

Abstract
The vanillyl‐alcohol oxidase (VAO) family is a rich source of biocatalysts for the oxidative bioconversion of phenolic compounds. Through genome mining and sequence comparisons, we found that several family members lack a generally conserved catalytic aspartate. This finding led us to study a VAO‐homolog featuring a glutamate residue in place of the common aspartate. This 4‐ethylphenol oxidase from Gulosibacter chungangensis (Gc4EO) shares 42 % sequence identity with VAO from Penicillium simplicissimum, contains the same 8α‐N3‐histidyl‐bound FAD and uses oxygen as electron acceptor. However, Gc4EO features a distinct substrate scope and product specificity as it is primarily effective in the dehydrogenation of para‐substituted phenols with little generation of hydroxylated products. The three‐dimensional structure shows that the characteristic glutamate side chain creates a closely packed environment that may limit water accessibility and thereby protect from hydroxylation. With its high thermal stability, well defined structural properties and high expression yields, Gc4EO may become a catalyst of choice for the specific dehydrogenation of phenolic compounds bearing small substituents.
Keywords: biocatalysis, dehydrogenation, enzyme structure, flavoprotein, genome mining
Gc4EO, a new member of the VAO/PCMH family from the actinobacteria Gulosibacter chungangensis was discovered. This oxidase is a very appealing biocatalyst for the oxidation of 4‐alkylphenols, exhibiting high conversion yields with an attractive chemoselectivity towards the production of alkenes. Gc4EO showed high activity to produce 4‐vinylphenol, an interesting building block for the synthesis of polyvinylphenol.

Introduction
The fungal vanillyl‐alcohol oxidase (VAO) and its bacterial homolog eugenol oxidase (EUGO) are valuable biocatalysts to perform the oxidation of aromatic substrates derived from lignin degradation through biological or chemical processes.[ 1 , 2 , 3 , 4 , 5 ] They both belong to the 4‐alkylphenol oxidases, a subgroup of the more extensive VAO/PCMH family.[ 6 , 7 ] 4‐alkylphenol oxidases act on a broad spectrum of para‐substituted phenols. Kinetic and spectroscopic studies have demonstrated that the catalytic mechanism proceeds via the oxidation of the substrate Cα atom resulting in the formation of a p‐methide quinone intermediate.[ 5 , 8 ] The reaction is started by a hydride transfer from the substrate to the N5 position of the FAD. The two‐electron reduced FAD is then re‐oxidized by molecular oxygen yielding hydrogen peroxide, whereas the p‐quinone methide intermediate undergoes either hydroxylation or deprotonation (Scheme 1).[ 9 , 10 ] Given the potential of 4‐alkylphenol oxidases, we have carried out a sequence‐driven genome mining analysis, searching for new EUGO homologs of biocatalytic interest. Our search highlighted a putative EUGO from Gulosibacter chungangensis (Gc4EO) that was considered of particular interest for the natural variation of a highly conserved catalytic residue. The amino acid characterizing Gc4EO is Glu152 that corresponds to Asp151 and Asp170 of EUGO from Rhodococcus sp. strain RHA1 and VAO from Penicillium simplicissimum, respectively. Previous studies on VAO have shown that this residue acts as an active site base for the deprotonation of the p‐quinone methide intermediate and/or water activation in the case of hydroxylation.[ 11 , 12 ] Site‐directed mutagenesis had further demonstrated the crucial role of the aspartate side chain in product specificity and enantioselectivity. [13] In this work, we investigated Gc4EO and its intriguing sequence signature by taking advantage of an excellent expression system. Bioconversion analysis together with the structure determination allowed us to rationalize the substrate/product specificity and brought to light important similarities and differences between Gc4EO and the other well‐characterized enzymes of the 4‐alkylphenol oxidase subfamily.
Scheme 1.

A VAO‐like oxidase can produce phenolic alcohols (top) or alkenes (bottom). The alcohol can be further oxidized to the respective ketone.
Results and Discussion
A genome mining approach to identify new 4‐alkylphenol oxidases
We performed a genome mining analysis[ 14 , 15 , 16 ] using the sequence of EUGO from Rhodococcus sp. strain RHA1 as a template to discover new 4‐alkylphenol oxidases with natural variations in the catalytic Asp151 position. A cutoff between 45 % and 95 % amino acid sequence identity was initially used. Subsequently, likely aberrant sequences were manually discarded (e. g. shorter or longer sequences). Additionally, the targets containing a tyrosine in position homologous to Tyr384 of p‐cresol methylhydroxylase [17] were also discarded to prevent the selection of dehydrogenases rather than the intended oxidases. This first search led to a total of 232 EUGO‐like proteins. Using a 90 % identity cutoff (between the sequences), 69 putative oxidases were finally selected (Figures 1 and S1, Supporting Information). The catalytically relevant Asp151 was modestly conserved, showing variations for alanine and to lesser extent for glutamic acid and glycine (Figure 1).
Figure 1.

Cladogram of the 69 sequences obtained by genome mining. The molecular phylogenetic analysis was inferred by using the NJ method. The template sequence of EUGO Rhodococcus sp. strain RHA1 is in blue (RjEUGO; Uniprot accession number: Q0SBK1). PsVAO is VAO from Penicillium simplicissimum (Uniprot: P56216). The conservation of Asp151 (Rhodococcus EUGO) and its neighboring residues is shown at the bottom (21 Asp, 44 Ala, 3 Glu and 1 Gly in the selected 69 sequences).
