

Summary
Background
Tofacitinib is an oral Janus kinase inhibitor for the treatment of ulcerative colitis (UC). Post‐marketing surveillance (PMS) is an important part of monitoring adverse events (AEs).
Aims
To report an analysis of PMS case safety reports for tofacitinib in patients with UC
Methods
Worldwide tofacitinib PMS reports received in the Pfizer safety database from 30 May 2018 (first regulatory approval) to 25 August 2020 were analysed. The type and estimated reporting rate (RR) of serious AEs of interest, including infection, gastrointestinal, vascular, respiratory, neoplasm and cardiac events, were reviewed. Patient‐years of exposure (PY) was estimated based on worldwide sales data and the calculated daily regimens of tofacitinib 5 or 10 mg twice daily, immediate‐ or extended‐release formulations.
Results
During the 27‐month reporting period, worldwide post‐marketing exposure to tofacitinib was 8916 PY. Overall, 4226 case reports were received and included 12 103 AEs, of which 1839 were serious AEs (SAEs). Among the cases reported, 1141 (27.0%) included an SAE and 18 (0.4%) were fatal. The RR (per 100 PY) for SAEs of interest by Medical Dictionary for Regulatory Activities System Organ Class were 3.28 for infections, 1.26 for vascular disorders, 0.74 for respiratory disorders, 0.55 for neoplasms and 0.50 for cardiac disorders.
Conclusions
The types of AEs were consistent with those reported in tofacitinib clinical trials. Most reported AEs were non‐serious. Limitations of PMS reports and reliance on estimated RRs due to lack of precise values for exposure, required for incidence rate calculation, should be considered when interpreting these results.
Worldwide tofacitinib post‐marketing safety reports received in the Pfizer safety database from 30 May 2018 (first regulatory approval) to 25 August 2020 were analysed.
The type and estimated reporting rate of serious adverse events of interest, including gastrointestinal, infection, vascular, respiratory, neoplasm and cardiac events, were reviewed.
Patient‐years of exposure was estimated based on worldwide sales data.
The types of adverse events and reporting rates were consistent with the known tofacitinib safety profile.

1. INTRODUCTION
Ulcerative colitis (UC) is a chronic inflammatory disease of the colon and rectum which follows a relapsing and remitting course. 1 Tofacitinib is an oral Janus kinase (JAK) inhibitor for the treatment of UC. The efficacy and safety of tofacitinib in adults with moderately to severely active UC has been evaluated in clinical trials, including a dose‐ranging phase 2 induction trial, 2 two 8‐week phase 3 induction trials (OCTAVE Induction 1 and 2), 3 a 52‐week phase 3 maintenance trial (OCTAVE Sustain) 3 , and an open‐label, long‐term extension trial (OCTAVE Open). 4
Tofacitinib received its first marketing authorisation in the United States on 30 May 2018 for the treatment of adults with moderately to severely active UC and was approved in the European Union on 1 August 2018. Initially, tofacitinib was only available in an immediate‐release formulation (5 and 10 mg tablets), and an extended‐release formulation (11 and 22 mg tablets) was approved in the United States for use in patients with moderately to severely active UC in December 2019. The recommended dosage for induction is tofacitinib 10 mg twice daily, or 22 mg once daily for 8 weeks. If needed, induction treatment can be continued for a maximum of 16 weeks. 5 For maintenance treatment, tofacitinib 5 mg twice daily or 11 mg once daily is recommended. 5 For patients with a loss of response during maintenance treatment, tofacitinib 10 mg twice daily or 22 mg once daily may be considered and limited to the shortest duration, with careful consideration of the benefits and risks for the individual patient. 5
The safety profile of tofacitinib in patients with UC has been characterised in a pooled analysis of tofacitinib‐treated patients from phase 2 and 3 trials, and open‐label, long‐term extension studies (as of May 2019). 6 Adverse events (AEs) of special interest included serious infections, herpes zoster, opportunistic infections, malignancies (excluding non‐melanoma skin cancer [NMSC]), NMSC, major adverse cardiovascular events (MACE) and gastrointestinal perforations. In this integrated safety analysis, the AE incidence rate remained generally consistent over time. 6 , 7 Compared with the general population, patients with inflammatory bowel disease have been shown to be at increased risk for herpes zoster, 8 and this risk is elevated in patients with UC treated with tofacitinib. 9 Post‐marketing surveillance (PMS), the practice of monitoring drug safety following market release, is important for pharmacovigilance and complements data from clinical trials to establish a real‐world safety profile. PMS includes spontaneous and voluntary reporting of AEs, post‐marketing observational studies and active surveillance.
