

Abstract
The migration of CD4+ effector/memory T cells across the blood–brain barrier (BBB) is a critical step in MS or its animal model, EAE. T‐cell diapedesis across the BBB can occur paracellular, via the complex BBB tight junctions or transcellular via a pore through the brain endothelial cell body. Making use of primary mouse brain microvascular endothelial cells (pMBMECs) as in vitro model of the BBB, we here directly compared the transcriptome profile of pMBMECs favoring transcellular or paracellular T‐cell diapedesis by RNA sequencing (RNA‐seq). We identified the atypical chemokine receptor 1 (Ackr1) as one of the main candidate genes upregulated in pMBMECs favoring transcellular T‐cell diapedesis. We confirmed upregulation of ACKR1 protein in pMBMECs promoting transcellular T‐cell diapedesis and in venular endothelial cells in the CNS during EAE. Lack of endothelial ACKR1 reduced transcellular T‐cell diapedesis across pMBMECs under physiological flow in vitro. Combining our previous observation that endothelial ACKR1 contributes to EAE pathogenesis by shuttling chemokines across the BBB, the present data support that ACKR1 mediated chemokine shuttling enhances transcellular T‐cell diapedesis across the BBB during autoimmune neuroinflammation.
Keywords: atypical chemokine receptor 1, blood–brain barrier, transcellular diapedesis, T cell
Expression of high levels of the atypical chemokine receptor 1 (ACKR1) promote transcellular T‐cell diapedesis across the blood–brain barrier (BBB) in vitro possibly by shuttling inflammatory chemokines from the abluminal to the luminal side of the BBB.
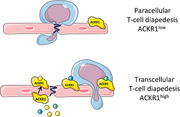
Introduction
The blood‐brain barrier (BBB) is a functional and physical barrier mainly established by highly specialized central nervous system (CNS) microvascular endothelial cells. As such, the BBB is one of the barriers between blood and the CNS parenchyma [1, 2]. The main function of the BBB is to maintain CNS homeostasis, a requirement for proper neuronal activity. This is achieved by the unique set of properties of the brain endothelial cells (review in [3]), such as the lack of fenestrae, the low pinocytotic activity, the expression of specific transporters, and the presence of continuous and complex adherence and tight junctions (AJs and TJs) between BBB endothelial cells, which inhibit the paracellular diffusion of potentially harmful molecules.
In its function to maintain CNS homeostasis, the BBB also strictly controls immune cell entry into the CNS parenchyma [4, 5]. Under physiological conditions, immune surveillance of the CNS is characterized by controlled lymphocyte trafficking, which does not affect CNS homeostasis [4]. However, in neuroinflammatory conditions — such as MS or its animal model, EAE — high numbers of CD4+ T cells cross the BBB at the level of postcapillary venules, causing CNS inflammation. CD4+ T‐cell extravasation across the BBB has been extensively studied and is characterized by a multistep cascade of consecutive molecular interactions between CD4+ T cells and BBB endothelial cells (summarized in [6]). First, T‐cell arrest on the BBB is mediated by the interaction between endothelial VCAM‐1 and ICAM‐1 with their integrin ligands on the T cells – α4β1 (VLA‐4) and αLβ2 (LFA‐1) integrins, respectively. Subsequent polarization and crawling of CD4+ T cells on the BBB in search of permissive sites of diapedesis is mediated by endothelial ICAM‐1 and ICAM‐2 [7]. In this context extended crawling of T‐cells against the direction of blood flow is characteristic for the BBB, highlighting the unique barrier properties of this vascular bed [8, 9].
T‐cell diapedesis across the BBB occurs via two different pathways: paracellular, via the endothelial cell‐to‐cell contacts between adjacent endothelial cells, or transcellular, via a pore through the endothelial cell body itself [6]. In peripheral vascular beds immune cell diapedesis occurs almost exclusively via the paracellular pathway through the endothelial junctions. It was originally thought that the complexity of the BBB tight junctions would prohibit paracellular T‐cell diapedesis, thus favoring transcellular T‐cell diapedesis across the BBB [10]. Recent observations have however challenged this view, as at least under low inflammatory conditions T cells preferentially cross the BBB via the paracellular pathway [11, 12], while more severe inflammatory conditions result in a significant increase in transcellular diapedesis [10, 11].
The active role of the BBB endothelial cells in directing the cellular pathway of T‐cell diapedesis prompted us to further explore the cellular and molecular mechanisms involved. To this end, we made use of our well‐established in vitro BBB model, in which primary mouse brain microvascular endothelial cells (pMBMECs) form a tight and polarized monolayer and favor transcellular or paracellular T‐cell diapedesis under physiological flow depending on the cytokine stimulus applied prior to the assay. We directly compared the transcriptome profile of pMBMECs favoring transcellular or paracellular T‐cell diapedesis by RNA sequencing (RNA‐seq) analysis to identify potential endothelial target genes involved in directing transcellular or paracellular T‐cell diapedesis across the BBB. Among the final candidate genes, we identified the atypical chemokine receptor 1 (Ackr1) significantly upregulated in pMBMECs favoring transcellular T‐cell diapedesis. As we have previously observed an active role for ACKR1 in the pathogenesis of EAE, we next explored the role of ACKR1 in mediating transcellular T‐cell diapedesis across the BBB. While pMBMECs lacking ACRK1 supported T‐cell arrest and crawling indistinguishable to WT pMBMECs, we observed reduced transcellular T‐cell diapedesis across ACKR1–/– pMBMECs under physiological flow by in vitro live‐cell imaging. Our data identify ACKR1 as one molecule contributing to transcellular T‐cell diapedesis across our in vitro BBB model.
Results
Transcriptome profiling of cytokine‐stimulated pMBMECs
To identify endothelial target genes potentially involved in directing T cells to paracellular or transcellular sites of diapedesis across the BBB, we profiled the transcriptome of differentially cytokine‐stimulated pMBMECs by RNA‐seq. To this end, we made use of our previous observation that stimulating pMBMECs specifically with either 10 ng/mL TNF‐α or 20 ng/mL IL‐1β induces low or high cell surface levels of ICAM‐1 [11]. While low cell surface levels of ICAM‐1 maintain T‐cell crawling preferentially to paracellular sites of diapedesis, high endothelial cell surface levels of ICAM‐1 reduce T‐cell crawling and favor transcellular T‐cell diapedesis across pMBMECs under physiological flow, respectively [11] (Fig. 1A).
Figure 1.
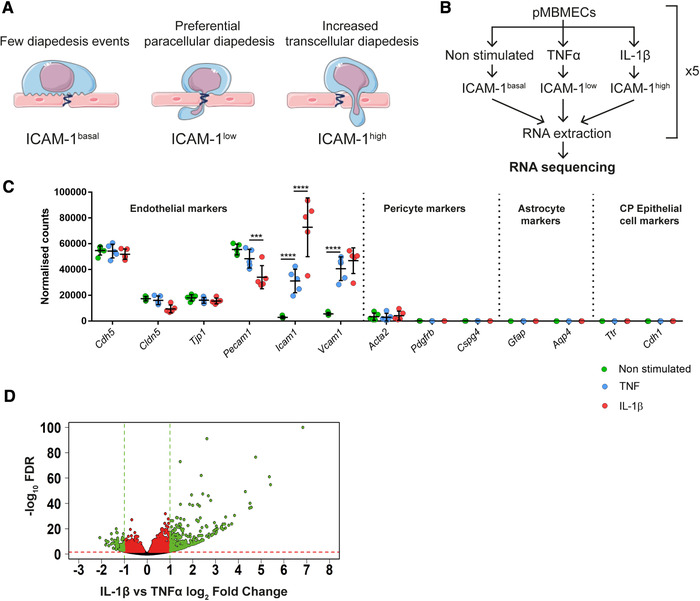
Workflow and RNA‐seq analysis of cytokine‐stimulated pMBMECs. (A) Schematic representation of T‐cell diapedesis pathway across pMBMECs in association with the endothelial ICAM‐1 levels. T‐cell, endothelial cells, and junctions were adapted from Servier Medical Art (http://smart.servier.com/), licensed under a Creative Common Attribution 3.0 Generic License. (B) Diagram of RNA‐seq study design. pMBMECs were stimulated for 4 h with 10 ng/ml TNF‐α or 20 ng/ml IL‐1β. RNA was isolated at five different timepoints in parallel producing five biological replicates per experimental group and the samples were sequenced. (C) RNA‐seq expression level of the chosen genes shown as normalized reads. CP, Choroid Plexus. Data shown as mean ± SD of the respective five biological replicates. Statistical analysis was performed using the Benjamini‐Hochberg algorithm on the p‐values in order to obtain the False Discovery Rate (FDR). ***FDR ≤ 0.001; ****FDR ≤ 0.0001. (D) Volcano plot of IL‐1β versus TNF‐α‐stimulated pMBMECs gene expression. Differentially expressed genes between IL‐1β and TNFα stimulation were identified and further selected according to selected criteria described in detail in Results and Materials and methods. Volcano plot shows gene expression as log2Fold Change between normalized mean of reads from IL‐1β versus TNF‐α stimulated pMBMECs. FDR is shown as ‐log10 for visualization purposes; 1.3 on the Y‐axis is the equivalent of ‐log10 of 0.05 and was considered as a threshold for positive selection. Green and red dotted lines represent the selection thresholds. Black dots: genes with adjusted FDR < 1.3; Red dots: genes with log2‐fold change between ‐1 and 1 and FDR ≥ 1.3; Green dots: genes with simultaneous FDR ≥ 1.3 and log2‐fold change ≤ ‐1 or ≥ 1. Genes labeled in green were selected for further steps.
Transcriptome profiling of non‐stimulated pMBMECs was included as a baseline control. The RNA from five independent biological samples per condition was isolated and sequenced (Fig. 1B). Principal component analysis (PCA) showed three clearly distinct clusters of samples for TNF‐α, IL‐1β, and non‐stimulated (Supplementary Figure S1A). Although one IL‐1β sample did not cluster with the other four samples, we decided against excluding this sample to avoid the introduction of any hypothesis‐driven data manipulation. Interestingly, principal component 1 (X‐axis), which explained the vast majority of the variability in our samples (82%), showed non‐stimulated and IL‐1β stimulated samples most distanced, with TNF‐α‐stimulated samples in between both, confirming the diverse transcriptomic profile of pMBMECs depending on the respective cytokine stimulation. The purity of the samples and presence of possible contaminants was assessed by checking the gene expression of known markers [1, 13]. (http://betsholtzlab.org/VascularSingleCells/database.html) for endothelial cells, pericytes, astrocytes, and choroid plexus epithelial cells (Fig. 1C). As expected, the pMBMECs transcriptomes showed high expression of endothelial cell‐specific markers and low to absent expression of markers for pericytes, astrocytes, or epithelial cells. We found significantly higher ICAM‐1 mRNA expression levels in IL‐1β versus TNF‐α and non‐stimulated pMBMECs (Fig. 1C). This observation nicely corroborated that the chosen timepoint of 4 h after cytokine stimulation for transcriptome profiling of pMBMECs reflects the differential ICAM‐1 protein expression levels as previously observed in TNF‐α versus IL‐1β‐stimulated pMBMECs at 16 h after stimulation [7, 11, 14]. The significant Icam1 mRNA upregulation upon TNF‐α stimulation (∼2.9 log2‐fold increase compared to non‐stimulated pMBMECs) and upon IL‐1β stimulation (∼3.9 log2‐fold increase compared to non‐stimulated pMBMECs), resulting in ∼1 log2 fold increase of Icam1 mRNA in IL‐1β‐ vs TNFα‐stimulated pMBMECs, confirmed that the pMBMECs used in our transcriptomic study responded to the inflammatory stimuli used in our experimental design.