Out of the 69 targets, the putative enzymes encoded by KAB1645308, RLA09811, PZN69483 and AMK59458 (NCBI accession codes) were chosen because their sequences contain natural variations in the catalytic aspartate position: Glu152 in KAB1645308, Gly150 in PZN69483 and Ala150 in RLA09811 and AMK59458 (Table S1). These putative oxidases showed high sequence identities with Rhodococcus EUGO and Penicillium VAO (37–52 %), conservation of the catalytic Tyr‐Tyr‐Arg triad (Tyr471, Tyr91, Arg472 in Rhodococcus EUGO), [18] and conservation of the His covalently bound to the flavin (His390 in Rhodococcus EUGO; Figure S2). We attempted the expression of all four putative enzymes in E. coli. RLA09811 and AMK59458 were soon discarded because it was not possible to obtain soluble proteins under any tested condition. Although successfully expressed, PZN69483 was discarded because of its low thermostability. The oxidase from Gulosibacter chungangensis (KAB1645308; named Gc4EO) was instead chosen for further studies given its excellent expression level and thermostability. Moreover, the protein seemed to be particularly interesting due to its genomic context (Figure S3). Downstream of its gene, two more genes of putative redox enzymes are present: i) 4‐hydroxyphenylacetate 3‐monooxygenase, predicted to be involved in the hydroxylation of 4‐phenolic compounds and ii) the gene for a putative styrene monooxygenase requiring an alkenylbenzene substrate, which could be the product of activity by the upstream Gc4EO. Such a genomic context indicates that the enzyme is involved in the metabolism of aromatic compounds. This notion is consistent with published evidence suggesting that Gulosibacter genus is involved in the aerobic biodegradation of phenols. [19]
Properties and stability of Gc4EO
Gc4EO can be expressed at high levels in E. coli NEB10β cells. From a 1 L culture, about 150–160 mg of yellow‐colored recombinant Gc4EO was purified. The expression levels of Gc4EO were the same of Rhodococcus EUGO (160 mg L−1) [4] and higher than expression of the fungal VAO (50 mg L−1).[ 4 , 20 ] The purified enzyme migrated as a single band on SDS‐PAGE. The protein showed a typical flavoprotein absorbance spectrum with absorption maxima at 350 nm and 436 nm (Figure 2). Covalent flavination was confirmed by SDS‐PAGE analysis resulting in a highly UV‐fluorescent band indicative of a covalently tethered flavin cofactor. SEC‐MALLS experiments were performed to determine the oligomeric state of the protein, in 10 mM Tris‐HCl, pH 7.5. The results suggested that tag‐less Gc4EO is a monodisperse dimer in solution (2×58,991 Da; Figure S4). Additionally, Gc4EO demonstrated to be a stable protein showing an apparent Tm of 65 °C in potassium phosphate buffer pH 7.5, as measured by the ThermoFAD method. [21] Such pronounced homogeneity and the high thermostability are obviously promising features for biocatalytic purposes.
Figure 2.

Purification and spectral characterization of Gc4EO. (A) Visible absorbance spectra of native Gc4EO (21 μM) and after incubating with 0.1 % w/v SDS (dashed line).
Substrate specificity and product characterization of Gc4EO
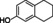
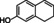
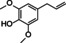
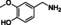
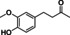
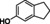
The substrate profile was investigated by analyzing the reactions by HPLC (Figure S5). 24‐Hour small‐scale conversions were carried out using 4.0 μM of Gc4EO and various 4‐alkylphenolic compounds at 1.0 mM concentration (Table 1). Gc4EO exhibited activity (conversion 50–99 %) on 12 out of 19 tested substrates. Gc4EO showed a poor conversion (<5 %) for 4‐alkylphenols with a para‐methyl substituent (e. g. p‐cresol) or a long aliphatic chain (e. g. 4‐n‐heptylphenol). Additionally, also α‐aryl ketones (4‐hydroxy‐3‐methoxy phenylacetone), di‐ortho methoxyphenols (4‐allyl‐2,6‐dimethoxyphenol) and medium‐chain amines (tyramine) were not efficiently converted by the enzyme. To confirm the identity of the reaction products, the samples were analyzed by H1‐NMR, HPLC and/or GC‐MS (Table 1, Figures S6–S8). As previously reported for Penicillium VAO, oxidation of vanillylamine and vanillyl alcohol by Gc4EO produced vanillin as final product. [18] As for Rhodococcus EUGO and Penicillium VAO, incubation of Gc4EO with eugenol yielded coniferyl alcohol although only 50 % conversion was achieved. All the other identified substrates (zingerone, 5‐indanol, 4‐ethylguaiacol, 4‐ethylphenol, 4‐cyclohexylphenol, 4‐cyclopentylphenol, 2‐methoxy‐4‐propylphenol, 4‐propylphenol, 5,6,7,8‐tetrahydro‐2‐naphtol) were mostly or exclusively converted to the corresponding vinylphenols. For instance, zingerone and 5‐indanol were converted to the α,β‐unsaturated phenolic ketone and cycloalkene products, respectively. Hydroxylated products were identified only in the conversions of 2‐methoxy‐4‐ethyllphenol and 4‐ethylguaiacol (2‐methoxy‐4‐propylphenol) but in both reactions they were present in small amounts (Figures S5, S7 and S8). This feature clearly differentiates Gc4EO from VAO, EUGO and p‐cresol methyl hydroxylase that preferentially catalyze the oxidation of p‐substituted phenols into the corresponding α‐hydroxy benzyl phenols. The marked preference towards dehydrogenation featured by Gc4EO can be very attractive to produce phenolic alkenes.
Table 1.