We evaluated the safety profile of tofacitinib in patients with UC in the post‐marketing setting and compared with previously reported PMS data in patients with rheumatoid arthritis (RA). 10
2. METHODS
This analysis reviewed worldwide tofacitinib PMS data in the Pfizer safety database from 30 May 2018 to 25 August 2020. The sources of these PMS reports included spontaneous reports from patients, healthcare professionals and regulatory authorities, solicited reports from patient support and market research programmes, and reports extracted from the literature. The PMS data used in this analysis were not collected as part of a clinical study and were non‐interventional; therefore, ethics approval was not required.
2.1. Estimated worldwide post‐marketing exposure
Worldwide exposure estimates are based on audited unit sales of tofacitinib from the IQVIA Health Multinational Integrated Data Analysis System (MIDAS) database. Data were extrapolated for the first and second quarter of 2020 from the average of the previous four quarters and adjusted pro‐rata to the end of the reporting period. Using the average daily dose, audited unit sales were converted into patient‐days (days of therapy) and divided by 365.25 (days in a year) to obtain patient‐years of exposure.
2.2. Data analysis
For each case report, the type and frequency of AEs and serious AEs of interest by Medical Dictionary for Regulatory Activities (MedDRA) System Organ Class were reviewed. MedDRA is a standardised terminology with pre‐defined terms arranged into a hierarchy of five levels (Figure 1).
FIGURE 1.

MedDRA hierarchy. From reported AEs, verbatim information is linked to a Lowest Level Term. Each Lowest Level Term is linked to a parent Preferred Term that is a distinct descriptor for a symptom, sign, disease diagnosis, therapeutic indication, investigation, surgical or medical procedure, or medical, social or family history characteristic. Related Preferred Terms are grouped into High Level Terms based upon anatomy, pathology, physiology, aetiology or function. High Level Terms, related by anatomy, pathology, physiology, aetiology or function are linked to High Level Group Terms. High Level Groups Terms are grouped into System Organ Classes based on aetiology, manifestation site or purpose. AE, adverse event; MedDRA, Medical Dictionary for Regulatory Activities
For safety data reporting purposes, all events reported within a case report were coded to a MedDRA Lowest Level Term regardless of what might have been perceived as the focus of the report. All cases reported at least one AE, some cases reported more than one AE. Not all cases reported a serious AE and some cases reported more than one serious AE. The seriousness of AEs was assessed using International Council on Harmonisation criteria: an event was considered serious if it resulted in death, hospitalisation or caused permanent disability. The reporting rate for serious AEs of special interest per MedDRA System Organ Class was estimated by dividing the number of serious AEs by 100 patient‐years of exposure. Multiple events could be reported in the same case report and were considered separate events. Serious AEs of clinical interest in patients receiving tofacitinib included infections, neoplasms, gastrointestinal disorders, cardiac events, vascular events and respiratory events. Results are reported descriptively.
Findings in the PMS reported from patients with UC were contextualised against previously reported PMS data from patients with RA received in the Pfizer safety database from 6 November 2012 to 5 November 2015. 10
3. RESULTS
3.1. Patient characteristics
During the reporting period, the worldwide post‐marketing exposure to tofacitinib based on estimated worldwide sales, was 8916 patient‐years. Overall, 4226 case reports were received and included 12 103 AEs, of which 1839 were serious AEs. Among the 4226 cases reported, 1141 (27.0%) included a serious AE. The proportion of cases with a serious AE was similar for the immediate‐release and extended‐release formulations (Table 1).
TABLE 1.
Summary of patient characteristics and adverse events, overall and by tofacitinib formulation
| Tofacitinib overall | Tofacitinib formulation | |||
|---|---|---|---|---|
| Immediate release (5 and 10 mg b.i.d.) | Extended release, XR (11 and 22 mg q.d.) | Not reported a | ||
| Total number of case reports | 4226 | 3616 | 98 | 512 |
| Gender, b (%) | ||||
| Male | 46.5 | 49.3 | 37.8 | 27.9 |
| Female | 41.6 | 44.1 | 59.2 | 21.1 |
| Not reported | 11.9 | 6.6 | 3.1 | 51.0 |
| Age (median), c years (range) | 45.0 (9.0‐93.0) | 45.0 (9.0‐93.0) | 52.0 (18.0‐80.0) | 43.5 (14.0‐80.0) |
| Number of AEs d | 12 103 | 11 184 | 228 | 692 |
| Number of cases with an SAE (%)e e | 1141 (27.0) | 1025 (28.3) | 25 (25.5) | 92 (18.0) |
| Number of fatal cases (%) | 18 (0.4) | 15 (0.4) | 1 (1.0) | 2 (0.4) |
Abbreviations: AE, adverse event; b.i.d., twice daily; q.d., once daily; SAE, serious adverse event.