In order to select candidate genes potentially directing transcellular versus paracellular T‐cell diapedesis across pMBMECs, we first identified differentially regulated transcripts between IL‐1β and TNF‐α‐stimulated pMBMECs, favoring transcellular and paracellular diapedesis, respectively [11]. From the whole pMBMECs transcriptome, consisting of a total of 47069 transcripts, 3703 transcripts were found to be significantly upregulated or downregulated in IL‐1β stimulated pMBMECs when compared to TNF‐α‐stimulated pMBMECs. To identify the most relevant candidates, we performed a series of consecutive selections (summarized in Supporting Information Figure S1B). As a first step, we applied the following selection criteria to the whole transcriptome of IL‐1β versus TNF‐α‐stimulated pMBMECs: base mean of normalized reads between all the conditions ≥100; log2‐fold change ≥ 1 or ≤‐1 and false discovery rate (FDR) < 0.05. By applying these parameters, we found 196 differentially regulated transcripts of genes that met our selection criteria (Fig. 1D) and were therefore positively selected for further consideration. The most significantly upregulated genes upon IL‐1β stimulation (green dots on the right side of Fig. 1D) encode for the cytokine Csf3 and for various chemokines such as Cxcl5, Cxcl1, Cxcl2, and Cxcl3 ( Supporting Information Table 1). Since the receptors of these chemokines have been reported to be mostly expressed on myeloid cells [15, 16], we decided to not favor these genes in our candidate selection. Next, we investigated the function of each of the remaining 196 selected genes. Taking advantage of the repository Ingenuity Pathway Analysis (IPA), transcripts of genes with known functions related to cell movement, cytoskeleton remodeling, membrane trafficking, cell adhesion, and other functions or pathways potentially associated with T‐cell diapedesis were positively selected, for a total of 67 transcripts of genes, with varying expression levels among the different stimulations (Fig. 2A and B). As functional testing of 67 candidate genes was not possible, in the next step 3 of the authors performed independently and exactly at the same time in‐depth literature searches on these 67 candidate genes and categorized them on known gene functions (cell movement, cytoskeleton remodeling, membrane trafficking, cell adhesion, endothelial expression, known roles in immune cell diapedesis in general) but also considering the availability of reagents to verify a potential function of the target gene product in transcellular diapedesis (availability of antibodies, function‐blocking reagents, knock‐out mice, zebrafish models). The three selection tables were subsequently combined and discussed. With this approach, eight genes were finally selected with the majority of them (6/8; Nhs, Asap3, PLscr1, Lmo2, Ackr1, Rab8b) being upregulated at the RNA level upon IL‐1β stimulation in pMBMECs, while the others (2/8; Vamp1, Fam65b) were downregulated (Table 1 and Supporting Information Fig. S2A). In conclusion, our transcriptomic approach proved to be a powerful tool to select candidate genes potentially involved in directing the cellular pathway of T‐cell diapedesis across the BBB.
Figure 2.
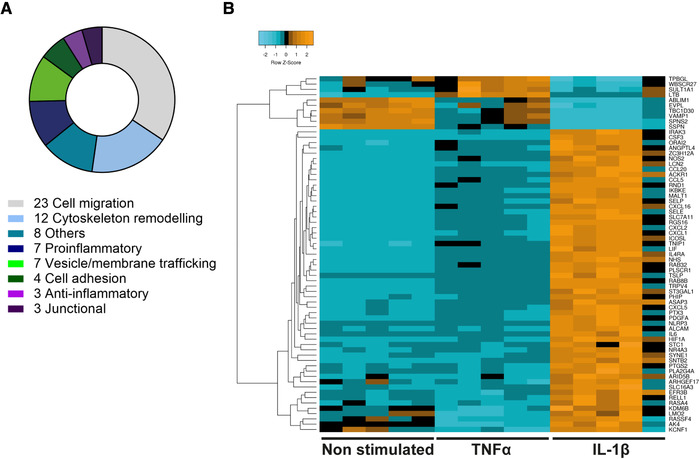
Functions and relative expression levels of 67 selected candidate genes. (A) Number of genes per functional category for the 67 selected genes. Each gene was manually assigned to one category according to information collected from IPA or UniProt repositories. (B) Heatmap for relative gene expression level (shown as Z‐score) for each of the selected 67 genes among all the different replicates and stimulations. Relative expression levels are shown in shades of blue, black, and orange. As Icam1 expression levels were used as a reference for transcellular T‐cell diapedesis, Icam1 is not included in this analysis.
Table 1.
Candidate genes selected from RNAseq
| Gene name | Ensembl Gene ID | Entrez gene ID | Normalized reads basemean | Normalized reads NS | Normalized reads TNF‐α | Normalized reads IL‐1β |
|---|---|---|---|---|---|---|
| Fam65b | ENSMUSG00000036006 | 193385 | 127.2 | 300.7 | 67.4 | 13.6 |
| Nhs | ENSMUSG00000059493 | 195727 | 541.6 | 150.4 | 292.2 | 1182.1 |
| Asap3 | ENSMUSG00000036995 | 230837 | 785.9 | 505.9 | 598.5 | 1253.2 |
| Plscr1 | ENSMUSG00000032369 | 22038 | 910.6 | 256.0 | 659.4 | 1816.3 |
| Vamp1 | ENSMUSG00000030337 | 22317 | 1139.1 | 1813.0 | 1183.7 | 420.7 |
| Lmo2 | ENSMUSG00000032698 | 16909 | 1298.0 | 1292.0 | 766.2 | 1835.7 |
| Ackr1 | ENSMUSG00000037872 | 13349 | 2000.7 | 84.5 | 848.2 | 5069.3 |
| Rab8b | ENSMUSG00000036943 | 235442 | 3295.6 | 1876.0 | 2601.4 | 5409.3 |
Selection of the most promising candidate gene contributing to transcellular T‐cell diapedesis
Based on our RNA‐seq analysis, we have selected eight candidate genes as potential mediators of transcellular T‐cell diapedesis across the BBB. To investigate if their RNA expression was confirmed at the protein level, we performed immunofluorescence stainings on cytokine‐stimulated pMBMECs. The observed staining patterns gave us an indication of the cellular distribution of each protein product of the respective candidate genes (Table 2; Supporting Information Fig. 3). While most proteins showed a subcellular distribution that could not be readily associated with a potential role in transcellular T‐cell diapedesis (Table 2; Supporting Information Fig. 3), ACRK1 showed reproducible cell surface staining on single pMBMECs. Based on our RNA‐seq data (Fig. 3A) and our previous observation of the involvement of endothelial ACKR1 in chemokine shuttling across the BBB and EAE pathogenesis [17], we thus selected Ackr1 as the main candidate gene to further explore its role in directing transcellular T‐cell diapedesis across the BBB.
Table 2.
Reagents used for immunofluorescence staining of candidate genes
| Antigen | Company | Cat number | Clonality | Host | Reactivity | Isotype | Dilution used | Observations |
|---|---|---|---|---|---|---|---|---|
| FAM65B | Santa Cruz | 134289 | 3J7 | Mouse | Human | IgG2b | 1/50 | Not detectable |
| NHS | Novus | NBP2‐56313 | Polyclonal | Rabbit | Human, Mouse (91%), Rat (91%) | IgG | 1/50 | Dotty staining, junctional accumulation |
| ASAP3 | Santa Cruz | sc‐376479 | Monoclonal (G‐12) | Mouse | Mouse | IgG1 | 1/100 | Not detectable |
| PLSCR1 | Proteintech | 11582‐1‐AP | Polyclonal | Rabbit | Mouse, Human, Rat | IgG | 1/100 | Nuclear staining |
| VAMP1 | Proteintech | 13115‐1‐AP | Polyclonal | Rabbit | Mouse, Human, Rat | IgG | 1/100 | Faint dotty perinuclear staining |
| LMO2 | Santa Cruz | sc‐65736 | Monoclonal (1A9‐1) | Mouse | Mouse, Human, Rat | IgG1 | 1/100 | Dotty perinuclear staining |
| ACKR1 | Homemade (Ulrich von Adrian) [19] | Monoclonal (6B7) | Rat | Mouse | IgG2a | 1/250 | Single cells show positive staining | |
| RAB8B | Proteintech | 11792‐1‐AP | Polyclonal | Rabbit | Mouse, Human, Dog | IgG | 1/100 | Dotty perinuclear and cytoplasmatic staining |
Figure 3.
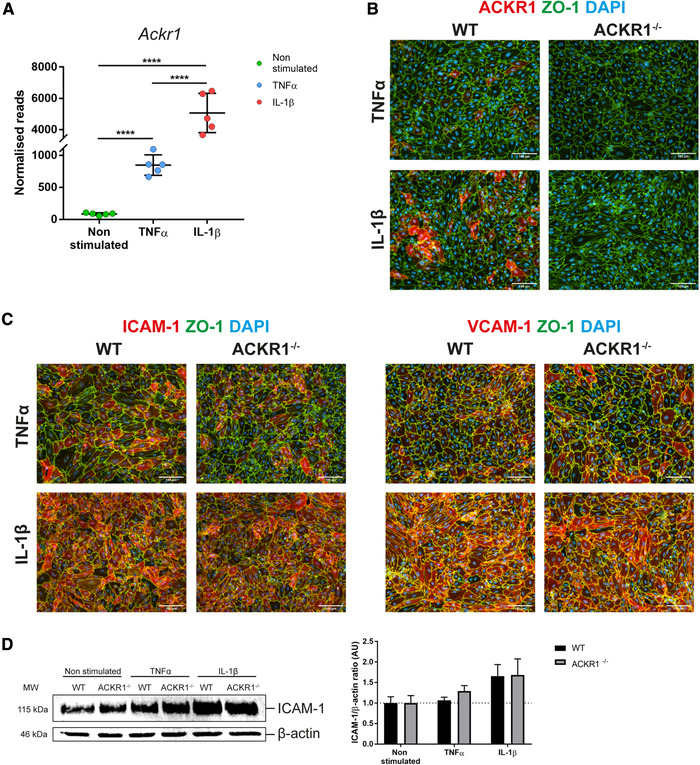
ACKR1 is expressed at the BBB in vitro and its absence does not affect adhesion molecule expression. (A) Ackr1 RNA levels shown as normalized reads of the five biological replicates per conditions used for the RNA‐seq analysis. Data shown as mean ± SD. Statistical analysis was performed using the Benjamini‐Hochberg algorithm on the p‐values in order to obtain the FDR. ****FDR ≤ 0.0001. (B) Immunofluorescence staining for ACKR1 (red), ZO‐1 (green), and DAPI (blue) on WT or ACKR1–/– pMBMECs stimulated with 10 ng/ml TNF‐α or 20 ng/ml IL‐1β for 16 h. Magnification: 20×. Representative images from three independent experiments where ACKR1 and ZO‐1 were analyzed in one well per assay compared to the specific isotype control staining in a second well. (C) Immunofluorescence staining for ICAM‐1 or VCAM‐1 (red), ZO‐1 (green), and DAPI (blue) on WT or ACKR1–/– pMBMECs stimulated with 10 ng/ml TNFα or 20 ng/ml IL‐1β for 16 h. Magnification: 20×. Representative images from three independent experiments where staining for ICAM1, VCAM‐1, and ZO‐1 was analyzed in one well per assay and compared to the specific isotype control staining in a second well. (D) Immunoblot analysis of ICAM‐1 levels in WT and ACKR1–/– pMBMECs upon cytokine stimulation and in non‐stimulated conditions. For each pMBMEC preparation 6 to 8 WT and ACKR1–/– mice were used. On the left is shown a representative blot while on the right, data are quantified as ICAM‐1/β‐actin ratio, normalized to the non‐stimulated condition for each genotype, and shown as mean ± SD from four independent experiments.