Substrate and product profile of Gc4EO.[a]
|
Substrate |
Structure |
Conv. [%] |
Products |
||
|---|---|---|---|---|---|
|
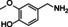
vanillylamine |
|
99 % |
|
|
|
|
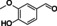
zingerone |
|
99 % |
|
|
|
|
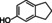
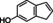
5‐indanol |
|
99 % |
|
|
|
|
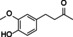
4‐ethylguiacol |
|
99 % |
|
|
|
|
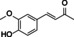
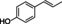
eugenol |
|
50 % |
|
|
|
|
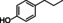
vanillyl alcohol |
|
99 % |
|
|
|
|
4‐ethylphenol |
|
99 % |
|
|
|
|
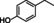
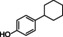
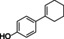
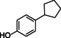
4‐cyclohexylphenol |
|
95 % |
|
|
|
|
2‐methoxy‐4‐propylphenol |
|
99 % |
|
|
|
|
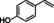
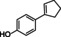
4‐cyclopentylphenol |
|
99 % |
|
|
|
|
4‐propylphenol |
|
99 % |
|
|
|
|
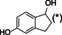
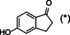
5,6,7,8‐tetrahydro‐2‐naphtol |
|
99 % |
|
|
|
|
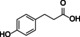
3‐(4‐hydroxyphenyl) propionic acid |
|
n.c. |
|
|
|
|
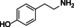
tyramine |
|
n.c. |
|
|
|
|
4‐hydroxy‐3‐methoxyphenylacetone |
|
n.c. |
|
|
|
|
4‐n‐heptylphenol |
|
n.c. |
|
|
|
|
p‐cresol |
|
n.c. |
|
|
|
|
p‐creosol |
|
n.c. |
|
|
|
|
4‐allyl‐2,6‐dimethoxyphenol |
|
n.c. |
|||
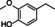
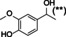
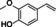
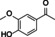
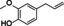
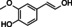
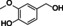
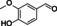
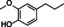
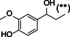
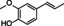
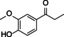
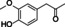
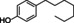
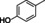
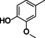
[a] For conversions, 4.0 μM Gc4EO was incubated with 1.0 mM substrate in air‐saturated 50 mM potassium phosphate buffer, pH 8.0 at 25 °C for 24 h. Product analysis was done by HPLC and GC‐MS (Figure S8). (*) Indicates likely side products (Figure S5C), not verified by GC‐MS. Conversions lower than 5 % are indicated as n.c. (**) The alcohols were produced in lower abundancy (Figures S6 and S7) and their chirality was not further investigated.
To complement the bioconversion data, we evaluated the initial oxidase rates of Gc4EO on a set of substrates (Table 2, Figure S9). The highest rate was found towards 4‐ethylphenol, showing a k cat of 4.1 s−1 and a catalytic efficiency of 45.5 s−1 mM−1. For the other 4‐alkylphenols, Gc4EO exhibited good specific activities towards vanillyl alcohol, 5‐indanol, 4‐ethyl guaiacol and eugenol (2.9, 2.3, 0.9 and 0.7 s−1, respectively). For vanillylamine and zingerone, Gc4EO showed poor catalytic activities (0.01–0.02 s−1). The data indicate that Gc4EO is optimally active with a broad range of para‐substituted phenolic compounds bearing short alkyl/cycloalkyl‐side chains in para‐position.
Table 2.
Steady‐state kinetic parameters for Gc4EO.[a]
|
Substrate |
k cat [s−1] |
K M [μM] |
k cat/K M [s−1 mM−1] |
|
|---|---|---|---|---|
|
vanillylamine |
|
0.017 |
350 |
0.05 |
|
zingerone |
|
0.012 |
30 |
0.4 |
|
5‐indanol |
|
2.3 |
425 |
5.4 |
|
4‐ethylguaiacol |
|
0.87 |
60 |
14.5 |
|
eugenol |
|
0.75 |
9.5 |
73.6 |
|
vanillyl alcohol |
|
2.9 |
220 |
20.4 |
|
4‐ethylphenol |
|
4.1 |
90 |
45.5 |
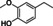
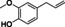
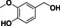
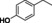
[a] The kinetic parameters were measured at 25 °C in 50 mM potassium phosphate buffer pH 8.0. The errors are within 10 % of the measured values (Figure S9).
Effect of the temperature on Gc4EO conversion efficiency
Since 4‐ethylphenol behaved as the best substrate of Gc4EO, we monitored 4‐vinylphenol production under varying conditions by increasing substrate concentrations (1–100 mM) and temperatures (25 °C, 37 °C and 50 °C) (Table S2). The oxidase achieved full conversion of 4‐ethylphenol at all three temperatures for substrate concentrations up to 5 mM (TON=1,250). By increasing the concentration of 4‐ethylphenol to 100 mM, the conversion dropped to 50 % (TON=16,000). In general, the catalyst demonstrated to easily operate in a broad range of temperatures whereas conversions at high substrate concentrations conditions will probably require further optimization such a more dedicated reactor setup to support the reaction with enough oxygen supply.