Some case reports were missing information.
3721 cases reported gender.
3436 cases reported age.
All cases reported at least one AE. Some cases reported >1 AE; therefore, the number of AEs exceeds the number of cases.
A total of 1839 SAEs were reported; not all cases reported an SAE, and some cases reported >1 SAE.
Of the 4226 case reports, 3573 (84.5%) were spontaneous reports, 632 (15.0%) were from non‐interventional solicited sources, and 21 (0.5%) were non‐study literature cases (ie cases not reported in a study but published as a case report). Over half of the case reports originated in the United States (51.1%), and 27.9% were from Canada.
Of cases with reported gender (n = 3721; 88.1%) or age (n = 3436; 81.3%), 46.5% occurred in men, and the median age was 45 years (range 9‐93 years).
3.2. Serious AEs and non‐serious AEs
Table 2 presents a summary of AEs and serious AEs by MedDRA System Organ Class. Overall, the MedDRA System Organ Class with the most reported AEs was general disorders and administration site conditions. When analysed by tofacitinib formulation, the MedDRA System Organ Class with the most reported AEs was general disorders and administration site conditions for immediate release, and injury, poisoning and procedural complications for extended release (Table S1).
TABLE 2.
Overall AEs and SAEs by MedDRA System Organ Class
| AEs | SAEs | |||
|---|---|---|---|---|
| Number of AEs (N = 12 103) | Number of cases with an AE (%) (N = 4226) | Number of SAEs (N = 1839) | Estimated RR a (PY = 8916) | |
| General disorders and administration site conditions b | 3199 | 2179 (51.6) | 233 | 2.61 |
| Injury, poisoning and procedural complications | 1629 | 1269 (30.0) | 60 | 0.67 |
| GI disorders | 1994 | 1109 (26.2) | 621 c | 6.97 |
| Infections and infestations | 934 | 709 (16.8) | 292 d | 3.28 |
| Investigations | 842 | 592 (14.0) | 73 | 0.82 |
| Nervous system disorders | 651 | 530 (12.5) | 69 | 0.77 |
| Psychiatric disorders | 486 | 389 (9.2) | 17 | 0.19 |
| Skin and subcutaneous tissue disorders | 512 | 377 (8.9) | 23 | 0.26 |
| Musculoskeletal and connective tissue disorders | 511 | 362 (8.6) | 43 | 0.48 |
| Respiratory, thoracic and mediastinal disorders | 474 | 334 (7.9) | 66 e | 0.74 |
| Vascular disorders | 166 | 152 (3.6) | 112 f | 1.26 |
| Metabolism and nutrition disorders | 112 | 108 (2.6) | 11 | 0.12 |
| Renal and urinary disorders | 102 | 89 (2.1) | 28 | 0.31 |
| Blood and lymphatic system disorders | 76 | 72 (1.7) | 19 | 0.21 |
| Eye disorders | 82 | 67 (1.6) | 25 | 0.28 |
| Immune system disorders | 64 | 62 (1.5) | 17 | 0.19 |
| Neoplasms benign, malignant and unspecified | 56 | 52 (1.2) | 49 g | 0.55 |
| Cardiac disorders | 67 | 51 (1.2) | 45 h | 0.50 |
MedDRA System Organ Classes with <1% of AEs are not shown.
Abbreviations: AE, adverse event; GI, gastrointestinal; MedDRA, Medical Dictionary for Regulatory Activities; N, total number of cases/events; PY, patient‐years of exposure; RR, reporting rate; SAE, serious AE; UC, ulcerative colitis.
RR is calculated by dividing the number of SAEs per MedDRA System Organ Class by the estimated PY (per 100 PY).
General disorders and administration site conditions is the System Organ Class term verbatim and captures numerous Preferred Terms such as infusion site reaction and injection site pain which may not be applicable to an oral drug.
Serious GI events (n ≥ 50) included haematochezia (200), UC (159) and rectal haemorrhage (76).
Serious infection events (n ≥ 8) included C difficile infection (43), pneumonia (36), COVID‐19 (12), C difficile colitis (8), cytomegalovirus (8) and herpes zoster (8).
Most serious respiratory events were pulmonary embolism (28).
Serious vascular events (n ≥ 10) included haemorrhage (55), thrombosis (23) and deep vein thrombosis (18).
Serious neoplasm events (n ≥ 3) included breast cancer female (4), colon cancer (4), breast cancer (3), lymphoma (3), malignant melanoma (3), neoplasm malignant (3) and prostate cancer (3).