To investigate its role in mediating T‐cell diapedesis across the BBB, we made use of C57BL/6 mice lacking expression of ACKR1 [18]. Immunofluorescence staining using a novel ACKR1‐specific monoclonal antibody [19] showed that ACKR1 protein expression was not detectable on non‐stimulated pMBMECs ( Supporting Information Fig. 4) but readily upregulated on the cell surface of pMBMECs upon cytokine stimulation, as previously described by us using a polyclonal antibody (Fig. 3B) [17]. Moreover, ACKR1 immunostaining was observed on the surface of individual pMBMECs stimulated with TNF‐α, while IL‐1β stimulation significantly increased the number of pMBMECs staining positive for ACKR1. As expected, pMBMECs isolated from ACKR1–/– C57BL/6 mice did not show any immunostaining for ACKR1, irrespective of the pro‐inflammatory stimulus applied (Fig. 3B).
Caveolae have been suggested to serve as a vesicular transport system in ACKR1 mediated chemokine shuttling across endothelial cells [20, 21] and to mediate transcellular T‐cell diapedesis across the BBB [12]. Expression of caveolin‐1 mRNA was detected in stimulated and non‐stimulated pMBMECs in our RNA‐seq dataset and expression levels did not change with cytokine stimulation (Supporting Information Table 1). To confirm constitutive expression of caveolin‐1 in pMBMECs at the protein level, immunostainings for caveolin‐1 in non‐stimulated as well as IL‐1β‐stimulated pMBMECs monolayers (Supporting Information Fig. 4) confirmed caveolin‐1 detection in all brain endothelial cells. Caveolin‐1 is thus constitutively expressed in pMBMECs and expression levels are not increased by pro‐inflammatory cytokine stimulation.
To ensure that lack of ACKR1 does not affect expression levels of VCAM‐1 and ICAM‐1, which mediate T‐cell arrest and crawling on pMBMECs, we next investigated their protein levels in WT and ACKR1–/– pMBMECs under inflammatory conditions. Side‐by‐side immunofluorescence stainings for ICAM‐1 and VCAM‐1 on WT and ACKR1–/– pMBMECs did not show any differences in the staining pattern (Fig. 3C). In contrast to VCAM‐1, cell surface levels of ICAM‐1 are critical in regulating the cellular pathway of T‐cell diapedesis across pMBMECs [11, 22]. Thus, we specifically confirmed equal protein expression levels of ICAM‐1 in WT and ACKR1–/– pMBMECs by Western Blot (WB) (Fig. 3D). Altogether, our data confirmed increased expression of ACKR1 in pMBMECs under IL‐1β‐stimulated conditions and that lack of ACKR1 does not impact on VCAM‐1 and even more importantly on ICAM‐1 expression. Thus, ACKR1–/– pMBMECs are a suitable model to investigate the role of ACKR1 in transcellular T‐cell diapedesis across the BBB.
Absence of ACKR1 reduces transcellular T‐cell diapedesis across pMBMECs
To investigate the contribution of ACKR1 in transcellular versus paracellular T‐cell diapedesis across pMBMECs, we studied the migration of CD4+ effector/memory T cells across WT and ACKR1–/– pMBMECs under physiological flow by live‐cell imaging. Junctional visualization was achieved by labeling PECAM‐1 on pMBMECs with a non‐blocking anti‐PECAM‐1 antibody [23], directly conjugated to Alexa Fluor 555. To confirm that this methodology is equally reliable as studying the cellular pathway of T‐cell diapedesis across pMBMECs isolated from vascular endothelial cadherin (VE‐Cadherin)‐GFP knock‐in C57BL/6J mice [11, 14, 24], we stained VE‐Cadherin‐GFP pMBMECs with the anti‐PECAM‐1 Alexa Fluor 555 antibody. Imaging under physiological flow over a period of 30 min showed no loss of signal of the junctional PECAM‐1 staining over time (Supporting Information Fig. 5A) and confirmed strong co‐localization of the PECAM‐1 Alexa Fluor 555 and the VE‐cadherin‐GFP fluorescence signals (Supporting Information Fig. 5B). This successfully validated the use of the anti‐PECAM‐1 antibody for visualization of endothelial junctions on pMBMECs during live‐cell imaging in vitro and therefore allowed to discriminate between paracellular and transcellular T‐cell diapedesis events (Fig. 4C and Supporting Information Video S1, details in Materials and Methods). First, we investigated if the absence of ACKR1 in pMBMECs affects T‐cell arrest and post‐arrest T‐cell behavior. No differences were found in the number of T cells able to arrest on TNF‐α or IL‐1β‐stimulated WT and ACKR1–/– pMBMECs under physiological flow (Fig. 4A). The absence of endothelial ACKR1 did not affect T‐cell detachment, stationary behavior, probing, and crawling when compared to WT pMBMECs (Fig. 4B). Importantly, the fraction of arrested T cells that completed diapedesis across the pMBMECs monolayers in the observation time was also not affected by the lack of ACKR1 (Fig. 4B). In accordance with our previous study [11], IL‐1β stimulation of pMBMECs increased T‐cell diapedesis across the endothelial monolayer when compared to TNF‐α stimulated pMBMECs (Fig. 4D). Next, we identified the T‐cell diapedesis pathway across WT or ACKR1–/– pMBMECs based on the integrity of junctional PECAM‐1 localization during the diapedesis event (Fig. 4C). Lack of ACKR1 significantly reduced transcellular T‐cell diapedesis across IL‐1β‐stimulated pMBMECs (Fig. 4D). At the same time absence of ACKR1 did not affect crawling speed, crawling distance, or crawling directionality of the T cells that successfully completed paracellular or transcellular diapedesis across WT or ACKR1–/– pMBMECs (Fig. 4E and F; Supporting Information Fig. 6A). Also, the lack of ACKR1 did neither delay the start nor the overall duration of transcellular or paracellular T‐cell diapedesis on pMBMECs underflow, suggesting that T cells can normally crawl to sites permissive for diapedesis in the absence of ACKR1 (Supporting Information Fig. S6C and D). Furthermore, no differences in the T‐cell diapedesis pore size were found between WT and ACKR1–/– pMBMECs (Supporting Information Fig. 6B), suggesting that the remodeling capacities of the endothelial cell cytoskeleton are not affected by the absence of ACKR1. Taken together, the lack of ACKR1 in our in vitro BBB model selectively reduced transcellular T‐cell diapedesis across pMBMECs under flow in vitro without affecting any other step of the multi‐step T cell extravasation cascade.
Figure 4.
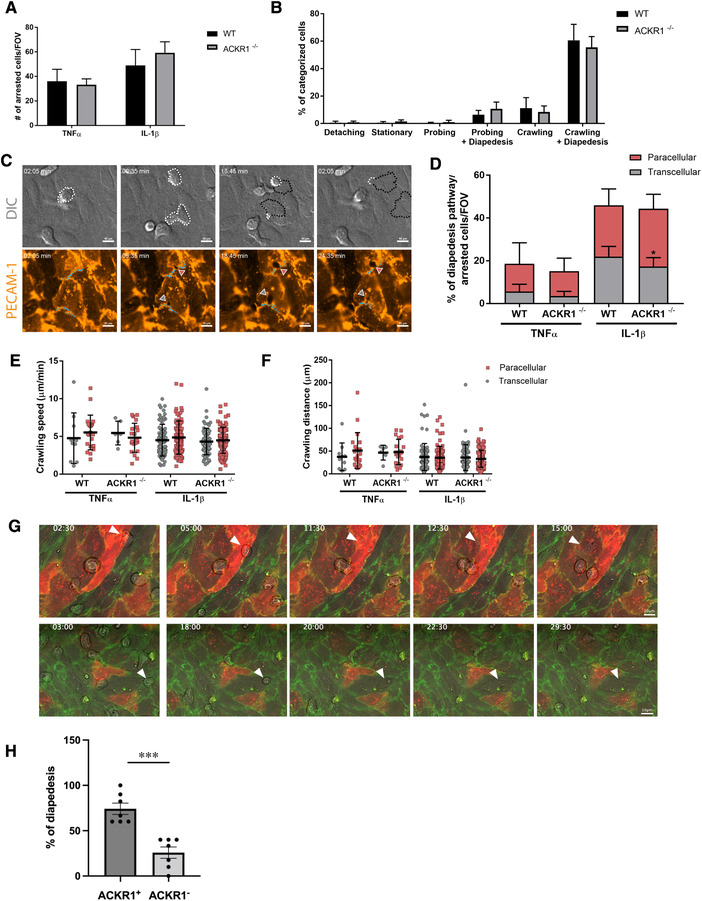
Absence and presence of ACKR1 reduced and enhanced transcellular T‐cell diapedesis across pMBMECs. (A) Number of arrested T‐cells/Field of view (FOV) on TNFα or IL‐1β‐stimulated WT or ACKR1–/– pMBMECs under flow conditions. Data shown as mean ± SD from four to five independent experiments. (B) T‐cell post‐arrest behavior on IL‐1β‐stimulated WT or ACKR1–/– pMBMECs under flow conditions. Each category is shown as a fraction of the sum of the categorized cells. Incomplete diapedesis events with prior probing or crawling accounted for <10% and are not shown for visualization reasons. Data shown as mean ± SD from four to five independent experiments. (C) Representative snapshots of live cell imaging under flow conditions of T cells undergoing paracellular or transcellular diapedesis across pMBMECs. White dotted line marks T cells area on top of the endothelium monolayer; black dotted line marks T cells area underneath the endothelium monolayer; light blue dotted line marks endothelial cell junctions based on PECAM‐1 AlexaFluor 555 fluorescence signal. Note the interruption of junctional integrity during paracellular diapedesis (red arrowhead), while during transcellular diapedesis (grey arrowhead) the junctions remained intact. DIC, differential interference contrast. Cropped from 63× magnification images. (D) T‐cell diapedesis pathway across cytokine‐stimulated WT or ACKR1–/– pMBMECs under flow conditions. Data are shown as percentage of diapedesis pathway normalized to the sum of the arrested cells/FOV for each condition. Data shown as mean ± SD from four to five independent experiments. Statistical analysis performed using unpaired t‐test with Welch's correction. *p‐value < 0.05. (E) Crawling speed of T‐cells that successfully completed paracellular or transcellular diapedesis events on cytokine‐stimulated WT or ACKR1–/– pMBMECs under flow conditions shown in μm/minutes. Data shown as mean ± SD from crawling tracks obtained from four to five independent experiments. (F) Crawling distance of T‐cells that successfully completed paracellular or transcellular diapedesis events on cytokine‐stimulated WT or ACKR1–/– pMBMECs under flow conditions shown in μm. Data shown as mean ± SD from crawling tracks obtained from four to five independent experiments with seven and eight videos analyzed for ACKR1–/– and WT pMBMECs, respectively. (G) Representative snapshots of live cell imaging under flow conditions of T cells undergoing transcellular diapedesis across ACKR1+ and ACKR1neg VE‐CadGFP pMBMECs. ACKR1+ pMBMECs are visible by the red immunofluorescence signal based on positive staining with the rat‐anti‐mouse ACKR1 Ab directly labeled with Alexa Fluor 647. The green fluorescence signal shows the endothelial junctions marked by VE‐cadherin–GFP. The T cells are visible in gray (phase‐contrast signal). White arrowheads highlight T cells performing transcellular diapedesis across an ACKR1+ (upper panel) or an ACKR1neg (lower panel) pMBMEC at the respective time points (minutes:seconds) indicated at the top. Images of the entire pMBMECs monolayer were acquired in eight tiles, which were subsequently stitched together for analysis of the entire field of view. The representative images were cropped from 63x magnification images (Supporting Information Videos 2 and 3). (H) Quantification of transcellular T‐cell diapedesis events across IL‐1β‐stimulated pMBMECs staining positive (ACKR1+) or not (ACKR1–) under physiological flow. Seven videos with a total number of 465 arrested T cells from which 69.1 ± 10.5% performed diapedesis were analyzed. Data show percentage of transcellular diapedesis rates normalized to total number of transcellular diapedesis events. Data are shown as mean ± SEM.
