2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yokomizo T, Izumi T, Shimizu T. Leukotriene B4: metabolism and signal transduction. Arch Biochem Biophys. 2001;385(2):231‐241. [DOI] [PubMed] [Google Scholar]
- 36. Fabre JE, Goulet JL, Riche E, et al. Transcellular biosynthesis contributes to the production of leukotrienes during inflammatory responses in vivo. J Clin Invest. 2002;109(10):1373‐1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hammer M, Mages J, Dietrich H, et al. Dual specificity phosphatase 1 (DUSP1) regulates a subset of LPS‐induced genes and protects mice from lethal endotoxin shock. J Exp Med. 2006;203(1):15‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhao Q, Wang X, Nelin LD, et al. MAP kinase phosphatase 1 controls innate immune responses and suppresses endotoxic shock. J Exp Med. 2006;203(1):131‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Salojin KV, Owusu IB, Millerchip KA, Potter M, Platt KA, Oravecz T. Essential role of MAPK phosphatase‐1 in the negative control of innate immune responses. J Immunol. 2006;176(3):1899‐1907. [DOI] [PubMed] [Google Scholar]
- 40. Chi H, Barry SP, Roth RJ, et al. Dynamic regulation of pro‐ and anti‐inflammatory cytokines by MAPK phosphatase 1 (MKP‐1) in innate immune responses. Proc Natl Acad Sci USA. 2006;103(7):2274‐2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Izumi T, Yokomizo T, Obinata H, Ogasawara H, Shimizu T. Leukotriene receptors: classification, gene expression, and signal transduction. J Biochem. 2002;132(1):1‐6. [DOI] [PubMed] [Google Scholar]
- 42. Corhay JL, Henket M, Nguyen D, Duysinx B, Sele J, Louis R. Leukotriene B4 contributes to exhaled breath condensate and sputum neutrophil chemotaxis in COPD. Chest. 2009;136(4):1047‐1054. [DOI] [PubMed] [Google Scholar]
- 43. Vachier I, Bonnans C, Chavis C, et al. Severe asthma is associated with a loss of LX4, an endogenous anti‐inflammatory compound. J Allergy Clin Immunol. 2005;115(1):55‐60. [DOI] [PubMed] [Google Scholar]
- 44. Higham A, Cadden P, Southworth T, et al. Leukotriene B4 levels in sputum from asthma patients. ERJ Open Res. 2016;2(4):00088‐2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sze E, Bhalla A, Nair P. Mechanisms and therapeutic strategies for non‐T2 asthma. Allergy. 2020;75(2):311‐325. [DOI] [PubMed] [Google Scholar]
- 46. Fitzpatrick AM, Chipps BE, Holguin F, Woodruff PG. T2‐"Low" Asthma: overview and management strategies. J Allergy Clin Immunol Pract. 2020;8(2):452‐463. [DOI] [PubMed] [Google Scholar]
- 47. Valdes AM, Andrew T, Gardner JP, et al. Obesity, cigarette smoking, and telomere length in women. Lancet. 2005;366(9486):662‐664. [DOI] [PubMed] [Google Scholar]
- 48. Mena S, Ortega A, Estrela JM. Oxidative stress in environmental‐induced carcinogenesis. Mutat Res. 2009;674(1–2):36‐44. [DOI] [PubMed] [Google Scholar]
- 49. Butterfield DA, Hensley K, Cole P, et al. Oxidatively induced structural alteration of glutamine synthetase assessed by analysis of spin label incorporation kinetics: relevance to Alzheimer's disease. J Neurochem. 1997;68(6):2451‐2457. [DOI] [PubMed] [Google Scholar]
- 50. Fisher MT, Stadtman ER. Oxidative modification of Escherichia coli glutamine synthetase. Decreases in the thermodynamic stability of protein structure and specific changes in the active site conformation. J Biol Chem. 1992;267(3):1872‐1880. [PubMed] [Google Scholar]
- 51. Levine RL. Oxidative modification of glutamine synthetase. II. Characterization of the ascorbate model system. J Biol Chem. 1983;258(19):11828‐11833. [PubMed] [Google Scholar]
- 52. Kirkham PA, Barnes PJ. Oxidative stress in COPD. Chest. 2013;144(1):266‐273. [DOI] [PubMed] [Google Scholar]
- 53. Hoenderdos K, Condliffe A. The neutrophil in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2013;48(5):531‐539. [DOI] [PubMed] [Google Scholar]
- 54. Quante T, Ng YC, Ramsay EE, et al. Corticosteroids reduce IL‐6 in ASM cells via up‐regulation of MKP‐1. Am J Respir Cell Mol Biol. 2008;39(2):208‐217. [DOI] [PubMed] [Google Scholar]
- 55. King EM, Holden NS, Gong W, Rider CF, Newton R. Inhibition of NF‐kappaB‐dependent transcription by MKP‐1: transcriptional repression by glucocorticoids occurring via p38 MAPK. J Biol Chem. 2009;284(39):26803‐26815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Grimes JM, Grimes KV. p38 MAPK inhibition: a promising therapeutic approach for COVID‐19. J Mol Cell Cardiol. 2020;144:63‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ripon MAR, Bhowmik DR, Amin MT, Hossain MS. Role of arachidonic cascade in COVID‐19 infection: a review. Prostaglandins Other Lipid Mediat. 2021;154:106539. [DOI] [PMC free article] [PubMed] [Google Scholar]