Serious cardiac events (n ≥ 4) included acute myocardial infarction (4), angina pectoris (4), myocardial infarction (4) and pericarditis (4).
The most commonly reported AEs by Preferred Term (Figure 2A) were related to lack of or incomplete efficacy of the drug (drug ineffective [18.5%], condition aggravated [13.7%] and therapeutic product effect incomplete [7.4%]). When analysed by tofacitinib formulation, there were differences in the most commonly reported AEs by Preferred Term. In patients treated with tofacitinib immediate‐release formulation, the three most frequently reported Preferred Terms were drug ineffective, condition aggravated and product dose omission, whereas in patients treated with tofacitinib extended‐release formulation, product use in unapproved indication, condition aggravated and off‐label use were the three most frequently reported Preferred Terms (Figure 2B,C).
FIGURE 2.

Most frequently reported AEs based on MedDRA Preferred Terms (reporting proportion ≥2%): (A) tofacitinib overall, (B) tofacitinib immediate‐release formulation and (C) tofacitinib extended‐release formulation. The percentage was calculated by dividing the number of cases per Preferred Term by the total number of cases. AE, adverse event; MedDRA, Medical Dictionary for Regulatory Activities
Overall, 1839 serious AEs were reported. Reporting rates per 100 patient‐years for serious AEs by System Organ Class are presented in Table 2. The majority of reported serious AEs were within the MedDRA System Organ Class of gastrointestinal disorders, infections and infestations, and general disorders and administration site conditions (Table 2).
3.3. Gastrointestinal disorders
Cases with gastrointestinal AEs accounted for 26.2% of all cases reported, with a total of 1994 AEs in patients with UC. The five most frequently reported gastrointestinal AEs were diarrhoea (241), haematochezia (200), UC (159), nausea (156) and abdominal pain (122). In total, 621 serious gastrointestinal disorder events were reported, yielding a reporting rate of 6.97 per 100 patient‐years. The most frequent serious AEs (n ≥ 50) included haematochezia (200), UC (159) and rectal haemorrhage (76). There were 15 AEs within the High Level Group Term of gastrointestinal ulceration and perforation of which 14 were classed as serious AEs: duodenal perforation (1), gastric ulcer (1), gastrointestinal perforation (1), intestinal perforation (3), intestinal ulcer (2), large intestinal perforation (3), peptic ulcer (1), rectal perforation (1) and rectal ulcer (1). The reporting rate for gastrointestinal disorder events in the PMS data was observed to be higher in patients with UC than in patients with RA (Figure 3).
FIGURE 3.
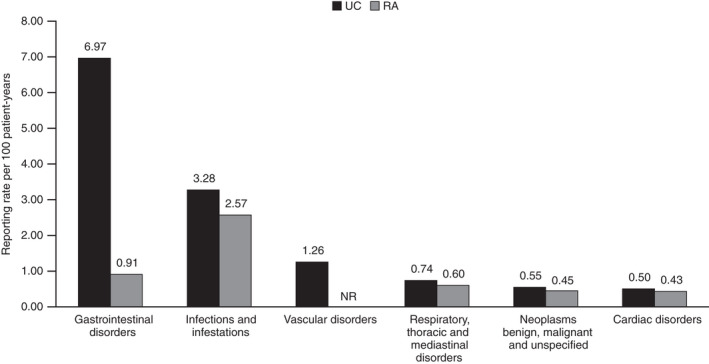
Reporting ratea of SAEs by MedDRA System Organ Class in the PMS data for tofacitinib in patients with UC and RAb. Post‐marketing exposure to tofacitinib based on estimated worldwide sales, was 8916 patient‐years for patients with UC and 34 223 patient‐years for patients with RA. MedDRA, Medical Dictionary for Regulatory Activities; NR, not reported at the time of analysis; PMS, post‐marketing surveillance; RA, rheumatoid arthritis; SAE, serious adverse event; UC, ulcerative colitis. aReporting rate is calculated by dividing the number of SAEs per MedDRA System Organ Class by the estimated patient‐years of exposure (per 100 patient‐years). bAs reported by Cohen et al. 10
3.4. Infections
Overall, 16.8% of cases reported an infection AE, with a total of 934 infection events of which 292 were serious. The estimated reporting rate for serious infections and infestations in patients with UC (3.28 per 100 patient‐years) was observed to be higher than that reported from patients with RA (2.57 per 100 patient‐years; Figure 3). The most frequently reported infections in patients with UC were nasopharyngitis (134) and herpes zoster (127). The most commonly reported serious infection events in patients with UC (MedDRA Preferred Term n ≥ 8) were two Preferred Terms within the High Level Term of Clostridia infections (C difficile colitis [8]/infection [43]), pneumonia (36), COVID‐19 (12), cytomegalovirus (8) and herpes zoster (8).