ACKR1 expressing pMBMECs favor transcellular over paracellular T‐cell diapedesis in vitro
Having shown that the absence of ACKR1 selectively reduces transcellular T‐cell diapedesis across IL‐1β‐stimulated pMBMECs, we next asked if those pMBMECs that upregulated ACKR1 in the IL‐1β‐stimulated pMBMECs monolayer are those that specifically mediate transcellular T‐cell diapedesis events. Visualization of cell surface ACKR1 on IL‐1β‐stimulated VE‐cadherin‐GFP pMBMECs monolayer was achieved by labeling with the anti‐ACKR1 antibody directly conjugated to Alexa 647. This allowed for simultaneous visualization of the endothelial junctions and ACKR1 cell surface expression (Fig. 4G). Next, we studied T‐cell diapedesis across ACKR1+ and ACKR1– VE‐cadherin‐GFP pMBMECs under physiological flow by live‐cell imaging and quantified transcellular T‐cell diapedesis events that occurred through ACKR1+ versus ACKR1neg VE‐cadherin‐GFP pMBMECs. We observed that ACRK1+ pMBMECs promoted significantly more transcellular T‐cell diapedesis events when compared to ACKR1neg pMBMECs in the same pMBMECs monolayer (Fig. 4G and H; Supporting Information Videos 2 and 3). These data further support that ACKR1 mediates transcellular T‐cell diapedesis across our in vitro model of the BBB.
ACKR1 is induced on the inflamed BBB in vivo and localized on the endothelial cell surface rather than restricted to BBB junctions
Recent studies suggested ACKR1 to be exclusively localized to endothelial junctions in peripheral vascular beds [19, 25]. This observation is in apparent contradiction to its role in favoring transcellular T‐cell diapedesis across pMBMECs in vitro. To thus specifically explore protein expression and subcellular localization of ACKR1 protein on the BBB in vivo, we made use of the novel monoclonal antibody directed against ACKR1 [19]. Confocal imaging of cerebellum and spinal cord sections of healthy mice showed low immunostaining of ACKR1 in meningeal venules but the absence of immunostaining on venules in the CNS parenchyma according to our previous observations using a polyclonal anti ACKR1 antibody (Supporting Information Fig. 7) [17]. During neuroinflammation in EAE, we readily observed ACKR1 immunostaining in both meningeal and CNS parenchymal venules (Fig. 5A; Supporting Information Fig. 7). In accordance with our previous observations, we found CD45+ immune cell infiltrates surrounding ACKR1+ but never ACKR1neg CNS microvessels, supporting the role of ACKR1 in T‐cell diapedesis across the BBB in vivo (Supporting Information Fig. 7). To understand if ACKR1 is available on the brain endothelial surface to support transcellular T‐cell diapedesis in vivo, we next explored the subcellular distribution of ACKR1 on the CNS endothelium. To this end, we specifically investigated the co‐localization of ACKR1 immunostaining with the junctional VE‐Cadherin‐GFP signal in VE‐cadherin knock‐in mice suffering from EAE. Confocal imaging of thick vibratome brain sections as well as of spinal cord frozen sections showed ACKR1 immunostaining to partially co‐localize with the VE‐cadherin‐GFP signal in brain venules in mice suffering from EAE (Fig. 5B and C; Supporting Information Fig. 7). At the same time, strong ACKR1 immunostaining was detected on brain and spinal cord endothelial cells outside of endothelial cell‐to‐cell contacts (Fig. 5C; Supporting Information Fig. 7). Taken together our observations show that unlike in peripheral tissues, ACKR1 protein is only expressed at the inflamed BBB and its subcellular distribution is not restricted to BBB junctions. By shuttling inflammatory chemokines across the BBB, ACKR1 may thus contribute to transcellular T‐cell diapedesis across the inflamed BBB during EAE.
Figure 5.
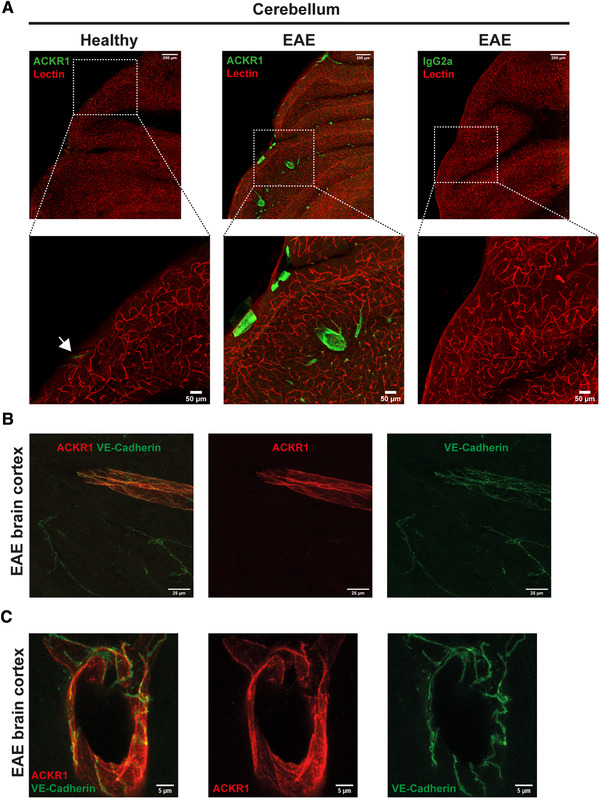
Protein expression of ACKR1 on inflamed CNS venules in vivo outside of BBB junctions. (A) Confocal imaging of 100 μm thick sections of healthy or EAE mouse cerebellum. Mice were injected i.v. with 33 μg of DyLight594 lectin (red) and after 15 min, perfused with 2% PFA in PBS. After post‐fixation, sections were prepared and staining for ACKR1 or rat IgG2a (green) was performed. EAE score: 1, day 31 post‐immunization. Maximal intensity projections are shown. White arrow in the magnified area of the healthy cerebellum indicates an ACKR1+ meningeal vessel. Magnification 40x. Representative images from two independent experiments. (B and C) Confocal imaging of 100 μm thick sections of VE‐Cadherin‐GFP (green) knock‐in mouse brain suffering from EAE. Mice were perfused with 2% PFA in PBS. After post‐fixation, sections were prepared and staining for ACKR1 (red) was performed. EAE score:1, day 28 post‐injection. Magnification: 63×, in (C) is shown a digital zoom from 63× magnification image. Representative images from three independent experiments that analyzed tissue sections from three individual mice suffering from EAE.
Discussion
Immune cell trafficking into the CNS is a major hallmark of EAE and MS [26, 27]. In particular, T‐cell migration across the BBB is a multistep cascade of events that has been analyzed by us and others. After their initial VCAM‐1 mediated arrest on the BBB, T‐cells crawl over long distances against the direction of the blood flow [8] in an ICAM‐1 and ICAM‐2‐dependent manner [7, 11, 22]. Finally, T cells cross the BBB by transcellular or paracellular diapedesis using molecular mechanisms incompletely understood. Using pMBMECs as a BBB in vitro model, we have previously reported the active role played by BBB endothelial cells in directing T cells to sites permissive for paracellular or transcellular diapedesis [11]. Differential cytokine stimulation of pMBMECs with either TNF‐α or IL‐1β allows to induce low or high cell surface levels of endothelial ICAM‐1, and preferential paracellular or transcellular T‐cell diapedesis across pMBMECs under flow, respectively. Here, we took advantage of this observation to perform a transcriptomic analysis of TNF‐α versus IL‐1β ‐stimulated pMBMECs favoring paracellular versus transcellular T‐cell diapedesis to identify novel molecular mediators regulating the cellular pathway of T‐cell diapedesis across the BBB.
Candidate gene selection based on our RNA‐seq analysis of IL‐1β vs TNFα‐stimulated pMBMECs consisted of sequential filtration steps, ultimately leading us to define Ackr1 as the main candidate gene able to direct the T‐cell diapedesis pathway across the BBB. This observation is in accordance with a recent study that specifically identified a role for IL‐1 signaling at the BBB in EAE pathogenesis by upregulating ICAM‐1, VCAM‐1, and ACKR1 [28].
It is well accepted that chemokines play a major role in regulating immune cell migration in general [29, 30]. Chemokines bind to G‐protein coupled receptors (GPCR) initiating intracellular signaling through heterotrimeric G proteins, resulting in downstream regulation of cell migration processes [15, 31, 32]. In the context of immune cell extravasation, immobilized chemokines on the luminal side of endothelial cells are recognized by GPCRs on immune cells, triggering “inside‐out” integrin activation resulting in integrin conformational change and immune cell arrest on the endothelium. Immune cell polarization is followed by their crawling on the endothelium in search of sites permissive for diapedesis [33]. Strikingly, blocking GPCR‐signaling in effector/memory T cells also prohibits their diapedesis across endothelial cells including those of the BBB under flow in vitro [34, 35], suggesting a yet less appreciated involvement of chemokines in immune cell diapedesis across endothelial barriers.