The three‐dimensional structure of Gc4EO
To shed light on the characteristic sequence fingerprint of this new oxidase, we determined the crystal structure of Gc4EO in the native state and in complex with isoeugenol at 1.7 Å and 2.8 Å resolution, respectively (Figure 3A and Table S3; PDB codes 7PBG and 7PBI). As observed in solution, the enzyme is dimeric (Figures 3B and S4). The dimer interface creates a funnel‐shaped path that likely represents the accession route for the diffusion of the substrates into the catalytic pocket (Figure 3C). The structure of the enzyme with isoeugenol shows that the ligand is buried inside a closed and round cavity of approximately 412 Å3[22–23] located on the si‐side of the flavin ring (Figure 3C). As a result, the ligand is mostly embedded within the active site with the Cα atom sitting right in front of the N5 atom of the flavin ring (∼3.1 Å) as expected for a redox mechanism that involves a hydride transfer between the substrate and the cofactor. [23] The hydroxy group of the ligand is hydrogen bonded with Tyr92, Tyr476 and Arg477 (Figure 3D). This Tyr‐Tyr‐Arg triad represents an anion binding cage, stabilizing the phenolate form of the substrate. Binding of isoeugenol involves Van der Waals interactions with the protein residues delimiting the ligand binding site. The aromatic ring of the ligand sits on top of the isoalloxazine ring of FAD and establishes a T‐shaped π‐π stacking with Phe397 (Figure 3E). Furthermore, the methoxy substituent interacts with the side chains of Val167 and Ile432 whereas the propenyl side chain establishes contacts with Phe283 and Phe397. It is interesting to observe that ligand binding induces a localized conformational change resulting in the movement of Tyr476 that shifts in order to form a hydrogen bond with the hydroxy group of isoeugenol (Figure 3D).
Figure 3.

The crystal structure of Gc4EO. (A) Weighted 2Fo−Fc electron density map showing the covalent bond between the C8 M of FAD and His395 (subunit A). The electron‐density map of the native enzyme shows a strong peak at ∼3.3 Å from the re‐side of the flavin. We have interpreted this feature as due to a bound Cl− ion. The contour level of the map is 1.0 σ. The carbon atoms of FAD are represented in yellow, oxygen atoms in red and phosphorus atoms in orange. The chloride ion is shown in green. (B) The two subunits of the Gc4EO dimer are shown in white‐blue and sand, respectively. The FAD carbons are shown in yellow. (C) Sliced view of the Gc4EO surface in the same orientation as in A. Isoeugenol is represented with the same color code used for the cofactor, except for the carbon atoms shown in green. (D–E) Side chains in direct contact with isoeugenol. Tyrosine 476 shifts its position as compared with the unligated structure to form a hydrogen bond with the hydroxy group of isoeugenol. Hydrogen bonds are shown as dashed lines. Protein, ligand, and FAD carbon atoms are colored in deep teal, green, and yellow, respectively, oxygen atoms are in red, nitrogen atoms are in blue, and phosphorus atoms are in orange.
The overall conformation of Gc4EO is very similar to that of EUGO and VAO with rsmd values of 1.1 Å and 1.3 Å for the Cα atoms and 52 % and 42 % sequence identity, respectively. The main difference is the quaternary structure of VAO that forms a stable octamer due to the presence of a loop at the dimer‐dimer interface that is lacking in both Gc4EO and EUGO (Figure S2).[ 25 , 26 , 27 ] Gc4EO, VAO and EUGO share the same covalent bond occurring between the C8 M atom of FAD and the side chain of His395 (equivalent of His390 in EUGO and His422 in VAO; Figures 3A and S2). However, several critical differences in the active sites of these enzymes can be noted. Gc4EO shares the same ligand position and orientation with VAO whereas isoeugenol bound to Gc4EO appears to be flipped when compared to the orientation observed in EUGO (Figure 4). The different orientation of the ligand is caused by a steric effect due to the presence of the bulkier Phe397 (Phe424 in VAO; Figure 4E) in place of Gly392 of EUGO (Figure 4B). The presence of Phe397 further influences the substrate specificity of Gc4EO. As seen for VAO, [25] the substrates featuring a double methoxy substitution as 4‐allyl‐2,6‐dimethoxyphenol are not accepted by the enzyme (Table 1). Conversely, EUGO is able to promote catalysis on these di‐methoxy‐substituted substrates, though with modest catalytic efficacy. [23] Looking at the backbone conformation of Gc4EO, we noticed an additional feature promoting the orientation of the ligand: the β‐strand around the active site is shifted in Gc4EO compared to EUGO and VAO structures with a 2.2 Å shift at the level of Ile432 (Ile427 in EUGO and Thr459 in VAO) (Figure 4C and 4F). The shift of the enzyme backbone creates more space for the flipped methoxy group of isoeugenol in Gc4EO. A common feature between Gc4EO and EUGO is their preference for short‐chain alkyl phenols substrates. Consistently, the residues facing the substrate alkyl side chain in Gc4EO are generally more like those of EUGO than VAO (Figure 4B and 4E). Specifically, the residues are less bulky in VAO, creating more space for longer alkyl side chains. In summary, the aromatic‐binding niche of the Gc4EO active site is similar to that of VAO whereas the alkyl side chain binding site more closely resembles that of EUGO.
Figure 4.

Active site comparison. (A–C) The substrate‐binding site of Gc4EO in complex with isoeugenol (green) is superposed onto that of EUGO in complex with the same ligand (dark grey; PDB ID: 5FXD). Residues are labeled following the scheme Gc4EO/EUGO. (A) Tyr92‐Arg477‐Tyr476 facing the ligand hydroxy group and Val167‐Tyr169‐Ile432 facing the methoxy group of isoeugenol are conserved. (B) The natural variation F397/G392 promotes the 180° flipped orientation of the ligand. (C) The backbone shift (∼2.2 Å) at position Ile432 of Gc4EO. Carbon atoms of Gc4EO and EUGO are represented in deep tail and grey, respectively. (D–F) The substrate‐binding site of Gc4EO in complex with isoeugenol (green) is superposed onto that of VAO in complex with the same ligand (purple; PDB ID: 2VAO). Residues are labeled following the scheme Gc4EO/VAO. (D) The superposition of the Gc4EO/VAO residues around the isoeugenol aromatic ring. (E) The binding sites for the propenyl side chain of isoeugenol show a largely different amino acidic composition. (F) The backbone shift of Ile432 of Gc4EO (Thr459 in VAO) that directly interacts with the methoxy group of isoeugenol as shown in panel D. Carbon atoms of Gc4EO and VAO are represented in deep tail and salmon, respectively.