Serious AEs with Preferred Terms indicative of potential opportunistic infections were identified including fungal oesophagitis (1), Pneumocystis jirovecii pneumonia (1), unspecific opportunistic infection (1) and tuberculosis (1).
3.5. Neoplasms
In patients with UC receiving tofacitinib, 52 cases (1.2%) reported 56 neoplasm AEs, including 49 serious AEs with an estimated reporting rate of 0.55 per 100 patient‐years, which was similar to that observed in PMS data from patients with RA (Figure 3). The most commonly reported serious neoplasm events (n ≥ 3) were two Preferred Terms within the High Level Term of breast and nipple neoplasms malignant (breast cancer female [4]/breast cancer [3]), colon cancer (4), lymphoma (3), malignant melanoma (3), neoplasm malignant (3) and prostate cancer (3). There were four serious AEs of NMSC including one event each of basal cell carcinoma and squamous cell carcinoma, and two events of unspecified skin cancer.
3.6. Vascular events
Overall, 166 vascular AEs from 152 (3.6%) cases were recorded with 112 classified as serious AEs (estimated reporting rate: 1.26 per 100 patient‐years). The most commonly reported serious vascular events (n ≥ 10) included haemorrhage (55), thrombosis (23) and deep vein thrombosis (18).
3.7. Respiratory events
Of the 66 serious respiratory events (estimated reporting rate: 0.74 per 100 patient‐years), the majority were pulmonary embolism (28). Other serious respiratory events of interest were dyspnoea (8), interstitial lung disease (2) and pulmonary fibrosis (1). The estimated reporting rate for respiratory events was similar in patients with UC and patients with RA (Figure 3).
3.8. Cardiac events
A total of 67 cardiac AEs were reported of which 45 were serious (estimated reporting rate: 0.50 per 100 patient‐years). The most commonly reported serious cardiac events (n ≥ 4) were three Preferred Terms within the High Level Term of ischaemic coronary artery disorders (acute myocardial infarction [4]/myocardial infarction [4] and angina pectoris [4]) and pericarditis (4). The estimated reporting rate for cardiac events was similar in patients with UC and patients with RA (Figure 3).
3.9. Deaths
Among the cases reported, 18 (0.4%) were fatal, giving an overall mortality rate of 0.20 per 100 patient‐years. Of the 18 fatal cases, three were determined to be related to tofacitinib by a medically qualified person who assessed the case, and four were determined to be unrelated to tofacitinib. For the three cases related to tofacitinib, the Preferred Terms recorded with a fatal outcome were: cerebrovascular accident (one case); acute myocardial infarction (one case); and acute respiratory failure, pneumonia streptococcal, pneumonia and tuberculosis (one case). For 10 cases, neither a cause of death nor an assessment of relatedness to tofacitinib was reported. In the one remaining case, the cause of death was reported but no assessment of relatedness was reported. In those 11 cases where no assessment of relatedness was reported, by default these cases were assessed by Pfizer as being related to tofacitinib treatment.
4. DISCUSSION
This analysis evaluated PMS reports for tofacitinib in patients with UC over a 27‐month period after first marketing authorisation. Based on this review of PMS data for tofacitinib in UC received in the Pfizer safety database from 30 May 2018 to 25 August 2020, the types of AEs were consistent with the known tofacitinib safety profile, 5 however, the safety profile of a drug following approval is dynamic.
Post‐marketing studies are an important aspect of monitoring safety following drug approval. A recently completed phase 3b/4 randomised, open‐label safety study in patients with RA (Study A3921133; NCT02092467) who were aged ≥50 years, had ≥1 additional cardiovascular risk factor and were receiving methotrexate. This study, an FDA post‐approval requirement, found that the non‐inferiority criterion was not met comparing tofacitinib (doses of 5 mg twice daily and 10 mg twice daily combined) plus methotrexate with tumour necrosis factor inhibitor (TNFi; adalimumab or etanercept) plus methotrexate, for the co‐primary endpoints of adjudicated MACE and malignancies excluding NMSC. 11 Given that the underlying mechanism(s) for these AEs remain unknown, the effect of JAK inhibitors on cardiovascular disease and malignancy risk requires further research.