During EAE, expression of inflammatory chemokines is mainly upregulated in astrocytes localized in the CNS parenchyma [36, 37, 38]. To reach the luminal side of the BBB and thus contribute to immune cell recruitment across the BBB, these chemokines will have to breach the BBB. In a previous study, we have identified ACKR1 to shuttle inflammatory chemokines across the BBB [17].
In contrast to classical GPCRs, the atypical chemokine receptors (ACKRs) fail to couple to G‐proteins due to the lack of the DRYLAIV consensus motif in the second intracellular loop resulting in non‐canonical GPCRs responses [39, 40]. Instead, ACKRs regulate chemokine bioavailability, by acting as decoy or scavenging receptors or by transporting chemokines [41]. ACKR1 is one of four ACKRs known to date. ACKR1 binds 20 proinflammatory but not homeostatic chemokines of both CC and CXC families and is expressed in erythrocytes and endothelial cells [41, 42]. While on erythrocytes ACKR1 acts as a chemokine reservoir, buffering the available levels of chemokines in the plasma, on endothelial cells, it actively transports inflammatory chemokines from the basolateral to the luminal side of the endothelial cell [41, 43], including the BBB [17]. Supporting its important role in mediating leukocyte migration across the vascular wall, localization of ACKR1 is restricted to endothelial cells of postcapillary venules [19], the vascular segment allowing for the extravasation of circulating leukocytes into tissues [44].
In accordance with our previous observation [17], we here report the expression of ACKR1 in mouse brain microvascular endothelial cells under inflammatory conditions at the RNA and protein level. Importantly, ACKR1 expression was significantly higher in IL‐1β versus TNF‐α‐stimulated pMBMECs making it an attractive candidate to contribute to transcellular T‐cell diapedesis across the BBB. Analyzing the cellular pathway of T‐cell diapedesis across ACKR1–/– pMBMECs under physiological flow in fact showed a significant reduction in the numbers of T‐cell using the transcellular pathway to cross the in vitro BBB model. Furthermore, those pMBMECs expressing ACKR1 in IL‐1β‐stimulated conditions supported significantly more transcellular T‐cell diapedesis events under physiological flow when compared to ACKR1neg pMBMECs in the very same monolayer.
We have previously shown that ACKR1 shuttles the inflammatory chemokines CCL2 and CCL5 from the basolateral to the luminal side of pMBMECs [17]. Our RNA‐seq analysis showed increased expression of CCL2 and CCL5 in cytokine‐stimulated pMBMECs (Supporting Information Table 1) suggesting that in the current setup these chemokines when released to the abluminal side could be shuttled by ACKR1 to the luminal side of pMBMECs, creating a high local concentration of chemokines that contributes to transcellular T‐cell diapedesis. These local “hot‐spots” of chemokines might contribute to enhancing GPCR‐mediated activation of the integrin LFA‐1 on T cells. LFA‐1 mediated clustering of the high levels of endothelial ICAM‐1 present on IL‐1β‐stimulated pMBMECs could then initiate transcellular T‐cell diapedesis as observed before [44]. At the same time our observations that T‐cell crawling prior to paracellular or transcellular diapedesis across pMBMECs was not affected by the absence of ACKR1 challenge this hypothesis, which may however be due to a dominant role of cell surface levels of endothelial ICAM‐1 previously observed by us in this context [34]. Furthermore, duration of T‐cell diapedesis, as well as the endothelial pore size during T cell diapedesis was unaltered by the absence of ACKR1, leaving unanswered the question of how lack of ACKR1 exactly leads to reduced transcellular T‐cell diapedesis across pMBMECs.
One possible explanation is that ACKR1 besides shuttling chemokines from the basolateral to the luminal side of pMBMECs also affects their intracellular distribution. Indeed, it has been suggested that upon chemokine binding, ACKR1 translocates intracellularly, where it colocalizes with caveolin in intracellular vesicles in Madin‐Darby canine kidney (MDCK) cells [43], suggesting caveolae as an intracellular mechanism of transport of ACKR1. In addition, it has been shown that CCL2 can be transported from the abluminal to the luminal side of brain microvascular endothelial cells by a transcellular mechanism that at least to a certain degree involves binding of CCL2 to CCR2 and caveolae [45, 46]. Our pMBMECs constitutively express caveolin‐1 at the mRNA and protein level, with caveolin‐1 expression however not further upregulated upon cytokine stimulation of the pMBMECs. Interestingly however, all brain endothelial cells in a pMBMECs monolayer stain positive for caveolin‐1. Previous studies have colocalized ACKR1 to caveolin 1 and therefore to caveolae as a vesicular transport compartment [20, 21]. T cells extend numerous protrusions into and through the pMBMEC monolayers when searching for sites permissive for diapedesis in the process of probing or crawling [11]. As caveolae have also been shown to play a role in transcellular, but not paracellular, T‐cell diapedesis across the BBB [12], it is tempting to speculate that caveolae transporting ACKR1 with bound chemokines may trigger membrane fusion events enhancing transcellular T‐cell diapedesis across BBB endothelial cells. In this scenario, chemokines transported by ACKR1 in the next proximity of T‐cell protrusions could potentially establish a polarized gradient able to enhance transcellular T‐cell diapedesis across BBB endothelial cells. The precise modalities by which ACKR1 translocates in BBB endothelial cells and thus influences subcellular chemokine distribution are not known and therefore, the precise mechanisms of how ACKR1 enhances transcellular T‐cell diapedesis across the BBB will need further investigations.
Making use of a novel monoclonal antibody directed against mouse ACKR1 we here confirmed our previous observation [17] that, in contrast to peripheral tissues, ACKR1 is not constitutively expressed in BBB forming venules in the CNS parenchyma. In brain and spinal cord sections from healthy mice, ACKR1 immunostaining was only detected in meningeal venules, but absent from venules in the CNS parenchyma. This ACKR1 immunostaining pattern differentiating meningeal from CNS parenchymal endothelial cells is similar to that observed for P‐selectin. Under physiological conditions, expression of P‐selectin is absent in CNS parenchymal endothelial cells but constitutively present in meningeal endothelial cells, where it is stored in Weibel‐Palade bodies and can be rapidly redistributed to the luminal membrane, allowing for immune cell recruitment [47]. This observation underscores that in the absence of neuroinflammation, the CNS parenchymal vasculature does strictly control immune cell trafficking, in contrast to the meningeal vasculature, where immunosurveillance in the CNS takes place [4]. During EAE, expression of ACKR1 is upregulated in meningeal venules and induced in parenchymal venules making its function available for immune cell recruitment across the BBB. Interestingly, ACKR1 immunostaining was also observed in brain sections of MS patients [17], supporting a role for ACKR1 in immune cell recruitment to the CNS also in MS. Increased transcellular immune cell trafficking across the BBB in neuroinflammation thus correlates with the expression pattern of ACKR1 on the BBB, so does its subcellular distribution, which we here show to be localized to the endothelial cell surface rather than limited to the BBB endothelial junctions. Cell surface localization of ACKR1 on the BBB is thus different when compared to peripheral vascular beds, where ACKR1 was shown to localize to endothelial junctions and, by presenting CXCL2, mediate paracellular diapedesis of neutrophils [25].
Taken together, our study identifies a potential role for endothelial ACKR1 in contributing to transcellular T‐cell diapedesis across the inflamed BBB. Therapeutic targeting of molecular mechanisms mediating specifically this cellular pathway of T cell trafficking increased in neuroinflammation bears the hope to restore physiological immune cell trafficking across the BBB via paracellular pathways. In general, therapeutic targeting of ACKR1 in this context may be considered as despite its expression on erythrocytes, the absence of ACKR1 on erythroid cells is compatible with life [48]. On the other hand, erythrocyte ACKR1 buffers circulating chemokines, and a systemic therapeutic blockage of ACKR1 may result in dysregulation of chemokine plasma levels, with either chemokine overstimulation of circulating immune cells or affecting the release of neutrophils from the bone marrow, as suggested by the neutropenia observed in individuals lacking erythrocytic ACKR1 [49]. Nevertheless, our results encourage further studies to explore the precise mechanisms of how ACKR1 directs transcellular T‐cell diapedesis across the BBB. Indeed, a better understanding of the molecular mechanisms regulating paracellular and transcellular diapedesis across the BBB is of crucial importance as it represents a key step in the migration of autoaggressive T cells into the CNS parenchyma during neuroinflammation.
Materials and methods
Mice
Mice were housed in individually ventilated cages under specific pathogen‐free conditions at 22°C with free access to chow and water. Animal procedures executed were approved by the Veterinary Office of the Canton Bern (permit no. BE31/17 and BE77/18 and BE98/20) and are in keeping with institutional and standard protocols for the care and use of laboratory animals in Switzerland. C57BL/6J mice were obtained from Janvier (Saint‐Berthevin Cedex, France). VE‐Cadherin GFP knock‐in mice were provided by D. Vestweber (Münster, Germany) [24, 50]. ACKR1–/– mice were provided by A. Rot (London, UK) [18].
Antibodies
Primary antibodies used for immunofluorescence staining were rabbit anti‐mouse ZO‐1 (Thermo Fisher Scientific, Cat No 61–7300), rat anti‐mouse VCAM‐1 (in house, clone 9DB3), rat anti‐mouse ICAM‐1 (in house, clone 25ZC7), rabbit anti‐caveolin‐1 (BD Biosciences, Cat N° 610060), and biotinylated rat‐anti‐mouse CD45 (Biolegend, CatN° 405237). Primary antibodies used for testing RNA‐seq candidate genes protein levels are listed in Table 2. Secondary antibodies used for immunofluorescence staining were goat anti‐rat IgG (H+L) Cy3 (Jackson ImmunoResearch, Cat. No 112‐165‐003), donkey anti‐rat IgG (H+L) Cy3 (Jackson ImmunoResearch, Cat No 712‐165‐153), goat anti‐rabbit IgG (H+L) Cy3 (Jackson ImmunoResearch, Cat No 111‐165‐144), goat anti‐mouse IgG (H+L) Cy3 (Jackson ImmunoResearch, Cat No 115‐166‐006) donkey anti‐rabbit IgG (H+L) AlexaFluor 488 (Jackson ImmunoResearch, Cat No 711‐546‐152), goat anti‐rat IgG (H+L) AlexaFluor 488 (Thermo Fisher Scientific, Cat No A11006); donkey‐anti‐rabbit Cy5 (Jackson ImmunoResearch, Cat No 711‐175‐152), and Streptaviding‐AlexaFluor 647 (Biolgegend, CatN° 103104).
In vitro BBB model – pMBMECs
pMBMECs were isolated from 7 to 9 weeks old C57BL/6 mice or VE‐cadherin‐GFP knock‐in mice and cultured exactly as described before [2, 7]. Where indicated, cytokine stimulation was performed with 10 ng/ml TNF‐α (Peprotech, London, UK) or 20 ng/ml IL‐1β (Peprotech, London, United Kingdom) for 16–20 h prior to the experiment. For RNA‐seq experiment, cytokine stimulation was performed for 4 h prior RNA extraction.