A key difference between Gc4EO and EUGO/VAO is the natural variation of a crucial residue involved in catalysis: Glu152 of Gc4EO corresponds to an aspartate in VAO and EUGO (Asp170 and Asp151, respectively) (Figure S2). One of the main goals of our study was to evaluate if the presence of this unusual residue was associated with characteristic structural features that might have an implication in tuning the enzyme reactivity. The Glu152 side chain of Gc4EO directly interacts with Gln430 creating a more densely packed environment compared to EUGO where the conserved Gln425 interacts with the smaller Asp151 resulting in a looser and more opened niche around the Cα atom of the substrate (Figure 5) This effect is even more pronounced in VAO where Asp170 is associated to a shorter Thr457 compared to the homologous Gln430/Gln425 of Gc4EO/EUGO. From these structural features, our hypothesis is that Glu152 selectively reduces the water accessibility to the propenyl Cα atom of the p‐quinone methide intermediate due to the bulkiness of its side chain. With its longer side chain, Glu152 may also better function as a base to de‐protonate the Cβ of the p‐quinone methide being at ∼3.8 Å distance (Figure 5). The presence of Glu152 may not be the only element that mitigates intermediate hydroxylation in Gc4EO. Phe397, close to the substrate ring edge, may create a closer environment when combined with the Gln430‐Glu152 pair on top of the substrate (Figure 4B and 4E). The reduced water accessibility, caused by these structural features, is regioselective for the Cα carbon because water can access the Cγ as it happens in eugenol‐to‐coniferyl alcohol oxidation catalyzed by Gc4EO (Table 1). Indeed, the crystallographic structure of Gc4EO shows that Cγ can be accessible to water, due to the presence of the small Ser383 side chain (Figures 4B, 4E and 5). Collectively, these findings reinforce the idea that Gc4EO is tailored for small phenolic substrates to mainly produce dehydrogenated products.
Figure 5.
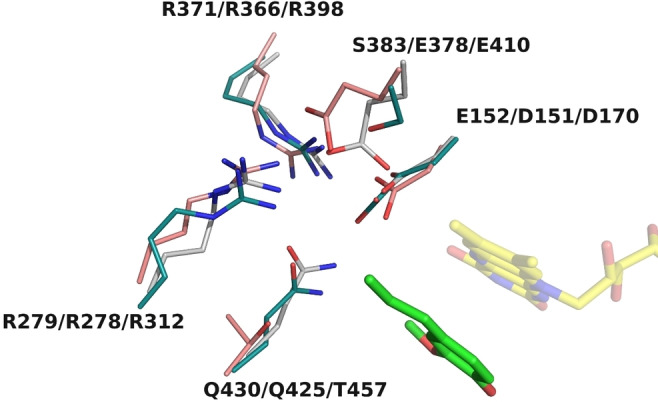
The characteristic of Glu152 of Gc4EO. The highly conserved Asp151/Asp170 in EUGO/VAO is replaced by a bulkier Glu152 in Gc4EO creating a more densely packed environment around at the Cα carbon of the ligand.
Conclusion
Our work shows that Gc4EO can be well expressed as recombinant protein using E. coli. In addition to the straightforward expression and purification, the enzyme is thermostable (Tm app=65 °C), readily crystallizable and harbors a covalently attached flavin cofactor (eliminating the risk of cofactor loss). We demonstrate that the enzyme can perform conversions at 55 °C with medium substrate concentrations (1‐50 mM). Substrate profiling revealed that Gc4O is active on a broad spectrum of short‐chain phenolic compounds mainly acting as a dehydrogenase. The specificity of the enzyme is in line with the active site architecture. Particularly, the presence of the Glu152 in place of the more usual aspartate modulate substrate and product selectivity. Therefore, Gc4EO represents an excellent biocatalyst for the dehydrogenation of 4‐ethylphenol and similar small‐chain alkyl phenols such as those derived from the degradation of lignin‐containing biomasses. Therefore, it may develop as a valuable easy‐to‐use biocatalyst to modify lignin‐derived compounds.
Experimental Section
Sequence analysis: The sequence of Rhodococcus sp. strain RHA1 EUGO (Uniprot accession code Q0SBK1) was used as template for a sequence‐driven genome mining using the NCBI server for BLAST searches. After initial screening, 232 sequences were obtained by selecting targets with an amino acid sequence identity between 45–90 % with Rhodococcus EUGO. Subsequently, redundancy within the targets was reduced by using the CD‐HIT package. [27] The sequences of the putative flavin‐dependent oxidases were clustered and filtered using a sequence identify cutoff of 90 %. Multiple sequence alignments and the phylogenetic analyses were performed for the resulting 69 sequences. Both data analyzes were performed using CLC Sequence Viewer software V. 8.0. The phylogenetic tree was reconstructed using Neighbor Joining algorithm and Jukes‐Cantor for distance measures (500 bootstrap replications). Searches using NCBI and KEGG servers were performed for the identification and prediction of the biological function of the genetic context around the putative oxidase found on G. chungangensis.