AEs were consistent with the safety profile observed during randomised clinical trials. 12 The most frequently reported AEs were Preferred Terms within the System Organ Class of general disorders with some relating to a loss of efficacy rather than an adverse drug reaction, for example “drug ineffective” was the most reported AE. Headache, diarrhoea and fatigue were among the most frequently reported AEs by Preferred Term, all known adverse drug reactions for tofacitinib. The most frequently reported AEs by Preferred Term in the PMS data for tofacitinib were similar in patients with UC and patients with RA. 10
In the PMS data from patients with UC, the most frequently reported infections were nasopharyngitis and herpes zoster, which is consistent with infection AEs reported in tofacitinib clinical trials. 13 Compared with the general population, patients with inflammatory bowel disease are at increased risk of herpes zoster, 8 and this risk is increased further by treatment with immunosuppressive therapies. 14 Consistent with herpes zoster events reported in tofacitinib clinical trials, 13 the majority of herpes zoster infections in the PMS dataset were categorised as not serious.
Integrated safety analysis of tofacitinib from five clinical trials in patients with UC with treatment durations up to 6.8 years showed that malignancy events occurred infrequently, with no apparent clustering of type. 15 The types of malignancies reported in the PMS data were generally consistent with those reported in the UC tofacitinib clinical programme. 15
UC is a known risk factor for venous thromboembolism (including deep vein thrombosis and pulmonary embolism). 16 , 17 , 18 In this PMS analysis in which AEs were analysed using MedDRA classification, events of venous thromboembolism were captured under two different System Organ Classes; events of deep vein thrombosis were analysed as part of the vascular disorders and pulmonary embolism as respiratory, thoracic and mediastinal disorders. Here, the majority of serious respiratory, thoracic and mediastinal disorder events were of pulmonary embolism, confirming the need for prescribers to individualise treatment by considering risk factors for venous thromboembolism. Previously reported data from an ad hoc safety analysis of Study A3921133 in 2019, showed an increased frequency of venous thromboembolic events for tofacitinib relative to TNFi. 19 Venous thromboembolism has been determined to be an important identified risk of tofacitinib treatment, and local labelling has been updated accordingly. 5 , 20 Further research is needed to better understand the relationship between thromboembolic events and JAK inhibitors, including tofacitinib.
Relative to post‐marketing data collected over 36 months (6 November 2012 to 25 November 2015) from patients with RA, patients with UC were younger (median age of patients with UC 45 years vs median age of patients with RA 60 years), more likely to be male (patients with UC 46.5% vs patients with RA 21.8%), and AEs were more likely to be reported from countries other than the United States (51.1% of reports in patients with UC originated from the US vs 73.9% of reports from the US in patients with RA). 10 The proportion of cases that were fatal was lower in patients with UC compared with patients with RA (0.4% vs 1.1% respectively). 10 In the two datasets, there were modest differences in terms of System Organ Classes and the most frequently reported AEs. The reporting rates for MedDRA System Organ Classes of infections, neoplasms, respiratory events and cardiac events from the PMS data for tofacitinib in patients with UC were similar to those reported from PMS data in patients with RA. 10 Although, Cohen et al did not include the reporting rate for vascular events in patients with RA, it cannot be assumed that vascular events in patients with RA did not occur and it is possible that at the time of reporting, vascular events were few and had not been identified as an event of interest. 10 As expected, the reporting rate for gastrointestinal disorders was higher in the PMS data for tofacitinib in patients with UC compared with patients with RA. 10
This study has a number of strengths and weaknesses. Case reports are submitted voluntarily, and the magnitude of under‐reporting is unknown. Data from PMS reports are limited by the availability of details for each case, with case reports often missing important clinical information or being incompletely recorded. In this study, patient exposure to tofacitinib was calculated using commercial records and yielded reporting rates as opposed to incidence rates which are calculated using actual exposure (ie exposure time is censored at the time of the event) in clinical trials and thereby limits the comparison of these data. Additionally, the patient‐years metric does not represent unique patient counts. Worldwide exposure estimates are based on audited unit sales of tofacitinib from the IQVIA Health Multinational Integrated Data Analysis System database which may not capture all sales of tofacitinib due to its availability in specialty pharmacies, and data are not captured from all markets where tofacitinib is available, potentially leading to under‐reporting of units sold. Furthermore, unit data from the MIDAS audit reflect units sold but not whether the drug was prescribed/taken by a patient. The low numbers of case reports for extended‐release formulation should be considered when interpreting these data. The Pfizer safety database in which these PMS reports are captured includes only AE information and does not capture information on patient follow‐up or details from patients who did not experience AEs while receiving tofacitinib.