RNA sequencing
RNA was extracted from pMBMECs with the High Pure RNA Isolation Kit (Hoffman‐La Roche, Basel, Switzerland). Each sample consisted of pMBMECs isolated from 24 female C57BL/6 mice at 7 weeks of age. RNA libraries for five replicates per condition (non‐stimulated, TNFα‐stimulated, IL‐1β‐stimulated pMBMECs) were prepared according to the manufacturer's instruction using TruSeq stranded mRNA Library Prep Kit (Illumina, San Diego, USA) with polyA selection. RNA sequencing was performed using Illumina Hiseq 2500 at the Next Generation Sequencing (NGS) Platform of the University of Bern. The quality of the samples was assessed using a Fragment analyzer (Applied Biosystems, Foster City, USA) with the samples having RIN values ≥ 8 and an average of 9.2.
RNA‐seq analysis
Following sequencing, the data was demultiplexed and the, generated fastq files were checked for quality measures using FASTQC software (version 0.11.2)(Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc). Depth of sequencing was on average ∼25 M reads for each of the 15 replicates. The few rRNA fragments that were still present despite the polyA selection were removed using Trimmomatic (version 0.33) [51] followed by an additional round of quality control. The fastq files were then aligned to the Ensembl mm10 reference genome using TopHat2 (version 2.0.13) [52] after which Qualimap (version 2.2) [53] was used for quality control and IGV (version 2.3.69) [54] for visualization of the aligned reads. Counts by transcript were evaluated using HTSeq‐count (version 0.6.1) [55] and inter‐sample normalization and group differential expression analysis was performed with the DESeq 2 package (Version 1.12.4) [56] in R (Version 3.2.2).
Comparison between normalized reads of each transcript from each condition was performed and differentially regulated transcripts of genes between every possible design group comparison (non‐stimulated vs. TNF‐α; non‐stimulated vs. IL‐1β and IL‐1β vs. TNF‐α) were identified. For the purpose of this project, we focused on the differentially regulated transcripts of genes between IL‐1β versus TNF‐α‐stimulated pMBMECs. Multiple testing correction was conducted on the p‐values using the Benjamini‐Hochberg algorithm in order to obtain the FDR. Selection criteria to reduce the number of differentially regulated transcripts of genes were defined as: base mean of normalized reads between all the conditions ≥ 100; log2‐fold change ≥ 1 or ≤ ‐1 and FDR < 0.05. Analysis of functions and pathways involvement of each of the transcripts that were selected was performed on IPA (Version 2.1, Qiagen, Hilden, Germany). The final selection step (from 67 to 8 genes) was performed by screening available literature that already investigated the function of the genes, possibly linked to cell migration context. Moreover, considering the timeframe of this study, in the selection process we gave priority to genes for which knock‐out mice and/or blocking antibodies are available.
Immunofluorescence staining of pMBMECs
Confluent pMBMECs monolayers grown on on coverslips for sticky‐Slides ibiTreat (Ibidi, Munich, Germany) mounted on 12 wells μ‐Chamber (Ibidi, Munich, Germany) were gently washed with warm DPBS and subsequently fixed with either 1% paraformaldehyde (PFA, MERCK, Darmstadt, Germany) in DPBS at RT for 10 min or with −20°C methanol (for ACKR1 staining) for 30 s. After fixation, cells were washed 2× with DPBS. Blocking buffer (5% skimmed milk (Rapilait, Migros, Switzerland), 0.3% Triton‐X‐100 (Schweizerhall, Basel, Switzerland Batch Nr. A11021), 0.04% NaN3 (Fluka Chemie, Buchs, Switzerland) in TBS pH 7.4) was applied and incubated for 20 min at RT. Subsequently blocking buffer containing the primary antibody was added for 1 h at RT. The primary antibody was removed from the cells by 2× washing steps with DPBS and blocking buffer containing the secondary antibody and 1 μg/ml 4′,6‐diamidin‐2‐phenylindol (DAPI, AppliChem, Darmstadt, Germany) was added to the cells and incubated light protected for 1 h at RT. After washing three times with DPBS, the 12 well μ‐Chamber were removed, and the cells on the coverslips for sticky‐Slides ibiTreat were mounted on glass‐slides with embedding medium Mowiol (Sigma‐Aldrich, Steinheim, Germany) prior to fluorescence microscopy. Images were acquired using a Nikon Eclipse E600 microscope connected to a Nikon Digital Camera DXM1200F with the Nikon NIS‐Elements BR3.10 software (Nikon, Tokyo, Japan). Images were processed and assembled using Adobe Illustrator software.
Western blot
pMBMECs from WT and ACKR1–/– mice were lysed in RIPA buffer (10 mM Tris‐Cl (pH 8.0), 1 mM EDTA, 1% Triton X‐100, 0.1% sodium deoxycholate, 0.1% SDS, 140 mM NaCl, 1 mM PMSF), with protease inhibitors cOmplete ULTRA Tablets, Mini, EDTA‐free, EASYpack (1 tablet/10 ml) (Hoffman‐La Roche, Basel, Switzerland). Protein concentration was measured using the Pierce™ BCA Protein Assay Kit (Thermo Scientific™ Pierce™ Protein Biology, Waltham, USA), according to the manufacturer's instructions. Twenty micrograms of each sample was boiled for 5 min at 95°C and loaded onto a 10% SDS‐polyacrylamide gel. After running, the gel was transferred to a nitrocellulose membrane (Amersham Protan, GE Healthcare, United Kingdom), using a Trans‐Blot Turbo transfer system (BioRad Laboratories, Hercules, CA, USA), according to the manufacturer's instructions. Membranes were blocked with Rockland Buffer (Rockland, Limerick, PA, USA) for 1 h at RT and incubated overnight at 4°C with rabbit anti‐mouse ICAM‐1 (Eurogentec, custom made, polyclonal [14, 57]). Membranes were then washed and incubated with mouse anti‐mouse β‐actin (Merck, Darmstadt, Germany, cat. No A5316) for 1 h at RT. After washing steps, membranes were incubated with the secondary antibodies goat anti‐rabbit Alexa Fluor® 680 (Thermo Fisher Scientific, cat. No A21109) and goat anti‐mouse IRDye® TM 800 (Rockland Immunochemicals, cat. No 605‐732‐125), for 1 h at RT. Protein bands detection was made with the Odyssey near‐infrared imaging system and software (LI‐COR Biotechnology, Lincoln, NE, USA). Bands intensity was quantified using the ImageJ software (NIH, Bethesda, MD, USA) and normalized against β‐actin.
T cells
For in vitro experiments, we used the encephalitogenic CD4+ Th1 effector/memory proteolipid protein (PLP) peptide aa139–153‐specific T‐cell line SJL.PLP7 (CD4+ TEM cells) [58] as previously described [7].
Experimental autoimmune encephalomyelitis
EAE was induced in 8–12 weeks old female C57BL/6J WT and ACKR1–/‐ C57BL/6J mice as described before [59, 60]. Pertussis toxin (List Biological Laboratories, Campbell, US) (300 ng in 100 μl PBS/mouse) was injected intraperitoneally (i.p.) on day 0 (immunization day) and day 2 post‐immunization. Weights and clinical severity were assessed twice daily and scored as [61]: 0, asymptomatic; 0.5, limb tail; 1, hind leg weakness; 2, hind leg paraplegia; 3, hind leg paraplegia and incontinence.
Immunofluorescence staining of tissue sections
Where indicated, mice were injected i.v. with 33 μg of DyLight594 lectin (Vector Laboratories, Burlingame, US). 15 minutes after injection, mice were perfused with 1% PFA in DPBS (Merck, Darmstadt, Germany), and brains were removed and placed in 1% PFA/DPBS overnight at 4°C. Brains were then moved to DPBS and stored at 4°C until sectioning. Sectioning was performed using the VT1000s vibratome (Leica, Wetzlar, Germany). One hundred micrometer thick sections were blocked with blocking buffer (5% skimmed milk (Rapilait, Migros, Switzerland), 0.3% Triton‐X‐100 (Schweizerhall, Basel, Switzerland Batch Nr. A11021), 0.04% NaN3 (Fluka Chemie, Buchs, Switzerland) in TBS pH 7.4) for 2 h at RT under shaking conditions. Blocking buffer was removed and sections were incubated with blocking buffer containing the primary antibody overnight at 4°C under shaking conditions. After three times washing with DPBS, sections were incubated with blocking buffer containing the secondary antibody for 2 h at RT under shaking conditions and protected from light. After 3x washing with DPBS, sections were mounted on glass‐slides and allowed to dry overnight. Sections were then rehydrated with DPBS and cover slides were mounted with embedding medium Mowiol (Sigma‐Aldrich, Steinheim, Germany) prior to confocal microscopy. Images were acquired using a LSM800 (Carl Zeiss, Oberkochen, Germany), Leica TCS SP8 (Leica, Wetzlar, Germany), or an LSM 880 (Carl Zeiss, Oberkochen, Germany) confocal microscope. Images were processed and mounted using Adobe Illustrator software.
In vitro live‐cell imaging
In vitro live‐cell imaging of activated SJL.PLP7 T‐cell diapedesis across pMBMECs was performed as previously described [7, 11, 62]. SJLP.LP7 T cells were resuspended at 1 × 106 cells/ml in T‐cell restimulation medium (RPMI‐1640, 10% FBS, 2 mM l‐glutamine, 1 mM sodium pyruvate, 1 mM nonessential amino acids, 100 U penicillin–streptomycin, 0.05 mM 2‐mercaptoethanol). Prior the experiment, WT and ACKR1–/– pMBMECs were stained with 1 μg/ml of nonblocking anti‐PECAM‐1 antibody [23] directly conjugated to Alexa Fluor 555 for 2 hours at 37°C, 5% CO2. Accumulation of SJLPLP7 cells on WT or ACKR1–/– pMBMECs in the flow chamber was allowed for 5 min at a low shear (0.1 dyn/cm2), followed by physiological shear (1.5 dyn/cm2) for additional 25 min, for a total recording time of 30 min. Image acquisition was performed at 63× magnification with an inverted microscope (AxioObserver, Carl Zeiss, Oberkochen, Germany) with differential interference contrast and fluorescence illumination using the camera Evolve Delta (Teledyne Photometrics, Tucson, USA). Image frames were recorded every 25 s. Image analysis was performed using ImageJ software (ImageJ software, National Institute of Health, Bethesda, USA). The number of arrested T cells/FOV was manually counted on the frame following the onset of physiological shear. T‐cell post‐arrest behavior was defined and expressed as fractions of categorized T cells set to 100% as follows: T cells that detached during the observation time (“Detaching”); T cells that remained immobile with a non‐polarized shape (“Stationary”); stationary T cells that actively probed the endothelium (“Probing”); T cells that continuously crawled on the endothelial surface (“Crawling”), T cells that crossed pMBMECs with prior probing or crawling (“Probing+Diapedesis” and “Crawling+Diapedesis”); T cells that did not fully complete diapedesis events with prior probing or crawling (“Probing+Incomplete diapedesis” and “Crawling+Incomplete diapedesis”) accounted for <10% of the categorized cells and are not shown for visualization reasons. T cells that crawled out of the FOV were excluded from every category. T‐cell crawling tracks were evaluated after manual tracking of individual T cells using the manual tracking plug‐in of ImageJ. Distance, speed, and forward migration index of crawling tracks were evaluated using chemotaxis and migration tool (version 2.0, Ibidi, Martinsried, Germany). T‐cell diapedesis events were classified as paracellular or transcellular based on the appearance of the junctional signal: diapedesis events with interruption of junctional integrity were classified as paracellular; diapedesis events without interruption of junctional integrity were classified as transcellular. Due to unclear junctional signal, an average of ∼7% of the T‐cell diapedesis events/arrested cells/FOV were not categorized in TNF‐α‐stimulated WT and ACKR1–/– pMBMECs. For the same reason, an average of ∼18% of the T‐cell diapedesis events/arrested cells/FOV were not categorized in IL‐1β‐stimulated WT and ACKR1–/– pMBMECs. Maximum diapedesis pore size opened by the T cells during the diapedesis events was manually measured in the Zen software (Blue version, Carl Zeiss, Oberkochen, Germany) based on the absence of diffused PECAM‐1 signal in the diapedesis spot. Diapedesis starting time and total duration were manually measured based on the first and last frame in which T cells initiated and completed the diapedesis event, respectively.