Expression and purification of Gc4EO: The sequences of the selected genes (Table S1) were codon optimized for E. coli and ordered for synthesis with flanking BsaI sites. The genes were cloned into pBAD‐SUMO by Golden Gate assembly and transformed in chemocompetent E. coli NEB 10β cells. The plasmids were isolated and sequenced for confirmation. For expression of the enzyme candidates, single colonies harboring the respective vectors were grown in LB medium supplemented with ampicillin (50 μg mL−1) at 37 °C overnight. For expression and purification of Gc4EO, single colonies harboring the pBAD‐His6‐SUMO‐Gc4EO vector were grown in TB medium supplemented with 50 μg mL−1 ampicillin at 37 °C and 200 rpm until the O.D600 was 0.6–0.7. Protein expression was induced adding 0.02 % w/v of L‐arabinose and cells were grown for 20 h at 24 °C. Cells were harvested by centrifugation (5,000×g, 15 min, 10 °C) and the pellet was resuspended in lysis buffer (50 mM Tris‐HCl pH 8.0, 150 mM NaCl, 5 mM imidazole, 1 mg mL−1 lysozyme, 10 μM FAD) including additional protease inhibitors: phenylmethylsulfonyl fluoride (1 mM), leupeptin (10 μM), pepstatin (10 μM), and 1 mg DNase I per 50 mL. Cell lysis was conducted using sonication using the following condition: pulse 5 s on, 25 s off, with a total sonication time of 2 min and 30 % amplitude. Lysed cells were centrifuged (56,000×g, 1 h, 4 °C) and the supernatant was collected and filtered (0.45 μm) prior to be loaded onto the HisTrap HP column (5 mL of resin, Cytiva) pre‐equilibrated with Buffer A (50 mM Tris‐HCl pH 8.0, 150 mM NaCl, 5 mM imidazole). The His‐tagged protein was eluted with elution buffer (50 mM Tris‐HCl pH 8.0, 150 mM NaCl, 300 mM imidazole) and concentrated down to a final volume of 1–2 mL. Subsequently, the sample was incubated with HisX6‐tagged SUMO protease (1.1 mg mL−1) to a volume ratio of 1 : 100 and dialyzed overnight using 10k dialysis cassette (Thermofisher) to remove the imidazole. After buffer exchange, the protein was then load onto a HisTrap column (5 mL, Cytiva) to perform a reverse‐nickel purification. The column was pre‐equilibrated with Buffer A with the protein eluting immediately after one column volume. The tag‐less protein was concentrated to a final volume of 500 μL and loaded onto a gel filtration column (Superdex 200 10/300, Cytiva) pre‐equilibrated with 10 mM Tris‐HCl, pH 7.5 at 4 °C. The protein eluted with a very high purity and homogeneity and an elution volume of 10–10.5 mL.
UV‐visible spectra: UV‐visible spectra of Gc4EO were recorded using a V‐660 Jasco spectrophotometer instrument. To determine the molar extinction coefficient of the protein‐bound FAD, the protein sample was supplemented with SDS 0.1 % w/v (final concentration) and incubated for 10’ (same volume of MQ water was added to the control solution). After centrifugation (1’ at 14,000 g), the UV‐visible spectrum of the unfolded protein was recorded and the FAD concentration was estimated using nm=11,300 M−1 cm−1. [29] The calculated value for the molar extinction coefficient for Gc4EO was 12,800 M−1 cm−1 at 450 nm and 14,440 M−1 cm−1 at 435 nm.
Steady‐state kinetics: Solution mixtures of 300 μL with increasing concentrations of substrates (final 2.5 % v/v DMSO) in 50 mM potassium phosphate buffer pH 8.0 and 150 mM NaCl were measured in the presence of 30–100 nM of enzyme. Reactions were followed spectrophotometrically using a SynergyMX micro‐plate reader (BioTek) at 25 °C. UV compatible plates were used when necessary. Activity with vanillyl alcohol and vanillylamine was determined by measuring the formation of vanillin ( 340nm=14,000 M−1 cm−1). Coniferyl alcohol formation was followed at 296 nm ( 296nm=6,800 M−1 cm−1), while activity against 4‐ethylguaiacol and 5‐indanol was determined by measuring the increase of absorption at 255 nm ( 250nm=50,000 M−1 cm−1) and 300 nm ( 300nm=11,500 M−1 cm−1), respectively. Zingerone consumption was followed at 336 nm ( 336nm=18,300 M−1 cm−1). [4] 4‐Vinylphenol formation was measured using a calibration curve from product enzymatically produced by Gc4EO (254 nm).
Thermostability determination of Gc4EO: Analysis of thermostability was determined by the ThermoFAD method. [4] Samples (20 μL) with purified Gc4EO were prepared in a 96‐well PCR plate. The samples contained 1 mg mL−1 enzyme in 50 mM potassium phosphate buffer pH 8.0 and 150 mM NaCl, adjusted at different cosolvents concentrations. Then, the plate was heated from 20 to 95 °C, increasing the temperature by 0.5 °C every 10 s, using an RT‐PCR instrument (CFX96‐Touch, Bio‐Rad). The apparent melting temperature for each condition was determined as the maximum of the derivative of the sigmoidal curve of the obtained thermogram (450–490 nm excitation filter and a 515–530 nm emission filter).
Small‐scale bioconversions: For the determination of the biocatalytic profile of Gc4EO, small‐scale conversions of 500 μL in 20 mL vials were carried out in air‐saturated 50 mM potassium phosphate buffer pH 8.0. Formulations were prepared with 4 μM Gc4EO and different 4‐alkylphenol substrates at final 2 % v/v DMSO. Reactions were incubated at 24 °C for 24 h or 48 h with constant shaking. For 4‐ethylphenol and 4‐ethylguaiacol, higher substrate concentrations were tested (1–100 mM), DMSO was adjusted up to 10 %. Mixtures were incubated for 24 h or 48 h at 24, 37 or 55 °C with constant agitation.