A known phenomenon in drug reporting is the Weber effect, which predicts higher AE reporting rates within the first 2 years of drug approval, decreasing thereafter, possibly due to reduced reporting of mild AEs. The impact of this effect is less clear when use for a new indication is approved for a drug already marketed for other indications. The data reported here were recorded within 27 months of market authorisation; therefore, the Weber effect should be considered when interpreting these estimated reporting rates. An accumulation of AE reports may not necessarily indicate causation by the drug and may relate to underlying diseases, comorbidities or other factors. In 2019, tofacitinib product labelling 5 , 20 was updated regarding risk factors for thromboembolism and along with communications to the public, healthcare professionals, and media, there is the potential for increased reporting of these events in patients receiving tofacitinib as a result of increased awareness.
Based on this review of PMS data for tofacitinib in UC, the types of AEs and reporting rates were consistent with the known tofacitinib safety profile, with no new risks identified. Pharmacovigilance surveillance is ongoing and is important not only for monitoring known risks associated with treatment with tofacitinib but also for identifying new potential risks.
AUTHORSHIP
Guarantor of the article: Irene Modesto.
Author contributions: All authors interpreted the data and drafted and edited the manuscript. All authors approved the final draft of the manuscript for submission, including the authorship list.
Supporting information
Table S1
ACKNOWLEDGEMENT
This study was sponsored by Pfizer Inc. The authors thank the patients, investigators and study teams involved in OCTAVE Induction 1 and 2. This work was supported by Pfizer Inc. Writing support, under the guidance of the authors, was provided by Helen Findlow, PhD, CMC Connect, McCann Health Medical Communications and was funded by Pfizer Inc, New York, NY, USA in accordance with Good Publication Practice (GPP3) guidelines (Ann Intern Med 2015;163:461–464).
Declaration of personal interests: David T. Rubin has received grant support from Takeda; and has served as a consultant for AbbVie, AbGenomics, Allergan Inc, Boehringer Ingelheim, Bristol‐Myers Squibb, Celgene Corp/Syneos, Check‐cap, Dizal Pharmaceuticals, Eli Lilly, GalenPharma/Atlantica, Genentech/Roche, Gilead Sciences, GSK, Ichnos Sciences S.A, Janssen, Narrow River Management, Pfizer Inc, Prometheus Laboratories, Reistone, Shire, Takeda, and Techlab Inc. Séverine Vermeire has received grant support from AbbVie, MSD, Pfizer Inc, and Takeda; served as a speaker for AbbVie, Dr. Falk Pharma, Ferring, Hospira, MSD, Takeda, and Tillotts; and served as a consultant for AbbVie, Celgene, Ferring, Galapagos, Genentech/Roche, Hospira, Janssen, MSD, Mundipharma, Pfizer Inc, Second Genome, Shire, and Takeda. Silvio Danese has served as a consultant for AbbVie, Allergan, Amgen, AstraZeneca, Biogen, Boehringer Ingelheim, Celgene, Celltrion, Ferring, Gilead Sciences, Hospira, Janssen, Johnson & Johnson, MSD, Mundipharma, Pfizer Inc, Roche, Sandoz, Takeda, TiGenix, UCB, and Vifor. Siew C. Ng has received research grant support from AbbVie, Ferring, and Olympus; served as a speaker for AbbVie, Ferring, Janssen, Menarini, and Takeda; and holds a directorship with Microbiota I Center. Irene Modesto, Kenneth K. Kwok, Nana Koram, and Thomas V. Jones are employees of Pfizer Inc. Irene Modesto, Kenneth K. Kwok, Nana Koram, and Thomas V. Jones own stocks and shares in Pfizer Inc.
Rubin DT, Modesto I, Vermeire S, et al. Worldwide post‐marketing safety surveillance experience with tofacitinib in ulcerative colitis. Aliment Pharmacol Ther. 2022;55:302–310. 10.1111/apt.16619
The Handling Editor for this article was Dr Nicholas Kennedy, and it was accepted for publication after full peer‐review.
DATA AVAILABILITY STATEMENT
Upon request, and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions and exceptions, Pfizer may also provide access to the related individual anonymized participant data. See https://www.pfizer.com/science/clinical‐trials/trial‐data‐and‐results for more information.