For the set of experiments dedicated to analyze if ACKR1+ pMBMECs specifically favor transcellular T cell diapedesis, pMBMECs were prepared from VE‐cadherin‐GFP knock‐in mice [24], and IL‐1β‐stimulated pMBMECs were stained with 38 μg/ml of the rat‐anti‐mouse ACKR‐1 monoclonal antibody directly conjugated to Alexa Fluor 647 for 30 minutes at 37°C and 5% CO2 prior to the imaging. The assays were performed exactly as outlined above with image frames recorded every 30 seconds for 30 min. Transcellular T cell diapedesis was identified as outlined above including as additional category positive or negative cell surface staining for ACKR1.
Statistics
Statistical analysis was performed using GraphPad Prism 6.0 software. To compare two groups, unpaired t‐test with Welch's correction was performed. Results are shown as mean ± SD and a p‐value < 0.05 was considered significant.
Conflict of interest
The authors declare no conflict of interest.
Statements on ethics and integrity policies
Animal procedures executed were approved by the Veterinary Office of the Canton Bern (permit no. BE31/17 and BE77/18) and are in keeping with institutional and standard protocols for the care and use of laboratory animals in Switzerland.
Author contribution
L.M. performed experiments, analyzed data, provided figures, and wrote the first draft of the manuscript. D.F. performed bioinformatic analysis including figures and contributed to manuscript writing; S.S. performed experiments and analyzed data; N.H.J. performed experiments, analyzed data, and made figures; J.R.P. performed experiments and analyzed data and made figures; S.B. performed experiments and documented and analyzed data; I.G. established methodology and performed experiments; A.T. developed and provided reagents; U.v.A. developed and provided reagents; U.D. performed quality control and supervision of genetic mouse lines; R.L. supervised experiments and edited the manuscript; R.B. supervised the bioinformatics analysis; B.E. was associated with conceptualization and supervision of the project, funding acquisition, and finalization of the manuscript.
Peer review
The peer review history for this article is available at https://publons.com/publon/10.1002/eji.202149238
Abbreviations
- Ackr1
atypical chemokine receptor 1
- AJ
adherence junctions
- BBB
blood‐brain barrier
- DAPI
4′,6‐diamidin‐2‐phenylindol
- FDR
false discovery rate
- GPCR
G‐protein coupled receptor
- MDCK
Madin‐Darby canine kidney
- PCA
principal component analysis
- pMBMEC
primary mouse brain microvascular endothelial cell
- TJ
tight junctions
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgments
We gratefully acknowledge cand. med. Fabio Bösch for experimental help and Federico Saltarin for his support in confocal imaging. This project has received funding from the European Union Horizon 2020 research and innovation programme under the Marie Skłodowska‐Curie Grant Agreement No. 675619 BtRAIN to L.M., D.V., R.B., and B.E.; SNSF grants 310030_189080 and 310030E_189312 to B.E. and NIH AR068383 and AI155865 grants to U.v.A.
Open Access Funding provided by Universitat Bern.
[Correction added on April 13th 2022, after first online publication: CSAL funding statement has been added.]
Data availability statement
RNA sequence data supporting the findings of this study were deposited in the Gene Expression Omnibus (GEO) of the National Center for Biotechnology Information (NCBI) with primary accession code GSE141980.
References
- 1. Tietz, S. and Engelhardt, B. , Brain barriers: Crosstalk between complex tight junctions and adherens junctions. J. Cell Biol. 2015. 209: 493–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Coisne, C. , Dehouck, L. , Faveeuw, C. , Delplace, Y. , Miller, F. , Landry, C. , Morissette, C. et al., Mouse syngenic in vitro blood‐brain barrier model: a new tool to examine inflammatory events in cerebral endothelium. Lab. Invest. 2005. 85: 734–746. [DOI] [PubMed] [Google Scholar]
- 3. Daneman, R. and Prat, A. , The blood‐brain barrier. Cold Spring Harb. Perspect. Biol. 2015. 7: a020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Engelhardt, B. , Vajkoczy, P. and Weller, R. O. , The movers and shapers in immune privilege of the CNS. Nat. Immunol. 2017. 18: 123–131. [DOI] [PubMed] [Google Scholar]
- 5. Engelhardt, B. , Immune cell entry into the central nervous system: involvement of adhesion molecules and chemokines. J. Neurol. Sci. 2008. 274: 23–26. [DOI] [PubMed] [Google Scholar]
- 6. Marchetti, L. and Engelhardt, B. , Immune cell trafficking across the blood‐brain barrier in the absence and presence of neuroinflammation. Vasc Biol 2020. 2: H1–H18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Steiner, O. , Coisne, C. , Cecchelli, R. , Boscacci, R. , Deutsch, U. , Engelhardt, B. and Lyck, R. , Differential roles for endothelial ICAM‐1, ICAM‐2, and VCAM‐1 in shear‐resistant T cell arrest, polarization, and directed crawling on blood‐brain barrier endothelium. J. Immunol. 2010. 185: 4846–4855. [DOI] [PubMed] [Google Scholar]
- 8. Bartholomäus, I. , Kawakami, N. , Odoardi, F. , Schläger, C. , Miljkovic, D. , Ellwart, J. W. , Klinkert, W. E. F. , Flügel‐Koch, C. , Issekutz, T. B. , Wekerle, H. , Flügel, A. , Effector T cell interactions with meningeal vascular structures in nascent autoimmune CNS lesions. Nature. 2009. 462: 94–98. [DOI] [PubMed] [Google Scholar]
- 9. Steiner, O. , Coisne, C. , Engelhardt, B. and Lyck, R. , Comparison of immortalized bEnd5 and primary mouse brain microvascular endothelial cells as in vitro blood‐brain barrier models for the study of T cell extravasation. J. Cereb. Blood Flow Metab. 2011. 31: 315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wolburg, H. , Wolburg‐Buchholz, K. and Engelhardt, B. , Diapedesis of mononuclear cells across cerebral venules during experimental autoimmune encephalomyelitis leaves tight junctions intact. Acta Neuropathol. 2005. 109: 181–190. [DOI] [PubMed] [Google Scholar]
- 11. Abadier, M. , Haghayegh Jahromi, N. , Cardoso Alves, L. , Boscacci, R. , Vestweber, D. , Barnum, S. , Deutsch, U. et al., Cell surface levels of endothelial ICAM‐1 influence the transcellular or paracellular T‐cell diapedesis across the blood‐brain barrier. Eur. J. Immunol. 2015. 45: 1043–1058. [DOI] [PubMed] [Google Scholar]
- 12. Lutz, S. E. , Smith, J. R. , Kim, D. H. , Olson, C. V. L. , Ellefsen, K. , Bates, J. M. , Gandhi, S. P. et al., Caveolin1 Is Required for Th1 Cell Infiltration, but Not Tight Junction Remodeling, at the Blood‐Brain Barrier in Autoimmune Neuroinflammation. Cell Rep. 2017. 21: 2104–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vanlandewijck, M. , He, L. , Mae, M. A. , Andrae, J. , Ando, K. , Del Gaudio, F. , Nahar, K. et al., Author Correction: A molecular atlas of cell types and zonation in the brain vasculature. Nature 2018. 560: E3. [DOI] [PubMed] [Google Scholar]
- 14. Wimmer, I. , Tietz, S. , Nishihara, H. , Deutsch, U. , Sallusto, F. , Gosselet, F. , Lyck, R. et al., PECAM‐1 Stabilizes Blood‐Brain Barrier Integrity and Favors Paracellular T‐Cell Diapedesis Across the Blood‐Brain Barrier During Neuroinflammation. Front. Immunol. 2019. 10: 711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Griffith, J. W. , Sokol, C. L. and Luster, A. D. , Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu. Rev. Immunol. 2014. 32: 659–702. [DOI] [PubMed] [Google Scholar]
- 16. Hughes, C. E. and Nibbs, R. J. B. , A guide to chemokines and their receptors. FEBS J. 2018. 285: 2944–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Minten, C. , Alt, C. , Gentner, M. , Frei, E. , Deutsch, U. , Lyck, R. , Schaeren‐Wiemers, N. et al., DARC shuttles inflammatory chemokines across the blood‐brain barrier during autoimmune central nervous system inflammation. Brain 2014. 137: 1454–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dawson, T. C. , Lentsch, A. B. , Wang, Z. , Cowhig, J. E. , Rot, A. , Maeda, N. and Peiper, S. C. , Exaggerated response to endotoxin in mice lacking the Duffy antigen/receptor for chemokines (DARC). Blood 2000. 96: 1681–1684. [PubMed] [Google Scholar]
- 19. Thiriot, A. , Perdomo, C. , Cheng, G. , Novitzky‐Basso, I. , McArdle, S. , Kishimoto, J. K. , Barreiro, O. et al., Differential DARC/ACKR1 expression distinguishes venular from non‐venular endothelial cells in murine tissues. BMC Biol. 2017. 15: 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Luo, H. , Chaudhuri, A. , Johnson, K. R. , Neote, K. , Zbrzezna, V. , He, Y. and Pogo, A. O. , Cloning, characterization, and mapping of a murine promiscuous chemokine receptor gene: homolog of the human Duffy gene. Genome Res. 1997. 7: 932–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Middleton, J. , Neil, S. , Wintle, J. , Clark‐Lewis, I. , Moore, H. , Lam, C. , Auer, M. et al., Transcytosis and surface presentation of IL‐8 by venular endothelial cells. Cell 1997. 91: 385–395. [DOI] [PubMed] [Google Scholar]
- 22. Haghayegh Jahromi, N. , Marchetti, L. , Moalli, F. , Duc, D. , Basso, C. , Tardent, H. , Kaba, E. et al., Intercellular Adhesion Molecule‐1 (ICAM‐1) and ICAM‐2 Differentially Contribute to Peripheral Activation and CNS Entry of Autoaggressive Th1 and Th17 Cells in Experimental Autoimmune Encephalomyelitis. Front. Immunol. 2019. 10: 3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Baldwin, H. S. , Shen, H. M. , Yan, H. C. , DeLisser, H. M. , Chung, A. , Mickanin, C. , Trask, T. et al., Platelet endothelial cell adhesion molecule‐1 (PECAM‐1/CD31): alternatively spliced, functionally distinct isoforms expressed during mammalian cardiovascular development. Development 1994. 120: 2539–2553. [DOI] [PubMed] [Google Scholar]