HPLC analysis: Samples from the small‐scale conversion were analyzed by HPLC at 280 nm (Jasco UV‐2075 UV/Vis detector) using MQ (0.1 % v/v formic acid) and acetonitrile as eluents. Separation was carried out using a C18 column (Alltima HP 5 μm C18). Prior the injection, sample were diluted 1 : 4 with acetonitrile and vigorously vortexed. Subsequently, samples were centrifuged (5’ at 14,000 g) and diluted with acetonitrile at final concentrations below 1.0 mM. Reactions using vanillylamine and vanillyl alcohol were compared with a standard solution of vanillin.
GC‐MS analysis: Products from small‐scale conversions were analyzed by GC‐MS. The reaction mixtures were extracted by mixing three times one volume of ethyl acetate containing 0.1 % v/v mesitylene as an external standard for 45 s (with or without previous supplementation with 10 M HCl). Then, to remove residual water, anhydrous sulfate magnesium was added to the organic solution. Separation and product identification was carried out using a GCMS‐QP2010 Ultra instrument (Shimadzu) (equipped with electron ionization and quadrupole separation) with a HP‐1 column and helium as mobile phase. The temperature method was a gradient from 50 to 250 °C at 10 °C min−1.
1 H‐NMR spectroscopy: For 1H‐NMR analysis, small‐scale conversions of 15 mL were performed using 2 mM 4‐ethylphenol or 4‐ethylguaiacol as substrate. Extraction was performed three times with ethyl acetate, dried over anhydrous sulfate magnesium and concentrated by rota‐evaporation. The extracts were suspended in 0.5 mL CDCl3 and NMR analysis was performed in a 600 MHz Varian Unity Plus spectrometer.
Statistical analysis: Kinetic parameters were obtained by fitting the obtained data to the Michaelis–Menten equation using GraphPad Prism v6.05 for Windows (GraphPad Software, La Jolla, CA, United States). Chromatograms from HPLC were analyzed using ChromNav. GC chromatograms and MS spectra were analyzed using GCMSsolution Postrun Analysis 4.11 (Shimadzu).
Protein crystallization, X‐ray data collection, and structure determination: Purified Gc4EO was concentrated to 25 mg mL−1 in 10 mM Tris‐HCl buffer pH 7.5 at 4 °C. Crystallization was performed using the vapor‐diffusion sitting‐drop technique at 20 °C by mixing equal volumes of protein and precipitant solution consisting of 0.1 M Trizma pH 8.0 (Sigma‐Aldrich), 24 % w/v PEG 6000 (Merck) which led to big yellow crystals after 4 weeks. Crystals of Gc4EO in combination with 5 mM isoeugenol were obtained in vapor‐diffusion sitting‐drop at 20 °C using the protein sample concentrated to 15 mg mL−1 in 10 mM Tris‐HCl pH 7.5 and mixed with the precipitant solution containing 0.2 M NaCl and 2.2 M (NH4)2SO4. The Gc4EO‐isoeugenol complex was prepared by soaking crystals (45 min) in cryoprotectant solutions consisting of 0.2 M NaCl, 2.4 M (NH4)2SO4, 20 % v/v glycerol and 5 mM isoeugenol. X‐ray diffraction data used for structure determination and refinement were collected at the PXI and PXIII beamlines of the Swiss Light Source in Villigen (SLS), Switzerland. Data were scaled using the XDS [30] program and CCP4 [31] package to process the data. The crystal structure of Gc4EO was solved by molecular replacement (Phaser MR) using the coordinates of Eugenol Oxidase from Rhodococcus jostii RHA1 (PDB entry5FXD) [22] as a search model excluding the ligand and water molecules. Manual building, the addition of water molecules, and crystallographic refinement were performed with COOT [32] and REFMAC5 [33] from the CCP4 suite. Figures were created with PyMOL (DeLano Scientific; www.pymol.org) and Chimera. [34]
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
This research was supported by the Bio Based Industries Joint Undertaking under the European Union's Horizon 2020 research and innovation program under Grant Agreement No. 837890 (SMARTBOX). We thank the European Synchrotron Radiation Facility (ESRF) and the Swiss Light Source (SLS) for providing beamtime and assistance. Open Access Funding provided by Universita degli Studi di Pavia within the CRUI‐CARE Agreement.
L. Alvigini, A. Gran-Scheuch, Y. Guo, M. Trajkovic, M. Saifuddin, M. W. Fraaije, A. Mattevi, ChemBioChem 2021, 22, 3225.
Contributor Information
Prof. Dr. Marco W. Fraaije, Email: m.w.fraaije@rug.nl.
Prof. Dr. Andrea Mattevi, Email: andrea.mattevi@unipv.it.