REFERENCES
- 1. Ordás I, Eckmann L, Talamini M, Baumgart D, Sandborn W. Ulcerative colitis. Lancet. 2012;380:1606–1619. [DOI] [PubMed] [Google Scholar]
- 2. Sandborn WJ, Ghosh S, Panes J, et al. Tofacitinib, an oral Janus kinase inhibitor, in active ulcerative colitis. N Engl J Med. 2012;367:616–624. [DOI] [PubMed] [Google Scholar]
- 3. Sandborn WJ, Su C, Sands BE, et al. Tofacitinib as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2017;376:1723–1736. [DOI] [PubMed] [Google Scholar]
- 4. Lichtenstein GR, Loftus EV Jr, Wei SC, et al. Tofacitinib, an oral, small‐molecule Janus kinase inhibitor, in the treatment of ulcerative colitis: analysis of an open‐label, long‐term extension study with up to 5.9 years of treatment [abstract]. J Crohns Colitis 2020;14:S100‐101, abstract DOP61. [Google Scholar]
- 5. Pfizer Inc. Xeljanz® (tofacitinib): highlights of prescribing information. http://labeling.pfizer.com/ShowLabeling.aspx?id=959
- 6. Sandborn WJ, Panés J, D'Haens GR, et al. Tofacitinib for the treatment of ulcerative colitis: up to 6.8 years of safety data from global clinical trials [abstract]. Am J Gastroenterol 2020;115:S353‐S354, abstract S0703. [Google Scholar]
- 7. Sandborn WJ, Panes J, Panaccione R, et al. Tofacitinib for the treatment of ulcerative colitis: up to 5.4 years of safety data from global clinical trials [abstract]. Gastroenterology. 2019;156(Suppl 1):S1097;abstract Tu1717. [Google Scholar]
- 8. Yun H, Yang S, Chen L, et al. Risk of herpes zoster in auto‐immune and inflammatory diseases: implications for vaccination. Arthritis Rheumatol. 2016;68:2328–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Winthrop KL, Melmed GY, Vermeire S, et al. Herpes zoster infection in patients with ulcerative colitis receiving tofacitinib. Inflamm Bowel Dis. 2018;24:2258–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cohen S, Curtis JR, DeMasi R, et al. Worldwide, 3‐year, post‐marketing surveillance experience with tofacitinib in rheumatoid arthritis. Rheumatol Ther. 2018;5:283–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pfizer Inc. Pfizer shares co‐primary endpoint results from post‐marketing required safety study of XELJANZ® (tofacitinib) in subjects with rheumatoid arthritis (RA). https://www.pfizer.com/news/press‐release/press‐release‐detail/pfizer‐shares‐co‐primary‐endpoint‐results‐post‐marketing
- 12. Sandborn WJ, Panés J, D’Haens GR, et al. Safety of tofacitinib for treatment of ulcerative colitis, based on 4.4 years of data from global clinical trials. Clin Gastroenterol Hepatol. 2019;17:1541–1550. [DOI] [PubMed] [Google Scholar]
- 13. Winthrop KL, Loftus EV, Baumgart DC, et al. Tofacitinib for the treatment of ulcerative colitis: analysis of infection rates from the ulcerative colitis clinical programme. J Crohns Colitis. 2021;15:914–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Colombel JF. Herpes zoster in patients receiving JAK inhibitors for ulcerative colitis: mechanism, epidemiology, management, and prevention. Inflamm Bowel Dis. 2018;24:2172–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lichtenstein GR, Rogler G, Ciorba MA, et al. Tofacitinib, an oral Janus kinase inhibitor: analysis of malignancy (excluding nonmelanoma skin cancer) events across the ulcerative colitis clinical program. Inflamm Bowel Dis. 2021;27:816–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Weng M‐T, Park SH, Matsuoka K, et al. Incidence and risk factor analysis of thromboembolic events in East Asian patients with inflammatory bowel disease, a multinational collaborative study. Inflamm Bowel Dis. 2018;24:1791–1800. [DOI] [PubMed] [Google Scholar]
- 17. Kappelman MD, Horvath‐Puho E, Sandler RS, et al. Thromboembolic risk among Danish children and adults with inflammatory bowel diseases: a population‐based nationwide study. Gut. 2011;60: 937–943. [DOI] [PubMed] [Google Scholar]
- 18. Bernstein CN, Blanchard JF, Houston DS, Wajda A. The incidence of deep venous thrombosis and pulmonary embolism among patients with inflammatory bowel disease: a population‐based cohort study. Thromb Haemost. 2001;85:430–434. [PubMed] [Google Scholar]
- 19. Mease P, Charles‐Schoeman C, Cohen S, et al. Incidence of venous and arterial thromboembolic events reported in the tofacitinib rheumatoid arthritis, psoriasis and psoriatic arthritis development programmes and from real‐world data. Ann Rheum Dis. 2020;79:1400–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. European Medicines Agency . Xeljanz® (tofacitinib): summary of product characteristics. https://www.ema.europa.eu/en/documents/product‐information/xeljanz‐epar‐product‐information_en.pdf
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Data Availability Statement
Upon request, and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions and exceptions, Pfizer may also provide access to the related individual anonymized participant data. See https://www.pfizer.com/science/clinical‐trials/trial‐data‐and‐results for more information.