- 24. Winderlich, M. , Keller, L. , Cagna, G. , Broermann, A. , Kamenyeva, O. , Kiefer, F. , Deutsch, U. et al., VE‐PTP controls blood vessel development by balancing Tie‐2 activity. J. Cell Biol. 2009. 185: 657–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Girbl, T. , Lenn, T. , Perez, L. , Rolas, L. , Barkaway, A. , Thiriot, A. , Del Fresno, C. et al., Distinct Compartmentalization of the Chemokines CXCL1 and CXCL2 and the Atypical Receptor ACKR1 Determine Discrete Stages of Neutrophil Diapedesis. Immunity 2018. 49: 1062–1076 e1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Larochelle, C. , Alvarez, J. I. and Prat, A. , How do immune cells overcome the blood‐brain barrier in multiple sclerosis? FEBS Lett. 2011. 585: 3770–3780. [DOI] [PubMed] [Google Scholar]
- 27. Lopes Pinheiro, M. A. , Kooij, G. , Mizee, M. R. , Kamermans, A. , Enzmann, G. , Lyck, R. , Schwaninger, M. et al., Immune cell trafficking across the barriers of the central nervous system in multiple sclerosis and stroke. Biochim. Biophys. Acta 2016. 1862: 461–471. [DOI] [PubMed] [Google Scholar]
- 28. Hauptman, J. , Johann, L. , MArini, F. , Kitic, M. , Colombo, E. , Mufazalov, I. A. , Krueger, M. et al., Interleukin‐1 promotes autoimmune neuroinflammation by suppressing endothelial heme oxygenase‐1 at the blood‐brain barrier. Acta Neuropathol. 2020. 140: 549–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schall, T. J. and Bacon, K. B. , Chemokines, leukocyte trafficking, and inflammation. Curr. Opin. Immunol. 1994. 6: 865–873. [DOI] [PubMed] [Google Scholar]
- 30. Strazza, M. and Mor, A. , Consider the chemokines: a review of the interplay between chemokines and T cell subset function. Discov Med 2017. 24: 31–39. [PMC free article] [PubMed] [Google Scholar]
- 31. Lodowski, D. T. and Palczewski, K. , Chemokine receptors and other G protein‐coupled receptors. Current opinion in HIV and AIDS 2009. 4: 88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rot, A. and von Andrian, U. H. , Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu. Rev. Immunol. 2004. 22: 891–928. [DOI] [PubMed] [Google Scholar]
- 33. Nourshargh, S. and Alon, R. , Leukocyte migration into inflamed tissues. Immunity 2014. 41: 694–707. [DOI] [PubMed] [Google Scholar]
- 34. Rudolph, H. , Klopstein, A. , Gruber, I. , Blatti, C. , Lyck, R. and Engelhardt, B. , Postarrest stalling rather than crawling favors CD8(+) over CD4(+) T‐cell migration across the blood‐brain barrier under flow in vitro. Eur. J. Immunol. 2016. 46: 2187–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shulman, Z. , Cohen, S. J. , Roediger, B. , Kalchenko, V. , Jain, R. , Grabovsky, V. , Klein, E. et al., Transendothelial migration of lymphocytes mediated by intraendothelial vesicle stores rather than by extracellular chemokine depots. Nat. Immunol. 2011. 13: 67–76. [DOI] [PubMed] [Google Scholar]
- 36. Ransohoff, R. M. , Hamilton, T. A. , Tani, M. , Stoler, M. H. , Shick, H. E. , Major, J. A. , Estes, M. L. et al., Astrocyte expression of mRNA encoding cytokines IP‐10 and JE/MCP‐1 in experimental autoimmune encephalomyelitis. FASEB J. 1993. 7: 592–600. [DOI] [PubMed] [Google Scholar]
- 37. Miyagishi, R. , Kikuchi, S. , Takayama, C. , Inoue, Y. and Tashiro, K. , Identification of cell types producing RANTES, MIP‐1 alpha and MIP‐1 beta in rat experimental autoimmune encephalomyelitis by in situ hybridization. J. Neuroimmunol. 1997. 77: 17–26. [DOI] [PubMed] [Google Scholar]
- 38. Glabinski, A. R. , Krakowski, M. , Han, Y. , Owens, T. and Ransohoff, R. M. , Chemokine expression in GKO mice (lacking interferon‐gamma) with experimental autoimmune encephalomyelitis. J. Neurovirol. 1999. 5: 95–101. [DOI] [PubMed] [Google Scholar]
- 39. Nibbs, R. , Graham, G. and Rot, A. , Chemokines on the move: control by the chemokine “interceptors” Duffy blood group antigen and D6. Semin. Immunol. 2003. 15: 287–294. [DOI] [PubMed] [Google Scholar]
- 40. Mantovani, A. , Bonecchi, R. and Locati, M. , Tuning inflammation and immunity by chemokine sequestration: decoys and more. Nat. Rev. Immunol. 2006. 6: 907–918. [DOI] [PubMed] [Google Scholar]
- 41. Ulvmar, M. H. , Hub, E. and Rot, A. , Atypical chemokine receptors. Exp. Cell Res. 2011. 317: 556–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gardner, L. , Patterson, A. M. , Ashton, B. A. , Stone, M. A. and Middleton, J. , The human Duffy antigen binds selected inflammatory but not homeostatic chemokines. Biochem. Biophys. Res. Commun. 2004. 321: 306–312. [DOI] [PubMed] [Google Scholar]
- 43. Pruenster, M. , Mudde, L. , Bombosi, P. , Dimitrova, S. , Zsak, M. , Middleton, J. , Richmond, A. et al., The Duffy antigen receptor for chemokines transports chemokines and supports their promigratory activity. Nat. Immunol. 2009. 10: 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Muller, W. A , Mechanisms of leukocyte transendothelial migration. Annu Rev Pathol 2011. 6: 323–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dzenko, K. A. , Andjelkovic, A. V. , Kuziel, W. A. and Pachter, J. S. , The chemokine receptor CCR2 mediates the binding and internalization of monocyte chemoattractant protein‐1 along brain microvessels. J. Neurosci. 2001. 21: 9214–9223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ge, S. , Song, L. , Serwanski, D. R. , Kuziel, W. A. and Pachter, J. S. , Transcellular transport of CCL2 across brain microvascular endothelial cells. J. Neurochem. 2008. 104: 1219–1232. [DOI] [PubMed] [Google Scholar]
- 47. Barkalow, F. J. , Goodman, M. J. , Gerritsen, M. E. and Mayadas, T. N. , Brain endothelium lack one of two pathways of P‐selectin‐mediated neutrophil adhesion. Blood 1996. 88: 4585–4593. [PubMed] [Google Scholar]
- 48. Tournamille, C. , Colin, Y. , Cartron, J. P. and Le Van Kim, C. , Disruption of a GATA motif in the Duffy gene promoter abolishes erythroid gene expression in Duffy‐negative individuals. Nat. Genet. 1995. 10: 224–228. [DOI] [PubMed] [Google Scholar]
- 49. Nibbs, R. J. B. and Graham, G. J. , Immune regulation by atypical chemokine receptors. Nature Reviews Immunology 2013. 13: 815. [DOI] [PubMed] [Google Scholar]
- 50. Gorina, R. , Lyck, R. , Vestweber, D. and Engelhardt, B. , beta2 integrin‐mediated crawling on endothelial ICAM‐1 and ICAM‐2 is a prerequisite for transcellular neutrophil diapedesis across the inflamed blood‐brain barrier. J. Immunol. 2014. 192: 324–337. [DOI] [PubMed] [Google Scholar]
- 51. Bolger, A. M. , Lohse, M. and Usadel, B. , Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014. 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kim, D. , Pertea, G. , Trapnell, C. , Pimentel, H. , Kelley, R. and Salzberg, S. L. , TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013. 14: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Okonechnikov, K. , Conesa, A. and Garcia‐Alcalde, F. . Qualimap 2: advanced multi‐sample quality control for high‐throughput sequencing data. Bioinformatics 2016. 32: 292–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Robinson, J. T. , Thorvaldsdottir, H. , Winckler, W. , Guttman, M. , Lander, E. S. , Getz, G. and Mesirov, J. P. , Integrative genomics viewer. Nat. Biotechnol. 2011. 29: 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Anders, S. , Pyl, P. T. , Huber, W. , HTSeq‐a Python framework to work with high‐throughput sequencing data. Bioinformatics. 2015. 31: 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Love, M. I. , Huber, W. and Anders, S. , Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol. 2014. 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lyck, R. , Lecuyer, M. A. , Abadier, M. , Wyss, C. B. , Matti, C. , Rosito, M. , Enzmann, G. et al., ALCAM (CD166) is involved in extravasation of monocytes rather than T cells across the blood‐brain barrier. J. Cereb. Blood Flow Metab. 2017. 37: 2894–2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Laschinger, M. and Engelhardt, B. , Interaction of alpha4‐integrin with VCAM‐1 is involved in adhesion of encephalitogenic T cell blasts to brain endothelium but not in their transendothelial migration in vitro. J. Neuroimmunol. 2000. 102: 32–43. [DOI] [PubMed] [Google Scholar]
- 59. Doring, A. , Wild, M. , Vestweber, D. , Deutsch, U. and Engelhardt, B. , E‐ and P‐selectin are not required for the development of experimental autoimmune encephalomyelitis in C57BL/6 and SJL mice. J. Immunol. 2007. 179: 8470–8479. [DOI] [PubMed] [Google Scholar]
- 60. Engelhardt, B. , Kempe, B. , Merfeld‐Clauss, S. , Laschinger, M. , Furie, B. , Wild, M. K. and Vestweber, D. , P‐selectin glycoprotein ligand 1 is not required for the development of experimental autoimmune encephalomyelitis in SJL and C57BL/6 mice. J. Immunol. 2005. 175: 1267–1275. [DOI] [PubMed] [Google Scholar]
- 61. Tietz, S. M. , Zwahlen, M. , Haghayegh Jahromi, N. , Baden, P. , Lazarevic, I. , Enzmann, G. and Engelhardt, B. , Refined clinical scoring in comparative EAE studies does not enhance the chance to observe statistically significant differences. Eur. J. Immunol. 2016. 46: 2481–2483. [DOI] [PubMed] [Google Scholar]
- 62. Coisne, C. , Lyck, R. and Engelhardt, B. , Live cell imaging techniques to study T cell trafficking across the blood‐brain barrier in vitro and in vivo. Fluids Barriers CNS 2013. 10: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Data Availability Statement
RNA sequence data supporting the findings of this study were deposited in the Gene Expression Omnibus (GEO) of the National Center for Biotechnology Information (NCBI) with primary accession code GSE141980.