References
- 1. Liao Y., Koelewijn S. F., van den Bossche G., van Aelst J., van den Bosch S., Renders T., Navare K., Nicolaï T., van Aelst K., Maesen M., Matsushima H., Thevelein J. M., Van Acker K., Lagrain B., Verboekend D., Sels B. F., Science 2020, 367, 1385–1390. [DOI] [PubMed] [Google Scholar]
- 2. Van Aelst K., van Sinay E., Vangeel T., Cooreman E., van den Bossche G., Renders T., van Aelst J., van den Bosch S., Sels B. F., Chem. Sci. 2020, 11, 11498–11508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. de Jong E., van Berkel W. J., van der Zwan R. P., de Bont J. A., Eur. J. Biochem. 1992, 208, 651–657. [DOI] [PubMed] [Google Scholar]
- 4. Jin J., Mazon H., van den Heuvel R. H., Janssen D. B., Fraaije M. W., FEBS J. 2007, 274, 2311–2321. [DOI] [PubMed] [Google Scholar]
- 5. Fraaije M. W., Veeger C., van Berkel W. J. H., Eur. J. Biochem. 1995, 234, 271–277. [DOI] [PubMed] [Google Scholar]
- 6. Leferink N. G. H., Heuts D. P. H. M., Fraaije M. W., van Berkel W. J. H., Arch. Biochem. Biophys. 2008, 474, 292–301. [DOI] [PubMed] [Google Scholar]
- 7. Fraaije M. W., van Berkel W. J. H., Benen J. A., Visser J., Mattevi A., Trends Biochem. Sci. 1998, 23, 206–207. [DOI] [PubMed] [Google Scholar]
- 8. Fraaije M. W., van Berkel W. J. H., J. Biol. Chem. 1997, 272, 18111–18116. [DOI] [PubMed] [Google Scholar]
- 9. Chaiyen P., Fraaije M. W., Mattevi A., Trends Biochem. Sci. 2012, 37, 373–380. [DOI] [PubMed] [Google Scholar]
- 10. Mattevi A., Fraaije M. W., Mozzarelli A., Olivi L., Coda A., van Berkel W. J. H., Structure 1997, 5, 907–920. [DOI] [PubMed] [Google Scholar]
- 11. van den Heuvel R. H. H., Fraaije M. W., Mattevi A., van Berkel W. J. H., J. Biol. Chem. 2000, 275, 14799–14808. [DOI] [PubMed] [Google Scholar]
- 12. van den Heuvel R. H. H., Fraaije M. W., van Berkel W. J. H., FEBS Lett. 2000, 481, 109–112. [DOI] [PubMed] [Google Scholar]
- 13. van den Heuvel R. H. H., Fraaije M. W., Mattevi A., Ferrer M., van Berkel W. J. H., Proc. Natl. Acad. Sci. USA 2000, 97, 9455–9460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gran-Scheuch A., Trajkovic M., Parra L., Fraaije M. W., Frontiers Microbiol. 2018, 9, 1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jiang J., Tetzlaff C. N., Takamatsu S., Iwatsuki M., Komatsu M., Ikeda H., Cane D. E., Biochemistry 2009, 48, 6431–6440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.A. Zaparucha, V. de Berardinis, C. Vaxelaire-Vergne, Modern Biocatalysis: Advances Towards Synthetic Biological Systems 2018, 1–27.
- 17. Cunane L. M., Chen Z.-W., Shamala N., Matthews F. S., Cronin C. N., McIntire W. S., J. Mol. Biol. 2000, 295, 357–374. [DOI] [PubMed] [Google Scholar]
- 18. Ewing T. A., Gygli G., Fraaije M. W., van Berkel W. J. H., The Enzymes 2020, 47, 87–116. [DOI] [PubMed] [Google Scholar]
- 19. Zhai Z., Wang H., Yan S., Yao J., J. Chem. Technol. Biotechnol. 2012, 87, 105–111. [Google Scholar]
- 20. Benen J. A., Sánchez-Torres P., Wagemaker M. J., Fraaije M. W., van Berkel W. J. H., Visser J., J. Biol. Chem. 1998, 273, 7865–7872. [DOI] [PubMed] [Google Scholar]
- 21. Forneris F., Orru R., Bonivento D., Chiarelli L. R., Mattevi A., FEBS J. 2009, 276, 2833–2840. [DOI] [PubMed] [Google Scholar]
- 22. Nguyen Q. T., De Gonzalo G., Binda C., Rioz-Martínez A., Mattevi A., Fraaije M. W., ChemBioChem 2016, 17, 1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.N. R. Voss, M. Gerstein, Nucleic Acids Res. 2010, 38 (Web Server issue): W555–W562. [DOI] [PMC free article] [PubMed]
- 24. Fraaije M. W., van den Heuvel R. H. H., Roelofs J. C. A. A., van Berkel W. J. H., Eur. J. Biochem. 1998, 253, 712–719. [DOI] [PubMed] [Google Scholar]
- 25. Fraaije M. W., Veeger C., van Berkel W. J. H., Eur. J. Biochem. 1995, 234, 271–277. [DOI] [PubMed] [Google Scholar]
- 26. Holm L., Bioinformatics 2019, 35, 5326–5327. [DOI] [PubMed] [Google Scholar]
- 27. Ewing T. A., Gudrun G., van Berkel W. J. H., FEBS J. 2016, 283, 2546–2559. [DOI] [PubMed] [Google Scholar]
- 28. Huang Y., Niu B., Gao Y., Fu L., Li W., Bioinformatics 2010, 26, 680–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Macheroux P., Methods Mol. Biol. 1999, 1–7. [DOI] [PubMed] [Google Scholar]
- 30. Kabsch W., Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Collaborative Computational Project, Number 4, Acta Crystallogr. D Biol. Crystallogr. 1994, 50, 760–763.
- 32. Jung W. S., Singh R. K., Lee J. K., Pan C. H., PLoS One 2013, 8, 2–11. [Google Scholar]
- 33.G. N. Murshudov, P. Skubák, A. A. Lebedev, N. S. Pannu, R. A. Steiner, R. A. Nicholls, M. D. Winn, F. Long, A. A. Vagin, Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 355–367. [DOI] [PMC free article] [PubMed]
- 34. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E., J. Comput. Chem. 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- 35. Park M. H., Traiwan J., Jung M. Y., Kim W., Int. J. Syst. Evol. 2012, 62, 1055–1060. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information